

ABSTRACT
The human papillomavirus (HPV) E1 helicase promotes viral DNA replication through its DNA unwinding activity and association with host factors. The E1 proteins from anogenital HPV types interact with the cellular WD repeat-containing factor UAF1 (formerly known as p80). Specific amino acid substitutions in E1 that impair this interaction inhibit maintenance of the viral episome in immortalized keratinocytes and reduce viral DNA replication by up to 70% in transient assays. In this study, we determined by affinity purification of UAF1 that it interacts with three deubiquitinating enzymes in C33A cervical carcinoma cells: USP1, a nuclear protein, and the two cytoplasmic enzymes USP12 and USP46. Coimmunoprecipitation experiments indicated that E1 assembles into a ternary complex with UAF1 and any one of these three USPs. Moreover, expression of E1 leads to a redistribution of USP12 and USP46 from the cytoplasm to the nucleus. Chromatin immunoprecipitation studies further revealed that E1 recruits these threes USPs to the viral origin in association with UAF1. The function of USP1, USP12, and USP46 in viral DNA replication was investigated by overproduction of catalytically inactive versions of these enzymes in transient assays. All three dominant negative USPs reduced HPV31 DNA replication by up to 60%, an effect that was specific, as it was not observed in assays performed with a truncated E1 lacking the UAF1-binding domain or with bovine papillomavirus 1 E1, which does not bind UAF1. These results highlight the importance of the USP1, USP12, and USP46 deubiquitinating enzymes in anogenital HPV DNA replication.
IMPORTANCE Human papillomaviruses are small DNA tumor viruses that induce benign and malignant lesions of the skin and mucosa. HPV types that infect the anogenital tract are the etiological agents of cervical cancer, the majority of anal cancers, and a growing proportion of head-and-neck cancers. Replication of the HPV genome requires the viral protein E1, a DNA helicase that also interacts with host factors to promote viral DNA synthesis. We previously reported that the E1 helicase from anogenital HPV types associates with the WD40 repeat-containing protein UAF1. Here, we show that UAF1 bridges the interaction of E1 with three deubiquitinating enzymes, USP1, USP12, and USP46. We further show that these deubiquitinases are recruited by E1/UAF1 to the viral origin of DNA replication and that overexpression of catalytically inactive versions of these enzymes reduces viral DNA replication. These results highlight the need for an E1-associated deubiquitinase activity in anogenital HPV genome replication.
INTRODUCTION
Human papillomaviruses (HPVs) infect the stratified epithelium of the skin and mucosa. While the majority of infections remain subclinical or cause only benign lesions, infections by a subset of anogenital HPVs, known as high-risk types, have the potential to progress to cancer. It is now well established that these oncogenic types are at the root of cervical cancer and of a large proportion of anal and other genital cancers (reviewed in references 1 to 3). They are also responsible for a subset of head-and-neck cancers, in particular, those of the oropharynx (4).
The HPV genome is a circular double-stranded DNA molecule that is maintained in an episomal form in the nucleus of infected keratinocytes. Replication of the HPV episome is ensured by the viral proteins E1 and E2 at different phases of the differentiation-dependent viral life cycle (reviewed in references 1 and 2). Upon infection of basal keratinocytes, these two proteins help to amplify and establish the viral episome at approximately 50 to 100 copies per cell (establishment phase). It is believed that this copy number is then maintained at an approximately constant level in undifferentiated cells (maintenance phase), either through once-per-cell cycle replication of all episomes or by random replication of a subset of them, with the latter mechanism being favored at higher levels of E1 (reviewed in reference 2). As the infected cells migrate toward the upper layers of the epithelium and become increasingly differentiated, the viral genome is further replicated, reaching up to 1,000 copies per cell (amplification step). It is also at this productive stage of the life cycle that the capsid proteins L1 and L2 are synthesized, allowing the packaging of these episomes into new viral particles that are shed by desquamation of the terminally differentiated keratinocytes.
Replication of the papillomavirus episome is initiated by the cooperative binding of E1 and E2 at a specific region of the genome known as the viral origin (ori) of replication. E2 binds with a high affinity and a high specificity to the ori and can simultaneously interact with E1 through a protein-protein interaction. As such, E2 can function as a loading factor to recruit E1 monomers to the ori and promote their assembly into a double hexamer (5–12). This oligomeric complex is the replication-competent form of the E1 helicase that melts the ori, unwinds the viral DNA in a bidirectional manner, and interacts with several components of the host DNA replication machinery, such as the DNA polymerase α-primase, topoisomerase I, and the single-stranded DNA-binding protein RPA (13–18).
Apart from its highly conserved helicase and ori DNA-binding domains, E1 comprises a more divergent N-terminal region that is strictly required for DNA replication in vivo. Several motifs in this region mediate the nucleocytoplasmic shuttling of E1. These include a highly conserved bipartite nuclear localization signal (NLS) and, for most but not all HPV types, a Crm1-dependent nuclear export signal (NES). The activity of this NES is inhibited by cyclin A/E-Cdk2 phosphorylation to ensure the accumulation of E1 in the nucleus during S phase. Through proteomic studies, we previously showed that the N-terminal region of E1 from anogenital HPV types also contains a binding site for the cellular protein p80/UAF1 (USP1-associated factor 1), hereafter called UAF1, located between amino acids (aa) 10 and 40 in HPV11 and HPV31 E1 (19). Several lines of evidence support an important role for the E1-UAF1 interaction in replication of the viral genome. First, mutations in E1 that prevent its interaction with UAF1 reduce viral DNA replication by 70% in transient assays and abrogate the long-term maintenance of the viral episome in undifferentiated keratinocytes (19, 20). Second, E1 relocalizes UAF1 from the cytoplasm to the nucleus and, in the presence of E2, to specific nuclear foci (E2 foci) where viral DNA replication is thought to occur. Third, E1 and E2 recruit UAF1 to the viral origin, as evidenced by chromatin immunoprecipitation (ChIP) experiments (21). Fourth, inhibition of the E1-UAF1 interaction by overexpression in trans of an E1-derived UAF1-binding peptide (N40) precludes the recruitment of UAF1 to the ori and inhibits HPV DNA replication by 70%. Altogether, these observations suggest that E1 is recruited to the origin in association with UAF1 and that this complex is more active than E1 alone in supporting viral DNA replication.
The mechanism by which UAF1 increases HPV DNA replication could not be inferred from its primary amino acid sequence. Indeed, UAF1 does not have any known enzymatic activity, although it is comprised of several WD40 repeats and of two SUMO-like domains (SLDs), which are commonly involved in protein-protein interactions (22, 23). Thus, it is likely that UAF1 stimulates viral DNA replication through its interaction with cellular proteins. During the course of this study, it was reported that UAF1 interacts with three deubiquitinating enzymes (DUBs): the nuclear enzyme USP1 and the two cytoplasmic and related enzymes USP12 and USP46 (24, 25), findings that we have independently confirmed in C33A cervical carcinoma cells and that are described herein. Importantly, we also show in this study, for HPV31, that E1 does not compete with these enzymes for binding to UAF1 but, rather, forms a ternary complex with UAF1 and any one of its associated USPs. We also demonstrate that E1 relocalizes USP12 and USP46 from the cytoplasm to the nucleus and, furthermore, that it recruits all three USPs to the viral origin in a UAF1-dependent manner. Finally, we provide functional evidence that the enzymatic activity of these USPs is required for anogenital HPV DNA replication by showing that overexpression of dominant negative versions of these enzymes inhibits HPV31 DNA replication, with little to no effect on cellular DNA synthesis or on the DNA replication catalyzed by bovine papillomavirus 1 (BPV1) E1, which does not interact with UAF1. These findings suggest that USP1, USP12, and USP46 play an important role in anogenital HPV DNA replication through their association with UAF1 and E1.
MATERIALS AND METHODS
Plasmid construction and mutagenesis.
Short hairpin RNAs (shRNAs) against UAF1, USP1, USP12, and USP46 were expressed from the pLKO.1-Puro plasmid (Open Biosystems) and had the following targeting sequences: GTGCAGGTTTCCTATGTTATT for UAF1, GTGCAAGTTTCGTATGTCATT for UAF1-mut (mutated residues are underlined), TTTGATCTAGTTTGTCTCTGG for USP1-a, TAATGCCTGTTTGGTCACTGG for USP1-b, ATCAAACTCCAAACCACTAGC for USP1-c, TTTCACAAGTAAGACATCTGG for USP12, and CGCTTACCAATGAAACTCGATCTCGAG for USP46. A control shRNA was generated by randomizing the UAF1-targeting sequence to obtain an inactive shRNA with the same nucleotide composition (GCTATGAGAATAACGGTAACA). USP1, USP12, and USP46 open reading frames (ORFs) were obtained from Life Technologies. Fusions of these USPs to green fluorescent protein (GFP) were constructed by inserting PCR fragments encoding the corresponding ORFs between the SacI and BamHI sites of plasmid pGFP2-N1 (BioSignal Packard-PerkinElmer). Expression vectors for USP1 fused to red fluorescent protein (RFP-USP1), RFP-USP12, and RFP-USP46 were generated by cloning the PCR-amplified ORFs between the XhoI and XbaI sites of the pcDNA3-mRFP1 plasmid (19). Catalytically inactive versions of USP1 (with the C90S mutation), USP12 (with the C48S mutation), and USP46 (with the C44S mutation) were constructed by site-directed mutagenesis. All DNA constructs were verified by sequencing. Primer sequences and additional details on the construction of these plasmids are available upon request. The other plasmids used in this study have been described previously, namely, those encoding HPV31 E1 fused to GFP (GFP-E1) and to a triple-Flag (3F) epitope (3F-E1) (26), 3F-UAF1 and its truncations (21), codon-optimized 3F-E1 (p31E1) and 3F-E2 (p31E2) (20), and UAF1 fused to the red fluorescent protein (RFP-UAF1) (19).
Antibodies and Western blotting.
Commercially available antibodies were used to detect 3× Flag-tagged proteins (M2 monoclonal antibody; catalog no. F1804; Sigma-Aldrich), β-tubulin (monoclonal antibody; catalog no. T4026; Sigma-Aldrich), GFP (a mixture of two mouse monoclonal antibodies; catalog no. 11814460001; Roche), RFP (monoclonal antibody; catalog no. AB65856; Abcam), and hemagglutinin (HA)-tagged proteins (HA.11 antibody; catalog no. MMS-101P; Covance). Polyclonal antibodies against UAF1 were custom produced (Open Biosystems) by injecting rabbits with a recombinant C-terminal fragment of UAF1 (amino acids 400 to 677) purified from bacteria, as previously described (19). For Western blot analysis, proteins were transferred onto polyvinylidene difluoride membranes and detected with horseradish peroxidase-conjugated secondary antibodies from GE Healthcare, either sheep anti-mouse IgG (catalog no. NA931) or donkey anti-rabbit IgG (NA934V), using an enhanced chemiluminescence detection kit (GE Healthcare).
Cell culture and transfections.
Cells of the human cervical carcinoma cell line C33A were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 50 IU/ml of penicillin, and 50 μg/ml streptomycin (Wisent Bioproducts). For selection and growth of stably transfected cells, the culture medium was supplemented either with 15 μg/ml bleomycin (Bleocin; catalog no. 203408; EMD Millipore) for GFP-USP constructs or with 2 μg/ml puromycin (catalog no. P8833; Sigma-Aldrich) for shRNA plasmids. Transfection of C33A was performed with the Lipofectamine 2000 reagent (Life Technologies) according to the manufacturer's protocol.
Luciferase-based transient HPV31 and BPV1 DNA replication assays.
The HPV31 and BPV1 DNA replication assays were performed as described previously (20, 27). Briefly, C33A cells were seeded at a density of 25,000 cells per well in white flat-bottom 96-well plates and transfected 24 h later with a mix of four plasmids, an origin-containing plasmid with a firefly luciferase (FLuc) reporter in cis (pFLORI31 or pFLORI-BPV1), a Renilla luciferase (RLuc) plasmid as an internal control (pRL), and the E1 and E2 expression vectors, at the quantities indicated below. For all experiments, the total amount of transfected DNA was adjusted to 100 ng with pcDNA-mRFP1 as the carrier DNA. Firefly and Renilla luciferase activities were measured using a Dual-Glo luciferase assay system (Promega) at 48 h posttransfection. As a negative control, the assay was performed with a firefly luciferase plasmid lacking the viral origin to measure the expression of the luciferase reporter gene in the absence of viral DNA replication.
Coimmunoprecipitation (co-IP) assays.
C33A cells were grown on 100-mm plates and transfected with the indicated DNA. Cells were harvested at 48 h posttransfection in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 10 μg/ml antipain, 2 μg/ml leupeptin, 2 μg/ml aprotinin, 1 μg/ml pepstatin A, 1 mM phenylmethylsulfonyl fluoride). Cleared cellular extracts were then immunoprecipitated for 3 h with 40 μl of protein G-Sepharose (GE Healthcare) conjugated to 1 μg of anti-Flag or anti-GFP antibody. The resin was washed 3 times with Tris-buffered saline (50 mM Tris-HCl, pH 7.4, 150 mM NaCl), and the bound proteins were eluted in 5× Laemmli buffer prior to Western blotting.
Affinity purification of UAF1 and identification of interacting proteins by MS.
UAF1-containing protein complexes were purified from C33A cervical carcinoma cells stably expressing 3F-UAF1 or 3F alone as a control. Cell pellets from 10- by 100-mm dishes were lysed in 35 ml of lysis buffer (0.1% Triton X-100, 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 10 μg/ml antipain, 2 μg/ml leupeptin, 2 μg/ml aprotinin, 1 μg/ml pepstatin A, 1 mM phenylmethylsulfonyl fluoride). Lysates were cleared by centrifugation, and the supernatants were incubated for 1 h with 250 μl of anti-Flag M2 affinity gel (Sigma). Following extensive washes with high-salt buffer (50 mM Tris-HCl, pH 7.4, 500 mM NaCl), the coimmunoprecipitated proteins were eluted twice with 625 μl of a solution of 3F peptide (150 μg/ml), the two eluates were pooled, and the proteins were precipitated with trichloroacetic acid overnight. Proteins were resolved on a 4 to 15% gradient SDS-polyacrylamide gel and silver stained, and the entire gel was cut into slices that were sent for mass spectrometry (MS) analysis at the Institut de Recherches Cliniques de Montréal proteomic core facility. Tryptic peptides were identified by liquid chromatography-tandem MS (LC-MS/MS) with a microcapillary reversed-phase high-pressure liquid chromatograph coupled to an LTQ (ThermoElectron) quadrupole ion trap mass spectrometer with a nanospray interface. The resulting peptide MS/MS spectra were interpreted using MASCOT software (Matrix Science) and searched against the spectra for proteins in the National Center for Biotechnology Information (NCBI) nonredundant protein database or UniRef protein database.
ChIP.
The chromatin immunoprecipitation (ChIP) protocol was modified from a previously published procedure (21). C33A cells were plated at a density of 3.6 × 106 cells on 100-mm plates and transfected 24 h later with the following plasmids: 0.5 μg pFLORI31, 0.1 μg pRL, 1 μg p31E1, 2 μg RFP-E2, 4.4 μg RFP-UAF1, and 4 μg a GFP-USP construct (or GFP alone). In all experiments, the total amount of DNA was adjusted to 12 μg with pUC18 DNA as the carrier. At 24 h posttransfection, cells were cross-linked with 1% formaldehyde and lysed in lysis buffer and the DNA was sheared with a Covaris S2 sonicator. Each lysate was diluted 10-fold in ChIP dilution buffer and precleared using protein G-Sepharose (catalog no. 16-0618; GE Healthcare) that was previously blocked with 5 mg/ml bovine serum albumin and 0.5 mg/ml salmon sperm DNA. The lysates (1 million cells per immunoprecipitation) were incubated overnight with the indicated antibodies, after which protein G-Sepharose was added and the components were mixed for 3 h to capture protein-DNA complexes. The resin was then successively washed with 2 ml of the following buffers: low-salt buffer, high-salt buffer, LiCl buffer, and (twice) TE (Tris-EDTA) buffer. Immunoprecipitates were recovered by two successive elutions with 150 μl of elution buffer (1% SDS, 0.1 M NaHCO3) at 65°C and treated with RNase A, and the cross-links were reversed by overnight incubation in 0.2 M NaCl at 65°C. The eluates were treated with proteinase K for 1 h at 45°C, and DNA was purified using Qiagen spin columns. The eluates and corresponding input DNA were then analyzed by quantitative PCR (qPCR), as previously described (21).
Confocal fluorescence microscopy.
C33A cells were plated at a density of 6 × 105 cells/well on coverslips and transfected 24 h later with the indicated plasmids. At 24 h posttransfection, cells were fixed with 4% formaldehyde and permeabilized with 0.2% Triton X-100, and their DNA was stained with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; catalog no. D1306; Life Technologies). Slides were mounted using Vectashield mounting medium (Vector Laboratories). Images were collected with a Zeiss LSM710 laser scanning confocal microscope and analyzed using Zen 2009 LE software.
Cell cycle analysis.
Cell cycle profiles were obtained by staining live cells at 48 h posttransfection with 6.3 μg/ml Hoechst stain and 50 μM verapamil. Acquisitions were done using a BD LSR flow cytometer gated on the GFP-positive population.
Colony formation assay.
C33A cells (∼1.2 × 106) were transfected with 1.5 μg of the indicated plasmids in a 6-well plate. At 24 h posttransfection, cells were trypsinized and seeded on a new plate at a 1/15 dilution in fresh medium. Twenty-four hours later, medium containing G418 (500 μg/ml) or puromycin (2 μg/ml) was added, and the medium was changed every 3 to 4 days for a period of about 3 weeks or until fully resistant cells were selected. Colonies were fixed in cold methanol for 10 min and stained for 2 min at room temperature with methylene blue (1%, wt/vol, in 60% methanol-H2O).
RESULTS
UAF1 associates with the three deubiquitinating enzymes USP1, USP12, and USP46 in C33A cervical carcinoma cells.
We previously showed that the interaction of E1 with UAF1 stimulates viral DNA replication (19, 21). However, the function of UAF1 in this process has remained elusive. The fact that UAF1 is devoid of any known enzymatic activity but contains several WD40 repeats suggests that it functions through interaction with other cellular proteins. This prompted us to identify UAF1-interacting proteins from C33A cervical carcinoma cells. The C33A cell line was chosen because it supports transient HPV DNA replication and was used for most of our previous functional studies on E1 and UAF1. Stable cell populations expressing 3× Flag (3F)-tagged UAF1 or 3F alone as a negative control were generated and used to purify UAF1-containing protein complexes by a single immunoprecipitation step with the anti-Flag M2 antibody. Following extensive washes with high-salt buffer, protein complexes were eluted from the antibody by competition with a 3F peptide and analyzed by SDS-PAGE and silver staining (Fig. 1A). Proteins purified from cells expressing 3F-UAF1 but absent from the control purification were then identified by tandem mass spectrometry. This approach uncovered the three deubiquitinating enzymes USP1, USP12, and USP46 as the main UAF1-interacting proteins in C33A cells, based on spectral counting (Fig. 1A). We consistently observed that the three USPs copurified in substoichiometric amounts relative to the amount of UAF1, perhaps because these enzymes are expressed at lower levels than UAF1, as suggested by the protein abundance data available in the PaxDb database (28).These UAF1-USP interactions were then validated by coimmunoprecipitation (co-IP) experiments (Fig. 1B). Briefly, 3F-UAF1 was coexpressed in C33A cells with any one of the three USPs, tagged with GFP, and immunoprecipitated using an anti-Flag antibody. The presence of the USP in each precipitate was then analyzed by Western blotting using an anti-GFP antibody. The results shown in Fig. 1B confirmed that UAF1 interacts with USP1, UPS12, and USP46 but not with an irrelevant protein, USP14, which was used as a negative control. Note that the co-IP experiment involving USP1 was performed in the presence of the proteasome inhibitor lactacystin to increase the expression level of this highly unstable enzyme. While this work was in progress, two other groups also reported on the interaction of UAF1 with USP1, USP12, and USP46 in other cell lines (24, 25, 29), thus leaving little doubt on the validity of these interactions.
FIG 1.
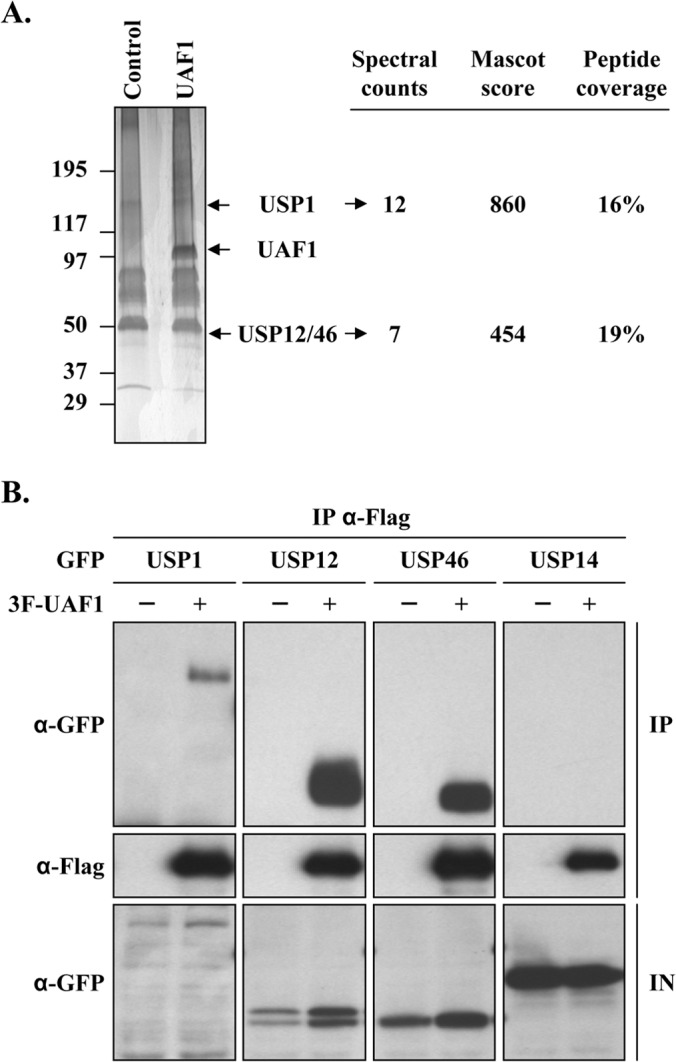
UAF1 interacts with USP1, USP12, and USP46 in C33A cervical carcinoma cells. (A) C33A stable cell lines expressing 3F-UAF1 (UAF1) or 3F alone (Control) were submitted to anti-Flag immunoprecipitation. Purified protein complexes were separated by SDS-PAGE and stained with silver nitrate; gel slices were then excised and their protein content was identified by LC-MS/MS. Major identified proteins are indicated on the right, along with their respective spectral counts, Mascot scores, and peptide coverage. USP12 and USP46, two proteins with high sequence identity, were often codetected, and their results are indicated together. (B) Co-IP of 3F-UAF1 with GFP-tagged USP1, USP12, or USP46 or with USP14 as a negative control. Cells transfected with the USP1 expression plasmid were treated for 24 h with 10 μM lactacystin prior to cell lysis. Results for input cell extracts (IN) are shown at the bottom.
The WD40 repeat region of UAF1 mediates its interaction with USPs.
We previously reported that the WD40 repeat region of UAF1 is required for its interaction with HPV E1. To investigate if this region of UAF1 also mediates binding to the USPs, truncated versions of 3F-UAF1 lacking its WD40 repeat region or C-terminal SUMO-like domains (SLD1 and SLD2) were generated (Fig. 2A) and used in co-IP experiments with RFP-tagged USP12 and USP46. The results presented in Fig. 2C show that these truncated UAF1 proteins are expressed at comparable levels but that only those containing the WD40 repeat region can interact with USP12 and USP46. Notably, only the longer UAF1 fragment (aa 1 to 573) could interact with USP12 and USP46 as efficiently as the full-length protein, with the shorter proteins (e.g., aa 1 to 400) displaying an apparent lower affinity for these USPs. Aside from weaker binding, a trivial reason for the apparent lower affinity of these shorter UAF1 fragments to USP12 and USP46 might be that they are toxic to transfected cells. This possibility was considered because it is shown, later in this study, that UAF1 is required for cellular proliferation (see Fig. 5). However, this concern was ruled out by showing that none of the truncated UAF1 proteins are deleterious to the growth of C33A cells in a colony formation assay (Fig. 2C). Thus, the results presented above indicate that UAF1 interacts with USP12 and USP46 primarily through its N-terminal WD40 repeat region (aa 1 to 400), although sequences in the C-terminal direction of this region (between residues 401 and 573, spanning SLD1) also contribute to the interaction. These findings are consistent with a previous report showing that the WD40 repeat region of UAF1 also mediates its association with USP1 (25). They are also reminiscent of what we previously observed for the binding of E1 to UAF1, which also requires the WD40 repeat region and sequences beyond this domain for full interaction (21) (summarized in Fig. 2A).
FIG 2.
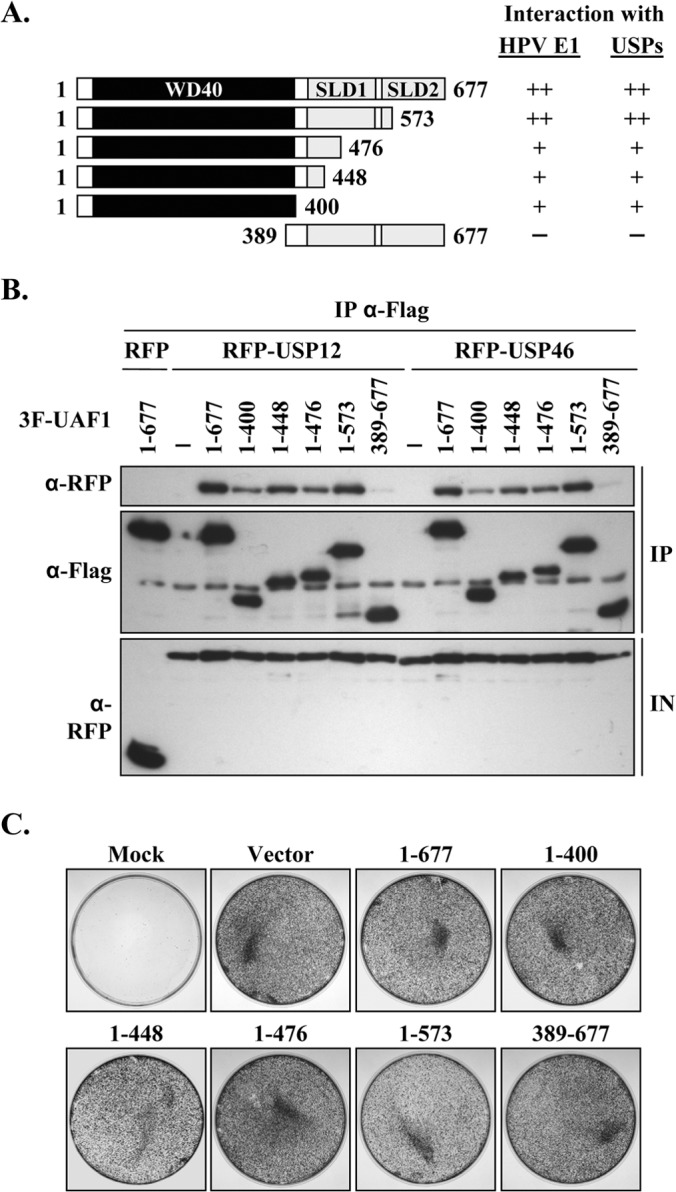
The WD40 repeat region of UAF1 is required for interaction with USP12 and USP46. (A) Schematic representation of truncated UAF1 proteins. The WD40 repeat region of UAF1 is indicated by a black box; and the two SUMO-like domains (SLD1 and SLD2) are indicated by gray boxes. The results obtained with USP12 and USP46 are summarized on the right (++, strong binding; +, weak binding; −, no binding). Also summarized are the results that we previously obtained with HPV E1 (21). (B) Coimmunoprecipitation mapping assay. The indicated 3F-UAF1 truncated proteins were expressed in C33A cells together with RFP-USP12 or RFP-USP46 and immunoprecipitated using an anti-Flag antibody. The presence of RFP-USPs in the immunoprecipitates was determined by Western blotting with an anti-RFP antibody. The results for input cell extracts (IN) are shown at the bottom. (C) Colony formation assay. C33A cells were transfected with expression vectors encoding the indicated UAF1 proteins and grown for approximately 3 weeks in G418-containing medium. Colonies were fixed with methanol and stained with methylene blue.
FIG 5.
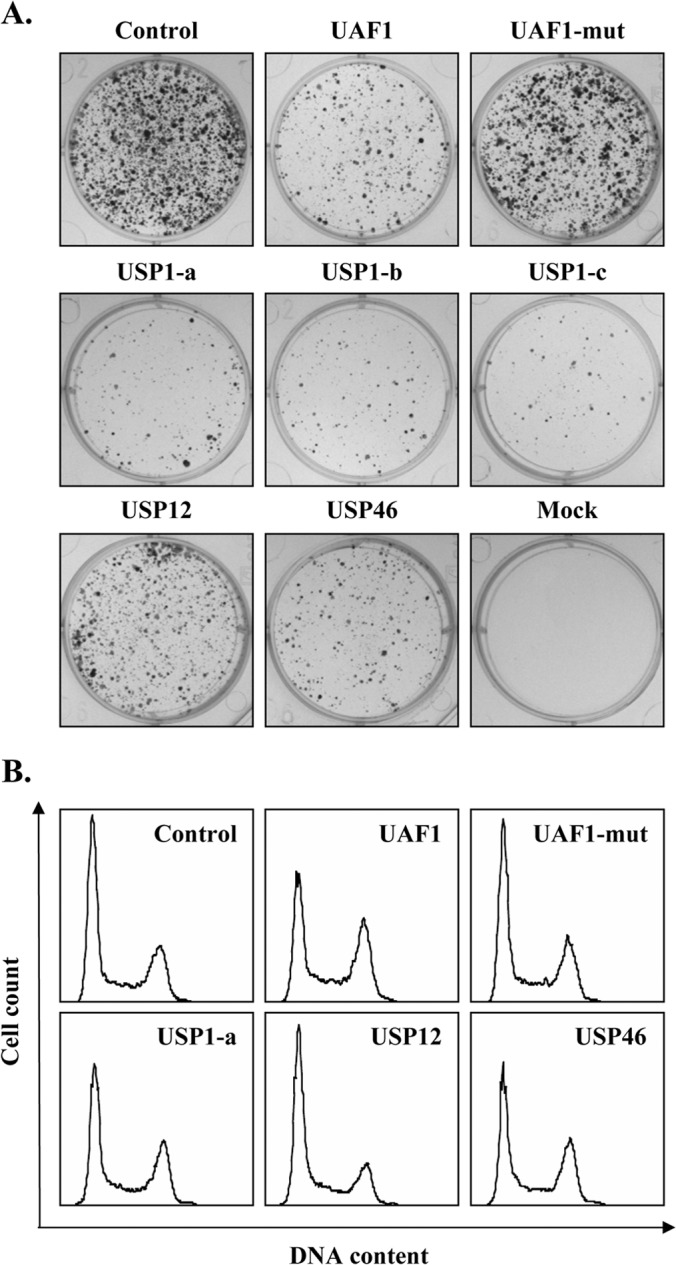
Depletion of USP1, USP12, USP46, or UAF1 by shRNAs impairs cellular proliferation. (A) Colony formation assay. C33A cells were transfected with the indicated shRNA expression vectors and selected for approximately 3 weeks in puromycin-containing medium. Drug-resistant colonies were fixed with methanol and stained with methylene blue. (B) Cell cycle analysis. C33A cells transiently expressing the indicated shRNA were trypsinized at 72 h posttransfection, and their DNA was stained with Hoechst and analyzed by flow cytometry.
HPV E1 forms a ternary complex with UAF1 and either USP1, USP12, or USP46.
The mapping results presented above indicated that USP1, USP12, and USP46 bind to a region of UAF1 similar to that to which HPV E1 binds. These findings raised the question as to whether USP1, USP12, and USP46 compete with E1 for binding to UAF1 or, alternatively, if these proteins assemble together into ternary complexes. To distinguish between these two possibilities, cells transiently expressing E1 (from HPV31) fused to yellow fluorescent protein (YFP-E1), 3F-UAF1, and either RFP-USP1, RFP-USP12, or RFP-USP46 were used in co-IP experiments to determine if precipitation of E1 would coprecipitate only UAF1 or both UAF1 and its associated USP. The results presented in Fig. 3A show that E1 does not compete with the USPs for binding to UAF1 but, rather, can assemble into a ternary complex with UAF1 and either USP1, USP12, or USP46. As a specificity control, these co-IP experiments included a mutant E1 protein carrying the V20A/E21A (VE) substitution that impairs UAF1 binding (19). As anticipated, E1 VE failed to efficiently associate with either UAF1 or the USPs (Fig. 3A). Collectively, these results indicate that E1 can assemble with UAF1 and either USP1, USP12, or USP46 into a ternary complex in which UAF1 bridges the interaction between E1 and the USP. In additional studies, we also determined, using catalytically inactive versions of USP1, USP12, and USP46, that their enzymatic activity is not required for complex formation with UAF1 and E1 (data not shown).
FIG 3.
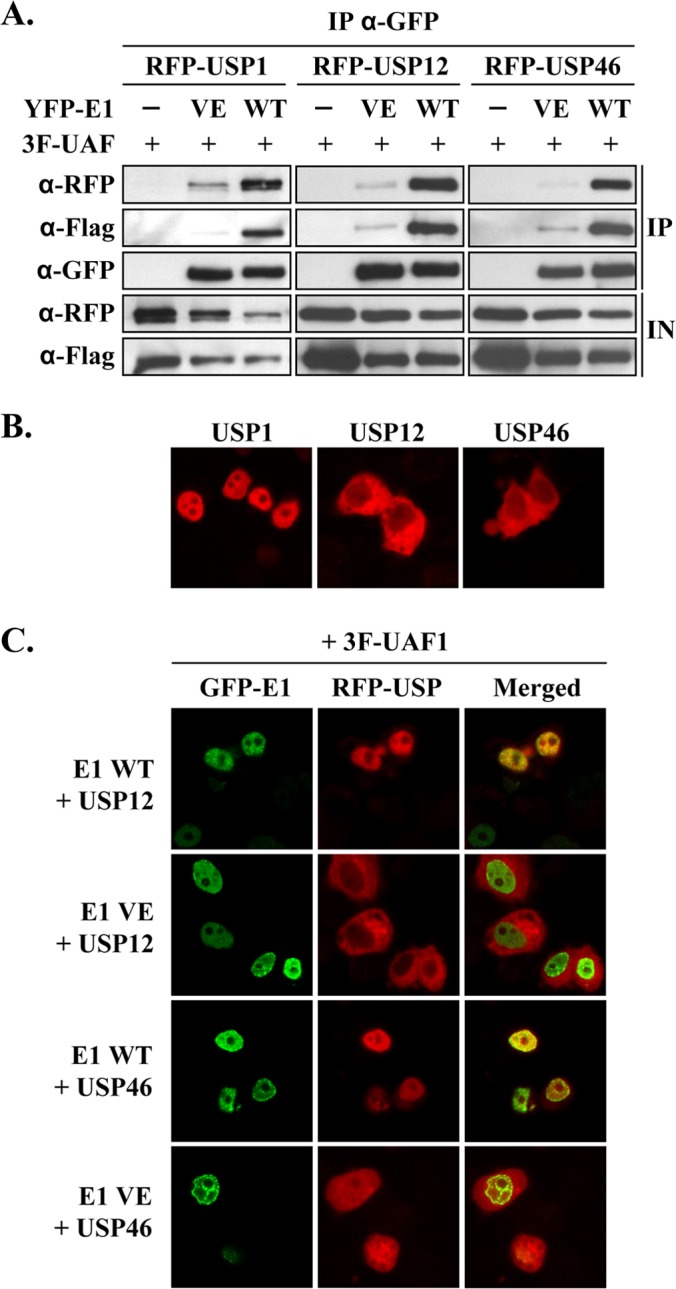
E1 assembles as a ternary complex with UAF1 and associated USPs. (A) Coimmunoprecipitation of YFP-E1 with 3F-UAF1 and RFP-USP1, RFP-USP12, or RFP-USP46. YFP-E1, as either the wild-type (WT) protein or the UAF1-binding-defective VE protein as a control, was immunoprecipitated at 48 h posttransfection using an anti-GFP antibody from cells transfected with the indicated expression vectors. Immunoprecipitates were analyzed by Western blotting with anti-RFP (for USPs), anti-Flag (for UAF1), or anti-GFP (for E1) antibodies. (B and C) Fluorescence confocal microscopy images showing the subcellular localization of the indicated RFP-USPs expressed either alone (B) or together with 3F-UAF1 and GFP-E1 (C). The UAF1-binding-defective E1 VE protein was used as a negative control. Nuclei were stained with DAPI.
Next, we investigated the subcellular localization of the three USPs as fusions with RFP by fluorescence confocal microscopy. USP1 was found to be localized primarily to the nucleus of transfected C33A cells, consistent with previous reports (30, 31), while USP12 and USP46 were present mostly in the cytoplasm (Fig. 3B). We previously determined that UAF1 is primarily a cytoplasmic protein that gets relocalized to the nucleus through its association with E1 (19). The cytoplasmic localization of USP12 and USP46 led us to test if these proteins would also be relocalized to the nucleus by E1. To do so, GFP-E1 was coexpressed with either RFP-USP12 or RFP-USP46, along with 3F-UAF1. As shown in Fig. 3C, both USP12 and USP46 were predominantly nuclear in E1-expressing cells. Relocalization of these two USPs from the cytoplasm to the nucleus was dependent upon the interaction of E1 with UAF1, as it was not observed with the E1 VE protein defective for UAF1 binding (Fig. 3C). Overall, these in vivo data further support the notion that E1 assembles into a ternary complex with UAF1 and either USP1, USP12, or USP46 and, moreover, that formation of this complex is dependent upon the capacity of E1 to bind UAF1.
USP1, USP12, and USP46 are recruited to the HPV origin by E1 and UAF1.
We previously demonstrated by ChIP that UAF1 is brought to the HPV origin of replication through its interaction with E1 (21). This prompted us to investigate if the three USPs associated with UAF1 could also be recruited to the viral DNA. To do so, ChIP assays were performed in cells transfected with an expression vector for GFP-tagged USP1, USP12, or USP46 and cotransfected with the four plasmids used in our luciferase-based HPV DNA replication assay (3F-E1 and 3F-E2 expression vectors, a plasmid expressing ori-FLuc, and the internal control plasmid expressing RLuc). Control experiments included the use of USP14 as an irrelevant deubiquitinase and of two mutant E1 proteins defective for origin binding (E1 OBD) and for interaction with UAF1 (E1 VE), respectively. The different GFP-USPs were then immunoprecipitated using an anti-GFP antibody or an isotype-matched irrelevant anti-HA antibody as a specificity control, and the amount of coprecipitated ori DNA was quantified by qPCR. Results presented in Fig. 4A show that the plasmid expressing ori was significantly enriched in immunoprecipitates from cells expressing USP12 and USP46, indicating that both enzymes are recruited to the HPV origin. USP1 was also detected at the viral origin but in smaller amounts, most likely because this protein is very unstable and poorly expressed. Recruitment of USP1, USP12, and USP46 to the ori was dependent on the integrity of the E1 OBD, consistent with the notion that these USPs are brought to the ori in association with E1 and UAF1. Accordingly, the interaction of E1 with UAF1 was also found to be important for the recruitment of USP1, USP12, and USP46 to the ori, as determined in parallel ChIP experiments conducted with the E1 VE protein defective for UAF1 binding (Fig. 4B). Finally, immunoprecipitation of E1 with an anti-Flag antibody revealed that its capacity to bind to the ori was not affected by overexpression of the GFP-USPs in these ChIP experiments (Fig. 4C). This was also true for the E1 VE protein, which was detected at the ori in amounts similar to those for the E1 wild type. The fact that this mutant protein can bind to the origin but is unable to recruit the USPs suggests that UAF1 and its associated USPs are not required for assembly of the E1-E2-ori complex. Altogether, the results presented above indicate that E1 specifically recruits USP1, USP12, and USP46 to the viral origin through its interaction with UAF1.
FIG 4.
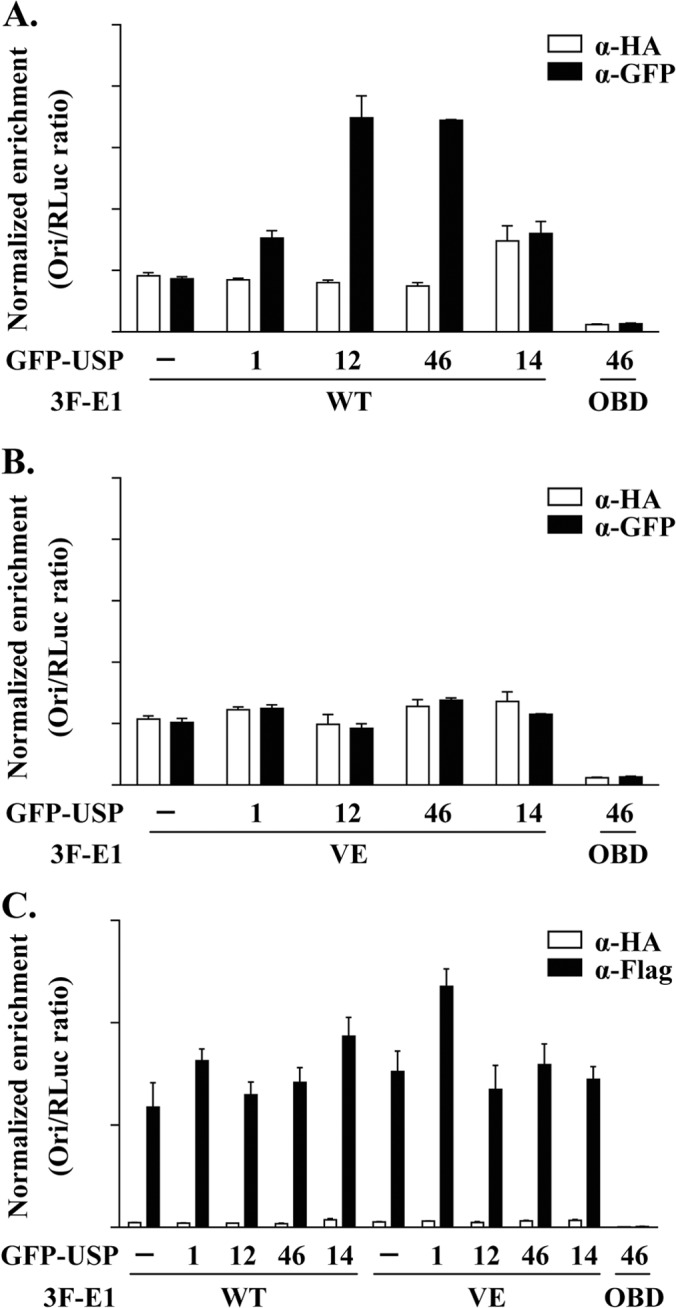
USP1, USP12, and USP46 are recruited to the HPV origin by E1 and UAF1. (A) ChIP assays were performed in C33A cells cotransfected with expression vectors for 3F-E1 (either the WT or OBD mutant protein, as indicated), RFP-E2, GFP-USP (USP1, USP12, USP46, or USP14), or GFP alone (−), together with an origin (ori)-containing plasmid and a Renilla luciferase (Rluc)-containing plasmid as an internal control. GFP-USP fusion proteins were immunoprecipitated with a GFP antibody or an HA antibody as a specificity control. The results of the ori enrichment levels determined by qPCR are shown after normalization to the amount of input DNA using the internal control (RLuc). Each value is the average of three replicates, with the standard deviations presented as error bars. (B) The same as for panel A but using the mutant E1 VE protein that is defective for binding UAF1. (C) The same as for panel A but using an anti-Flag antibody to immunoprecipitate the indicated E1 proteins.
Depletion of UAF1 and associated USP1, USP12, and USP46 by RNA interference impairs cellular proliferation and cell cycle progression.
To assess the requirement for USP1, USP12, and USP46 in HPV DNA replication, we wished to determine the effect of depletion of these proteins by RNA interference in our luciferase-based transient DNA replication assay. However, a prerequisite for these studies is that depletion of these proteins does not impair cellular proliferation, as any block in cell cycle progression outside of S phase would indirectly reduce viral DNA replication. To test the feasibility of these experiments, we screened a panel of shRNAs for those that could efficiently downregulate the expression of USP1, USP12, and USP46 (data not shown) and tested the most active ones for their effect on the proliferation of C33A cells in a colony formation assay. These studies led to the identification of three functional shRNAs against USP1 (USP1-a, -b, -c), one against USP12, and one against USP46, all of which, unfortunately, reduced the level of colony formation compared to that achieved with the control shRNA (Fig. 5A). We also included in this analysis a validated shRNA against UAF1 and, as a control, a defective version of this shRNA in which the targeting sequence was inactivated by three point mutations (UAF1-mut). Consistent with the results obtained by depletion of the USPs, downregulating the expression of UAF1 also impaired colony formation (Fig. 5A). To determine if the deleterious effect of these shRNAs was associated with a specific defect in cell cycle progression, they were transfected into C33A cells together with a GFP expression plasmid to facilitate identification of the transfected cells. At 48 h posttransfection, nuclear DNA was stained with Hoechst and the cell cycle distribution of GFP-positive cells was determined by flow cytometry. These experiments revealed that depletion of USP1, USP46, or UAF1 impairs cell cycle progression, as manifested by an increased number of cells in G2/M compared to the number of control cells in G2/M (Fig. 5B). Depletion of USP12 had less of an effect, although a small but reproducible increase in the number of cells in G2/M was also observed. This milder effect of USP12 is consistent with its lower growth-inhibitory activity observed in colony formation assays (Fig. 5A). Collectively, these data suggest that UAF1, USP1, USP46, and, to a lesser extent, USP12 are required for normal cell cycle progression, most likely to facilitate progression through G2/M. Although these findings revealed an important role for UAF1 and associated USPs in cellular proliferation, they also precluded the use of shRNAs against these proteins to assess their role in HPV DNA replication.
Overexpression of catalytically inactive USP1, USP12, and USP46 inhibits transient HPV DNA replication.
Like other cysteine proteases, USP1, USP12, and USP46 rely on a conserved cysteine at their active site for their deubiquitinase activity (Fig. 6A). Replacement of this catalytic residue by a serine was previously shown to inactivate these enzymes (24, 30, 32, 33). We surmised that overexpression of catalytically inactive USP1 (C90S), USP12 (C48S), and USP46 (C44S) could have a dominant negative effect on their endogenous counterparts and, as such, might be useful tools to probe the function of these enzymes in HPV DNA replication, provided, of course, that these inactivated enzymes do not impair cellular proliferation, as discussed above. To test this possibility, catalytically inactive (ci) versions of the USPs were created, tagged with RFP, and confirmed to be well expressed by Western blotting (Fig. 6B). These RFP-USPci proteins were then tested in a colony formation assay and found to have little to no effect on the proliferation of C33A cells (Fig. 6C). Consistent with this result, no significant effect on cell cycle progression was detected in cells transiently overexpressing these USPci enzymes (Fig. 6D). Note that this analysis was performed with fusions of the inactive USPs to GFP rather than RFP, because the increased brightness of GFP makes it a better reagent for flow cytometry. Similar results were also obtained with untagged USPci, thus ruling out the possibility that the RFP or GFP tag was preventing an effect of the USPci on cell cycle progression (data not shown). Having determined that overexpression of the USPci enzymes does not impair cellular proliferation, we then investigated their effect on HPV DNA replication. To do so, increasing amounts of each RFP-USPci expression vector were transfected in our luciferase-based HPV31 DNA replication assay (20) and the levels of luciferase activity were measured at 24 h posttransfection. Strikingly, all three USPci enzymes inhibited HPV DNA replication in a dose-dependent manner and with various potencies (Fig. 7A; inhibition efficiencies, USP1ci > USP46ci > USP12ci). Similar results were obtained with untagged USPci (data not shown). Despite being expressed at lower levels than the other two enzymes (Fig. 6B), USP1ci was the most inhibitory, as shown by the 70% reduction in viral DNA replication observed with the maximum amount of expression vector tested (Fig. 7A). Under the same conditions, USP12ci and USP46ci inhibited DNA replication by about 40% and 60%, respectively. We also tested the effect of a combination of all three USPci enzymes, achieved by transfecting cells with a 1:1:1 mixture of each USPci expression vector, and found that it was not appreciably more inhibitory than three equivalents of USP1ci (data not shown). To ascertain that these effects were due to a specific inhibition of viral DNA replication, these experiments were repeated using a luciferase plasmid lacking the HPV origin. As expected, expression of the three USPci enzymes had no effect in this context (Fig. 7B). As an additional specificity control, we took advantage of our previous observation that the E1 protein from BPV1 does not bind UAF1 (19) and, hence, that BPV1 DNA replication should be independent of USP1, USP12, and USP46. This was indeed found to be the case, as none of the USPci enzymes inhibited transient BPV1 DNA replication, also measured with a luciferase-based assay (Fig. 7C) (27). Collectively, these results suggest that the enzymatic activity of USP1, USP12, and/or USP46 facilitates HPV DNA replication.
FIG 6.
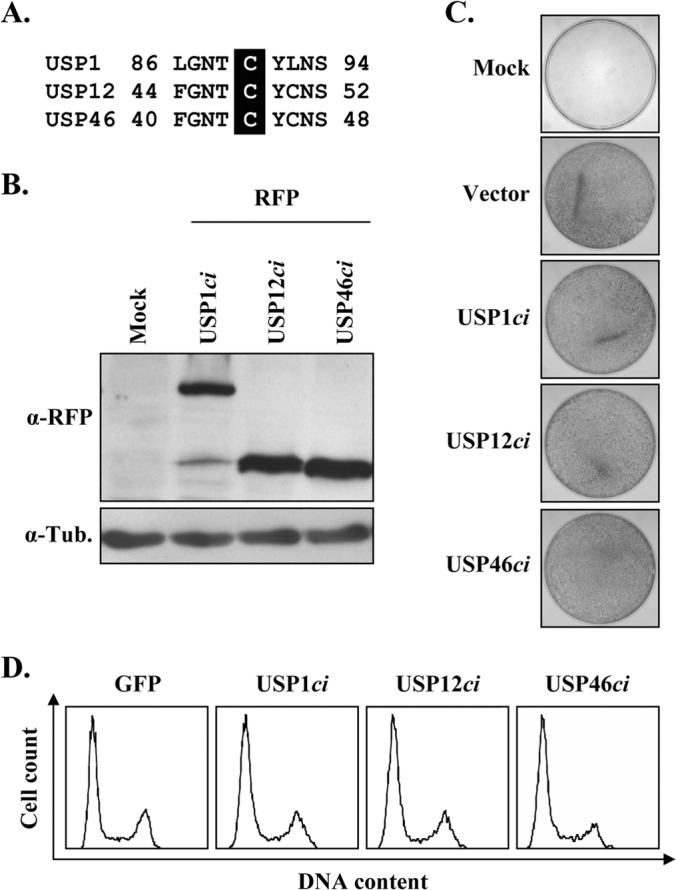
Catalytically inactive USP1, USP12, and USP46 do not inhibit cellular proliferation. (A) Amino acid sequence alignment of a short region of USP1, USP12, and USP46 surrounding the catalytic cysteine (highlighted). (B) Anti-RFP Western blot showing the expression of the different RFP-USP catalytically inactive (ci) enzymes. β-Tubulin (Tub.) was used as a loading control. (C) Colony formation assay. C33A cells were transfected with the indicated RFP-USPci expression plasmids and then selected for approximately 3 weeks in G418-containing medium. Colonies were fixed with methanol and stained with methylene blue. (D) Cell cycle analysis. C33A cells transiently expressing the indicated GFP-USPci enzymes were trypsinized at 48 h posttransfection, and their DNA was stained with Hoechst and analyzed by flow cytometry.
FIG 7.
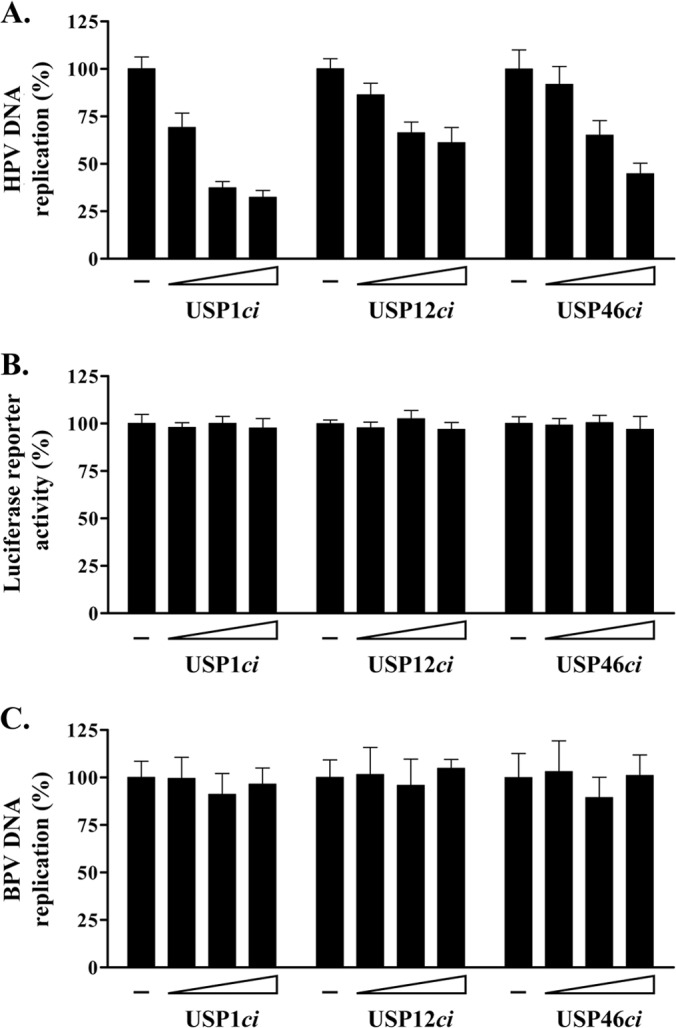
Specific inhibition of HPV DNA replication by catalytically inactive USP1, USP12, and USP46. (A) HPV DNA replication levels in cells expressing increasing amounts of RFP-tagged USP1ci, USP12ci, and USP46ci (8.75, 37.7, and 75 ng of the RFP-USPci expression vector). DNA replication activities are reported as a percentage of the signal obtained with cells cotransfected with the empty RFP vector as a control (−). Each value is the average of at least three independent experiments, each performed in triplicate, with the standard deviations presented as error bars. (B) The same as for panel A but using a firefly luciferase reporter plasmid lacking the viral origin. (C) Effect of the RFP-USPci enzymes on BPV1 DNA replication. The experiment was performed as described for panel A but using a BPV1 DNA replication assay.
Dominant negative inhibition of HPV DNA replication by catalytically inactive USP1, USP12, and USP46 requires the UAF1-binding site in E1.
From the results presented above, we surmised that the catalytically inactive USPs were inhibiting HPV DNA replication by competing with the endogenous enzymes for binding to UAF1. In support of this model, we found that the USPci enzymes could indeed bind to UAF1 in co-IP experiments similar to those whose results are presented in Fig. 1A (data not shown). In an attempt to test this model further, however, we wished to examine the effect of the USPci enzymes on HPV DNA replication catalyzed by a mutant E1 lacking the UAF1-binding site. We previously reported that UAF1 binds to the N-terminal 40 amino acids of E1 and that deletion of this region results in a truncated protein (E1Δ) whose DNA replication activity is about 25% that of wild-type E1 (the results are summarized in Fig. 8A) (20, 21). Since E1Δ does not interact with UAF1, we reasoned that its residual DNA replication activity should be independent of USP1, USP12, and USP46. This assumption was tested by measuring the effect of the USPci enzymes on HPV DNA replication catalyzed by E1Δ and by wild-type E1 in side-by-side comparisons. The results presented in Fig. 8B to D demonstrate that overexpression of inactive USP1, USP12, and USP46 has a more profound inhibitory effect on the replication activity of wild-type E1 than on E1Δ. Thus, the replication activity of E1Δ is largely independent of UAF1 and its associated USPs. Altogether, these results support the notion that USP1, USP12, and/or USP46 facilitates HPV DNA replication through the association of these enzymes with UAF1 bound at the N terminus of E1.
FIG 8.
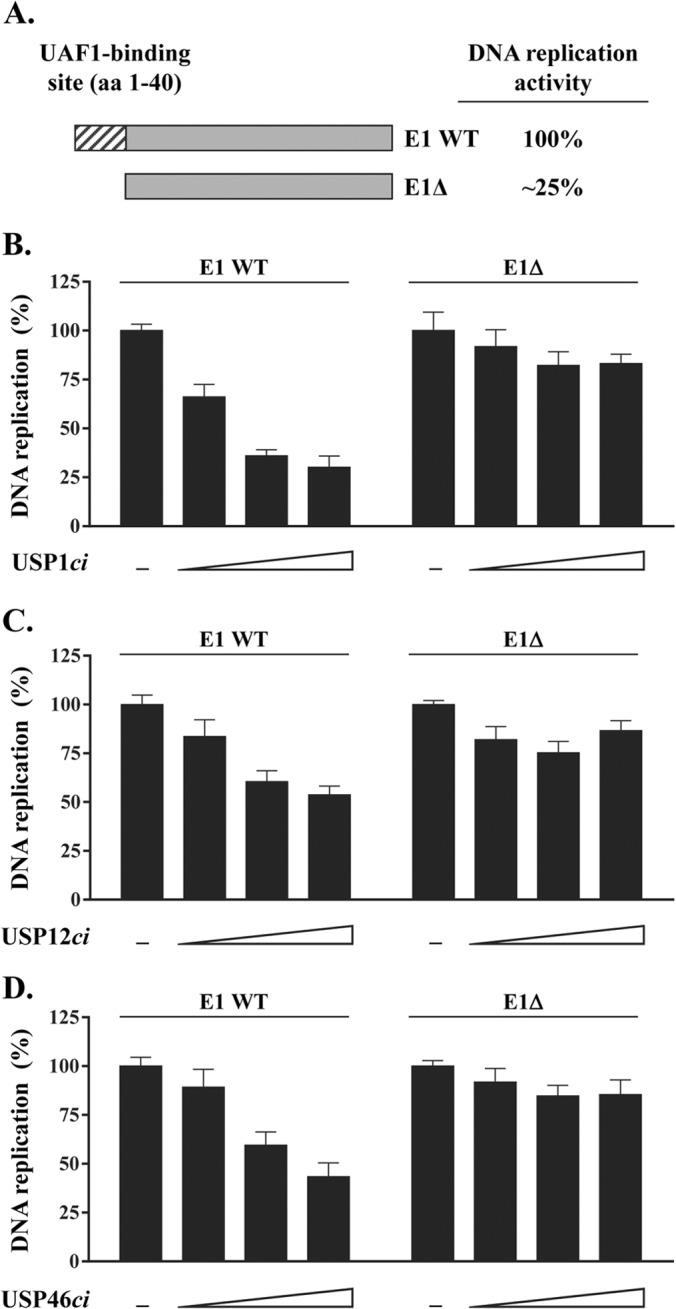
Inhibition of HPV DNA replication by catalytically inactive USP1, USP12, and USP46 requires the UAF1-binding site in E1. (A) Schematic representation of the HPV31 E1 wild-type protein (E1 WT) and of the truncated E1 derivative (E1Δ) lacking the N-terminal 40 amino-acid-long UAF1-binding site (hatched box). The relative DNA replication activity of these two proteins is indicated on the right. (B to D) Effect of overproducing RFP-tagged USP1ci (B), RFP-USP12ci (C), and RFP-USP46ci (D) on HPV DNA replication catalyzed by either the E1 wild-type protein or E1Δ. Increasing amounts of RFP-USPci expression vectors (8.75, 37.7, and 75 ng) were used. DNA replication activities are reported as a percentage of the signal obtained with cells cotransfected with the empty RFP vector as a control (−). Each value is the average of three independent experiments, each performed in triplicate, with the standard deviations presented as error bars.
DISCUSSION
We previously reported that the E1 helicase from anogenital HPV types binds to the cellular protein UAF1, an interaction that stimulates transient viral DNA replication and is required for the long-term maintenance of the HPV episome in undifferentiated keratinocytes (19, 21). It was previously reported that UAF1 associates with the deubiquitinating enzymes USP1, USP12, and USP46 (24, 25), findings that we have independently confirmed in this study in C33A cells. Importantly, we have also determined that E1 can form a ternary complex with UAF1 and any one of its associated USPs in co-IP experiments. Similarly, we observed that USP12 and USP46, which, like UAF1, are located predominantly in the cytoplasm, can be relocalized to the nucleus by E1, provided that the UAF1-binding site on E1 is intact. Similar experiments could not be done with USP1, as it is already a nuclear protein. ChIP experiments, however, demonstrated that all three USPs are recruited to the viral DNA by E1 in a UAF1-dependent manner. Collectively, these three lines of evidence indicate that E1-UAF1-USP ternary complexes can assemble in vivo and are loaded on the viral DNA, presumably by E2. As for the architecture of these ternary complexes, we surmise that UAF1 acts as a bridge between E1 and the USPs, since mutation of the UAF1-binding site in E1 abolishes its interaction with USP1, USP12, and USP46. It is also likely that only a single type of USP is present per complex, as it was previously reported that USP1, USP12, and USP46 bind to UAF1 in a mutually exclusive manner (24). However, it remains possible that all three types of UAF1-USP complexes could associate with E1 simultaneously in the context of an E1 double hexamer. Additional work will be required to elucidate the exact stoichiometry of these complexes.
The binding of UAF1 to USP1, USP12, or USP46 was previously shown to stimulate the deubiquitinase activity of these enzymes (24, 25, 34). In the case of USP1, this stimulation is thought to proceed through modulation of the enzyme active site, resulting in a higher catalytic efficiency (kcat) but no substantial change in substrate-binding affinity (Km) (32). To address the role of these deubiquitinase complexes in HPV DNA replication, we opted to downregulate the expression of USP1, USP12, and USP46 by RNA interference and, in separate experiments, to antagonize their function by overexpression of catalytically inactive enzymes. We observed that shRNA-mediated depletion of any one of the three USPs is deleterious to cellular proliferation and cell cycle progression, similar to what we obtained by depletion of UAF1. These results are consistent with previous reports indicating that UAF1 and USP1 knockout mice die as embryos and perinatally, respectively (35, 36). In contrast, overexpression of catalytically inactive versions of the USPs was well tolerated in C33A cells. It is intriguing that overexpression of the USPci enzymes was not growth inhibitory like depletion of the USPs by shRNA was. It is possible that USP1, USP12, and USP46 play essential roles in cellular proliferation that are independent of their enzymatic activity. Alternatively, the overexpressed USPci enzymes may not completely outcompete their endogenous counterparts, thus leaving sufficient levels of USP1, USP12, and USP46 activity to support cellular proliferation. Regardless of the explanation, the fact that the USPci enzymes were well tolerated made them useful tools to investigate the roles of USP1, USP12, and USP46 in transient HPV DNA replication. Remarkably, all three mutant enzymes inhibited HPV DNA replication in a dose-dependent manner, while they had little to no effect on viral DNA replication catalyzed by BPV E1 or by a mutant HPV E1 lacking its UAF1-binding site (E1Δ). Interestingly, in these experiments HPV DNA replication was never reduced by more than 70%, a level of inhibition similar to the levels obtained by mutating or truncating the UAF1-binding site in E1 or by overexpression of this domain in trans as an inhibitory peptide (19, 21). These results suggested that the effect of the USPci enzymes was mediated through their association with UAF1 bound at the N terminus of E1. Accordingly, we observed that the low levels of DNA replication supported by E1Δ, which lacks the UAF1-binding site, were resistant to USPci inhibition. Collectively, these findings provide strong evidence that one or more UAF1-USP deubiquitinase complexes are actively involved in HPV DNA replication through an interaction with the E1 N-terminal domain.
We repeatedly observed that the three USPci enzymes were not equally active at inhibiting HPV DNA replication, with USP1ci being the most potent, despite its lower expression levels. Because USP1 is a nuclear protein which encodes its own NLS (31), it is possible that USP1ci accumulates to higher levels than USP12 and USP46 in the nucleus, where viral DNA replication takes place. Alternatively, it is well established that USP1 is a very unstable enzyme regulated by autocleavage and, accordingly, that mutation of its catalytic cysteine to serine (as was done in this study) greatly increases its steady-state accumulation (37). As such, the more stable USP1ci may be particularly efficient at competing with the low levels of endogenous enzyme. Finally, it is also possible that USP1 is the preferred enzyme targeted by HPV. Note that because our functional studies were performed by overexpression of inactive USP1, USP12, and USP46 enzymes, all of which can bind to UAF1 in a mutually exclusive manner and compete with the endogenous USPs, we cannot distinguish at the moment if HPV DNA replication relies preferentially on USP1, USP12, USP46, or a combination of these enzymes. In co-IP experiments, E1 was equally capable of associating with all three USPs in complex with UAF1. We would like to determine which one or more of these three USPs are preferentially recruited to the viral DNA in cells maintaining HPV episomes, but such studies are currently limited by the lack of antibodies specific enough for ChIP or immunofluorescence experiments.
Although we believe that our data provide strong evidence that USP1, USP12, and/or USP46 stimulates HPV DNA replication, the exact mechanism by which these enzymes operate remains uncertain. Nevertheless, we can offer some possibilities based on our current knowledge of the endogenous functions of USP1, USP12, and USP46. These three USPs have been implicated in the regulation of the stability of various proteins, including the glutamate receptor (GLR-1), the inhibitors of DNA binding (IDs), and the protein phosphatase PHLPP1 (34, 38–40). On the basis of these observations, it is tempting to speculate that recruitment of these UAF1-USP complexes to the HPV origin serves to regulate the stability of one or more components of the viral replisome. The viral helicase E1 itself is a possible substrate. Regulation of E1 through ubiquitination/deubiquitination could serve as a mechanism to control the abundance of active double hexamers and/or facilitate their assembly or disassembly after one round of replication. It is equally possible that the UAF1-USP complexes target another component present at the viral DNA replication fork. Notably, cellular factors such as PCNA, FANCD2, and histone H2A or H2B have all been reported to be the substrates of the UAF1-USP complexes (25, 30, 37, 41). Identification of the target(s) of the UAF1-USP1, UAF1-USP12, and UAF1-USP46 complexes at the viral episome is the subject of ongoing investigations in our laboratory.
In this study, we have provided evidence that HPV DNA replication is facilitated by the cellular deubiquitinating enzyme USP1, USP12, and/or USP46. As such, our findings add to a growing body of literature on the requirement for deubiquitinase activity during the life cycle of many viruses. Some viruses even encode their own deubiquitinating enzyme, such as the UL36 protein of herpes simplex virus 1, while others hijack cellular enzymes for their own purposes, such as the recruitment of USP7 by the Epstein-Barr virus EBNA-1 protein (reviewed in reference 42). Many recent studies have highlighted the therapeutic potential of cellular deubiquitinases, which are increasingly recognized as highly druggable components of the ubiquitin proteasome system (reviewed in reference 43). We hope that our study will stimulate interest in targeting USP1, USP12, and/or USP46 for the treatment of HPV infections.
ACKNOWLEDGMENTS
We thank Fanny Bergeron-Labrecque for technical assistance. We also thank Mireille Cartier and members of the J. Archambault laboratory for critical readings of the manuscript.
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR) to J.A. M.L. was supported by a studentship from the Fonds de la Recherche en Santé du Québec (FRSQ) and a CIHR Frederick Banting and Charles Best doctoral scholarship award.
Footnotes
Published ahead of print 21 May 2014
REFERENCES
- 1.Hebner CM, Laimins LA. 2006. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev. Med. Virol. 16:83–97. 10.1002/rmv.488 [DOI] [PubMed] [Google Scholar]
- 2.Kadaja M, Silla T, Ustav E, Ustav M. 2009. Papillomavirus DNA replication—from initiation to genomic instability. Virology 384:360–368. 10.1016/j.virol.2008.11.032 [DOI] [PubMed] [Google Scholar]
- 3.Doorbar J. 2006. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. (Lond.) 110:525–541. 10.1042/CS20050369 [DOI] [PubMed] [Google Scholar]
- 4.Gillison ML, Alemany L, Snijders PJ, Chaturvedi A, Steinberg BM, Schwartz S, Castellsague X. 2012. Human papillomavirus and diseases of the upper airway: head and neck cancer and respiratory papillomatosis. Vaccine 30(Suppl 5):F34–F54. 10.1016/j.vaccine.2012.05.070 [DOI] [PubMed] [Google Scholar]
- 5.Androphy EJ, Lowy DR, Schiller JT. 1987. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature 325:70–73. 10.1038/325070a0 [DOI] [PubMed] [Google Scholar]
- 6.Frattini MG, Laimins LA. 1994. Binding of the human papillomavirus E1 origin-recognition protein is regulated through complex formation with the E2 enhancer-binding protein. Proc. Natl. Acad. Sci. U. S. A. 91:12398–12402. 10.1073/pnas.91.26.12398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frattini MG, Laimins LA. 1994. The role of the E1 and E2 proteins in the replication of human papillomavirus type 31b. Virology 204:799–804. 10.1006/viro.1994.1596 [DOI] [PubMed] [Google Scholar]
- 8.Mohr IJ, Clark R, Sun S, Androphy EJ, MacPherson P, Botchan MR. 1990. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science 250:1694–1699. 10.1126/science.2176744 [DOI] [PubMed] [Google Scholar]
- 9.Sanders CM, Stenlund A. 2001. Mechanism and requirements for bovine papillomavirus, type 1, E1 initiator complex assembly promoted by the E2 transcription factor bound to distal sites. J. Biol. Chem. 276:23689–23699. 10.1074/jbc.M101861200 [DOI] [PubMed] [Google Scholar]
- 10.Schuck S, Stenlund A. 2005. Assembly of a double hexameric helicase. Mol. Cell 20:377–389. 10.1016/j.molcel.2005.09.020 [DOI] [PubMed] [Google Scholar]
- 11.Schuck S, Stenlund A. 2005. Role of papillomavirus E1 initiator dimerization in DNA replication. J. Virol. 79:8661–8664. 10.1128/JVI.79.13.8661-8664.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson VG, West M, Woytek K, Rangasamy D. 2002. Papillomavirus E1 proteins: form, function, and features. Virus Genes 24:275–290. 10.1023/A:1015336817836 [DOI] [PubMed] [Google Scholar]
- 13.Clower RV, Fisk JC, Melendy T. 2006. Papillomavirus E1 protein binds to and stimulates human topoisomerase I. J. Virol. 80:1584–1587. 10.1128/JVI.80.3.1584-1587.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conger KL, Liu JS, Kuo SR, Chow LT, Wang TS. 1999. Human papillomavirus DNA replication. Interactions between the viral E1 protein and two subunits of human DNA polymerase alpha/primase. J. Biol. Chem. 274:2696–2705 [DOI] [PubMed] [Google Scholar]
- 15.Han Y, Loo YM, Militello KT, Melendy T. 1999. Interactions of the papovavirus DNA replication initiator proteins, bovine papillomavirus type 1 E1 and simian virus 40 large T antigen, with human replication protein A. J. Virol. 73:4899–4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loo YM, Melendy T. 2004. Recruitment of replication protein A by the papillomavirus E1 protein and modulation by single-stranded DNA. J. Virol. 78:1605–1615. 10.1128/JVI.78.4.1605-1615.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masterson PJ, Stanley MA, Lewis AP, Romanos MA. 1998. A C-terminal helicase domain of the human papillomavirus E1 protein binds E2 and the DNA polymerase alpha-primase p68 subunit. J. Virol. 72:7407–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park P, Copeland W, Yang L, Wang T, Botchan MR, Mohr IJ. 1994. The cellular DNA polymerase alpha-primase is required for papillomavirus DNA replication and associates with the viral E1 helicase. Proc. Natl. Acad. Sci. U. S. A. 91:8700–8704. 10.1073/pnas.91.18.8700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cote-Martin A, Moody C, Fradet-Turcotte A, D'Abramo CM, Lehoux M, Joubert S, Poirier GG, Coulombe B, Laimins LA, Archambault J. 2008. Human papillomavirus E1 helicase interacts with the WD repeat protein p80 to promote maintenance of the viral genome in keratinocytes. J. Virol. 82:1271–1283. 10.1128/JVI.01405-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fradet-Turcotte A, Morin G, Lehoux M, Bullock PA, Archambault J. 2010. Development of quantitative and high-throughput assays of polyomavirus and papillomavirus DNA replication. Virology 399:65–76. 10.1016/j.virol.2009.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehoux M, Fradet-Turcotte A, Lussier-Price M, Omichinski JG, Archambault J. 2012. Inhibition of human papillomavirus DNA replication by an E1-derived p80/UAF1-binding peptide. J. Virol. 86:3486–3500. 10.1128/JVI.07003-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang K, Moldovan GL, Vinciguerra P, Murai J, Takeda S, D'Andrea AD. 2011. Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev. 25:1847–1858. 10.1101/gad.17020911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanchez-Pulido L, Devos D, Sung ZR, Calonje M. 2008. RAWUL: a new ubiquitin-like domain in PRC1 ring finger proteins that unveils putative plant and worm PRC1 orthologs. BMC Genomics 9:308. 10.1186/1471-2164-9-308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohn MA, Kee Y, Haas W, Gygi SP, D'Andrea AD. 2009. UAF1 is a subunit of multiple deubiquitinating enzyme complexes. J. Biol. Chem. 284:5343–5351. 10.1074/jbc.M808430200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohn MA, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, D'Andrea AD. 2007. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol. Cell 28:786–797. 10.1016/j.molcel.2007.09.031 [DOI] [PubMed] [Google Scholar]
- 26.Fradet-Turcotte A, Bergeron-Labrecque F, Moody CA, Lehoux M, Laimins LA, Archambault J. 2011. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol. 85:8996–9012. 10.1128/JVI.00542-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gagnon D, Senechal H, D'Abramo CM, Alvarez J, McBride AA, Archambault J. 2013. Genetic analysis of the E2 transactivation domain dimerization interface from bovine papillomavirus type 1. Virology 439:132–139. 10.1016/j.virol.2013.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO, von Mering C. 2012. PaxDb, a database of protein abundance averages across all three domains of life. Mol. Cell. Proteomics 11:492–500. 10.1074/mcp.O111.014704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sowa ME, Bennett EJ, Gygi SP, Harper JW. 2009. Defining the human deubiquitinating enzyme interaction landscape. Cell 138:389–403. 10.1016/j.cell.2009.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R. 2005. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol. Cell 17:331–339. 10.1016/j.molcel.2005.01.008 [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Santisteban I, Zorroza K, Rodriguez JA. 2012. Two nuclear localization signals in USP1 mediate nuclear import of the USP1/UAF1 complex. PLoS One 7:e38570. 10.1371/journal.pone.0038570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villamil MA, Chen J, Liang Q, Zhuang Z. 2012. A noncanonical cysteine protease USP1 is activated through active site modulation by USP1-associated factor 1. Biochemistry 51:2829–2839. 10.1021/bi3000512 [DOI] [PubMed] [Google Scholar]
- 33.Zhang W, Tian QB, Li QK, Wang JM, Wang CN, Liu T, Liu DW, Wang MW. 2011. Lysine 92 amino acid residue of USP46, a gene associated with ‘behavioral despair' in mice, influences the deubiquitinating enzyme activity. PLoS One 6:e26297. 10.1371/journal.pone.0026297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dahlberg CL, Juo P. 2014. The WD40-repeat proteins WDR-20 and WDR-48 bind and activate the deubiquitinating enzyme USP-46 to promote the abundance of the glutamate receptor GLR-1 in the ventral nerve cord of Caenorhabditis elegans. J. Biol. Chem. 289:3444–3456. 10.1074/jbc.M113.507541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park E, Kim JM, Primack B, Weinstock DM, Moreau LA, Parmar K, D'Andrea AD. 2013. Inactivation of Uaf1 causes defective homologous recombination and early embryonic lethality in mice. Mol. Cell. Biol. 33:4360–4370. 10.1128/MCB.00870-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, D'Andrea AD. 2009. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev. Cell 16:314–320. 10.1016/j.devcel.2009.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D'Andrea AD. 2006. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 8:339–347. 10.1038/ncb1378 [DOI] [PubMed] [Google Scholar]
- 38.Li X, Stevens PD, Yang H, Gulhati P, Wang W, Evers BM, Gao T. 2013. The deubiquitination enzyme USP46 functions as a tumor suppressor by controlling PHLPP-dependent attenuation of Akt signaling in colon cancer. Oncogene 32:471–478. 10.1038/onc.2012.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kowalski JR, Dahlberg CL, Juo P. 2011. The deubiquitinating enzyme USP-46 negatively regulates the degradation of glutamate receptors to control their abundance in the ventral nerve cord of Caenorhabditis elegans. J. Neurosci. 31:1341–1354. 10.1523/JNEUROSCI.4765-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams SA, Maecker HL, French DM, Liu J, Gregg A, Silverstein LB, Cao TC, Carano RA, Dixit VM. 2011. USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell 146:918–930. 10.1016/j.cell.2011.07.040 [DOI] [PubMed] [Google Scholar]
- 41.Joo HY, Jones A, Yang C, Zhai L, Smith AD, IV, Zhang Z, Chandrasekharan MB, Sun ZW, Renfrow MB, Wang Y, Chang C, Wang H. 2011. Regulation of histone H2A and H2B deubiquitination and Xenopus development by USP12 and USP46. J. Biol. Chem. 286:7190–7201. 10.1074/jbc.M110.158311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Isaacson MK, Ploegh HL. 2009. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 5:559–570. 10.1016/j.chom.2009.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C. 2012. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene 31:2373–2388. 10.1038/onc.2011.443 [DOI] [PubMed] [Google Scholar]