

ABSTRACT
Hepatitis E virus (HEV) causes both the endemic and epidemic spread of acute hepatitis in many parts of the world. HEV open reading frame 3 (ORF3) encodes a 13-kDa multifunctional protein (vp13) that is essential for HEV infection of animals. The exact role of vp13 in HEV infection remains unclear. In this study, vp13 was found to enhance interferon (IFN) production induced by poly(I · C), a synthetic analog of double-stranded RNA. Poly(I · C) treatment induced a higher level of IFN-β mRNA in HeLa cells stably expressing vp13 than in control cells. Using a luciferase reporter construct driven by the IFN-β promoter, we demonstrated that vp13 enhanced retinoic acid-inducible gene I (RIG-I)-dependent luciferase expression. This enhancement was found to be due to both an increased level of RIG-I protein and its activation. The levels of both endogenous and exogenous RIG-I were increased by vp13 by extension of the half-life of RIG-I. Additionally, vp13 interacts with the RIG-I N-terminal domain and enhances its K63-linked ubiquitination, which is essential for RIG-I activation. Analysis of vp13 deletion constructs suggested that the C-terminal domain of vp13 was essential for the enhancement of RIG-I signaling. In HEV-infected hepatoma cells, wild-type HEV led to a higher level of RIG-I and more poly(I · C)-induced IFN-β expression than did ORF3-null mutants. Analysis of vp13 from four HEV genotypes showed that vp13 from genotype I and III strains boosted RIG-I signaling, while vp13 from genotype II and IV strains had a minimal effect. These results indicate that vp13 enhances RIG-I signaling, which may play a role in HEV invasion.
IMPORTANCE Hepatitis E virus (HEV) is a significant pathogen causing hepatitis in many parts of the world, yet it is understudied compared with other viral hepatitis pathogens. Here we found that the HEV open reading frame 3 product, vp13, enhances interferon induction stimulated by a synthetic analog of double-stranded RNA. This enhancement may play a role in HEV invasion, as vp13 is essential for HEV infection in vivo. The results of this study provide insights into virus-cell interactions during HEV infection. In addition to revealing its possible roles in HEV interference with cellular signaling, these results suggest that the second half of the vp13 sequence can be ligated into the genomes of attenuated live viruses to induce an innate immune response for better protective immunity, as well as a marker for differentiation of vaccinated animals from those infected with the corresponding wild-type viruses.
INTRODUCTION
Hepatitis E virus (HEV), a single-stranded, positive-sense RNA virus, is the sole member of the genus Herpesvirus in the Herpesviridae family (1). HEV infection causes acute hepatitis, and its unique feature is a high mortality rate (up to 20%) among pregnant women (2, 3). Cases of acute hepatitis due to HEV infection have been reported in industrialized countries in recent years (4). The prevalence of HEV in industrialized countries may be underestimated. HEV infection is zoonotic, with pigs and several other animal species serving as potential reservoirs (5). Chronic HEV infection of immunocompromised individuals, including organ transplant recipients, has been reported (6).
HEV genomic sequences are heterogenic among different strains. At least four genotypes exist among HEV strains worldwide. Genotypes I and II are restricted to humans, whereas genotypes III and IV are zoonotic with an expanded host range (7, 8). HEV propagation in cultured cells has been inefficient and limited. HEV replication occurs in cells that have been transfected with transcripts from an HEV replicon (pSK-E2) containing the full-length cDNA of the HEV genome (9, 10). HEV replicons containing either a green fluorescent protein (pSKE2-GFP) (11) or a luciferase (pE-LUC) (12) reporter gene were also constructed to facilitate the study of HEV biology. Recently, an HEV strain of genotype III isolated from a chronically infected patient was adapted to grow in human HepG2/C3A hepatoma cells and found to infect pig and deer cell lines (13).
The HEV genome is approximately 7.2 kb in length and consists of three open reading frames (ORFs) (14). ORF1 encodes a nonstructural polyprotein that includes the RNA-dependent RNA polymerase. ORF2 encodes the capsid protein, the major structural protein of virions. ORF3 encodes a phosphoprotein with a molecular mass of approximately 13 kDa (vp13 here) (14). A number of studies showed that vp13 plays roles in cellular signaling pathways (8) and interacts with microtubules (15). Moreover, vp13 is essential for the establishment of HEV infection in macaques and pigs under experimental conditions (16, 17). ORF3-null mutants of HEV failed to establish a productive infection in rhesus monkeys, suggesting an essential role for vp13 in vivo (16). These data indicate that vp13 may play an important role in HEV-cell interactions, yet the exact role of vp13 in HEV infection remains unknown. It is also not known whether vp13 has any effect on host innate immune responses.
Host pattern recognition receptors (PRRs) for RNA viruses include the retinoic-acid-inducible gene I (RIG-I)-like receptor (RLR) pathway and the Toll-like receptor (TLR) pathway. TLRs that can detect viral RNA are TLR3, TLR7, and TLR8 (18). All TLRs except TLR3 signal through the adaptor molecule myeloid differentiation factor 88 (MyD88) (19). TLR3 signals solely via the adaptor TRIF (TIR domain-containing adaptor inducing beta interferon [IFN-β]) (20). The RLR family of PRRs is composed of RIG-I and melanoma differentiation-associated gene 5 (MDA5) (21). Both RIG-I and MDA5 signal through the adaptor IPS-1 (also known as MAVS, Cardif, or VISA) on the mitochondrial outer membrane (22). Both RLR and TLR3 can recognize viral genomic double-stranded RNA (dsRNA) or the replication intermediates of RNA viruses. Activation of RLR and TLR signaling leads to activation of two IκB kinase (IKK)-related kinases, TBK1 and IKKε, which phosphorylate IFN regulatory factor 3 (IRF3) and IRF7 (23, 24). These transcription factors are translocated into the nucleus and result in the induction of type I IFNs, which not only lead to an antiviral state in neighboring uninfected cells but also serve as key regulators to evoke an adaptive immune response.
In this study, vp13 was found to enhance IFN expression induced by poly(I · C), a synthetic analog of dsRNA. vp13 expression led to an increased level of RIG-I via extension of the half-life of the protein. Immunoprecipitation assay results indicated that vp13 interacted with the RIG-I N-terminal domain and increased its ubiquitination. In HEV-infected hepatoma cells, wild-type HEV led to higher levels of RIG-I and greater expression of poly(I · C)-induced IFN-β than those caused by ORF3-null HEV mutants. These results indicate that vp13 enhanced IFN induction via RIG-I signaling.
MATERIALS AND METHODS
Cells, viruses, and replicons.
HEK293T cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). HeLa and S10-3 cells (13) were maintained in DMEM-reduced serum supplemented with 3% FBS. Transfection of HeLa, HEK293T, and S10-3 cells was performed with FuGeneHD reagent (Promega, Madison, WI) in accordance with the manufacturer's instructions. HeLa cells stably expressing vp13 were established by transfection of the cells with VenusN1-vp13 or VenusC1-vp13 (15) and selection for resistance to G418 (500 μg/ml). Cell cloning was done by limiting dilution. Poly(I · C) (InvivoGen, San Diego, CA) was used to induce IFN production at a concentration of 10 μg/ml for direct addition to cultured cells or at 1 μg/ml for transfection of cells. Avirulent LaSota Newcastle disease virus (NDV) with the inserted gene for green fluorescence protein (GFP) was used as an indicator of poly(I · C)-induced IFN production as previously described (31).
Full-length HEV, HEV-GFP, and HEV-luciferase RNAs were obtained by in vitro transcription from replicon plasmids pSK-E2, pSK-E2-GFP, and pE2-Luc (10–12), respectively, with the AmpliCap-Max T7 High Yield Message Maker kit (Cellscript, Madison, WI). Transfection of S10-3 cells with RNA was performed with DMRIE-C reagent (Invitrogen, Grand Island, NY).
Plasmids.
The HEV ORF3 plasmids, VenusN1-vp13, and VenusC1-vp13 were reported previously (15). HEV replicons, pSK-E2, pSK-E2-GFP, pE2-Luc, and ORF3-null pSK-E2 were described previously (10–12, 16). ORF3 sequences from strains of HEV genotypes II (GenBank accession number M74506) and IV (accession number AB074915) were synthesized (GenScript, Piscataway, NJ) and cloned into the XhoI and EcoRI sites of the VenusC1 vector as described previously (15). ORF3 of HEV genotype III strain Kernow (13) was similarly cloned into VenusC1 vectors with the primers listed in Table 1. ORF3 truncation mutants were constructed with the VenusC1 vector and the primers listed in Table 1.
TABLE 1.
Primers used in this study
| Primera | Sequences (5′ to 3′)b | Application |
|---|---|---|
| RIG-I-F1 | TTAGGTACCATGACCACCGAGCAGCGAC | Cloning of RIG-I |
| RIG-I-R1 | GGAGGTACCTCATTTGGACATTTCTGCTG | |
| H3F11 | CGCCTCGAGTGGGTTCGCGACCATGC | Cloning of vp13 D1 |
| H3R12 | CAGAATTCTTAGGTTGGTTGGATGAATAT | |
| H3R24 | CAGAATTCTTACCTGGTCACGCCAAGCGG | Cloning of vp13 D2 |
| H3F8 | GCGAATTCATGTTCATCCAACCAACCC | Cloning of vp13 D3 |
| H3R23 | CAGAATTCTTAGCGGCGCGGCCCCAGCTGTG | |
| T2H3F2 | GCCTCGAGGTTCGCCACCATGCGCCCTAG | Cloning of ORF3 of genotype II |
| T2H3R2 | GCGAATTCTCAGCGCCGCAGCCCCGGCTG | |
| KH3F2 | CGCTCGAGGATCACCATGTGCCCTAG | Cloning of ORF3 of HEV strain Kernow |
| KH3R3 | GCGAATTCTCAACGGCGCAGCCCCAGC | |
| T4H3F2 | GCCTCGAGAGATGCCACCATGCGCTCTCG | Cloning of ORF3 of genotype IV |
| T4H3R2 | GCGAATTCTCAACGGCGCAGCCCCAGCTG |
F, forward primer; R, reverse primer. H3 at the beginning of a primer name indicates the primer is based on sequences of HEV ORF3.
The restriction enzyme cleavage sites used for cloning are italicized.
Full-length RIG-I was cloned into the KpnI site of the pCMV-Flag-MAT-1 vector. The construction of Myc-RIG-I(N) (26), MDA5(N) (27), FLAG-TBK1, and FLAG-IKKε (23) plasmids was described previously. pCDNA3-TRIF-CFP (28) and pRK5-HA-Ubiquitin-K63 (29) were obtained from Addgene.
IFA and live-cell fluorescence microscopy.
An immunofluorescence assay (IFA) was carried out as reported previously (15), with a chimpanzee antibody against HEV. A cover glass with cells was mounted onto a slide with Fluoromount-G clear mounting medium containing 4′,6-diamidino-2-phenylinodole (DAPI; SouthernBiotech, Birmingham, AL) and observed by fluorescence microscopy. GFP expression in live cells transfected with RNA from a HEV replicon containing the GFP gene was similarly observed.
Western blot analysis.
Whole-cell lysates were analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and Western blotting as described previously (15, 30). Antibodies against GFP (Santa Cruz Biotechnology, Santa Cruz, CA), FLAG (Sigma, St. Louis, MO), hemagglutinin (HA; Thermo Fisher Scientific, Waltham, MA), Myc (Rockland Immunochemicals, Inc., Gilbertsville, PA), vp13, RIG-I (Santa Cruz), and tubulin (Sigma) were used for blotting. The chemiluminescence signal was recorded digitally with a ChemiDoc XRS imaging system (Bio-Rad Laboratories, Hercules, CA). Digital signal acquisition and densitometry analyses were conducted with the Quantity One Program, version 4.6 (Bio-Rad). To determine the half-life of RIG-I, cycloheximide (Sigma) was used at a final concentration of 100 μg/ml to inhibit protein translation. Cell lysate samples harvested at the indicated time points after cycloheximide treatment were subjected to Western blotting.
Reverse transcription and real-time quantitative PCR (RT-qPCR).
Total RNA was isolated from cells with TRIzol reagent (Invitrogen) according to the manufacturer's protocol. RNase-free DNase was used to remove carryover DNA from the RNA isolation procedure. Reverse transcription was carried out with avian myeloblastosis virus reverse transcriptase and a combination of a random primer and oligo(dT) as previously reported (31). The primers used for detection of IFN-β (31) and RIG-I (32) cDNA were described previously. Real-time PCR with SYBR green detection was done as described previously (33). Transcripts of housekeeping gene RPL32 (ribosomal protein L32) were also amplified from the same samples to serve as an internal control for normalization. Gene expression was quantified by the 2−ΔΔCT method (34) and is shown as the fold difference from a mock-treated control.
IP.
Immunoprecipitation (IP) was conducted as previously described (35, 36), with modifications. HEK293T cells were lysed with lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 0.2 mM EDTA, 2 mM EGTA, 0.5% IGEPAL CA-630, 10% glycerol, 1 mM sodium vanadate) supplemented with a protease inhibitor cocktail (Sigma). The lysate was clarified by centrifugation at 14,000 × g for 5 min at 4°C. Antibodies against Myc or GFP were added to the supernatant. IP with protein G agarose (KPL Inc., Gaithersburg, MD) was done by following the manufacturer's instructions. IP samples obtained with Myc antibody were subjected to Western blotting with vp13 antibody. IP samples obtained with GFP antibody were subjected to Western blotting with RIG-I antibody.
For detection of ubiquitinated RIG-I, N-ethylmaleimide (Thermo Scientific, Rockford, IL) was included in the lysis buffer at a final concentration of 50 μM. After lysate clarification, the supernatant was moved to a fresh tube and SDS was added to a final concentration of 1%. The supernatant was then boiled for 5 min and cooled on ice for IP with Myc antibody. The IP samples were subjected to Western blotting with ubiquitin antibody.
Luciferase reporter assay.
HEK293T cells were transfected with a firefly luciferase reporter plasmid expressing the IFN-β promoter and the plasmids indicated. Renilla luciferase vector pGL4.74 hRL-TK (Promega) was also included for normalization. Empty-vectors of test plasmids were included as controls. Cell lysates were prepared 48 h after transfection, and luciferase levels were determined by firefly and Renilla luciferase assays according to the manufacturer's instructions (Promega). Luciferase activity was quantified with a VICTOR3 Multilabel Counter (Perkin-Elmer Life and Analytical Sciences, Wellesley, MA). Firefly luciferase levels were normalized to Renilla luciferase expression. Values representing fold luciferase activity differences from control cells are shown.
Statistical analysis.
Differences in indicators between treatment samples, such as IFN-β mRNA level differences between the group in the presence of vp13 and the control sample, were assessed by Student t test. A two-tailed P value of <0.05 was considered significant.
RESULTS
vp13 enhances poly(I · C)-induced IFN production.
In our studies of vp13, we established HeLa cells stably expressing vp13 with plasmids encoding vp13 fused to the N- or C-terminal end of GFP, VenusN1-vp13, or VenuC1-vp13, respectively (15). HeLa-VenusN1-vp13 was first used in an assay to assess the effect of vp13 on poly(I · C) induction of type I IFNs. NDV is sensitive to type I IFNs and was used as an indicator of IFN induction. HeLa-VenusN1-vp13 cells were inoculated with NDV-GFP at 12 h posttreatment with poly(I · C) and monitored for GFP expression. The cell line had a low level of VenusN1-vp13 expression that did not interfere with observation of NDV-GFP replication. Stable HeLa-VenusN1-vp13 cells were expected to have a number of GFP-positive cells similar to or greater than that of control cells if vp13 had an inhibitory or no effect on IFN induction. However, substantially fewer cells with vp13 expression than control cells were GFP positive (Fig. 1A). The vp13 expression in cells without poly(I · C) did not affect NDV-GFP replication. This result suggested that vp13 enhanced poly(I · C)-induced IFN production.
FIG 1.

Enhancement of poly(I · C)-induced IFN production by vp13. (A) Reduced NDV-GFP replication in HeLa cells stably expressing vp13 (VenusN1-vp13). Cells were treated with poly(I · C) for 12 h and then inoculated with NDV-GFP. Cells were observed by fluorescence microscopy 24 h after NDV inoculation. (B) Increase in poly(I · C)-induced IFN-β mRNA in HeLa cells expressing vp13 as detected by RT-qPCR. HeLa cells stably expressing VenusN1-vp13 or VenusC1-vp13 and normal HeLa cells were treated with poly(I · C), which was added directly to cultured cells. Fold induction of the IFN-β mRNA level in comparison with that of nontreated control cells is shown. Error bars represent standard errors of three repeated experiments. Significant differences from control HeLa cells are shown by **, which indicates P < 0.01. (C) Increase in poly(I · C)-induced IFN-β mRNA in HeLa cells expressing vp13. HeLa cells stably expressing VenusC1-vp13 and normal HeLa cells were transfected with poly(I · C). Relative fold induction of IFN-β mRNA is shown. (D) Detection of Venus-vp13 fusion protein in stable HeLa-vp13 cells. N, VenusN1-vp13; C, VenusC1-vp13. Western blotting was done with an anti-vp13 antibody.
To confirm this observation, we examined IFN-β transcript levels by RT-qPCR. HeLa, HeLa-VenusN1-vp13, and HeLa-VenusC1-vp13 stable cells were treated with poly(I · C) for 12 h and harvested for RNA isolation. The direct addition of poly(I · C) to the cells was supposed to activate the TLR3 signaling pathway to induce IFN production. Both HeLa-VenusN1-vp13 and HeLa-VenusC1-vp13 cells had 5.6- and 10.2-fold more IFN-β mRNA than normal HeLa cells after direct poly(I · C) treatment (Fig. 1B). Because poly(I · C) treatment induced higher levels of IFN-β mRNA in HeLa-VenusC1-vp13 cells than in HeLa-VenusN1-vp13 cells, HeLa-VenusC1-vp13 cells were used in the following experiments. Similarly, VenusC1-vp13 plasmid was used for further analysis of the effect of vp13 on IFN signaling. To test if vp13 was also able to enhance the poly(I · C)-activated RLR pathway, we transfected cells with poly(I · C). The IFN-β mRNA level in HeLa-VenusC1-vp13 cells was 3.7-fold higher than that in HeLa cells after poly(I · C) transfection (Fig. 1C). These results indicated that vp13 enhanced poly(I · C)-induced IFN production in HeLa cells.
vp13 expression in stable HeLa cells was detected by Western blotting with an anti-vp13 antibody (Fig. 1D). Stable cells with VenusC1-vp13 had higher level of vp13 than did cells with VenusN1-vp13, which may be why poly(I · C) induced a higher level of IFN-β mRNA in the former cells.
vp13 enhances RIG-I-induced IFN-β expression in HEK293T cells.
Overexpression of signal molecules from the TLR3 and RLR pathways such as RIG-I, MDA5, TRIF, TBK1, and IKKε leads to activation of the IFN-β promoter (37–39). To find out which signal molecule in the IFN induction pathway was affected by vp13, we examined the induction of the IFN-β promoter with a luciferase reporter assay. HEK293T cells were transfected with RIG-I(N), MDA5(N), TRIF, TBK1, or IKKε along with vp13 or an empty-vector control. Among the molecules tested, IKKε overexpression induced the highest luciferase yield at 2,808-fold and TBK1 led to the lowest induction at 10-fold. Compared with the empty-vector control, the presence of vp13 significantly increased luciferase expression in cells transfected with N-terminal RIG-I or TBK1 by 5.94- or 6.82-fold, respectively (Fig. 2). Cotransfection of vp13 with the other signal molecules did not lead to a significant change in luciferase yield. This result suggested that vp13 may function at multiple steps within the RIG-I signaling pathway to enhance IFN promoter activation.
FIG 2.

vp13 enhances RIG-I-induced IFN-β expression. HEK293T cells were cotransfected with an IFN-β promoter luciferase reporter, vp13, and the stimulator RIG-I(N), TRIF, MDA5(N), TBK-1, or IKKε. An empty-vector (EV) vp13 plasmid was included as a control. At 24 h after transfection, cells were harvested for luciferase activity assay. Relative fold induction of luciferase activity is shown. Significant differences in IFN-β promoter activation between vp13 and EV are denoted by **, which indicates P < 0.01.
vp13 induces elevation of the endogenous RIG-I level.
To determine the mechanism by which vp13 enhances RIG-I-mediated IFN induction, we first tested whether vp13 affects RIG-I expression. While RIG-I was below the detection level in control HeLa cells, endogenous RIG-I was detected by immunoblot analysis in stable HeLa-VenusC1-vp13 cells (Fig. 3A). Likewise, transient expression of vp13 in HEK293T cells increased endogenous RIG-I expression to a detectable level (Fig. 3B). Similar results were obtained with S10-3, a cell line capable of supporting HEV replication. S10-3 cells transiently transfected with a VenusC1-vp13 plasmid had a considerably higher RIG-I level than cells transfected with the empty vector (Fig. 3C). These results indicated that vp13 expression led to an elevation of the basal level of RIG-I, which was likely one of the reasons for the enhancement of RIG-I-mediated IFN-β induction.
FIG 3.
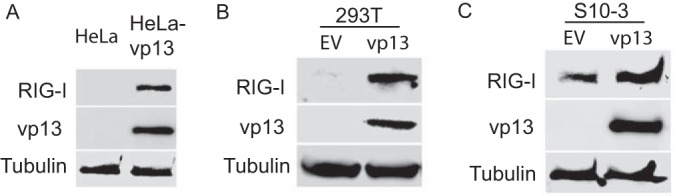
vp13 induces elevation of endogenous RIG-I protein. Western blotting with antibodies against RIG-I, vp13, and tubulin was conducted. (A) Highly elevated RIG-I protein level in stable HeLa-vp13 cells. Lysate of normal HeLa cells was included as a control. EV, empty vector. (B) Increased RIG-I protein level in HEK293T cells transiently transfected with a vp13 plasmid. Cells were harvested at 48 h after transfection. (C) Increased RIG-I protein level in S10-3 cells transiently transfected with a vp13 plasmid. Cells were harvested at 48 h after transfection.
vp13 expression extends the half-life of RIG-I.
The increased level of RIG-I in cells with vp13 expression could be due to either a higher level of transcription and/or translation or extension of the protein's half-life. To distinguish between these possibilities, HEK293T cells were transfected with VenusC1-vp13 or the empty vector and RIG-I mRNA levels were assessed by RT-qPCR. Similar RIG-I transcript levels were detected in the presence and absence of vp13 expression (Fig. 4A), which indicated that the RIG-I level elevation seen was not due to changes in mRNA levels.
FIG 4.
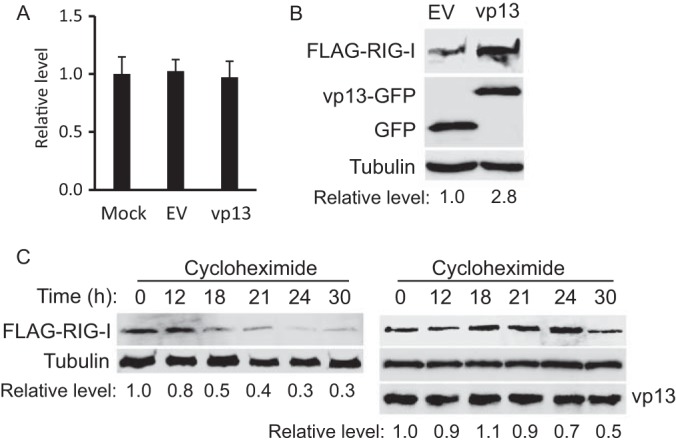
vp13 extends the half-life of RIG-I in HEK293T cells. (A) Stable RIG-I mRNA level in cells with vp13 expression. HEK293T cells were transfected with the empty vector (EV) or a vp13 plasmid and harvested at 24 h posttransfection. RT-qPCR was performed to detect RIG-I transcripts. (B) vp13 induces elevated expression of exogenous RIG-I. HEK293T cells were transfected with RIG-I and vp13 plasmids. Western blotting with antibodies against FLAG, GFP, and tubulin was conducted. (C) Extension of the half-life of RIG-I by vp13. HEK293T cells were transfected with FLAG-RIG-I and vp13 (right) or the empty vector (left). The cells were treated with cycloheximide 24 h after transfection and harvested at the times indicated above the lanes. Western blotting with antibodies against FLAG, tubulin, and vp13 was conducted.
We next assessed the effect of vp13 expression on the translation of RIG-I. HEK293T cells were transfected with a FLAG-RIG-I plasmid since endogenous RIG-I is barely detectable by Western blotting. The exogenous RIG-I protein level in cells with vp13 expression was 2.8-fold higher than that in cells transfected with the empty vector (Fig. 4B). These results suggest that vp13 acts at the protein level. We next examined the effect of vp13 on RIG-I protein stability. HEK293T cells were transfected with RIG-I and vp13 or the empty vector and treated with cycloheximide to block protein synthesis. This assay showed that the RIG-I level decreased 0.5-fold after 18 h of cycloheximide treatment in cells transfected with the empty vector but after 30 h of treatment in cells expressing vp13 (Fig. 4C). The half-life of RIG-I was extended from 18 h in cells with the empty vector to 30 h in cells with vp13 expression, indicating that vp13 extended the half-life of RIG-I.
vp13 interacts with the RIG-I N-terminal domain and increases its ubiquitination.
RIG-I is composed of two N-terminal caspase recruitment domains (CARDs), a central DexD/H box helicase/ATPase domain and a C-terminal regulatory domain (CTD) (40). N-terminal CARDs are responsible for binding to the adaptor molecule IPS-1 on mitochondria. In resting cells, the RIG-I CTD represses the interaction between CARDs of RIG-I and IPS-1 (41). As N-terminal CARDs of RIG-I interact with IPS-1 in the RLR pathway and ubiquitination leads to RIG-I activation (42), we tested whether vp13 would interact with RIG-I(N) or alter the ubiquitination status of RIG-I N-terminal CARDs. First we determined if vp13 increases RIG-I(N) expression in a way similar to that in which it increases full-length RIG-I protein expression. HEK293T cells were cotransfected with Myc-RIG-I(N) and vp13 plasmids. Western blotting results showed that cells expressing vp13 had a level of RIG-I(N) similar to that of cells with the empty vector (Fig. 5A), indicating that vp13 did not affect RIG-I(N) expression or stability.
FIG 5.
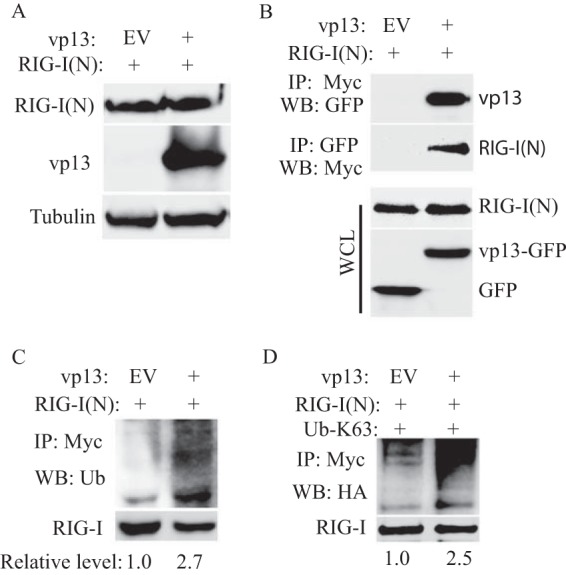
vp13 interacts with the RIG-I N-terminal domain and enhances its ubiquitination. (A) vp13 does not enhance the expression of RIG-I(N). HEK293T cells were transfected with vp13 and RIG-I(N) plasmids. Western blotting (WB) with antibodies against Myc, GFP, and tubulin was conducted. (B) IP indicates interaction of vp13 with RIG-I(N). HEK293T cells were transfected with vp13 and RIG-I(N) plasmids. The upper image shows the presence of vp13 in the RIG-I(N) IP complex. The second image from top shows the presence of RIG-I(N) in the vp13 IP complex. The lower two images shows the blotting of whole-cell lysate (WCL) with antibodies against Myc and GFP to detect RIG-I(N) and vp13, respectively. (C) Increased RIG-I(N) ubiquitination in HEK293T cells with vp13 expression. IP of HEK293T cell lysate was done with a Myc antibody. The upper image shows the presence of ubiquitin (Ub) in the IP complex. The lower image shows the RIG-I(N) protein pulled down. The relative level of the ubiquitination signal in the upper image is shown after normalization with RIG-I. (D) Increase in RIG-I(N) K-63 ubiquitination in HEK293T cells with vp13 expression. HEK293T cells were transfected with HA-Ubiquitin-K63, RIG-I(N), and vp13. EV, empty vector.
Next we tested whether vp13 interacts directly with RIG-I(N). HEK293T cells were cotransfected with Myc-RIG-I(N) and vp13 plasmids. IP with Myc antibody was conducted to pull down Myc-RIG-I(N), and Western blotting with GFP antibody showed the presence of vp13 in RIG-I(N) IP samples (Fig. 5B). Similarly, IP with GFP antibody was conducted to pull down GFP-vp13 and Western blotting with Myc antibody showed the presence of RIG-I(N) in vp13 IP samples. The expression of RIG-I(N) and vp13 was verified in whole-cell lysate (Fig. 5B). These results indicate that vp13 is capable of interacting with RIG-I(N). We reasoned that the direct interaction of vp13 and RIG-I(N) could induce RIG-I activation. IP was performed to pull down RIG-I(N), and then Western blotting with antibody against ubiquitin was conducted. The result showed that in cells with vp13 expression, the ubiquitinated RIG-I(N) level was significantly 2.7-fold higher than that in cells transfected with the empty vector (Fig. 5C). These results suggested that vp13 enhances RIG-I(N) ubiquitination, which may lead to an increase in IFN-β promoter activation.
It is known that lysine 63 (K63)-linked polyubiquitination of RIG-I by ubiquitin ligase TRIM25 and Riplet causes RIG-I activation (43–45). We reasoned that the vp13-enhanced ubiquitination of RIG-I should be K63 linked. HEK293T cells were transfected with plasmids expressing vp13, RIG-I(N), and HA-tagged K63-linked ubiquitin (HA-K63-ubi). IP of RIG-I(N) and Western blotting with an antibody against HA were conducted. The K63-linked ubiquitination of RIG-I in cells with vp13 expression was 2.5-fold greater than that in control cells (Fig. 5D). The results suggest that the presence of vp13 promoted RIG-I activation, as shown by K63-linked polyubiquitination.
The C-terminal domain of vp13 is sufficient to enhance RIG-I expression and poly(I · C)-mediated IFN production.
To determine which domain of vp13 is associated with the enhancement of IFN production, we constructed three vp13 truncation constructs, D1, D2, and D3 (Fig. 6A). D1 and D3 cover the N- and C-terminal halves of vp13, respectively, and D2 carries most of vp13 with a deletion at the C-terminal end. HEK293T cells were cotransfected with plasmids expressing full-length RIG-I and full-length vp13 or these three vp13 truncation constructs. Compared with the RIG-I levels in an empty-vector control, those in cells transfected with vp13, D1, D2, and D3 plasmids were 3.2-, 1.4-, 1.2-, and 2.8-fold higher, respectively (Fig. 6B). Full-length vp13 and D3 was detected with a vp13-specific antibody that was generated with a C-terminal peptide and a GFP antibody. The RIG-I level in cells with D3 was similar to that in cells with full-length vp13, which indicates that the C-terminal half of vp13 contains the domain responsible for the enhancement of poly(I · C)-induced IFN expression.
FIG 6.

The C-terminal domain of vp13 appears to correlate with enhancement of poly(I · C)-induced IFN-β expression. (A) Schematic illustration of cloning of vp13 fragments into a VenusC1 vector. The numbers above the lines are amino acid positions in vp13 (amino acids 1 to 114). (B) The C-terminal domain of vp13 correlates with RIG-I elevation. HEK293T cells were cotransfected with RIG-I and vp13 truncation plasmids. The empty vector (EV) was included as a control. Western blotting (WB) with antibodies against RIG-I, GFP, vp13, and tubulin was done. D1, D2, and D3 are vp13 deletion constructs. The vp13 antibody is specific to a C-terminal peptide and thus does not react with D1 and D2. (C) IFN-β reporter assay. HEK293T cells were cotransfected with RIG-I(N), IFN-β reporter, and vp13 plasmids. Cells were harvested for a luciferase activity assay 24 h after transfection. Luciferase levels relative to those of control cells transfected with the reporter plasmid or the empty vector are shown. Significant differences in IFN-β promoter activation from that in cells with the empty vector are denoted by **, which indicates P < 0.01.
To confirm this observation, we conducted an IFN-β reporter assay of HEK293T cells. The results showed that D3 increased IFN-β promoter activation to a level similar to that induced by full-length vp13, while D1 and D2 had a minimum effect (Fig. 6C). This indicated that the C-terminal domain of vp13 enhanced RIG-I-induced IFN expression.
The presence of vp13 in HEV-infected S10-3 cells leads to higher levels of poly(I · C)-induced IFN-β expression than those caused by the presence of an ORF3-null mutant.
The results described above showed that vp13 enhanced IFN induction. Therefore, we assessed whether vp13 plays a similar role during HEV replication. To address this question, we used S10-3 cells, a subclone of Huh-7 cells that supports HEV replication (13). The cells were transfected with full-length RNAs from HEV replicon pSK-E2 or pSK-E2-GFP. The GFP insertion in pSK-E2 interrupted ORF2 and ORF3 expression but offered a convenient indicator for direct observation of HEV replication. To determine the RIG-I protein level in S10-3 cells in the presence of vp13 expression, Western blotting was conducted. The result showed that the endogenous RIG-I level in S10-3 cells with pSK-E2 was considerably higher than that in cells with pSK-E2-GFP or pE2-Luc (Fig. 7A). Densitometry analysis showed that the RIG-I level in cells with pSK-E2 was 2.3-fold higher than that in cells with pSK-E2-GFP or pE2-Luc.
FIG 7.

Presence of vp13 in HEV-infected S10-3 cells (a subclone of Huh-7 cells) leads to higher IFN-β expression induced by poly(I · C) than in cells with vp13-null HEV. (A) Elevation of RIG-I level in HEV-infected cells with full-length HEV RNA. S10-3 cells were transfected with RNAs from pSK-E2, pSK-E2-GFP, and pE2-Luc replicons. Western blotting with antibodies against RIG-I, GFP, and tubulin was conducted 10 days after transfection. Relative levels of RIG-I after normalization with tubulin are shown below the lanes. Mock-infected cells were included for comparison. (B) IFN-β transcript detection by RT-qPCR in HEV-infected S10-3 cells. Cells were transfected with poly(I · C) to induce IFN expression. Fold induction of IFN-β mRNA in comparison with control cells without HEV RNA and poly(I · C) is shown. Significant differences between test and control samples are shown by * and **, which indicate P < 0.05 and P < 0.01, respectively. (C) Absence of vp13 in HEV-infected S10-3 cells leads to lower IFN-β transcript levels after poly(I · C) stimulation. (D) Detection of HEV replication in S10-3 cells. The left side of each image is an IFA of cells transfected with full-length HEV genomic RNA from pSK-E2 or pSK-E2Δ3. The red fluorescence on the left indicates the presence of HEV capsid protein, and the blue on the right shows nuclear DNA counterstaining with DAPI.
Transfection of cells with poly(I · C), which was expected to activate the RLR pathway, was conducted to induce IFN expression. Cells were harvested 12 h after poly(I · C) treatment for RNA isolation and RT-qPCR. The result showed that S10-3 cells transfected with pSK-E2 RNA had a 1.9-fold higher level of IFN-β transcripts than cells transfected with pSK-E2-GFP RNA in response to poly(I · C) transfection (Fig. 7B). Cells not transfected with poly(I · C) had a very low IFN-β transcript level, as expected.
These results suggested that the presence of vp13 in S10-3 cells enhanced poly(I · C)-induced IFN expression. To confirm this observation, we used an ORF3-null HEV replicon, pSKE2Δ3, in which a termination codon was introduced to stop ORF3 expression and that therefore did not express vp13 (16). The vp13-null mutant had a replication rate similar to that of the wide-type virus in S10-3 cells (46). Compared to S10-3 cells transfected with the wild-type pSKE2 replicon, cells transfected with the vp13-null pSKE2Δ3 mutant had significantly lower IFN-β transcript levels after poly(I · C) stimulation (Fig. 7C). An IFA with an antibody against the capsid protein confirmed similar HEV replication in S10-3 cells transfected with either wild-type or ORF3-null mutant genomic RNA (Fig. 7D). These data suggest that the presence of vp13 in HEV-infected cells elevates endogenous RIG-I, which subsequently enhances poly(I · C)-induced IFN expression.
Enhancement of IFN induction by vp13 appears to be genotype specific.
HEV genomic sequences are divergent. There are at least four genotypes of HEV across the world. The above-described experiments were done with vp13 from Sar55, a strain of genotype I HEV that can cause acute hepatitis. We wondered whether vp13 from strains of the other three genotypes would similarly enhance IFN production. ORF3 from strains of genotypes II, III, and IV were cloned. To determine the effect of vp13 on RIG-I(N)-induced IFN production, we performed an IFN-β promoter reporter assay. HEK293T cells were transfected with IFN-β promoter reporter and vp13 plasmids. The result showed that genotype III vp13 produced a RIG-I(N)-induced luciferase expression level 2.1-fold higher than that of cells with the empty vector (Fig. 8A). vp13 from the genotype II and IV HEV strains had slightly lower levels of luciferase activity than the empty-vector control. vp13 of genotype I enhanced luciferase activity, as expected.
FIG 8.

vp13 from HEV genotypes I and III enhance IFN induction. (A) Higher IFN-β promoter expression induced by vp13 from strains of HEV genotypes I and III. HEK293T cells were cotransfected with vp13 and IFN-β promoter reporter plasmids for a luciferase reporter assay. Relative luciferase levels are shown. Significant differences from cells transfected with the empty vector (EV) are shown by *, which indicates P < 0.05. (B) RIG-I elevation induced by vp13 from strains of HEV genotypes I and III. HEK293T cells were cotransfected with vp13 and RIG-I plasmids. Western blotting with antibodies against FLAG, GFP, and tubulin was conducted. (C) Alignment of the vp13 amino acid sequences of the four genotypes. Genotype I, GenBank accession number AF444002; genotype II, accession number M74506; genotype III, accession number HQ709170; genotype IV, accession number AB074915. Residues that are the same as those in the consensus sequence are represented by dots.
RIG-I protein levels in HEK293T cells cotransfected with full-length RIG-I and one of the four vp13 plasmids were determined by Western blotting. The results showed that compared with cells with the empty vector, cells with vp13 of the genotype III strain had a 2.4-fold higher level of RIG-I expression, which was similar to that of cells transfected with vp13 from the genotype I strain (Fig. 8B). Blotting with a GFP antibody confirmed similar levels of expression of vp13 from the four genotypes in the cells. However, cells with vp13 from the genotype II and IV HEV strains led to minimal RIG-I level changes. The results indicate that vp13 from the strains of different genotypes had variable effects on the RLR pathway.
Amino acid sequence alignment of HEV vp13 of the four genotypes shows that there are more variations in the C-terminal domain than in the N-terminal domain (Fig. 8C). As the C-terminal domain correlated with enhancement of IFN expression, variation in the C-terminal sequence could mean functional divergence. The amino acid sequence identity is 86% between genotypes I and II, 79% between genotypes I and III, and 78% between genotypes I and IV. Together, these data show that vp13 enhancement of IFN induction appears to be genotype or strain specific.
DISCUSSION
This study demonstrated that HEV vp13 enhances poly(I · C)-induced IFN expression by increasing the RIG-I protein level, interacting with RIG-I(N), and increasing RIG-I(N) ubiquitination. The vp13-mediated elevation of the RIG-I protein level is possibly due to the extension of the half-life of RIG-I. The C-terminal domain of vp13 was found to be sufficient for the enhancement of IFN induction. A vp13-mediated enhancement of IFN induction was also observed in HEV-infected S10-3 liver cells.
We observed that poly(I · C) treatment of stable HeLa-vp13 cells induced stronger inhibition of NDV-GFP replication than poly(I · C)-treated normal HeLa cells. This result indicates that vp13 plays a role in enhancing the poly(I · C)-induced antiviral response. Direct addition of poly(I · C) to cultured cells activates the TLR3 pathway, and transfection of the cells activates the RLR pathway. In our experiment, direct addition of poly(I · C) induced an IFN-β expression level lower than that induced by transfection. Therefore, we used the transfection method in this study to delineate the mechanism of vp13 enhancement of IFN induction. In addition, our results also provide potential answers to the vp13 augmentation of the poly(I · C)-induced TLR3 pathway; the presence of vp13 enhances TBK1-induced IFN-β expression, as TLR3 activation induces phosphorylation of IRF3 by TBK1.
Our screening of signal molecules in the IFN induction pathways showed that vp13 could enhance IFN induction by RIG-I and TBK1. vp13-enhanced RIG-I signaling was examined in this study. The mechanism of vp13 enhancement of IFN induction via TBK1 is unknown. Upregulation of TBK1 expression by vp13 is a possible but unlikely reason because IFN induction by upstream molecules such as TRIF and MDA5(N) was not augmented by vp13 cotransfection.
The mechanism of vp13-mediated enhancement of RIG-I induction of IFN was delineated in this study. Elevation of the basal level of RIG-I was discovered in stable HeLa-vp13 cells and transiently transfected HEK293T and S10-3 cells. The increase in the level of RIG-I in S10-3 cells could be more meaningful, as they are liver-derived, HEV-susceptible cells.
The upregulation of RIG-I appears to be due mainly to vp13-mediated extension of the half-life of the protein. The presence of vp13 extends the half-life of exogenous RIG-I from 18 to 30 h. The possible mechanisms of this extension might be inhibition of RIG-I degradation by the ubiquitin-proteasome pathway or increased protein translation. The former speculation seems more reasonable, as cycloheximide treatment blocks protein translation. The observations that RIG-I(N) expression was not affected by vp13 and that the half-life of the full-length RIG-I protein was extended in the presence of vp13 are consistent with this speculation.
The other reason for vp13 enhancement of IFN induction could be that vp13 interacts with RIG-I(N) and enhances its ubiquitination. RIG-I(N) IP pulled down vp13, and vp13 IP pulled down RIG-I(N). As RIG-I(N) contains the CARDs to interact with IPS-1 to transmit the signal to induce IFN production, the upregulation of RIG-I(N) ubiquitination by vp13 further enhances signaling. Two ubiquitin ligases, TRIM25 (43, 44) and Riplet (45), mediate the K63-linked polyubiquitination of RIG-I, leading to its interaction with IPS-1. This study shows that vp13 increases K63-linked RIG-I(N) polyubiquitination. The increase in K63-linked polyubiquitination is consistent with vp13 enhancement of RIG-I(N) activation. As the RIG-I CTD interacts with N-terminal CARDs in resting cells, vp13 is not able to interact with the whole RIG-I protein. Once the N-terminal CARDs are exposed after RIG-I undergoes a conformational change when the helicase domain of the RIG-I protein senses viral RNA (41), vp13 would be able to interact with the CARDs and further augment activation signaling.
The active domain of vp13 in this function is located in the C-terminal portion of the protein, as identified by truncation analysis. The C-terminal part of vp13 contains a proline-rich sequence that interacts with the Src homology 3 domain of cellular proteins (47). The PSAP motif of avian HEV was found to play a role in virus release in vivo though not to be essential for virus infectivity (48). We constructed vp13 with PSAP mutations, and our preliminary study with these mutant proteins indicates that the PSAP motif appears not to correlate with vp13 enhancement of IFN induction (data not shown). Further study is needed to identify the active motif involved in the enhancement of IFN induction found in this study. Our finding is consistent with a multifunctional character of vp13.
The vp13-mediated enhancement of IFN induction appears to be true in HEV-infected hepatoma cells, as poly(I · C) induced higher levels of IFN-β mRNA in HEV-infected S10-3 cells with vp13 expression. The elevation of the RIG-I protein level in HEV-infected S10-3 cells expressing vp13 further substantiates this observation and is consistent with the finding that vp13 increases RIG-I protein levels in stably or transiently transfected cells. Compared to ORF3-null-mutant-infected S10-3 cells, in HEV-infected S10-3 cells, the presence of vp13 caused a significant increase in the IFN-β mRNA level induced by poly(I · C). The small magnitude of this change could be due to the low rate of HEV-positive cells and virus-mediated inhibition of IFN induction. We noticed that the HEV ORF1 product inhibited poly(I · C)-induced IFN production (submitted for publication). Therefore, there appears to be a balance between IFN induction and inhibition, such that the vp13-induced enhancement might be under the control of other viral products. In addition to enhancement of IFN induction, vp13 also enhances RIG-I-mediated NF-κB promoter activation and leads to the expression of NF-κB-activated cytokines in stable HeLa-vp13 cells stimulated with poly(I · C) (data not shown). Among the cytokines elevated, some are proinflammatory and may contribute to HEV-mediated inflammation and pathogenesis during HEV infection. A recent report showed that higher levels of TNF-α, IL-6, IFN-γ, and TGF-β1, as well as a higher HEV viral load, were seen in pregnant women with acute viral hepatitis and fulminant hepatic failure (25).
It is generally expected that a virus would inhibit IFN induction and signaling to gain time for its own replication. Why vp13, a viral protein, enhances RIG-I-mediated IFN production is an intriguing question. In addition to the possibilities explained above, vp13-mediated enhancement of IFN induction could possibly play a role in the context of HEV infection, for example, stages of virus replication. We hypothesize that during early stages of HEV infection, ORF1 is more strongly expressed and IFN induction is inhibited and that during late stages, viral RNA replication is completed and ORF3 expression is increased to promote HEV egress and spreading to other cells.
Our data also showed that vp13-mediated IFN enhancement is different among the four HEV genotypes. vp13 from a genotype III strain is also able to enhance IFN induction in our study, but a recent report showed that vp13 from a strain of genotype III inhibits IFN-α-activated signaling in stably HEV-infected A549 cells (49). vp13 of genotype I strains appears to have no such effect on IFN-activated signaling because NDV-GFP replication was inhibited in stable HeLa-vp13 cells after poly(I · C) treatment. This inconsistency might be due to the different strains tested and the different cells used in our study.
On the other hand, the finding that vp13 enhances IFN induction would be useful in different applications. For example, the C-terminal domain of vp13 can be inserted into the genomes of attenuated live viruses to induce a stronger innate immune response for better protective immunity. Insertion of the C-terminal domain of vp13 into the recombinant virus also makes vp13 act as a marker for differentiation from wild-type virus infection. Further characterization of vp13 and its active domain in this application is warranted.
ACKNOWLEDGMENTS
We thank Suzanne Emerson at NIH, X.-.J. Meng at Virginia Tech University, Takashi Fujita of Kyoto University, and Tom Maniatis of Columbia University for some of the plasmids and antibodies used in this study.
Y. Nan, R. Wang, Z. Xu, and Y. Yu were partially supported by the China Scholarship Council. This study was partially supported by the NIH (012893) and the University of Maryland.
Footnotes
Published ahead of print 21 May 2014
REFERENCES
- 1.Emerson S, Anderson D, Arankalle VA, Meng X-J, Purdy M, Schlauder GG, Tsarev S. 2004. Virus taxonomy, VIIIth report of the ICTV. ICTV, London, United Kingdom [Google Scholar]
- 2.Jameel S. 1999. Molecular biology and pathogenesis of hepatitis E virus. Expert Rev. Mol. Med. 1999:1–16 [DOI] [PubMed] [Google Scholar]
- 3.Purcell RH, Emerson SU. 2001. Hepatitis E virus, p 3051–3061 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 4th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 4.Nelson KE, Kmush B, Labrique AB. 2011. The epidemiology of hepatitis E virus infections in developed countries and among immunocompromised patients. Expert Rev. Anti Infect. Ther. 9:1133–1148. 10.1586/eri.11.138 [DOI] [PubMed] [Google Scholar]
- 5.Meng XJ. 2011. From barnyard to food table: the omnipresence of hepatitis E virus and risk for zoonotic infection and food safety. Virus Res. 161:23–30. 10.1016/j.virusres.2011.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wedemeyer H, Pischke S, Manns MP. 2012. Pathogenesis and treatment of hepatitis E virus infection. Gastroenterology 142:1388–1397 e1381. 10.1053/j.gastro.2012.02.014 [DOI] [PubMed] [Google Scholar]
- 7.Meng XJ. 2010. Recent advances in hepatitis E virus. J. Viral Hepat. 17:153–161. 10.1111/j.1365-2893.2009.01257.x [DOI] [PubMed] [Google Scholar]
- 8.Ahmad I, Holla RP, Jameel S. 2011. Molecular virology of hepatitis E virus. Virus Res. 161:47–58. 10.1016/j.virusres.2011.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panda SK, Ansari IH, Durgapal H, Agrawal S, Jameel S. 2000. The in vitro-synthesized RNA from a cDNA clone of hepatitis E virus is infectious. J. Virol. 74:2430–2437. 10.1128/JVI.74.5.2430-2437.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emerson SU, Zhang M, Meng XJ, Nguyen H, St Claire M, Govindarajan S, Huang YK, Purcell RH. 2001. Recombinant hepatitis E virus genomes infectious for primates: importance of capping and discovery of a cis-reactive element. Proc. Natl. Acad. Sci. U. S. A. 98:15270–15275. 10.1073/pnas.251555098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emerson SU, Nguyen H, Graff J, Stephany DA, Brockington A, Purcell RH. 2004. In vitro replication of hepatitis E virus (HEV) genomes and of an HEV replicon expressing green fluorescent protein. J. Virol. 78:4838–4846. 10.1128/JVI.78.9.4838-4846.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graff J, Nguyen H, Kasorndorkbua C, Halbur PG, St Claire M, Purcell RH, Emerson SU. 2005. In vitro and in vivo mutational analysis of the 3′-terminal regions of hepatitis E virus genomes and replicons. J. Virol. 79:1017–1026. 10.1128/JVI.79.2.1017-1026.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shukla P, Nguyen HT, Torian U, Engle RE, Faulk K, Dalton HR, Bendall RP, Keane FE, Purcell RH, Emerson SU. 2011. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus-host recombinant. Proc. Natl. Acad. Sci. U. S. A. 108:2438–2443. 10.1073/pnas.1018878108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tam AW, Smith MM, Guerra ME, Huang CC, Bradley DW, Fry KE, Reyes GR. 1991. Hepatitis E virus (HEV): molecular cloning and sequencing of the full-length viral genome. Virology 185:120–131. 10.1016/0042-6822(91)90760-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kannan H, Fan S, Patel D, Bossis I, Zhang YJ. 2009. The hepatitis E virus open reading frame 3 product interacts with microtubules and interferes with their dynamics. J. Virol. 83:6375–6382. 10.1128/JVI.02571-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graff J, Nguyen H, Yu C, Elkins WR, St Claire M, Purcell RH, Emerson SU. 2005. The open reading frame 3 gene of hepatitis E virus contains a cis-reactive element and encodes a protein required for infection of macaques. J. Virol. 79:6680–6689. 10.1128/JVI.79.11.6680-6689.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang YW, Opriessnig T, Halbur PG, Meng XJ. 2007. Initiation at the third in-frame AUG codon of open reading frame 3 of the hepatitis E virus is essential for viral infectivity in vivo. J. Virol. 81:3018–3026. 10.1128/JVI.02259-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heil F, Ahmad-Nejad P, Hemmi H, Hochrein H, Ampenberger F, Gellert T, Dietrich H, Lipford G, Takeda K, Akira S, Wagner H, Bauer S. 2003. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur. J. Immunol. 33:2987–2997. 10.1002/eji.200324238 [DOI] [PubMed] [Google Scholar]
- 19.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr 1998. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell 2:253–258. 10.1016/S1097-2765(00)80136-7 [DOI] [PubMed] [Google Scholar]
- 20.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. 2003. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424:743–748. 10.1038/nature01889 [DOI] [PubMed] [Google Scholar]
- 21.Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131–137. 10.1038/ni1303 [DOI] [PubMed] [Google Scholar]
- 22.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988. 10.1038/ni1243 [DOI] [PubMed] [Google Scholar]
- 23.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–496. 10.1038/ni921 [DOI] [PubMed] [Google Scholar]
- 24.Sharma S, ten Oever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300:1148–1151. 10.1126/science.1081315 [DOI] [PubMed] [Google Scholar]
- 25.Kumar A, Devi SG, Kar P, Agarwal S, Husain SA, Gupta RK, Sharma S. 2014. Association of cytokines in hepatitis E with pregnancy outcome. Cytokine 65:95–104. 10.1016/j.cyto.2013.09.022 [DOI] [PubMed] [Google Scholar]
- 26.Lei X, Liu X, Ma Y, Sun Z, Yang Y, Jin Q, He B, Wang J. 2010. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 84:8051–8061. 10.1128/JVI.02491-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Jr, Akira S, Yonehara S, Kato A, Fujita T. 2005. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 175:2851–2858. 10.4049/jimmunol.175.5.2851 [DOI] [PubMed] [Google Scholar]
- 28.Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT. 2003. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the Toll adapters TRAM and TRIF. J. Exp. Med. 198:1043–1055. 10.1084/jem.20031023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, Dawson VL, Dawson TM. 2005. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci. 25:2002–2009. 10.1523/JNEUROSCI.4474-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang YJ, Wang KY, Stein DA, Patel D, Watkins R, Moulton HM, Iversen PL, Matson DO. 2007. Inhibition of replication and transcription activator and latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus by morpholino oligomers. Antiviral Res. 73:12–23. 10.1016/j.antiviral.2006.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nan Y, Wang R, Shen M, Faaberg KS, Samal SK, Zhang YJ. 2012. Induction of type I interferons by a novel porcine reproductive and respiratory syndrome virus isolate. Virology 432:261–270. 10.1016/j.virol.2012.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, Si-Tahar M. 2007. Cutting edge: influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J. Immunol. 178:3368–3372. 10.4049/jimmunol.178.6.3368 [DOI] [PubMed] [Google Scholar]
- 33.Patel D, Opriessnig T, Stein DA, Halbur PG, Meng XJ, Iversen PL, Zhang YJ. 2008. Peptide-conjugated morpholino oligomers inhibit porcine reproductive and respiratory syndrome virus replication. Antiviral Res. 77:95–107. 10.1016/j.antiviral.2007.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 35.Patel D, Nan Y, Shen M, Ritthipichai K, Zhu X, Zhang YJ. 2010. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J. Virol. 84:11045–11055. 10.1128/JVI.00655-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, Volchkov VE, Nichol ST, Basler CF. 2006. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J. Virol. 80:5156–5167. 10.1128/JVI.02349-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallatin WM, Gale MJ, Jr, Hoffman PA, Willerford DM, Draves KE, Benveniste RE, Morton WR, Clark EA. 1989. Selective replication of simian immunodeficiency virus in a subset of CD4+ lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 86:3301–3305. 10.1073/pnas.86.9.3301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu LL, Puri M, Horvath CM, Sen GC. 2008. Select paramyxoviral V proteins inhibit IRF3 activation by acting as alternative substrates for inhibitor of kappaB kinase epsilon (IKKe)/TBK1. J. Biol. Chem. 283:14269–14276. 10.1074/jbc.M710089200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beura LK, Sarkar SN, Kwon B, Subramaniam S, Jones C, Pattnaik AK, Osorio FA. 2010. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J. Virol. 84:1574–1584. 10.1128/JVI.01326-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730–737. 10.1038/ni1087 [DOI] [PubMed] [Google Scholar]
- 41.Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M., Jr 2007. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. U. S. A. 104:582–587. 10.1073/pnas.0606699104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920. 10.1038/nature05732 [DOI] [PubMed] [Google Scholar]
- 43.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920. 10.1038/nature05732 [DOI] [PubMed] [Google Scholar]
- 44.Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang C, Cui S, Myong S, Ha T, Hopfner KP, Jung JU. 2008. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc. Natl. Acad. Sci. U. S. A. 105:16743–16748. 10.1073/pnas.0804947105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. 2009. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 284:807–817. 10.1074/jbc.M804259200 [DOI] [PubMed] [Google Scholar]
- 46.Emerson SU, Nguyen H, Torian U, Purcell RH. 2006. ORF3 protein of hepatitis E virus is not required for replication, virion assembly, or infection of hepatoma cells in vitro. J. Virol. 80:10457–10464. 10.1128/JVI.00892-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korkaya H, Jameel S, Gupta D, Tyagi S, Kumar R, Zafrullah M, Mazumdar M, Lal SK, Xiaofang L, Sehgal D, Das SR, Sahal D. 2001. The ORF3 protein of hepatitis E virus binds to Src homology 3 domains and activates MAPK. J. Biol. Chem. 276:42389–42400. 10.1074/jbc.M101546200 [DOI] [PubMed] [Google Scholar]
- 48.Kenney SP, Pudupakam RS, Huang YW, Pierson FW, LeRoith T, Meng XJ. 2012. The PSAP motif within the ORF3 protein of an avian strain of the hepatitis E virus is not critical for viral infectivity in vivo but plays a role in virus release. J. Virol. 86:5637–5646. 10.1128/JVI.06711-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dong C, Zafrullah M, Mixson-Hayden T, Dai X, Liang J, Meng J, Kamili S. 2012. Suppression of interferon-alpha signaling by hepatitis E virus. Hepatology 55:1324–1332. 10.1002/hep.25530 [DOI] [PubMed] [Google Scholar]