

ABSTRACT
Viruses are dependent on their host cells for replication and thus have evolved in intimate association with them. The identification of host factors required for viral infection has led to advances in both viral and cellular biology. Vesicular stomatitis virus (VSV), a negative-sense RNA virus, replicates in all eukaryotic cells in culture, suggesting that the host requirements for its replication are ubiquitous. In this study, we performed a genome-wide small interfering RNA screen of human cells in culture and identified multiple cellular genes that influence the entry and replication of VSV. From a list of >300 genes, we selected the most promising candidates to perform further analysis to assign their functions to either the entry or intracellular replication step of infection. We implicate 3 new factors in VSV entry and 20 new factors in viral gene expression. These proteins have diverse cellular roles, including S-adenosylmethionine synthesis, respiration, and host translation machinery, underscoring the intimate relationship between VSV and the host cell. Together, these results provide a curated list of genes required for VSV replication.
IMPORTANCE Replication of vesicular stomatitis virus (VSV) has long served as a model for understanding host-virus interactions and neuropathogenesis. We performed a genome-wide analysis of host factors and revealed genes critical for viral replication, including some involved in vesicular trafficking, cell cycling, and protein modification. Our results provide an enriched list of host factors that are required for specific stages of VSV entry and gene expression. This study may also potentially expand the repertoire of targets for antiviral therapy against negative-strand RNA viruses.
INTRODUCTION
As obligate intracellular parasites, viruses are dependent on host cell factors for each step of replication. Developing a mechanistic understanding of a viral life cycle reveals knowledge of both viral replication and the functions of specific cellular proteins. For example, studies of virus-host interactions have provided significant insights into essential cellular pathways, including splicing and translation (1). Vesicular stomatitis virus (VSV), an enveloped, nonsegmented, negative-strand (NNS) RNA virus in the family Rhabdoviridae, has an extremely broad host range. The high titers of VSV obtained from replication in cell culture helped to ensure VSV's position as an important prototype for studying the biology of other, less tractable NNS RNA viruses.
The VSV genome encodes five viral proteins: nucleocapsid (N), phosphoprotein (P), matrix (M), glycoprotein (G), and large polymerase (L). VSV entry begins with the attachment of the virion, which leads to internalization of the particle via an altered mode of clathrin-dependent endocytosis that depends upon actin polymerization (2, 3). In the endocytic pathway, the virus encounters an acidic pH, which triggers conformational rearrangements in G that drive fusion of the viral and cellular membranes. Fusion releases into the cytoplasm a ribonucleoprotein (RNP) core comprising the N-encased genomic RNA and the viral L-P polymerase complex, and this process is accompanied by the release of the M protein. Numerous questions remain, however, concerning the roles of host factors in the entry pathway, including the use of attachment or receptor molecules, such as phosphatidylserine or the low-density lipoprotein receptor (4, 5), the precise details of the endocytic route (2, 3, 6), and whether host factors are required for M release (7).
Once the RNP is released into the cytoplasm, the polymerase complex, consisting of L, P, and N-coated genomic RNA, begins the primary round of transcription, capping, methylation, and polyadenylation of viral mRNAs (8, 9). The viral mRNAs are translated, and the resulting proteins initiate genomic replication, which provides templates for secondary transcription. The viral proteins and genome are assembled at the plasma membrane, from which new virions bud. While transcription requires only viral components, how viral mRNAs are recruited to the translation machinery remains uncharacterized, as infection inactivates the canonical cap-binding complex (10). Furthermore, it is not understood how VSV is able to coexist in the cell in the context of antiviral and stress responses. Identifying host factors required for VSV replication may answer the numerous questions remaining about virus replication and also illuminate the cellular roles of these proteins.
With the development of genome-wide small interfering RNA (siRNA) libraries, it is possible to uncover host factors required for viral replication in a relatively unbiased way. Genome-wide siRNA screens have been performed with many viruses, including human immunodeficiency virus and influenza A virus. Aside from increasing our knowledge about virus biology, these screens have even uncovered new functions for previously uncharacterized proteins, such as the antiviral family of IFITM (interferon-induced transmembrane protein) proteins (11–14). Previously, we utilized a targeted siRNA screen in HeLa cells to identify ribosomal proteins required for viral translation (15). Extensive analysis of one of our top hits, the ribosomal protein rpL40, revealed an unprecedented role of a large-ribosomal-subunit protein in transcript-specific translation (15). We now report the results of a genome-wide siRNA screen that identifies additional host factors required for VSV replication. We validated selected hits and specifically isolated the roles of host factors in VSV entry or viral gene expression. Together, our results reveal the highly intricate virus-host dynamics and provide important insight into the mechanistic action of select host factors.
MATERIALS AND METHODS
siRNA screen.
SMART pools (Dharmacon), comprising four duplexes targeting a single human mRNA transcript, or single duplexes were individually arrayed into wells of black, clear-bottom 384-well plates (Costar; Corning) containing a 1:100 dilution of Lipofectamine 2000 (Invitrogen) in Opti-MEM medium (Invitrogen). Duplexes and lipids were incubated for 20 min at room temperature and mixed with HeLa cells to yield final concentrations of 5 × 104 cells ml−1 and 25 nM siRNA. Plates were seeded with 1,250 HeLa cells per well, and cells were centrifuged for 5 min at 700 × g. At 48 h posttransfection (hpt), the approximately 5,000 cells were inoculated with recombinant VSV expressing enhanced green fluorescent protein (rVSV-EGFP) at a multiplicity of infection (MOI) of 1. Cells were fixed 7 h later with 2% formaldehyde in phosphate-buffered saline (PBS), the nuclei were counterstained with 4 μg ml−1 Hoechst nuclear dye (Invitrogen) for 10 min at room temperature, and unincorporated dye was removed by washing once with 60 μl PBS per well. Individual wells were examined using a cellWoRX High Content cell analysis system (Applied Precision Inc.), and the cell-scoring module of MetaXpress software (Molecular Devices) was employed to quantify the total number of cells and the percentage of EGFP-positive cells. All transfections were performed in duplicate.
Cells and viruses.
HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (Tissue Culture Biologicals). VSV, rVSV-EGFP, and rVSV-Ren-P were amplified in BHK-21 cells (ATCC) and purified through a 10% (wt/vol) sucrose cushion prepared in NTE (10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA), and virus stocks were stored in NTE at −80°C (16).
siRNA transfection.
siGENOME nontargeting siRNA 3 was utilized as the control siRNA (Dharmacon). For siRNA transfection in a 24-well plate, 1 μl of Lipofectamine 2000 was mixed with 100 μl of Opti-MEM and incubated at room temperature for 5 min. One hundred microliters of Opti-MEM and 2 μl of 20 μM siRNA were added, mixed, and incubated at room temperature for 15 min. A total of 3 × 104 trypsinized HeLa cells in 400 μl of DMEM with 10% fetal bovine serum was added to the lipid-siRNA mix and then plated, and experiments were performed 48 h later. This protocol was scaled accordingly for different cell culture surface areas.
RNP transfection.
To isolate RNPs, purified rVSV-Ren-P was mixed with 12.5 mM Tris, pH 7.4, 5% glycerol, 5 mM EDTA, pH 8, 3.5 mM dithiothreitol (DTT), 0.1% Triton-X, and 500 mM CsCl in a total volume of 600 μl and incubated on ice for 1.25 h. Six hundred microliters of 20 mM Tris, pH 7.4, and 3.5 mM DTT was added and loaded onto a 30 to 50% (vol/vol) glycerol gradient made in NTE with 3.5 mM DTT. Gradients were spun for 3.5 h at 4°C at 45,000 rpm in an SW50.1 rotor, and the pellet was resuspended in NTE. A total of 0.4 μg of VSV-Ren-P RNPs was transfected into HeLa cells by use of Lipofectamine 2000, and luciferase activity was assayed at 7 hpt.
Protein analysis.
At the indicated times postinfection or post-mock infection, cells were lysed in Rose lysis buffer (1% [vol/vol] Nonidet P-40 alternative, 0.4% [vol/vol] sodium deoxycholate, 66 mM EDTA, 10 mM Tris, pH 7.4), and equal amounts of total cytoplasmic protein were separated in a low-bis 10% polyacrylamide gel. Western blot analysis was performed using anti-actin (Chemicon) (1:5,000) and anti-VSV matrix (23H12; 1:10,000) antibodies.
RESULTS
Genome-wide siRNA screen identifies host factors required for VSV replication.
To employ an unbiased approach to determine host factors that are required for vesicular stomatitis virus entry and replication, we developed an siRNA screen specifically optimized to monitor a single cycle of viral infection (Fig. 1). In this screen, a recombinant VSV that expressed the reporter EGFP was used to infect HeLa cells that were transfected 48 h earlier with siRNA. We chose to infect cells at an MOI of 1, which results in approximately 50% of cells being infected. At 7 h postinfection (hpi), cells were fixed in 2% paraformaldehyde and the nuclei were stained with Hoechst stain to permit measurement of the effect of siRNA transfection on cell number. Fluorescence from EGFP and the Hoechst-stained nuclei were imaged separately using an autoscope. The collected images were analyzed to derive the number of cells per well, the percentage of cells that were EGFP positive, and the average intensity of the EGFP signal.
FIG 1.
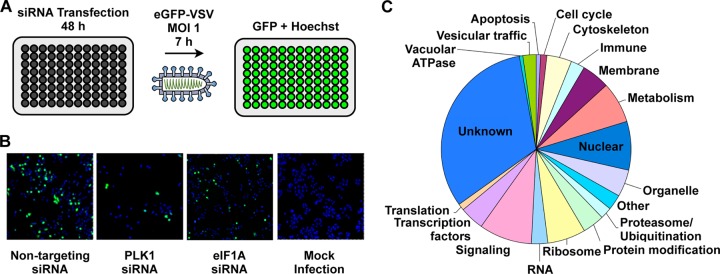
Genome-wide siRNA screen. (A) Schematic of siRNA screen. (B) Representative images of controls utilized during siRNA screen. Cells were treated with a nontargeting, PLK1-targeting, or eIF1A-targeting siRNA and infected with EGFP-VSV. Mock-infected cells are also shown. (C) Biological processes of candidate factors required for VSV replication.
As a positive control, we selected a pool of 4 siRNAs targeting eukaryotic translation initiation factor 1A, X-linked (eIF1AX). This essential protein is required for binding of the ribosomal 43S complex to the 5′ end of capped RNA. Transfection of cells with eIF1AX siRNA consistently diminished the percentage of EGFP-positive cells to 50% (± 9%) of the number of control treated cells that received an irrelevant siRNA that targeted firefly luciferase (Fig. 1). To determine the overall fraction of cells that received the siRNA, control wells were treated with an siRNA targeting polo-like kinase 1 (PLK1), a gene essential for cell viability, which reduced cell viability to 35% (± 10%) (Fig. 1).
Using this approach, we performed a genome-wide screen using an siRNA library that comprised 21,121 pools of four chemically synthesized siRNAs targeting each gene. Each gene was screened in duplicate. A gene was considered a candidate hit if the percentage of EGFP-positive cells or the mean fluorescence intensity was altered >3.0 standard deviations from the mean EGFP signal per plate (see Fig. S1 in the supplemental material). As controls, we eliminated from our list of candidate hits the siRNAs that altered cell viability >3.0 standard deviations, as judged by Hoechst staining of cell nuclei. Biological process analysis revealed that candidate genes fell into 18 primary categories, including ones that would likely be implicated in viral entry, such as vesicular trafficking, and ones involved in intracellular replication, such as protein-modifying factors (Fig. 1).
For subsequent analyses, we excluded all ribosomal genes and translation factors for a separate analysis (15), selected 500 of the remaining genes, and screened the four siRNAs of each pool separately. Of the 450 genes that when silenced reduced VSV replication, 305 were confirmed with 1 or more siRNAs (Fig. 2). Representative images are shown in Fig. 2. Specifically, 7 genes were confirmed with each of the 4 siRNAs, 9 were confirmed with 3 siRNAs, 73 with 2 siRNAs, and 216 with 1 siRNA. Thirty-two percent of the genes were not confirmed by this analysis (Fig. 2).
FIG 2.
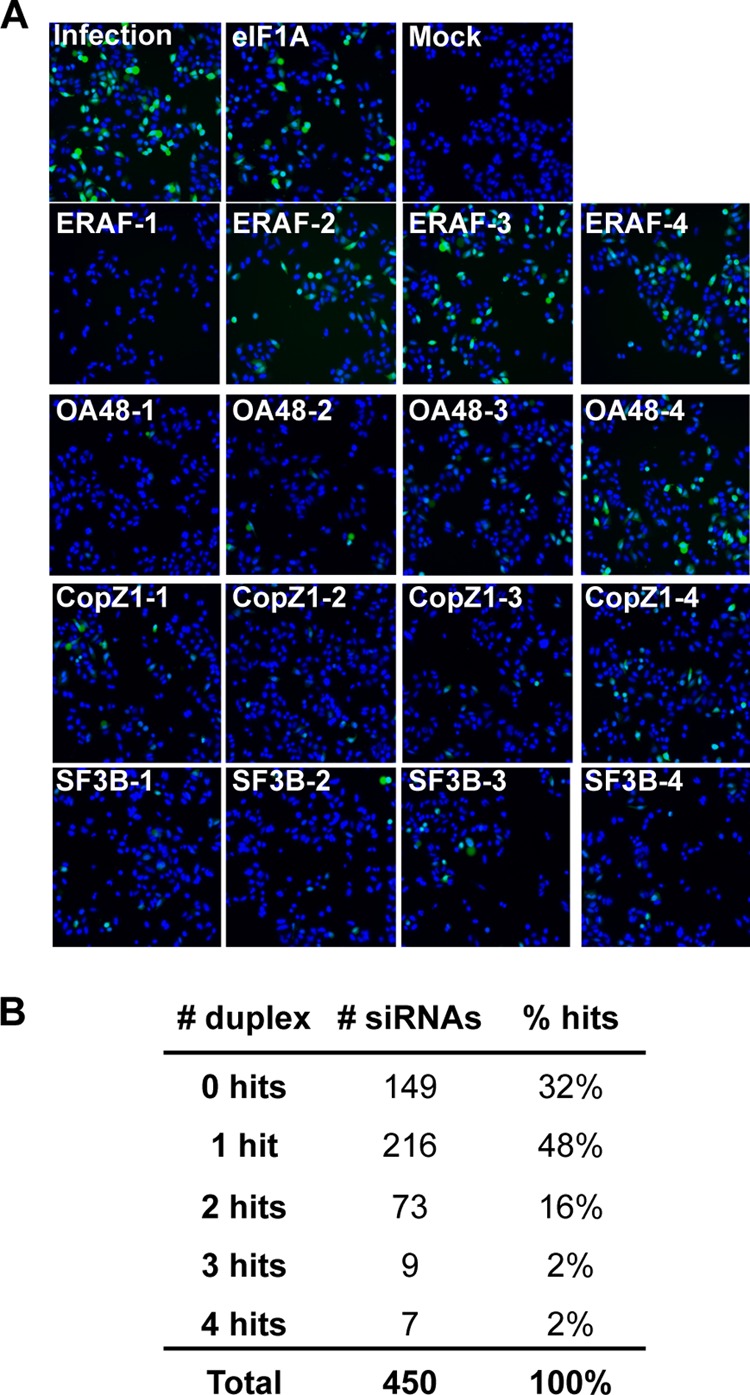
Secondary siRNA screen of candidate host proteins required for VSV replication. (A) Representative images of secondary siRNA screen. Cells were treated with four individual duplexes targeting each candidate and infected with EGFP-VSV. (B) Numbers of candidates (# siRNAs) whose knockdown by 0, 1, 2, 3, and 4 separate siRNAs inhibited VSV replication.
Impact of host gene silencing on endogenous gene expression.
To begin to understand the roles of host genes in VSV replication, we selected a subset of 29 genes for further investigation (see Fig. S2 in the supplemental material). We specifically focused on host factors whose knockdown had the largest effect on viral replication and the least effect on cell viability. In a 24-well format, cells were transfected with individual siRNA duplexes and subsequently infected with rVSV-EGFP at an MOI of 1. As a control, cells were transfected with a nontargeting siRNA designed to not target any human genes. In this format, 23 of the 29 genes selected were validated to be essential for VSV replication (Fig. 3).
FIG 3.
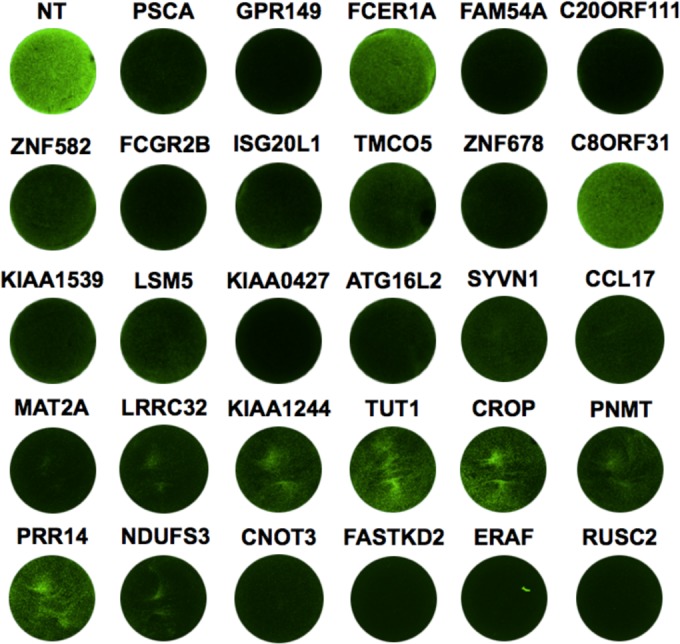
Tertiary screen for host genes involved in VSV replication. The images show VSV gene expression after siRNA transfection. HeLa cells were treated with a 50 nM concentration of a nontargeting (NT) siRNA or a single siRNA targeting the indicated gene. Forty-eight hours after transfection, cells were infected with rVSV-EGFP at an MOI of 1. Images were taken using a Typhoon fluorescence imager at 5 hpi.
As our initial and follow-up screens relied on EGFP fluorescence to indirectly measure viral replication, we next measured the direct effects of candidate gene silencing on endogenous viral gene expression. Following transfection with the indicated siRNA, cells were infected with wild-type VSV, cytoplasmic extracts were harvested at 7 hpi, and levels of M protein were determined by immunoblotting (Fig. 4A). For 23 of the 29 genes, we demonstrated that the individual siRNA led to a decrease in M protein expression. Through a genome-wide siRNA screen, secondary validation with individual duplexes, and measurements of M protein synthesis in cells infected with wild-type virus, we have therefore identified a high-confidence set of host genes required for optimal VSV infection.
FIG 4.
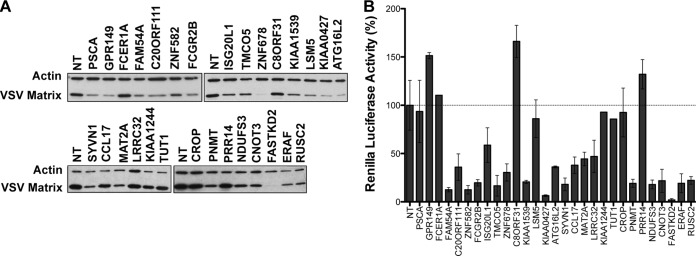
Identification of host genes required for VSV gene expression or entry. (A) Immunoblot of VSV matrix protein levels. Cells were infected at an MOI of 1, and cytoplasmic extracts were prepared at 7 hpi. VSV matrix was detected by immunoblotting using an antibody specific for VSV M (23H12), and immunoblotting of β-actin was used as a loading control. (B) VSV gene expression after transfection of RNPs. Cells were transfected with RNP cores isolated from rVSV-Ren-P, and levels of renilla luciferase activity were measured at 7 hpt. Renilla luciferase activity was normalized to the activity in cells treated with nontargeting (NT) siRNA, and results are means ± standard deviations (SD) for a representative experiment containing triplicate samples.
Identification of host factor requirements at different stages of infection.
To begin uncovering the functions of these genes in VSV replication, we employed an assay to segregate the host genes into those required for entry and those required for gene expression. The steps of viral entry can be bypassed by transfection of purified RNP cores into cells (17). We thus compared the effects of individual siRNAs on viral gene expression following RNP transfection to the effects on infection (6, 17–19). To do this, we transfected RNP cores from a reporter VSV that expresses renilla luciferase upon gene expression (rVSV-Ren-P) and measured luciferase activity at 7 hpt. Bypassing clathrin-dependent endocytosis rescued the viral gene expression defect for three cellular genes (encoding GPR149, PSCA, and LSM5), implicating these genes in VSV entry (Fig. 4B). GPR149 (G-protein-coupled receptor 149) is a transmembrane protein that is conserved in vertebrates, while PSCA (prostate stem cell antigen) is a glycosylphosphatidylinositol-anchored membrane glycoprotein that is overexpressed in prostate cancers (20, 21). The cell membrane localization of these two candidates is consistent with their assignment as potential entry factors. Unlike the former proteins, LSM5 (U6 snRNA-associated Sm-like protein) is not a membrane protein and instead is involved in pre-mRNA splicing; thus, its effects on entry may be indirect, through modulation of the expression of another protein (22). Conversely, knockdown of the remaining 20 host factors reduced gene expression following RNP core transfection. This result demonstrates that these 20 host factors influence one or more steps of viral gene expression.
DISCUSSION
Functional analysis of host genes identified by a genome-wide siRNA screen.
We identified 23 genes required for VSV replication by examining the results of the secondary screen of 450 host genes isolated from the genome-wide siRNA screen (Fig. 1 and 2). By narrowing the number of genes of interest, we could focus on mapping the functions of these host proteins in VSV infection. We identified 3 host proteins (GPR149, PSCA, and LSM5) required for VSV entry into host cells and 20 host factors required for VSV gene expression (Table 1). VSV is highly dependent on S-adenosylmethionine (SAM), as it binds directly to the L protein and is required for methylation of the viral mRNA cap (23–26). Notably, we identified MAT2A (methionine adenosyltransferase II, alpha), which catalyzes the formation of SAM, the essential methyl donor in cells, from methionine (27). The identification of SAM biogenesis factors by the genome-wide screen and the demonstration that these genes are required at the step of gene expression support the validity of our top hits.
TABLE 1.
Host gene functions in cells and during VSV replicationa
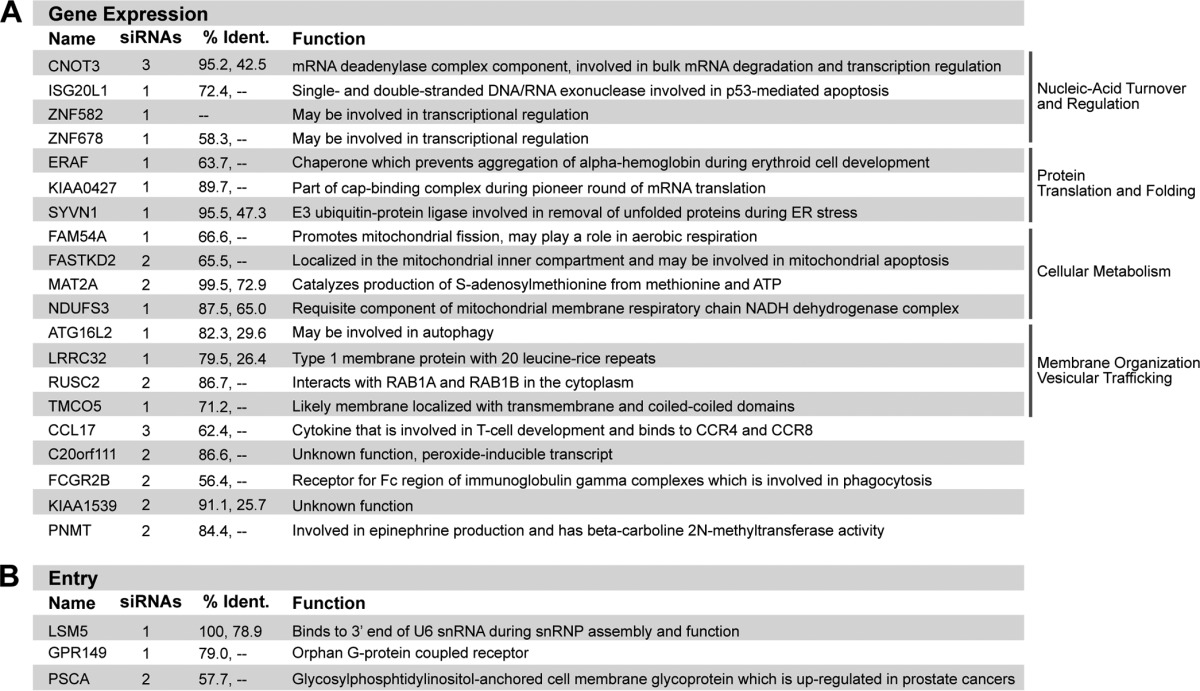
The top table (A) shows host genes required for VSV gene expression. The number of single duplexes that caused inhibition of VSV replication during rescreening, along with the known host function, is provided for each gene. % Ident., % amino acid identities between human and mouse (first number) and human and Drosophila (second number) homologous proteins. The bottom table (B) shows host genes required for VSV entry.
The majority of the host proteins identified in our follow-up screen are poorly characterized or of unknown function, and a greater understanding of their role in VSV infection may provide new insight into their natural cellular roles. While we do not yet fully understand the functions of the selected genes, a number of them have promising implications for VSV gene expression. Using information from existing gene ontology and related homologs, the human factors required for VSV replication can be assigned to functional classes that may indicate portions of the VSV life cycle specifically impaired by their depletion. Major classes of candidate host factors include proteins involved in nucleic acid turnover and regulation (CNOT3, ISG20L1, ZNF582, and ZNF678), protein translation and folding (ERAF, KIAA0427, and SYVN1), cellular metabolism (FAM54A, FASTKD2, MAT2A, and NDUFS3), and membrane organization and vesicular trafficking (LRRC32, RUSC2, and TMCO5).
One gene of particular interest is KIAA0427 (also known as CTIF [CBP80/20-dependent translation initiation factor]), a component of the cap-binding complex utilized during pioneer translation of mRNAs newly exported from the nucleus (28) (Fig. 4). During conventional host mRNA translation, the cap-binding complex containing KIAA0427 is replaced with eIF4E after the first round of translation. VSV infection induces dephosphorylation of eIF4E, resulting in a repressed canonical cap-binding complex (10). Despite eIF4E repression, the capped VSV mRNAs remain efficiently translated, indicating that a noncanonical cap-binding complex may be utilized. KIAA0427 represents an intriguing potential candidate member of this alternate complex.
Advantages and limitations of genome-wide siRNA screens.
Genetic screening is one tool for discovering new host factors required for virus infection. Limitations of siRNA screens include the potential for off-target effects or, conversely, incomplete knockdown due to long protein half-lives (29, 30). For six of the genes in our tertiary screen, for example, we were unable to replicate the phenotype discovered by using a large-scale screening approach that utilizes transfection of a pool of siRNAs (Fig. 3).
A similar genome-wide screen to identify factors required for VSV replication has also been reported (31). This screen differs from ours, as it was designed to examine phenotypes after several rounds of infection, with infection being performed at an MOI of 0.5 and assessed at 18 hpi. By imaging cells only at 7 hpi, our approach emphasizes host genes required during early steps in the pioneer round of infection that may be masked by multiple replication cycles. Additionally, we developed a sensitive assay using renilla luciferase-encoding VSV RNP cores, and we assigned an entry or gene expression defect for 29 of our hits. Notably, comparison of the results from the two screens identified eight hits in common, including ARCN1, COPB1, COPG, COPZ1, MAT2A, NHP2L1, SYVN1, and UTP6, suggesting that these are high-confidence host factors required for VSV replication and corroborating our screening methodology. Additional in-depth mechanistic analysis will be required to fully understand the roles of these host factors in viral replication. In prior work, we demonstrated that although silencing of the coatomer complex (ARCN1, COPB1, COPG, and COPZ1) results in an inhibition of the entry of VSV into cells, this inhibition likely reflects the prolonged knockdown of the coatomer complex associated with concordant changes in the composition of the plasma membrane.
An advantage of genome-wide siRNA screening for essential host-virus interactions is that it proffers previously uncharacterized proteins and host factor networks that cannot be identified by genetic knockouts. We performed a genome-wide siRNA screen in HeLa cells to identify cellular factors required for VSV infection. Here we broadly characterize the stage of viral replication affected following silencing of 23 host genes in VSV replication. These data present a useful initial point from which one can begin mechanistic studies of the host-virus networks coopted by VSV during infection.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge P. J. Kranzusch for critically reading the manuscript and the National Screening Laboratory for the Regional Centers of Excellence in Biodefense and Emerging Infectious Disease for assistance with the genome-wide siRNA screen.
This study was supported by NIH grants AI059371 and AI057159 to S.P.J.W. S.P.J.W. is a recipient of a Burroughs Wellcome Investigators in the Pathogenesis of Infectious Disease award. A.S.-Y.L. is supported by the Department of Defense through the National Defense Science and Engineering Graduate Fellowship Program and by the National Science Foundation through the Graduate Research Fellowship Program.
Footnotes
Published ahead of print 14 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00642-14.
REFERENCES
- 1.Panda D, Cherry S. 2012. Cell-based genomic screening: elucidating virus-host interactions. Curr. Opin. Virol. 2:784–792. 10.1016/j.coviro.2012.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, Whelan SP. 2009. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 5:e1000394. 10.1371/journal.ppat.1000394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cureton DK, Massol RH, Whelan SP, Kirchhausen T. 2010. The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis. PLoS Pathog. 6:e1001127. 10.1371/journal.ppat.1001127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlegel R, Willingham MC, Pastan IH. 1982. Saturable binding sites for vesicular stomatitis virus on the surface of Vero cells. J. Virol. 43:871–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. 2013. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. U. S. A. 110:7306–7311. 10.1073/pnas.1214441110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Blanc I, Luyet PP, Pons V, Ferguson C, Emans N, Petiot A, Mayran N, Demaurex N, Faure J, Sadoul R, Parton RG, Gruenberg J. 2005. Endosome-to-cytosol transport of viral nucleocapsids. Nat. Cell Biol. 7:653–664. 10.1038/ncb1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown JC, Newcomb WW, Lawrenz-Smith S. 1988. pH-dependent accumulation of the vesicular stomatitis virus glycoprotein at the ends of intact virions. Virology 167:625–629 [PubMed] [Google Scholar]
- 8.Baltimore D, Huang AS, Stampfer M. 1970. Ribonucleic acid synthesis of vesicular stomatitis virus. II. An RNA polymerase in the virion. Proc. Natl. Acad. Sci. U. S. A. 66:572–576. 10.1073/pnas.66.2.572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morin B, Kranzusch PJ, Rahmeh AA, Whelan SP. 2013. The polymerase of negative-stranded RNA viruses. Curr. Opin. Virol. 3:103–110. 10.1016/j.coviro.2013.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connor JH, Lyles DS. 2002. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J. Virol. 76:10177–10187. 10.1128/JVI.76.20.10177-10187.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254. 10.1016/j.cell.2009.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bushman FD, Malani N, Fernandes J, D'Orso I, Cagney G, Diamond TL, Zhou H, Hazuda DJ, Espeseth AS, Konig R, Bandyopadhyay S, Ideker T, Goff SP, Krogan NJ, Frankel AD, Young JA, Chanda SK. 2009. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog. 5:e1000437. 10.1371/journal.ppat.1000437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK. 2008. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135:49–60. 10.1016/j.cell.2008.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, Espeseth AS. 2008. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 4:495–504. 10.1016/j.chom.2008.10.004 [DOI] [PubMed] [Google Scholar]
- 15.Lee AS, Burdeinick-Kerr R, Whelan SP. 2013. A ribosome-specialized translation initiation pathway is required for cap-dependent translation of vesicular stomatitis virus mRNAs. Proc. Natl. Acad. Sci. U. S. A. 110:324–329. 10.1073/pnas.1216454109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whelan SP, Ball LA, Barr JN, Wertz GT. 1995. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc. Natl. Acad. Sci. U. S. A. 92:8388–8392. 10.1073/pnas.92.18.8388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cureton DK, Burdeinick-Kerr R, Whelan SP. 2012. Genetic inactivation of COPI coatomer separately inhibits vesicular stomatitis virus entry and gene expression. J. Virol. 86:655–666. 10.1128/JVI.05810-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rigaut KD, Birk DE, Lenard J. 1991. Intracellular distribution of input vesicular stomatitis virus proteins after uncoating. J. Virol. 65:2622–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carroll AR, Wagner RR. 1979. Role of the membrane (M) protein in endogenous inhibition of in vitro transcription by vesicular stomatitis virus. J. Virol. 29:134–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reiter RE, Gu Z, Watabe T, Thomas G, Szigeti K, Davis E, Wahl M, Nisitani S, Yamashiro J, Le Beau MM, Loda M, Witte ON. 1998. Prostate stem cell antigen: a cell surface marker overexpressed in prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 95:1735–1740. 10.1073/pnas.95.4.1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edson MA, Lin YN, Matzuk MM. 2010. Deletion of the novel oocyte-enriched gene, Gpr149, leads to increased fertility in mice. Endocrinology 151:358–368. 10.1210/en.2009-0760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salgado-Garrido J, Bragado-Nilsson E, Kandels-Lewis S, Seraphin B. 1999. Sm and Sm-like proteins assemble in two related complexes of deep evolutionary origin. EMBO J. 18:3451–3462. 10.1093/emboj/18.12.3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Ferra F, Baglioni C. 1983. Increase in S-adenosylhomocysteine concentration in interferon-treated HeLa cells and inhibition of methylation of vesicular stomatitis virus mRNA. J. Biol. Chem. 258:2118–2121 [PubMed] [Google Scholar]
- 24.Hefti E, Bishop DH. 1976. The 5′ sequences of VSV in vitro transcription product RNA (+/−SAM). Biochem. Biophys. Res. Commun. 68:393–400. 10.1016/0006-291X(76)91158-X [DOI] [PubMed] [Google Scholar]
- 25.Kotb M, Geller AM. 1993. Methionine adenosyltransferase: structure and function. Pharmacol. Ther. 59:125–143. 10.1016/0163-7258(93)90042-C [DOI] [PubMed] [Google Scholar]
- 26.Wong DL, Anderson LJ, Tai TC. 2002. Cholinergic and peptidergic regulation of phenylethanolamine N-methyltransferase gene expression. Ann. N. Y. Acad. Sci. 971:19–26. 10.1111/j.1749-6632.2002.tb04428.x [DOI] [PubMed] [Google Scholar]
- 27.Garcia-Trevijano ER, Latasa MU, Carretero MV, Berasain C, Mato JM, Avila MA. 2000. S-adenosylmethionine regulates MAT1A and MAT2A gene expression in cultured rat hepatocytes: a new role for S-adenosylmethionine in the maintenance of the differentiated status of the liver. FASEB J. 14:2511–2518. 10.1096/fj.00-0121com [DOI] [PubMed] [Google Scholar]
- 28.Kim KM, Cho H, Choi K, Kim J, Kim BW, Ko YG, Jang SK, Kim YK. 2009. A new MIF4G domain-containing protein, CTIF, directs nuclear cap-binding protein CBP80/20-dependent translation. Genes Dev. 23:2033–2045. 10.1101/gad.1823409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Echeverri CJ, Beachy PA, Baum B, Boutros M, Buchholz F, Chanda SK, Downward J, Ellenberg J, Fraser AG, Hacohen N, Hahn WC, Jackson AL, Kiger A, Linsley PS, Lum L, Ma Y, Mathey-Prevot B, Root DE, Sabatini DM, Taipale J, Perrimon N, Bernards R. 2006. Minimizing the risk of reporting false positives in large-scale RNAi screens. Nat. Methods 3:777–779. 10.1038/nmeth1006-777 [DOI] [PubMed] [Google Scholar]
- 30.Savas JN, Toyama BH, Xu T, Yates JR, 3rd, Hetzer MW. 2012. Extremely long-lived nuclear pore proteins in the rat brain. Science 335:942. 10.1126/science.1217421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panda D, Das A, Dinh PX, Subramaniam S, Nayak D, Barrows NJ, Pearson JL, Thompson J, Kelly DL, Ladunga I, Pattnaik AK. 2011. RNAi screening reveals requirement for host cell secretory pathway in infection by diverse families of negative-strand RNA viruses. Proc. Natl. Acad. Sci. U. S. A. 108:19036–19041. 10.1073/pnas.1113643108 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.