

ABSTRACT
The dietary exposure of the human population to the prions responsible for the bovine spongiform encephalopathy (BSE) epizooty has led to the emergence of variant Creutzfeldt-Jakob disease (vCJD). This fatal, untreatable neurodegenerative disorder is a growing public health concern because the prevalence of the infection seems much greater than the disease incidence and because secondary transmission of vCJD by blood transfusion or use of blood products has occurred. A current limitation in variant CJD risk assessment is the lack of quantitative information on the infectivity of contaminated tissues. To address this limitation, we tested the potential of a transgenic mouse line overexpressing human prion protein (PrP), which was previously reported to propagate vCJD prions. Endpoint titration of vCJD infectivity in different tissues was evaluated by two different methods: (i) the “classical” bioassay, based on the appearance of clinical symptoms and the detection of pathological prion protein in tissues of the inoculated mouse, and (ii) a shortened bioassay based on the detection of the protein in the mouse spleen at defined time points. The two methods proved equally sensitive in quantifying infectivity, even after very-low-dose inoculation of infected material, but the time schedule was shortened from ∼2.5 years to ∼1 year with the spleen bioassay. Compared to the “gold-standard” RIII model routinely used for endpoint titration of vCJD/BSE prions, either method improved the sensitivity by >2 orders of magnitude and allowed reevaluating the infectious titer of spleen from a vCJD individual at disease end stage to >1,000-fold-higher values.
IMPORTANCE Here, we provide key reevaluation of the infectious titer of variant CJD brain and spleen tissues. The highly sensitive, accelerated spleen-based assay should thus constitute a key advance for variant CJD epidemiological and risk assessment purposes and should greatly facilitate future titration studies, including, for example, those aimed at validating decontamination procedures. The overlooked notion that the lymphoid tissue exhibits a higher capacity than the brain to replicate prions even after low-dose infection raises new questions about the molecular and/or cellular determinant(s) involved, a key issue regarding potent silent carriers of variant CJD in the lymphoid tissue.
INTRODUCTION
Variant Creutzfeldt-Jakob disease (vCJD) is a fatal human infection originating from dietary exposure to bovine spongiform encephalopathy (BSE) prions. The number of definite cases has remained relatively low since its recognition in 1996, with fewer than 250 cases, mostly in the United Kingdom and France. However, the estimated number of asymptomatic carriers may be considerably higher (1), approaching, in the United Kingdom, 1/2,000 individuals born between 1941 and 1985, according to the most recent survey based on the screening of fixed lymphoid tissue libraries for the presence of disease-associated prion protein (PrPSc) (2).
Prions are thought to be essentially composed of PrPSc multimers. PrPSc is an abnormally folded conformer of the ubiquitously expressed, host-encoded prion protein PrPC. Upon infection, in certain tissues, PrPSc seeds would recruit and constrain PrPC to adopt its own abnormal, β-sheet-enriched conformation, through a so-called seeded-polymerization process (for a review, see reference 3). At variance with sporadic or genetic forms of CJD, vCJD PrPSc is readily detected in a number of lymphoid tissues in addition to the central nervous system (4–7). However, quantitative estimates of vCJD infectivity, which are critical to assess the risks for secondary transmission, are very limited. In addition, the quantitative relationships between infectivity and the forms of PrPSc routinely detected by immunoblot methods are uncertain and may differ among prion strain types (8, 9). Accurately quantifying prion infectivity requires inoculating permissive laboratory rodents with endpoint dilutions of the tissue extract of interest and estimating the disease attack rate to establish a 50% infectious dose (ID50) (10). One such bioassay in conventional RIII mice suggested that vCJD spleen contained a very low titer, in apparent contradiction with the prominent accumulation of PrPres in this tissue (7, 11) and the higher titers measured in some other prion-infected species (12–14). Interindividual transmission of vCJD has occurred through blood products from asymptomatic donors (15–17), suggesting that substantial levels of endogenous vCJD infectivity are present in blood at the preclinical stage of disease. Recently, vCJD blood fractions from a vCJD-confirmed patient were titrated in transgenic mice expressing bovine PrP. On the basis of the incubation period compared to that of a vCJD brain that was endpoint titrated, a low infectious titer was found (18). The value was consistent with that found for blood from several natural and experimental models of prion diseases (19–21). Titrations with a larger panel of vCJD patient samples would be needed to refine the infectivity values at the clinical stage of disease. Were a test-based, blood prevalence study to be initiated to detect vCJD asymptomatic carriers (22), relevant samples could be investigated by bioassay to provide estimates of infectivity levels at the preclinical stage.
There is significant concern that vCJD secondary transmission could happen through contaminated surgical and medical instruments. Prions are known to resist conventional inactivation methods, and accidental transmission has occurred during diagnostic or surgical procedures (reviewed in reference 23). While new inactivation strategies are constantly developed, their final effectiveness at eliminating prion infectivity mostly relies on inactivation of laboratory hamster prions instead of the human prions against which they are intended to be used. However, levels of prion inactivation efficacy differ greatly among the prion strain types and BSE and vCJD (BSE/vCJD) prions appear to be much more resistant than hamster prions (24). Thus, a standard bioassay to evaluate BSE/vCJD prion inactivation within reasonable time frames is urgently needed.
Rapid detection of minute amounts of PrPSc is now possible using cell-free assays such as protein misfolding cyclic amplification (PMCA) or quaking-induced conversion (QUIC). The sensitivity achieved allows detection of PrPSc present at low levels in biological tissues or fluid samples, including blood, urine, feces, or cerebrospinal fluid (for a review, see reference 25). These cell-free assays may positively impact the development of diagnostic tests but can also serve to determine whether a tissue contains any prion seeding activity. However, given the current uncertainties in the specificity of these techniques, it may be necessary to validate the result obtained in terms of infectivity by a highly sensitive bioassay.
Here, we have examined whether transgenic mice overexpressing human PrP (Met129 allele) could be used to quantify vCJD infectivity by endpoint titration by the following two different methods: (i) “classically,” by counting the number of mice developing clinical symptoms and accumulating PrPSc, and (ii) using shorter durations and defined time points to count the number of mice accumulating detectable levels of PrPSc in their spleens. The two methods proved equally sensitive in detecting infectivity, even after very-low-dose inoculation of infected material, but the time schedule was shortened from ∼2.5 year to ∼1 year with the spleen bioassay. Both methods proved >100-fold more sensitive than a RIII bioassay performed in parallel. Finally, using the spleen method, we reevaluated the infectious levels of vCJD-infected spleen and found values that were 1,000 times higher than those previous estimated (26).
MATERIALS AND METHODS
Ethics statement.
All animal experiments were approved by the Local Ethics Committee of the institutions of the authors (Comethea; permit number 12/034). Human tissue samples were selected from the tissue bank of the French National Neuropathology Network for CJD on the basis of the availability of autopsy-retained frozen brain material and informed consent from the relatives of patients for autopsy and research use, according to French regulations (L.1232-1 to L.1232-3, Code Santé Publique).
Mouse lines.
The human PrP tg650 line has been described previously (27). This line is homozygous with about 6-fold overexpression of human PrPC (Met129 allele) in brain. The RIII mouse line was kindly provided by P. Sarradin (INRA, Tours, France).
Prion-infected samples.
Pools of brain tissue from terminally sick tg650 mice (second passage from vCJD 4 as reported in reference 27) and frontal cortex or spleen extracts (approximately 3 g) from a vCJD clinical case (referred to as vCJD 2 in reference 27) were used as vCJD sources. The French BSE sample (Fr3 [28]) was kindly provided by O. Andréoletti (INRA, Toulouse, France), and the equivalent of 2 mg of cattle brain was injected intracerebrally into RIII mice to confirm the susceptibility of our mouse colony. The resulting mean incubation time (Table 1) was consistent with previous reports (29, 30).
TABLE 1.
Survival time of RIII mice and human-PrP-transgenic (tg650) mice inoculated with serial 10-fold dilutions of human vCJD brain, healthy brain, and BSE brain tissue
| Inoculum | Dilution | Mean no. of days of survival time ± SEM (n/n0)a |
|
|---|---|---|---|
| tg650 | RIII | ||
| vCJD | 10−3 | NDb | 568 ± 31 (6/6) |
| 10−4 | ND | 438c; 482 (2/7) | |
| 10−5 | 802 ± 35 (6/6) | 609–975 (0/7) | |
| 10−6 | 834 ± 51 (5/9) | 543–975 (0/7) | |
| 10−7 | 651c (1/9) | 538–975 (0/7) | |
| 10−8 | 687–1062 (0/8) | ND | |
| Healthy brain | 10−1 | 623–1098 (0/9) | 422–945 (0/7) |
| BSE (Fr3) | 10−1 | 753 ± 60 (3/5) | 413 ± 8 (9/9) |
Animal bioassay.
A strict protocol based on the use of disposable equipment and preparation of all inocula in a class II microbiological cabinet was followed to avoid any cross-contamination. Starting from 10% (wt/vol) brain homogenate (10−1 dilution), serial 10-fold dilutions of brain or spleen homogenates were prepared in 5% (wt/vol) glucose solution containing 5% (wt/vol) bovine serum albumin. Twenty microliters of each dilution (unless specified) was immediately inoculated into individually identified 6-to-8-week-old tg650 or RIII recipients by the intracerebral (IC) route. For the survival time bioassay, animals were supervised daily for the appearance of neurological signs associated with the development of a prion disease. Animals at the terminal stage of disease or at the end of life were euthanized. The brains and spleens of all animals (including those dying from intercurrent disease, when available) were analyzed for proteinase K-resistant PrPSc (PrPres) content using a Bio-Rad TsSeE detection kit before immunoblotting was performed as previously described (9, 31). For the spleen-based bioassay, tg650 mice were euthanized while still asymptomatic at defined time points, as indicated. Spleens and sometimes brains were removed and analyzed for PrPres content (cf. infra). For both methods, the number of positive mice was used to establish, by the Spearman-Karber method, the number of prion infectious units per gram of tissue leading to median mouse infection (ID50 per gram).
Detection of PrPres in the spleen bioassay.
Spleens were homogenized at 20% (wt/vol) in 5% (wt/vol) sterile glucose solution by use of a tissue homogenizer (Precellys 24 Ribolyzer; Bertin Technologies, France). PrPres was extracted from 200 μl of spleen homogenate by using a scrapie-associated fibril (SAF) protocol, as previously described (32). Proteinase K was used at 30 μg/ml. A 10-to-40-mg equivalent of tissue was migrated on 12% Bis-Tris NuPage gels (Invitrogen, France) and electrotransferred onto nitrocellulose membranes. Those were probed for PrP with 0.1 mg/ml biotinylated anti-PrP monoclonal antibody Sha31 as previously described (9, 31). Immunoreactivity was visualized by chemiluminescence (GE Healthcare). The amount of PrPres was determined by the use of GeneTools software after acquisition of chemiluminescent signals with a GeneGnome digital imager (Syngene, Frederick, MD) and was compared to that present in endpoint dilutions of vCJD-infected tg650 brain samples which were subjected to the same purification and immunoblotting protocol.
RESULTS
Titrating tg650-passaged vCJD prions by the survival time bioassay.
The vCJD material that was endpoint titrated consisted of two serial passages of vCJD in tg650 human-PrP-transgenic mice (27). The most relevant physiopathological hallmarks of vCJD (33), notably, the presence of numerous florid plaques in the brain, the prominent colonization of the lymphoid tissue, and the PrPres electrophoretic signature (27), were conserved in these mice. To determine the dose-response relationship, groups of 6 to 10 tg650 mice were injected intracerebrally with serial 10-fold dilutions of brain homogenate, as classically done (10). The mice were followed clinically until prion disease development, if any. The time intervals from inoculation to the terminal stage of disease were measured for each dilution. Brain and spleen of mice euthanized at the terminal stage of disease or at the end of life were collected and analyzed for PrPres content to confirm infection or disease. The results are summarized in Fig. 1A. At the 1/10 dilution, all the inoculated mice were diseased in ∼500 days (mean incubation time, 497 ± 18 days) and accumulated evidence of vCJD-like PrPres brain and spleen (Fig. 1C). This incubation time was comparable to that observed at the first and second passages (27), consistent with the absence of a transmission barrier for vCJD prions in tg650 mice. The mean survival times were prolonged with dilutions. Based on clinical signs and the presence of PrPres in brain and spleen, disease was seen with a 100% attack rate until the 10−5 dilution. At that dilution, the mean incubation time to disease approached 700 ± 30 days (range, 570 to 838 days). At higher dilutions, it was difficult to distinguish between clinical disease and the end of life. However, PrPres was detected in the brain and spleen of 2 of 10 mice at the 10−6 dilution and at the 10−7 dilution (last dilution injected) between 900 and 1,000 days postinoculation (Fig. 1A and C). Applying the Spearman-Karber method to data corresponding to the number of animals positive at each dilution provided a provisional infectious titer ≥ 108.8 intracerebral tg650 mouse ID50 U/g brain (ID50 IC in tg650/g).
FIG 1.
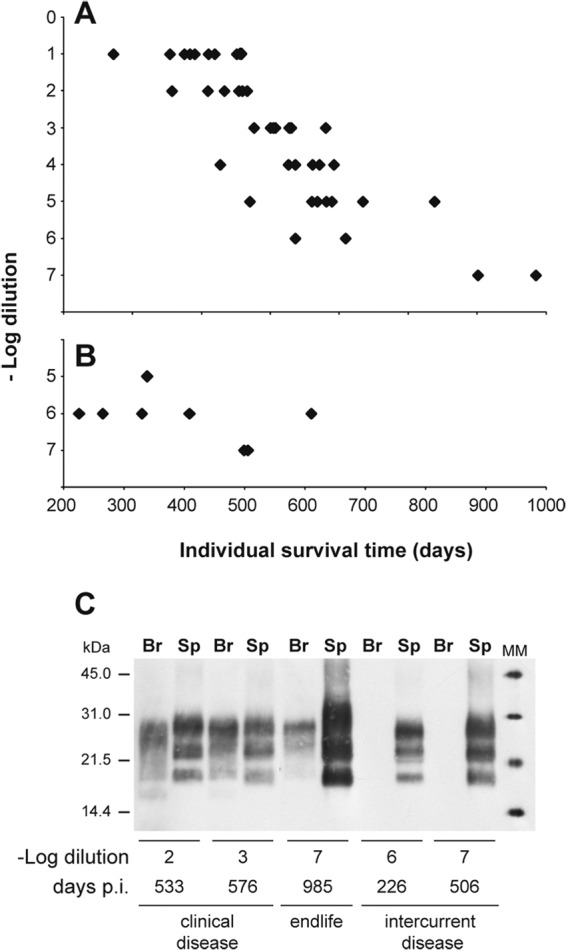
Endpoint titration of vCJD prions by the survival time bioassay in human-PrP-transgenic mice. (A and B) Individual survival time of human PrP transgenic (tg650) mice intracerebrally inoculated with serial 10-fold dilutions of brain homogenate from vCJD-infected tg650 mice. (A) All the mice included accumulated detectable levels of PrPres in their brain and spleen. They exhibited clinical signs of prion disease up to the 10−5 dilution. (B) Mice with intercurrent disease, which were not included in the titration but showed detectable levels of PrPres in their spleens but not in their brain. (C) PrPres immunoblot analysis of the brain (Br) and spleen (Sp) of tg650 mice upon inoculation with dilutions of vCJD-infected tg650 brain material. Analysis was performed at different time points postinoculation (p.i.), as indicated. The equivalent of 2 mg of tg650 spleen tissue was loaded for the SDS-PAGE analysis. For the brain, equivalents of 60 μg and 10 mg tissue were used when PrPres results were positive and negative, respectively. MM, molecular mass markers.
During this titration, some mice inoculated at the highest dilutions had to be euthanized for intercurrent disease and were not included in the titer quantification. The routine analysis of their tissues revealed detectable levels of PrPres in the spleen but not in the brain, from ∼200 days onward (Fig. 1B and C). This led us to reason that an endpoint titration based on a time course analysis of PrPres accumulation in the spleen could be as sensitive as survival time-based titration and might require less time to complete than the >2.5 years necessary to achieve a result by the latter method.
Titrating tg650-passaged vCJD prions by the spleen-based bioassay.
Reporter tg650 mice were inoculated intracerebrally with 10-fold dilution series of vCJD-infected tg650 brain (up to 10−11 dilution). Every ∼60 days, up to 500 days postinfection, three mice were euthanized and their spleens analyzed for PrPres content using a scrapie-associated fibril (SAF) protocol followed by a concentration step using acetone precipitation (32). This technique did not provide higher sensitivity than a Bio-Rad TsSeE kit routinely used to purify PrPres (see Materials and Methods), but it allowed proper migration on SDS-PAGE gels of up to 20-fold-larger amounts of spleen tissue equivalent and, in short, reliable detection of PrPres, even after low-dose inoculation (data not shown) (Fig. 2) (32). In other words, a half spleen could be immunoblotted for PrPres content, when necessary. To quantify and compare the signals obtained at the different time points, PrPres signal intensity was compared with that of serial dilutions of vCJD brain homogenates in experiments performed in parallel using the same PrPres purification process. The results obtained are summarized in Fig. 2. At 60 days postinoculation, PrPres was detected in all the spleens of mice inoculated up to the 10−3 dilution and in 1 of 3 mice inoculated with the 10−4 dilution. At 180 days postinoculation, PrPres was detected in all the analyzed spleens up to the 10−6 dilution. From 245 days postinfection onward, a major proportion (15/18) of spleens from mice inoculated with the 10−7 dilution were PrPres positive. The highest dilution able to induce PrPres accumulation in the spleen was 10−8. Remarkably, this was detected at approximately a year postinoculation, in all three spleens analyzed. In other words, infection still established in the spleen a year after inoculation of 200 pg brain tissue equivalent. At that stage, PrPres levels were nearly at maximum, i.e., equivalent to those observed after inoculation of lower dilutions (Fig. 2). In comparison, the brains were still negative at the 10−8 or 10−7 dilution (Fig. 2B and data not shown). PrPres was not detected in the spleen below the 10−8 dilution threshold at up to 500 days postinoculation. Applying the Spearman-Karber method to the proportion of animals positive at each dilution provided a provisional infectious titer of ∼1010.2 ID50 IC in tg650/g.
FIG 2.

Endpoint titration of vCJD prions by the spleen-based bioassay in human-PrP-transgenic mice. (A) Time course of PrPres accumulation in the spleen tissue of human-PrP-transgenic mice (tg650 line) inoculated with serial 10-fold dilutions of tg650-passaged vCJD brain. PrPres extracted from each individual spleen was detected by immunoblot analysis. For quantification, the levels were compared to those found in serially diluted vCJD tg650 mouse brains at the terminal stage of disease and are thus expressed as micrograms of brain equivalent. The black dots represent PrPres levels in each spleen and the black bars the mean levels for each time point analyzed. The positive threshold is indicated by the horizontal dotted line. (B) Immunoblots showing PrPres accumulation in the spleens and brains of tg650 mice inoculated with serial dilutions of tg650 vCJD prions and analyzed at 180, 245, and 362 days postinoculation (dpi). Digital acquisition was performed for 55, 30, and 5 min, respectively. The equivalent of 20 mg of tg650 spleen tissue was loaded on the SDS-PAGE gel up to the 10−5 dilution at 180 dpi and at the 10−6 dilution at 245 dpi. Otherwise, the equivalent of 40 mg of tissue equivalent was loaded. MM, molecular mass markers.
Together, these data indicated that a spleen PrPres-based endpoint titration assay was >10-fold more sensitive than the endpoint titration bioassay and required only 1 year to be completed.
Comparing the sensitivities of RIII and human-PrP-transgenic mice for titration of vCJD prions.
The infectious titer of vCJD prions in tg650 mice appeared to be between 103 and 105 times greater than that established in a RIII mouse bioassay performed with human vCJD brain (26). We therefore compared the relative sensitivities of the RIII and tg650 mouse models upon intracerebral inoculation of one vCJD brain source, first by using the same method, the survival time bioassay (partial endpoint titration; Table 1). In RIII mice, clinical disease and brain and spleen PrPres were detected in 6 of 6 mice inoculated at the 10−3 dilution and in 1 of 7 mice inoculated with the 10−4 dilution. At that dilution, another mouse that died of intercurrent disease at 438 days was PrPres positive in the spleen but not in the brain and was thus considered PrPres positive. Mice inoculated with the 10−5 to 10−7 dilutions were all PrPres negative in their brain and spleen tissues. The limiting dilution found here (10−4) thereby confirmed earlier observations (26). In marked contrast, disease was seen in tg650 mice with a 100% attack rate at the 10−5 dilution. At the 10−6 dilution, 4 of 9 mice were PrPres positive in brain and spleen (834 ± 51 days). At the 10−7 dilution, all the mice were clinically negative and no PrPres was detectable in the brain. However, one mouse euthanized at 651 days postinoculation has detectable levels of PrPres in the spleen. At higher dilutions, results for all of the mice remained negative.
To estimate the limiting dilution with the spleen-based tg650 assay, mice inoculated with serial 10-fold dilutions of vCJD brain material (starting dilution, 10−3) were euthanized at 120, 240, and 360 days postinoculation and their spleens analyzed for PrPres content (Fig. 3A and C). At 120 and 240 days postinoculation, PrPres was detected in all the spleens of mice inoculated with the 10−4 dilution. A total of 2/3 and 3/3 mice inoculated with the 10−5 dilution were PrPres positive at 240 days and 360 days, respectively. At the 10−6 dilution, 3 of 12 mice were PrPres positive at 240 days postinoculation and 0/12 at 360 days. At that stage, the 12 mice inoculated with the 10−7 dilution were all PrPres negative. The dilution limit of the vCJD brain extract tested was thus established at the 10−6 dilution, a value comparable to that obtained with the survival time bioassay, obtained in only 240 days.
FIG 3.

Titration of human brain and spleen tissues from a vCJD case by use of the spleen-based bioassay. (A and B) PrPres accumulation in the spleens of human-PrP-transgenic mice (tg650 line) inoculated with serial 10-fold dilutions of brain (A) and spleen (B) tissue extract from the same vCJD individual at the terminal stage of disease. The analysis was performed at the time indicated. PrPres extracted from each individual tg650 spleen was detected by immunoblot analysis. For quantification, the levels were compared to those found in serially diluted vCJD tg650 mouse brains at the terminal stage of disease and are thus expressed as micrograms of brain equivalent. The black dots represent PrPres levels in each spleen and the black bars the mean levels for each time point analyzed. (C) PrPres immunoblot analyses of tg650 mouse spleens at 240 and 360 days postinoculation (dpi) with vCJD brain or spleen material diluted 105-fold or 106-fold, as indicated. The equivalent of 15 mg of tg650 spleen tissue was loaded on the SDS-PAGE gel. Digital acquisition was performed for 55 min.
Applying the Spearman-Karber method to the proportion of animals testing positive at each dilution provided a provisional infectious titer of ∼106.4 ID50 IC in RIII/g and 108.1–8.77 ID50 IC in tg650/g (spleen-based bioassay and survival time bioassay, respectively). Collectively, these data indicate that the same human vCJD brain extract contained >100-fold-more detectable infectivity in tg650 than in RIII mice.
Reevaluating the amount of vCJD infectivity in human spleen.
Having found that the tg650 mouse model was much more sensitive than the RIII mouse bioassay, we next examined, by using the tg650 spleen-based bioassay, whether the very low infectious titer of vCJD prions in human spleen found with RIII mice (26) truly reflected the infectivity of this tissue. Tg650 mice were inoculated with serial dilutions of the spleen extract from the same vCJD individual whose brain was titrated (starting dilution, 10−3 [12 to 18 mice at a high dilution to increase the sensitivity]). The mice were euthanized at 360 days postinfection and analyzed for PrPres content in their spleen (Fig. 3B and C). At the 10−3 dilution, PrPres was detected in the spleens of 6 of 6 animals. At the 10−4 dilution, PrPres was detected in the spleens of 14 of 18 animals. At the 10−5 dilution, 3 of 15 mice analyzed were PrPres positive. At the 10−6 dilution, all the spleens analyzed were PrPres negative. The proportion of individuals testing positive allowed establishing an infectious titer of ∼106.1 ID50 IC in tg650/g in human vCJD spleen compared to 102.7 ID50 IC in RIII/g (26).
DISCUSSION
Using the spleen of “humanized” transgenic animals, we report here on the detection of vCJD infectivity with sensitivity similar to that of the survival time-based bioassay but within a markedly reduced time scale and independently of any clinical outcome. Functionally, instead of counting the number of mice developing a prion disease upon inoculation of endpoint dilutions of vCJD-infected material, we counted, at predetermined, shorter time durations, the number of spleens that scored positive for prion infection by detection of PrPres. The two methods proved equivalent in terms of detection limit and in providing an ID50. Accurately determining an ID50 by the survival time method with “slow” prion agents such as vCJD is difficult when incubation times are prolonged, as disease appearance and senescence can be confounded. Compared with the ∼2.5 years required for completion of the survival time bioassay, a full titration can be achieved within a year with the so-called spleen bioassay. Prolonging the incubation of the mice beyond this period may not provide a gain of sensitivity, as PrPres accumulation in spleen, even at the highest vCJD dilutions, could be maximal. Additionally, spleens of mice over 1.5 years of age may show impaired capacities to replicate prions, due to declines in the functions of follicular dendritic cells where PrPSc accumulate or alteration in the microarchitecture of the germinal center where these cells are located (34, 35).
The spleen-to-brain PrPC expression level ratios of the tg650 and conventional C57BL/6 mouse lines appear to be similar (28), thus excluding the possibility of aberrant quantitative expression of the human PrP transgene in tg650 mouse spleen that would explain the faster accumulation of PrPres in the spleen than in the brain during the disease incubation. Preliminary data indicate that titration of lymphocompetent ovine prions (natural sources or adapted to ovine-PrP-transgenic mice) can be made in the spleen of ovine-PrP-overexpressing mice (tg338 line; [31]) with higher sensitivity than in brain (Fig. 4 and data not shown). Thus, the observations made are not restricted to human PrP, vCJD prions, and the tg650 line. The capacity of the spleen to replicate prions with high sensitivity may have been overlooked in the context of the neurodegenerative nature of prion diseases, and the concept may even seem counterintuitive, as the most infectious tissue remains the brain at the terminal stage of disease (29, 36–38), as confirmed here with human-variant CJD. What could contribute to such capacity? The spleen is exposed to incoming prions as early as the brain, due to the spillover at the time of intracerebral inoculation, which provokes a rapid intravenous transport of an appreciable part of the inoculum to the spleen (32, 39). Besides, the greater celerity of the spleen versus brain in replicating certain prion strains has been known for some time (40, 41). The spleen “response time” (with regard to PrP detection) remained much faster than that of the brain at the dilution limit (240 versus >900 days), suggesting an efficient addressing and/or retention of the low-dose inoculum, allowing PrPC conversion. We previously reported that the prion species barrier was more readily crossed in the spleen than in the brain (28). Intrinsically, there might be, in the lymphoid tissue, a better conformational compatibility between the invading prion—be it homologous or not—and the cellular prion protein or a favored environment for prion conversion. Collectively, these data clearly indicate that vCJD prions could efficiently colonize “humanized” lymphoid tissue, even after very-low-dose infection. This has implications for public health risk assessment, with regard to the real prevalence of subclinically infected vCJD individuals (2).
FIG 4.
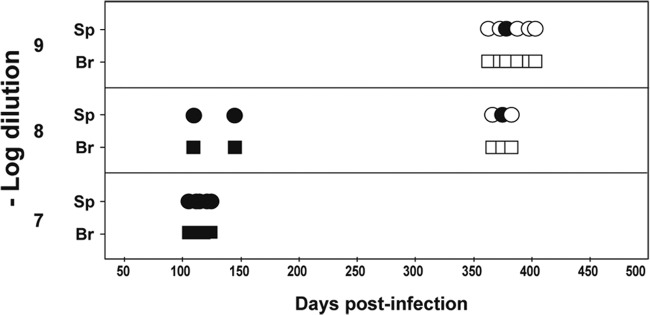
PrPres detection in the brain and spleen of ovine PrP transgenic mice inoculated with endpoint dilutions of sheep scrapie prions. Data represent PrPres detection in spleen (Sp; circles) and brain (Br; squares) of ovine PrP transgenic mice (tg338 line) euthanized at disease end stage (<160 days) or healthy after intracerebral inoculation of LA21K sheep scrapie prions (full titration in reference 9). Empty symbols indicate PrPres-negative tissue.
In the past, numerous bioassays aimed at titrating the infectivity of tissues from animals infected with BSE/vCJD prions have been performed with inbred RIII mice (26, 42). After inoculation with these agents, RIII mice developed the disease faster and at a higher attack rate than other, conventional mice. For cattle BSE, the dose necessary to kill RIII mice appeared to be 1,000- and 10,000-fold higher than for cows (43) and transgenic mice expressing bovine PrP (29), respectively. Very recently, bovine PrP mice were also shown to be highly susceptible to vCJD brain prions (18). Here, following inoculation with a human vCJD brain extract, tg650 mice revealed a sensitivity similar to and >100-fold greater than the sensitivities of bovine-PrP and RIII mice, respectively. The reevaluated infectious titer found (>108 ID50 IC in tg650/g of brain) is within the range seen in the brain of other prion-infected species (8–10, 44), including the most frequent sporadic form of CJD in human (27). A species barrier could exist between RIII mice and cattle or human BSE prions, which would limit their susceptibility at a low infectious dose and which would be abrogated by transgenic expression of bovine or human PrPC. Several lines of evidence indicate that the species barrier to the transmission of prions is essentially determined by the strain of prion and the PrP primary sequence identity between the invading prion and the recipient species (for a review, see reference 45). Alternatively, there might be no species barrier between vCJD/BSE prions and RIII mice but human- and bovine-PrP-transgenic mice would gain sensitivity due to PrP overexpression. Unfortunately, only a partial titration of BSE prions was done in transgenic mice overexpressing mouse PrP approximately 10-fold (tga20 mice) in comparison with RIII mice (29). At a dilution where 50% of the RIII mice were affected, 100% of the Tga20 mice developed disease. In contrast, sheep and transgenic mice overexpressing ovine PrP provided similar estimates of sheep prion infectious titers (46). Thus, whether PrP overexpression impacts the susceptibility to infection and the determination of the infectious titer remains uncertain. In practical terms, RIII-based bioassays are likely to underestimate the amount of BSE/vCJD prion infectivity present in the assayed tissue. This is patently clear with tissues exhibiting a smaller amount of infectivity than the central nervous system. vCJD spleen extract was previously shown to infect approximately 50% of the RIII mice at the highest concentration (26), suggesting traces of infectivity in this tissue. Here we showed that inoculation of 10,000-fold-diluted spleen material was sufficient to initiate vCJD prion replication in tg650 mouse spleen, providing an infectious titer > 106 ID50 IC in tg650/g. This measurement needs to be extended to a larger panel of samples. However, the relative levels compared to the brain of the same individual are consistent with those observed in other natural or experimental prion-infected species (12–14). That spleen would exhibit such high levels of infectivity throughout a long life makes this tissue a reservoir that could constantly spill infectivity to blood.
Although full endpoint titration of vCJD prions can be completed within a year, the spleen-based bioassay is obviously too slow to help in diagnosing vCJD. However, the level of sensitivity achieved would be compatible with detection of prionemia (18), at variance with RIII mice (26). Such an assay could complement cell-free (47–50) or standard (7) biochemical assays, when proving or confirming the presence of bona fide infectivity becomes an issue, due, for example, to inconsistencies among the biochemical tests and/or tissue resampling (51) or to the use of fixation treatments known to alter prion infectivity (11). This will be key to ensuring the accuracy of vCJD prevalence estimates in the United Kingdom (2).
ACKNOWLEDGMENTS
We thank the staff of Animalerie Rongeurs (INRA, Jouy-en-Josas, France) for excellent animal care and Annick Le Dur for excellent technical help.
This work was supported by grants from INRA-Transfert (Paris, France), Institut National de Veille Sanitaire (Paris, France), and the Alliance Biosecure Foundation (PrionBloodPrimate and PrionBloodConfirm projects).
Footnotes
Published ahead of print 21 May 2014
REFERENCES
- 1.Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Ritchie D, Penney M, Hegazy D, Ironside JW. 2004. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J. Pathol. 203:733–739. 10.1002/path.1580 [DOI] [PubMed] [Google Scholar]
- 2.Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L, Linehan J, Simmons M, Webb P, Bellerby P, Andrews N, Hilton DA, Ironside JW, Beck J, Poulter M, Mead S, Brandner S. 2013. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ 347:f5675. 10.1136/bmj.f5675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colby DW, Prusiner SB. 2011. Prions. Cold Spring Harb. Perspect. Biol. 3:a006833. 10.1101/cshperspect.a006833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wadsworth JD, Hill AF, Beck JA, Collinge J. 2003. Molecular and clinical classification of human prion disease. Br. Med. Bull. 66:241–254. 10.1093/bmb/66.1.241 [DOI] [PubMed] [Google Scholar]
- 5.Haïk S, Faucheux BA, Sazdovitch V, Privat N, Kemeny JL, Perret-Liaudet A, Hauw JJ. 2003. The sympathetic nervous system is involved in variant Creutzfeldt-Jakob disease. Nat. Med. 9:1121–1123. 10.1038/nm922 [DOI] [PubMed] [Google Scholar]
- 6.Heikenwalder M, Julius C, Aguzzi A. 2007. Prions and peripheral nerves: a deadly rendezvous. J. Neurosci. Res. 85:2714–2725. 10.1002/jnr.21246 [DOI] [PubMed] [Google Scholar]
- 7.Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J. 2001. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 358:171–180. 10.1016/S0140-6736(01)05403-4 [DOI] [PubMed] [Google Scholar]
- 8.Andréoletti O, Orge L, Benestad SL, Beringue V, Litaise C, Simon S, Le Dur A, Laude H, Simmons H, Lugan S, Corbiere F, Costes P, Morel N, Schelcher F, Lacroux C. 2011. Atypical/Nor98 scrapie infectivity in sheep peripheral tissues. PLoS Pathog. 7:e1001285. 10.1371/journal.ppat.1001285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tixador P, Herzog L, Reine F, Jaumain E, Chapuis J, Le Dur A, Laude H, Beringue V. 2010. The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLoS Pathog. 6:e1000859. 10.1371/journal.ppat.1000859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prusiner SB, Cochran SP, Groth DF, Downey DE, Bowman KA, Martinez HM. 1982. Measurement of the scrapie agent using an incubation time interval assay. Ann. Neurol. 11:353–358. 10.1002/ana.410110406 [DOI] [PubMed] [Google Scholar]
- 11.Wadsworth JD, Dalmau-Mena I, Joiner S, Linehan JM, O'Malley C, Powell C, Brandner S, Asante EA, Ironside JW, Hilton DA, Collinge J. 2011. Effect of fixation on brain and lymphoreticular vCJD prions and bioassay of key positive specimens from a retrospective vCJD prevalence study. J. Pathol. 223:511–518. 10.1002/path.2821 [DOI] [PubMed] [Google Scholar]
- 12.Hadlow WJ, Kennedy RC, Race RE. 1982. Natural infection of Suffolk sheep with scrapie virus. J. Infect. Dis. 146:657–664. 10.1093/infdis/146.5.657 [DOI] [PubMed] [Google Scholar]
- 13.Eklund CM, Kennedy RC, Hadlow WJ. 1967. Pathogenesis of scrapie virus infection in the mouse. J. Infect. Dis. 117:15–22. 10.1093/infdis/117.1.15 [DOI] [PubMed] [Google Scholar]
- 14.Dickinson AG, Fraser H. 1969. Genetical control of the concentration of ME7 scrapie agent in mouse spleen. J. Comp. Pathol. 79:363–366. 10.1016/0021-9975(69)90051-6 [DOI] [PubMed] [Google Scholar]
- 15.Hewitt PE, Llewelyn CA, Mackenzie J, Will RG. 2006. Creutzfeldt-Jakob disease and blood transfusion: results of the UK Transfusion Medicine Epidemiological Review study. Vox Sang. 91:221–230. 10.1111/j.1423-0410.2006.00833.x [DOI] [PubMed] [Google Scholar]
- 16.Wroe SJ, Pal S, Siddique D, Hyare H, Macfarlane R, Joiner S, Linehan JM, Brandner S, Wadsworth JD, Hewitt P, Collinge J. 2006. Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: a case report. Lancet 368:2061–2067. 10.1016/S0140-6736(06)69835-8 [DOI] [PubMed] [Google Scholar]
- 17.Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. 2004. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 364:527–529. 10.1016/S0140-6736(04)16811-6 [DOI] [PubMed] [Google Scholar]
- 18.Douet JY, Zafar S, Perret-Liaudet A, Lacroux C, Lugan S, Aron N, Cassard H, Ponto C, Corbiere F, Torres JM, Zerr I, Andreoletti O. 2014. Detection of infectivity in blood of persons with variant and sporadic Creutzfeldt-Jakob disease. Emerg. Infect. Dis. 20:114–117. 10.3201/eid2001.130353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andréoletti O, Litaise C, Simmons H, Corbière F, Lugan S, Costes P, Schelcher F, Vilette D, Grassi J, Lacroux C. 2012. Highly efficient prion transmission by blood transfusion. PLoS Pathog. 8:e1002782. 10.1371/journal.ppat.1002782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lacroux C, Bougard D, Litaise C, Simmons H, Corbiere F, Dernis D, Tardivel R, Morel N, Simon S, Lugan S, Costes P, Weisbecker JL, Schelcher F, Grassi J, Coste J, Andreoletti O. 2012. Impact of leucocyte depletion and prion reduction filters on TSE blood borne transmission. PLoS One 7:e42019. 10.1371/journal.pone.0042019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cervenakova L, Yakovleva O, McKenzie C, Kolchinsky S, McShane L, Drohan WN, Brown P. 2003. Similar levels of infectivity in the blood of mice infected with human-derived vCJD and GSS strains of transmissible spongiform encephalopathy. Transfusion 43:1687–1694. 10.1046/j.0041-1132.2003.00586.x [DOI] [PubMed] [Google Scholar]
- 22.Jackson GS, Burk-Rafel J, Edgeworth JA, Sicilia A, Abdilahi S, Korteweg J, Mackey J, Thomas C, Wang G, Mead S, Collinge J. 2014. A highly specific blood test for vCJD. Blood 123:452–453. 10.1182/blood-2013-11-539239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor DM. 1999. Inactivation of prions by physical and chemical means. J. Hosp. Infect. 43(Suppl):S69–S76. 10.1016/S0195-6701(99)90067-1 [DOI] [PubMed] [Google Scholar]
- 24.Giles K, Glidden DV, Beckwith R, Seoanes R, Peretz D, DeArmond SJ, Prusiner SB. 2008. Resistance of bovine spongiform encephalopathy (BSE) prions to inactivation. PLoS Pathog. 4:e1000206. 10.1371/journal.ppat.1000206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kraus A, Groveman BR, Caughey B. 2013. Prions and the potential transmissibility of protein misfolding diseases. Annu. Rev. Microbiol. 67:543–564. 10.1146/annurev-micro-092412-155735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruce ME, McConnell I, Will RG, Ironside JW. 2001. Detection of variant Creutzfeldt-Jakob disease infectivity in extraneural tissues. Lancet 358:208–209. 10.1016/S0140-6736(01)05411-3 [DOI] [PubMed] [Google Scholar]
- 27.Béringue V, Le Dur A, Tixador P, Reine F, Lepourry L, Perret-Liaudet A, Haik S, Vilotte JL, Fontes M, Laude H. 2008. Prominent and persistent extraneural infection in human PrP transgenic mice infected with variant CJD. PLoS One 3:e1419. 10.1371/journal.pone.0001419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Béringue V, Herzog L, Jaumain E, Reine F, Sibille P, Le Dur A, Vilotte JL, Laude H. 2012. Facilitated cross-species transmission of prions in extraneural tissue. Science 335:472–475. 10.1126/science.1215659 [DOI] [PubMed] [Google Scholar]
- 29.Buschmann A, Groschup MH. 2005. Highly bovine spongiform encephalopathy-sensitive transgenic mice confirm the essential restriction of infectivity to the nervous system in clinically diseased cattle. J. Infect. Dis. 192:934–942. 10.1086/431602 [DOI] [PubMed] [Google Scholar]
- 30.Green R, Horrocks C, Wilkinson A, Hawkins SA, Ryder SJ. 2005. Primary isolation of the bovine spongiform encephalopathy agent in mice: agent definition based on a review of 150 transmissions. J. Comp. Pathol. 132:117–131. 10.1016/j.jcpa.2004.08.002 [DOI] [PubMed] [Google Scholar]
- 31.Langevin C, Andreoletti O, Le Dur A, Laude H, Beringue V. 2011. Marked influence of the route of infection on prion strain apparent phenotype in a scrapie transgenic mouse model. Neurobiol. Dis. 41:219–225. 10.1016/j.nbd.2010.09.010 [DOI] [PubMed] [Google Scholar]
- 32.Beringue V, Adjou KT, Lamoury F, Maignien T, Deslys JP, Race R, Dormont D. 2000. Opposite effects of dextran sulfate 500, the polyene antibiotic MS-8209, and Congo red on accumulation of the protease-resistant isoform of PrP in the spleens of mice inoculated intraperitoneally with the scrapie agent. J. Virol. 74:5432–5440. 10.1128/JVI.74.12.5432-5440.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wadsworth JD, Collinge J. 2007. Update on human prion disease. Biochim. Biophys. Acta 1772:598–609. 10.1016/j.bbadis.2007.02.010 [DOI] [PubMed] [Google Scholar]
- 34.Brown KL, Gossner A, Mok S, Mabbott NA. 2012. The effects of host age on the transport of complement-bound complexes to the spleen and the pathogenesis of intravenous scrapie infection. J. Virol. 86:25–35. 10.1128/JVI.05581-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown KL, Wathne GJ, Sales J, Bruce ME, Mabbott NA. 2009. The effects of host age on follicular dendritic cell status dramatically impair scrapie agent neuroinvasion in aged mice. J. Immunol. 183:5199–5207. 10.4049/jimmunol.0802695 [DOI] [PubMed] [Google Scholar]
- 36.Kimberlin RH, Walker CA. 1986. Pathogenesis of scrapie (strain 263K) in hamsters infected intracerebrally, intraperitoneally or intraocularly. J. Gen. Virol. 67(Pt 2):255–263. 10.1099/0022-1317-67-2-255 [DOI] [PubMed] [Google Scholar]
- 37.Race R, Jenny A, Sutton D. 1998. Scrapie infectivity and proteinase K-resistant prion protein in sheep placenta, brain, spleen, and lymph node: implications for transmission and antemortem diagnosis. J. Infect. Dis. 178:949–953. 10.1086/515669 [DOI] [PubMed] [Google Scholar]
- 38.Brown P, Gibbs CJ, Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. 1994. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann. Neurol. 35:513–529. 10.1002/ana.410350504 [DOI] [PubMed] [Google Scholar]
- 39.Millson GC, Kimberlin RH, Manning EJ, Collis SC. 1979. Early distribution of radioactive liposomes and scrapie infectivity in mouse tissues following administration by different routes. Vet. Microbiol. 4:89–99. 10.1016/0378-1135(79)90045-2 [DOI] [Google Scholar]
- 40.Kimberlin RH, Walker CA. 1979. Pathogenesis of mouse scrapie: dynamics of agent replication in spleen, spinal cord and brain after infection by different routes. J. Comp. Pathol. 89:551–562. 10.1016/0021-9975(79)90046-X [DOI] [PubMed] [Google Scholar]
- 41.Muramoto T, Kitamoto T, Tateishi J, Goto I. 1992. The sequential development of abnormal prion protein accumulation in mice with Creutzfeldt-Jakob disease. Am. J. Pathol. 140:1411–1420 [PMC free article] [PubMed] [Google Scholar]
- 42.Wells GA, Hawkins SA, Austin AR, Ryder SJ, Done SH, Green RB, Dexter I, Dawson M, Kimberlin RH. 2003. Studies of the transmissibility of the agent of bovine spongiform encephalopathy to pigs. J. Gen. Virol. 84:1021–1031. 10.1099/vir.0.18788-0 [DOI] [PubMed] [Google Scholar]
- 43.Wells GA, Hawkins SA, Green RB, Austin AR, Dexter I, Spencer YI, Chaplin MJ, Stack MJ, Dawson M. 1998. Preliminary observations on the pathogenesis of experimental bovine spongiform encephalopathy (BSE): an update. Vet. Rec. 142:103–106. 10.1136/vr.142.5.103 [DOI] [PubMed] [Google Scholar]
- 44.Tamgüney G, Miller MW, Wolfe LL, Sirochman TM, Glidden DV, Palmer C, Lemus A, DeArmond SJ, Prusiner SB. 2009. Asymptomatic deer excrete infectious prions in faeces. Nature 461:529–532. 10.1038/nature08289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Béringue V, Vilotte JL, Laude H. 2008. Prion agent diversity and species barrier. Vet. Res. 39:47. 10.1051/vetres:2008024 [DOI] [PubMed] [Google Scholar]
- 46.Douet JY, Lacroux C, Corbiere F, Litaise C, Simmons H, Lugan S, Costes P, Cassard H, Weisbecker JL, Schelcher F, Andreoletti O. 26 February 2014. PrP expression level and sensitivity to prion infection. J. Virol. 10.1128/JVI.00369-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Edgeworth JA, Farmer M, Sicilia A, Tavares P, Beck J, Campbell T, Lowe J, Mead S, Rudge P, Collinge J, Jackson GS. 2011. Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet 377:487–493. 10.1016/S0140-6736(10)62308-2 [DOI] [PubMed] [Google Scholar]
- 48.Elder AM, Henderson DM, Nalls AV, Wilham JM, Caughey BW, Hoover EA, Kincaid AE, Bartz JC, Mathiason CK. 2013. In vitro detection of prionemia in TSE-infected cervids and hamsters. PLoS One 8:e80203. 10.1371/journal.pone.0080203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moudjou M, Sibille P, Fichet G, Reine F, Chapuis J, Herzog L, Jaumain E, Laferriere F, Richard CA, Laude H, Andreoletti O, Rezaei H, Beringue V. 2013. Highly infectious prions generated by a single round of microplate-based protein misfolding cyclic amplification. mBio 5:e00829–13. 10.1128/mBio.00829-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orrú CD, Wilham JM, Raymond LD, Kuhn F, Schroeder B, Raeber AJ, Caughey B. 2011. Prion disease blood test using immunoprecipitation and improved quaking-induced conversion. mBio 2:e00078–11. 10.1128/mBio.00078-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peden A, McCardle L, Head MW, Love S, Ward HJ, Cousens SN, Keeling DM, Millar CM, Hill FG, Ironside JW. 2010. Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia 16:296–304. 10.1111/j.1365-2516.2009.02181.x [DOI] [PubMed] [Google Scholar]
- 52.Béringue V, Herzog L, Reine F, Le Dur A, Casalone C, Vilotte JL, Laude H. 2008. Transmission of atypical bovine prions to mice transgenic for human prion protein. Emerg. Infect. Dis. 14:1898–1901. 10.3201/eid1412.080941 [DOI] [PMC free article] [PubMed] [Google Scholar]