

ABSTRACT
The RV144 HIV-1 vaccine trial demonstrated partial efficacy of 31% against HIV-1 infection. Studies into possible correlates of protection found that antibodies specific to the V1 and V2 (V1/V2) region of envelope correlated inversely with infection risk and that viruses isolated from trial participants contained genetic signatures of vaccine-induced pressure in the V1/V2 region. We explored the hypothesis that the genetic signatures in V1 and V2 could be partly attributed to selection by vaccine-primed T cells. We performed a T-cell-based sieve analysis of breakthrough viruses in the RV144 trial and found evidence of predicted HLA binding escape that was greater in vaccine versus placebo recipients. The predicted escape depended on class I HLA A*02- and A*11-restricted epitopes in the MN strain rgp120 vaccine immunogen. Though we hypothesized that this was indicative of postacquisition selection pressure, we also found that vaccine efficacy (VE) was greater in A*02-positive (A*02+) participants than in A*02− participants (VE = 54% versus 3%, P = 0.05). Vaccine efficacy against viruses with a lysine residue at site 169, important to antibody binding and implicated in vaccine-induced immune pressure, was also greater in A*02+ participants (VE = 74% versus 15%, P = 0.02). Additionally, a reanalysis of vaccine-induced immune responses that focused on those that were shown to correlate with infection risk suggested that the humoral responses may have differed in A*02+ participants. These exploratory and hypothesis-generating analyses indicate there may be an association between a class I HLA allele and vaccine efficacy, highlighting the importance of considering HLA alleles and host immune genetics in HIV vaccine trials.
IMPORTANCE The RV144 trial was the first to show efficacy against HIV-1 infection. Subsequently, much effort has been directed toward understanding the mechanisms of protection. Here, we conducted a T-cell-based sieve analysis, which compared the genetic sequences of viruses isolated from infected vaccine and placebo recipients. Though we hypothesized that the observed sieve effect indicated postacquisition T-cell selection, we also found that vaccine efficacy was greater for participants who expressed HLA A*02, an allele implicated in the sieve analysis. Though HLA alleles have been associated with disease progression and viral load in HIV-1 infection, these data are the first to suggest the association of a class I HLA allele and vaccine efficacy. While these statistical analyses do not provide mechanistic evidence of protection in RV144, they generate testable hypotheses for the HIV vaccine community and they highlight the importance of assessing the impact of host immune genetics in vaccine-induced immunity and protection. (This study has been registered at ClinicalTrials.gov under registration no. NCT00223080.)
INTRODUCTION
Vaccines are the most cost-effective form of public health intervention and have greatly reduced the global burden of infectious disease (1). While many licensed vaccines, such as those for smallpox, measles, and polio, are highly effective and have led to dramatic reductions in disease, others, such as the seasonal influenza vaccine and the Mycobacterium bovis BCG tuberculosis vaccine, offer only partial or heterogeneous protection (2, 3). The mechanisms underlying heterogeneous efficacy are often challenging to identify, though they may be partially due to variability in the host immune response to vaccination (4), which can vary with individual characteristics, such as age, gender, and major histocompatibility complex (MHC) group (5–9). Trials of HIV-1 vaccines, including the Step trial, in which prior immunity to adenovirus decreased innate and HIV-specific cellular immune responses to the adenovirus vectored vaccine (10–12), and the VaxGen Vax004 trial, in which race was associated with vaccine-induced neutralizing antibody responses (13), have elicited heterogeneous immune responses. Most recently, the RV144 trial of a canarypox vector prime (ALVAC-HIV) and bivalent rgp120 boost (AIDSVAX B/E) vaccine regimen was the first trial to show partial efficacy in reducing the risk of HIV-1 infections (14) and several studies have followed these results, attempting to understand the mechanisms of partial and potentially heterogeneous protection (15–24, 84). Specifically, Haynes et al. (15) designed a study intended to identify the immune correlates of risk (CoR) of infection, by comparing the rates of HIV-1 infection over time among vaccinated subgroups defined by their levels of vaccine-induced immune responses. The study identified two V2-specific immune response variables that significantly inversely correlated with the risk of infection: (i) levels of IgG binding to a gp70-scaffolded V1 and V2 (V1/V2) antigen and (ii) levels of IgG binding to a linear V2 peptide “hot spot.” These findings, together with more recent studies of V2-specific antibodies isolated from trial participants (18, 21, 22), suggest that vaccine-induced immune responses to the V1/V2 region of envelope may have had antiviral functions and predicted the efficacy observed in the trial.
Informed by these findings, which implied that the vaccine exerted immune pressure on the V1/V2 region, Rolland et al. (17) conducted a V1/V2-focused sieve analysis of the envelope sequences of viruses that infected RV144 participants. The sieve analysis—a statistical comparison of the viruses isolated from HIV-infected trial participants in the vaccine group versus the placebo group—showed genetic differences relative to the vaccine sequences at sites 169 and 181 in the V2 loop (HXB2 numbering). This was consistent with the hypothesis that vaccine-induced antibodies targeting the V1/V2 region selectively blocked specific genetic variants from infection. However, as with any sieve analysis, the immune mechanism applying pressure could not be discerned from the viral sequences alone; nor could it eliminate the possibility that the sieve effect was induced by postacquisition pressure that was present prior to viral sequencing (25, 26). Given the growing evidence that CD8+ and CD4+ T-cell-induced viral escape can happen in the acute phase of infection (27–31), additional, not necessarily mutually exclusive hypotheses could be that cytolytic T cells selectively targeted HIV-1 prior to the establishment of infection or that, after infection, they induced viral escape mutations in vaccine epitopes prior to viral sequencing. These hypotheses are analogous to those proposed in the analysis of the Step HIV vaccine trial (25), in which sieve effects were attributed mainly to selective pressure from T cells. Though the CD8+ T-cell response rate was low overall in the RV144 trial, the envelope-specific CD4+ T-cell response rate was 63% (14, 19, 32) and there are preliminary data suggesting that the RV144 vaccine regimen may have induced envelope-specific responses in CD8+ T cells of the mucosa (Schuetz et al., unpublished).
Therefore, we conducted a CD4+ and CD8+ T-cell-based sieve analysis focused on the V1/V2 region of the envelope protein (HXB2 positions 143 to 185), hypothesizing that the sieve effects identified in this region could be partially attributed to selective pressure from cytotoxic T cells. Specifically, we tested for a sieve effect associated with “HLA binding escape,” as predicted using computational HLA-peptide binding algorithms (33–35). We defined “binding escape” as any variation in HIV peptide sequences from infected participants, relative to predicted vaccine epitopes, that substantially reduced the HLA binding affinity. Our analysis identified A*02- and A*11-restricted CD8+ T-cell epitopes that were found only in the subtype B (MN) component of the protein boost, in which the number of predicted viral HLA binding escapes was greater in vaccine compared to placebo recipients. Although we hypothesized that such an effect was indicative of postacquisition selection pressure, we also found that vaccine efficacy (VE) was significantly increased in participants carrying the class I HLA allele A*02. While associations of HLA genotype with vaccine-induced immune responses have been previously noted (36–40), this is the first report of a potential association with vaccine efficacy. We also found evidence suggesting that the HLA A*02 allele may have modulated two vaccine-induced antibody responses and their associations with infection risk in the trial. Taken together, the results of this exploratory statistical analysis generate the hypothesis that HLA A*02 may have played a role in the partial efficacy that was observed in RV144. Importantly, they suggest that additional studies of T-cell responses to the subtype B protein component of the RV144 vaccine should be conducted in both blood and mucosal samples.
MATERIALS AND METHODS
Ethics statement.
The RV144 protocol was approved by the Institutional Review Boards of the Ministry of Public Health, the Royal Thai Army, Mahidol University, and the U.S. Army Medical Research and Materiel Command (ClinicalTrials.gov no. NCT00223080) (14). Written informed consent was obtained from all participants.
Study design and vaccine sequences.
Details about the cohort of volunteers enrolled in the RV144 HIV-1 vaccine trial and the subsequent study of case-control immune correlates are presented by Rerks-Ngarm and Pitisuttithum (14) and Haynes et al. (15) in the respective primary articles. Briefly, the vaccine regimen consisted of four injections (at 0, 1, 3, and 6 months) of the canarypox vector ALVAC-HIV (vCP1521), which expresses gp120 of CRF01_AE (92TH023), and two injections (at 3 and 6 months) of AIDSVAX B/E, which is composed of two gp120 proteins truncated at the amino terminus (start at amino acid 42): MN (subtype B) and CM244 (CRF01_AE). Following the same approach to screening amino acid sites taken by Rolland et al. (17), we conducted a focused analysis of the alignable portions of the V1/V2 region from amino acid residue 143 to residue 185 (HXB2 numbering).
HIV-1 sequencing.
Viral genomes from infected RV144 participants were reported previously (17, 25). Briefly, from each participant we obtained 2 to 14 (median, 10) near-full-length-genome sequences from plasma specimens collected at the time of HIV-1 diagnosis, typically between 1 and 6 months after infection. Intrasubject sequence diversity was low, with an average of 1.08 ± 0.02 unique amino acid residues per person at each site within the V1/V2 region. At any given site, at least 80% of participants had a single amino acid residue in all of their breakthrough sequences. Therefore, we limited our analysis to one representative sequence per individual, using the sequence with the smallest Hamming distance to the subject's consensus sequence. The data set analyzed here contained sequences from 109 infected participants (all CRF01_AE strains), after excluding participants who were infected with non-CRF01 AE viruses and 1 participant who was found to have been infected by another RV144 trial participant.
T-cell-based sieve analysis.
A sieve effect can be understood as a statistical difference between the genetic sequences of viruses that infected vaccine recipients and the genetic sequences of viruses that infected placebo recipients in a treatment-randomized, double-blind placebo-controlled vaccine trial (41). Here we describe a “Binding Escape Count” sieve method, a T-cell-based sieve analysis method that estimates the number of predicted HLA binding escapes in the amino acid sequences of viruses that infected trial participants. Escapes were identified by comparing the vaccine immunogen sequences to the sequences of viruses isolated from trial participants. For this comparison, we used all three vaccine immunogen sequences independently, 92TH023 in ALVAC-HIV (vCP1521) and CM244 and MN in the bivalent AIDSVAX B/E rgp120 boost, where 92TH023 and CM244 are CRF01_AE and MN is subtype B. Analysis was restricted to the V1/V2 region of envelope protein (HXB2 positions 143 to 185).
To count binding escapes in the viral sequence from a single participant, we first predicted likely vaccine-induced T-cell epitopes using a computational predictor of either class I HLA binding to 9-mer peptides (ADT [adaptive double threading]) (33, 42) or class II HLA binding to 15-mer peptides (NetMHCIIpan) (35). Using the vaccine sequence and the participant's HLA-A, -B, and -DRB alleles, we identified as putative epitopes any peptides that were predicted to bind to one of their HLA alleles with a level of affinity (i.e., half-maximal inhibitory concentration [IC50]) lower than a specific “binder” threshold. We then considered the sequence of the corresponding peptide in the aligned sequence of the virus isolated from the participant. If changes in the amino acids within an epitope weakened the binding affinity beyond the “escape” threshold, it was then counted as an escape. The number of binding escapes was summed for each participant, and the resulting sums were compared between the vaccine and placebo groups using a t-statistic. Within some predicted epitopes, a viral isolate may differ from the vaccine immunogen at several residues whereas another isolate may differ at only a single residue; however, the determination of which isolate is counted as an escape is based on the change in the predicted HLA binding affinity which considers all residues within the epitope. For this reason, the use of the term “escape” may differ from its typical usage to refer to single point mutations that result in abrogation of a T-cell epitope.
The Binding Escape Count T-cell-based sieve method has two adjustable parameters: (i) a binder threshold and (ii) an escape threshold. Typically, peptide-HLA interactions are classified according to two binding thresholds as either “strong” (<50 nM) or “weak” (50 to 500 nM) binders (34). However, since binding affinity is a dynamic process and is only one of several factors that determine the set of T-cell responses in a given individual, we considered binding thresholds that spanned a biologically relevant range (50 to 1,100 nM). Each binding threshold determines the set of epitopes that are used for comparing the vaccine sequence to the participants' viral sequences, with lower IC50s corresponding to stronger binding affinities and fewer predicted epitopes. Similarly, we considered a range of thresholds in defining HLA binding escape (IC50 = 500 to 8,000 nM). Each escape threshold defines nonbinding peptides. We tested for a sieve effect over the entire range of parameters and used an absolute maximum t-statistic test to calculate an overall P value. Conceptually, rather than testing if there was a sieve effect using each pair of binder and escape thresholds, the test asks if there is any pair of thresholds that yields a significant sieve effect. To calculate the P value for this test, we permuted participant treatment labels and, with each permuted data set, we computed the absolute maximum t-statistic over the range of all pairs of binding and escape thresholds. By repeating this procedure 20,000 times, we obtained a distribution of t-statistics under the null hypothesis that the vaccine had no effect. The overall P value was defined as the fraction of permuted data sets in which the absolute maximum t-statistic was greater (corresponding to a stronger sieve effect) than the one observed in the actual (unpermuted) data set. The results of this sieve method, including predicted epitopes and binding escapes, are presented using the optimal binder and escape thresholds that led to the maximum t-statistic, but the reported P value is always the overall P value that accounts for multiple testing over the spectrum of thresholds.
The “site-specific” sieve methods employed in Rolland et al. (17) differ from the Binding Escape Count method. The site-specific sieve methods analyze the distribution of residues at individual amino acid sites one at a time and count all mutations relative to the vaccine residues. In contrast, the Binding Escape Count T-cell-based sieve method tests whether the observed genetic differences can be attributed to vaccine-induced T-cell immune responses by assessing short peptides. This method is sensitive only to mutations in predicted T-cell epitopes that alter HLA binding affinity. Moreover, each vaccine epitope is considered only within the subset of participants who carry the restricting HLA allele. Due to these differences, it is possible that a T-cell-driven sieve effect that is missed by a site-specific method could be identified by the Binding Escape Count method. It is also possible that sieve effects mediated by other immune responses could be detected by a site-specific method and yet not be found to be attributable to T-cell selection by this method. Importantly, a sieve effect may be the result of multiple mechanisms of selection, and neither this method nor the site-specific sieve analysis method can rule out any single mechanism of immune pressure.
HLA genotyping of volunteers.
High-resolution typing of class I HLA-A, -B, and -C loci was performed on samples obtained from all the infected cases in the trial and the uninfected control participants selected for the study of the case-control immune correlates (15). Typing was performed using both DNA sequence-based typing (SBT) and the sequence-specific oligonucleotide probe (SSOP) method, with fully concordant results. SBT was carried out by PCR amplification and sequencing of exons 2 and 3, with ambiguous types being resolved to four digits (43) using the dbMHC SBT interpretation interface (http://www.ncbi.nlm.nih.gov/projects/gv/mhc/). The SSOP method was performed using a LABType SSO Class I HD system (One Lambda, Canoga Park, CA), which is based on Luminex xMAP technology, and results were interpreted using the accompanying HLA Fusion 2.0.0 software. HLA types are reported according to the IMGT/HLA nomenclature (version 3.7.0; http://www.ebi.ac.uk/imgt/hla/ambig.html). The HLA haplotype analysis was completed using the HLA I and HLA II genotypes of uninfected RV144 volunteers and the PyPop software package (44). The T-cell-based sieve analysis uses the four-digit HLA types; however, subsequent associations with VE and immune measurements were studied using two-digit HLA types to increase statistical power.
Vaccine efficacy and immune correlates of risk of HIV infection.
Overall vaccine efficacy and viral genotype-specific vaccine efficacy were estimated in HLA A*02-positive and A*02-negative (A*02+/−) subgroups using the “case-only” method (45), which uses data only from the infected subjects. The case-only method is valid only if the trial is randomized and has a rare study outcome, and the RV144 trial meets both criteria. The method is optimally powerful (adding host genetic information from uninfected vaccine recipients would not increase statistical power) and cost-effective because only data from infected cases are needed. Immune correlates of risk were assessed in HLA+ and HLA− vaccinated subgroups using the same method as that used in the original study of RV144 immune correlates (15)—logistic regression with subject weighting to account for the two-phase sampling case-control design. To test if each response variable was differentially correlated with infection risk in the A*02+ versus A*02− vaccinated subgroups, an A*02 interaction variable was included in the regression model. Immune measurements in the IgA-ConsensusA and IgA-C1 biotin assays were treated as positive or negative responses, and because of small numbers of vaccine recipients in some HLA × immune response × infection status strata, we used Zelen's exact test of homogeneity of odds ratios (OD) (46) to determine whether the response variable was differentially correlated with infection risk in A*02+ versus A*02− vaccine recipients. This test gives a P value for the null hypothesis that the odds ratios of infection risk for a positive immune response versus a negative immune response are identical in the A*02+/− vaccinated subgroups.
Experimental validation of the A*02-KMQKEYALL epitope.
The A*02-KMQKEYALL epitope was validated using peripheral blood mononuclear cell (PBMC) samples from subjects in a long-term nonprogressor HIV-1-infected cohort (47). The subjects were recruited and enrolled at the U.S. NIH-sponsored HIV Vaccine Trials units. The appropriate Institutional Review Boards approved the studies, and volunteers provided written consent. Details of the experiments are included in the supplemental material. Briefly, we measured functional T-cell responses in cryopreserved peripheral blood mononuclear cells (PBMCs) using a gamma interferon enzyme-linked immunosorbent spot (ELISpot) assay. We used 15-mer and 9-mer peptides from the V1/V2 region of envelope, including variants from the vaccine and breakthrough strains (see Table SA2 and Table SA3 in the supplemental material).
MHC purification and peptide-binding assays.
Purification of MHC molecules by affinity chromatography has been detailed elsewhere (48). Briefly, Epstein-Barr virus (EBV)-transformed homozygous cell lines or single MHC allele-transfected 721.221 or C1R lines are utilized as sources of HLA class I MHC molecules. HLA molecules are purified from cell pellet lysates by repeated passage over protein A Sepharose beads conjugated with the W6/32 (anti-HLA-A, -B, and -C) antibody. In some cases, HLA-A molecules may be separated from HLA-B and -C molecules by prepassage over a B1.23.2 (anti-HLA-B, -C, and some -A) column. Protein purity and concentration and the effectiveness of depletion steps are monitored by SDS-PAGE and bicinchoninic acid (BCA) assay.
Assays to quantitatively assess peptide binding to class I MHC molecules are based on the inhibition of binding of a high-affinity radiolabeled peptide to purified MHC molecules and are performed essentially as detailed elsewhere (48–50). Briefly, 0.1 to 1 nM radiolabeled peptide is coincubated at room temperature with 1 μM to 1 nM purified MHC in the presence of a cocktail of protease inhibitors and 1 μM β2-microglobulin. Following a 2-day incubation, MHC-bound radioactivity is determined by capturing MHC/peptide complexes on W6/32 (anti-class I) antibody-coated Lumitrac 600 plates (Greiner Bio-one, Frickenhausen, Germany) and measuring bound cpm using a TopCount (Packard Instrument Co., Meriden, CT) microscintillation counter. In the case of competitive assays, the concentration of peptide yielding 50% inhibition of the binding of the radiolabeled peptide is calculated. Under the conditions utilized, where [label] < [MHC] and IC50 ≥ [MHC], the measured IC50s are reasonable approximations of the true dissociation constant (Kd) values (51, 52). Each competitor peptide is tested at six different concentrations covering a 100,000-fold dose range and in three or more independent experiments. As a positive control, the unlabeled version of the radiolabeled probe is also tested in each experiment.
RESULTS
T-cell-based sieve analysis finds more HLA binding escapes in viruses isolated from vaccine recipients than in viruses isolated from placebo recipients.
To address the hypothesis that the sieve effects previously identified in the V1/V2 region of envelope (17) could be attributed to vaccine-induced selective pressure from cytolytic T cells (27–31), we performed a CD4+ and CD8+ T-cell-based sieve analysis of the V1/V2 region (HXB2 143 to 185) of viruses that were isolated from infected trial participants. This statistical method compares the sequences of the vaccine immunogens to those of the isolated viruses and identifies amino acid differences in predicted vaccine epitopes that abrogate HLA binding. In some cases, a viral isolate may differ from the vaccine immunogen at several residues within an epitope and the determination of whether these differences constitute a “binding escape” depends on the effect of these differences on the predicted HLA binding affinity, relative to that of the vaccine immunogen. A sieve effect was detected if the distributions of these viral “binding escapes” indicated significantly greater numbers in viruses isolated from vaccine than in viruses isolated from placebo recipients. In this analysis, we considered the sequences of each of the three vaccine immunogens independently, including the ALVAC-HIV (vCP1521) 92TH023 CRF01_AE sequence and the AIDSVAX B/E protein boost with CM244 CRF01_AE and MN subtype B sequences (see Materials and Methods for details).
Of the three vaccine immunogens that were tested, only epitopes in the MN protein boost showed evidence of viral binding escape that was greater in vaccine recipients than in placebo recipients (P value = 0.018). Though the sieve analysis was a test for a vaccine treatment effect summing the whole V1/V2 region, once the effect was detected, the epitopes and HLA alleles driving the effect were examined. Based on the HLA alleles that were present in infected RV144 participants, there were 12 predicted CD8+ T-cell epitopes in the MN sequence V1/V2 region (Fig. 1A; see also Table SA1 in the supplemental material). These epitopes, all characterized by HLA-peptide binding affinities below 80 nM, were evenly distributed among the treatment groups (Fig. 1B), with 91% of vaccine recipients and 83% of placebo recipients having at least one predicted epitope (P = 0.37). Only three of these epitopes, restricted by alleles expressed by 72% of vaccine and 65% of placebo recipients, were associated with viral HLA binding escape (Fig. 1C): A*11-GTIKGGEMK (HXB2 147 to 155), A*11-TSIGDKMQK (163 to 171), and A*02-KMQKEYALL (168 to 176). The number of HLA binding escapes detected in the corresponding viral 9-mers was significantly higher in vaccine recipients (Fig. 1D) (average escapes per participant of 0.72 in the vaccine group versus 0.29 in the placebo group; P value = 0.018). This evidence of a T-cell-based sieve effect is specific to epitopes that were predicted in the sequence of the MN strain rgp120 immunogen. Due to differences in the sequences (Fig. 1A) of the 92TH023 strain immunogen and the CM244 strain immunogen compared to the MN strain immunogen, some peptides are predicted epitopes in only the MN immunogen. These differences account for the lack of evidence for a sieve effect for the CRF01_AE immunogens (92TH023 P value = 0.28; CM244 P value = 0.25). Though 89% of the infections in the trial were CRF01_AE, it is possible that an MN-derived epitope may have applied cross-reactive pressure on the viruses (see Discussion for details).We performed an identical analysis using the class II HLA alleles of participants to test for binding escape in predicted CD4+ T-cell epitopes. Though vaccine peptides in V1/V2 were predicted to bind many class II alleles carried by RV144 participants (see Fig. SA1 in the supplemental material), we did not detect a difference in the numbers of predicted binding escapes in vaccine recipients versus placebo recipients.
FIG 1.

CD8+ T-cell-associated sieve effect. (A) Twelve 9-mer peptides of MN gp120 were identified as potential vaccine epitopes (horizontal bars and boxed residues) based on their predicted binding affinities (IC50) with the HLA alleles of RV144 participants. Their locations in the amino acid sequence of V1/V2 are shown relative to those in the MN strain (subtype B) and the 92TH023 strain (CRF01_AE) gp120 sequences. Three of these 9-mers were identified as peptides in which HLA “binding escapes” were predicted in at least one infected participant (boxed amino acid residues), with the restricting allele appearing below each epitope. (B) The percentage of infected participants with an allele that was predicted to bind each of the 12 potential epitopes is shown, with the epitope identified by its N-terminal starting amino acid. (C) The percentage of each treatment group that had predicted binding escapes in the three 9-mer peptides within which any escape was predicted. These epitopes are indicated at the bottom of the figure with their starting position, designation, and predicted restricting allele. (D) The two bars show the percentage of infected participants in each group whose viral isolate had 0, 1, or 2 predicted binding escapes in the V1/V2 region.
HLA A*02 allele modifies overall vaccine efficacy.
We hypothesized that if the predicted HLA binding escapes in the V1/V2 region were indicative of vaccine-induced anamnestic responses, then vaccine efficacy (VE) should not have been different in the subgroups of participants with and without each of the HLA alleles implicated in the sieve analysis. To test this hypothesis, we first estimated VE in A*11+ and A*11− participants and found no difference (“Case-only” method [45] testing A*11 and VE interaction, P = 0.45) (Fig. 2A and Table 1). For this reason, we did not further consider the effects of A*11 in this study. We then estimated VE in the A*02+/− subgroups and found that estimated VE in participants with A*02 was 54% (P = 0.006 for nonzero VE; 95% confidence interval [CI], 21% to 73%), which was higher than the estimated overall VE (31%) and significantly higher than that in participants without the A*02 allele (3%; P = 0.90 for nonzero VE) (A*02+/− interaction P = 0.050) (Fig. 2A and Table 1). These estimates of VE were based on the 125 infections in the modified intent-to-treat (mITT) cohort, whereas the sieve analysis was based on a subset of 109 CRF01_AE infections. We recomputed VE in A*02+/− subgroups within this subset of infections, thereby estimating the VE against CRF01_AE viruses. The results were similar to the overall results: VE for the A*02+ participants was 60% (CI, 26% to 78%, P = 0.004), which was significantly greater than that for the A*02− participants, for whom VE was 3% (CI, −62% to 43%, P = 0.90) (A*02+/− interaction P = 0.034). Together, these findings establish A*02 as a borderline-significant effect modifier of VE and suggest that the A*02 allele and the predicted A*02 vaccine epitope in V2 may be related to protective vaccine-induced immune responses.
FIG 2.
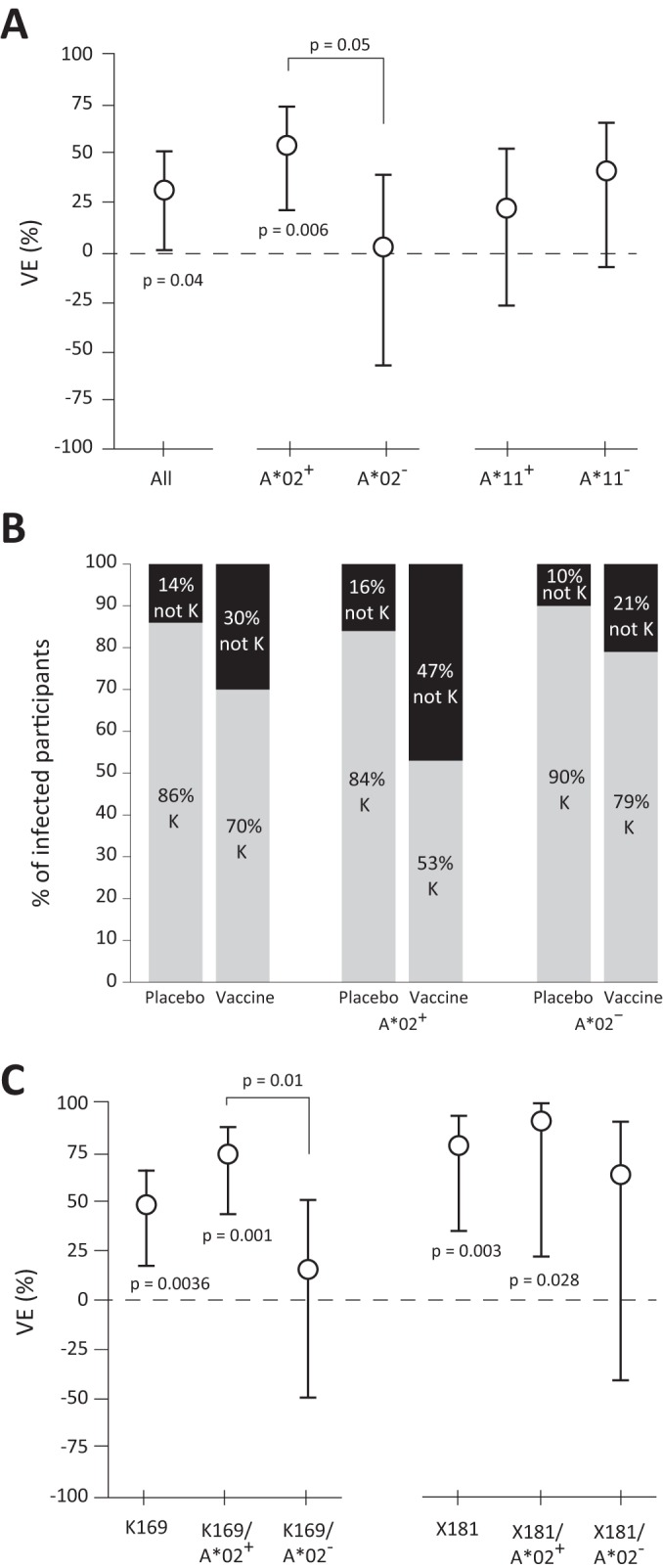
Greater VE in A*02+ participants. (A) Estimated VE in the RV144 trial for the entire cohort and also for participants with and without the HLA A*02 allele. Error bars indicate 95% confidence intervals. For each group along the x axis, significant P values are given for the hypothesis test that VE = 0. The P value shown between the A*02+/− subgroups indicates a significant difference in VE between the two groups. (B) The percentage of infected participants whose isolated virus contained the vaccine-matched lysine at position 169 is reported as a percentage of the treatment group and/or A*02+/− subgroup indicated along the x axis. The pair of columns on the left refer to the entire cohort, whereas the next two sets divide the cohort into A*02+/− subgroups. (C) Estimated genotype-specific VE against K169 and X181 viruses. Estimates are reported for the whole cohort and also for A*02+/− subgroups, as indicated on the x axis. Error bars show the 95% confidence interval for each VE estimate.
TABLE 2.
HLA A*02 modifies genotype-specific VE against K169 virusesa
| HLA type | Site 169 | No. of infections |
Est. VE (%) | 95% CI (%) |
P value (H0: VE = 0) | A*02+/− interaction P value | ||
|---|---|---|---|---|---|---|---|---|
| Vaccine | Placebo | Lower | Upper | |||||
| Overall | Match (K) | 30 | 57 | 48 | 18 | 66 | 0.0036 | — |
| A*02+ | 8 | 31 | 74 | 44 | 88 | 0.001 | 0.02 | |
| A*02− | 22 | 26 | 15 | −49 | 52 | 0.56 | ||
| Overall | Mismatch (X) | 14 | 9 | −55 | −258 | 33 | 0.3 | — |
| A*02+ | 8 | 6 | −33 | −284 | 54 | 0.59 | 0.65 | |
| A*02− | 6 | 3 | −100 | −700 | 50 | 0.32 | ||
—, not applicable.
TABLE 1.
HLA A*02 modifies VEa
| HLA type | No. of infections |
Est. VE (%) | 95% CI (%) |
P value (H0: VE = 0) | HLA+/− interaction P value | ||
|---|---|---|---|---|---|---|---|
| Vaccine | Placebo | Lower | Upper | ||||
| Overall | 51 | 74 | 31 | 1 | 51 | 0.04 | — |
| A*02+ | 19 | 41 | 54 | 21 | 73 | 0.006 | 0.050 |
| A*02− | 32 | 33 | 3 | −58 | 40 | 0.9 | |
| A*11+ | 29 | 37 | 22 | −27 | 52 | 0.33 | 0.45 |
| A*11− | 22 | 37 | 41 | −0.8 | 65 | 0.053 | |
Est., estimated; —, not applicable.
HLA A*02 modifies genotype-specific vaccine efficacy at site 169 in the V2 loop.
Previously, Rolland et al. (17) reported a sieve effect in the sequences of viruses that were isolated from infected RV144 participants. Specifically, sieve effects were identified at two positions, 169 and 181 (HX2B numbering), in the V2 loop of envelope. The effect at site 169 was evidenced by a relative enrichment of the amino acid lysine (K) in viruses that infected placebo recipients compared to those that infected vaccine recipients. The 92TH023 and CM244 vaccine sequences also contain K at site 169, while the MN sequence has methionine (M). Though the majority of infections were type CRF01_AE and none of the viral isolates had M at site 169 and since we observed a T-cell-based sieve effect that depended on the A*02 epitope KMQKEYALL (168 to 176), in which position 169 was an anchor residue, we hypothesized that the K169 sieve effect was related to the A*02 epitope and would be stronger in the A*02+ subgroup. To test this, we computed genotype-specific VE against viruses matching K169 in the A*02+/− subgroups. While the overall VE against viruses matching K169 was 48% (17), we found that VE against K169 viruses in the A*02+ participants was 74% (P = 0.001 for nonzero VE; 95% CI, 44% to 88%) (Fig. 2A and Table 2) and was significantly higher than in A*02− participants, estimated at 15% (P = 0.56 for nonzero VE; 95% CI, −49% to 52%) (A*02+/− interaction P = 0.01). These estimates of genotype-specific VE fundamentally depend on a difference in the numbers of viruses matching K169 in vaccine recipients versus placebo recipients (70% versus 86%, respectively; Fig. 2B). While this difference was evident in the A*02+ subgroup, with 53% versus 84% matching the K169 genotype, the vaccine and placebo recipients in the A*02− subgroup were more similar (79% versus 90%). This finding suggested that the immune responses in the A*02+ vaccine recipients—compared to all other study participants—preferentially blocked infection by or selected against viruses matching the 92TH023 and CM244 vaccine sequences at position 169.
Since Rolland et al. (17) also reported a sieve effect at position 181, we repeated the above analysis on the basis of the virus genotypes at position 181. For the entire cohort, VE against viruses not matching the ALVAC vaccine at position 181 (X181) was 78% (P = 0.0028 for nonzero VE; Fig. 2C) whereas the VE against matched viruses (I181) was 17% (P = 0.38 for nonzero VE) (Table 3). Our analyses showed that whereas VE against X181 viruses in A*02+ participants was 90% (P = 0.028 for nonzero VE; 95% CI, 22% to 99%), it was not significantly different from VE against the same X181 viruses in A*02− participants (63%; P = 0.15 for nonzero VE) (A*02+/− interaction P = 0.29). Therefore, there was no statistical evidence that the A*02 allele modified genotype-specific VE against X181 viruses.
TABLE 3.
HLA A*02 does not modify genotype-specific VE against I181 or I181X virusesa
| HLA type | Site 181 | No. of infections |
Est. VE (%) | 95% CI (%) |
P value (H0: VE = 0) | A*02+/− interaction P value | ||
|---|---|---|---|---|---|---|---|---|
| Vaccine | Placebo | Lower | Upper | |||||
| Overall | Match (I) | 40 | 48 | 17 | −26 | 45 | 0.38 | — |
| A*02+ | 15 | 27 | 44 | −4 | 70 | 0.068 | 0.08 | |
| A*02− | 25 | 21 | −19 | −113 | 33 | 0.56 | ||
| Overall | Mismatch (X) | 4 | 18 | 78 | 35 | 93 | 0.0028 | — |
| A*02+ | 1 | 10 | 90 | 22 | 99 | 0.028 | 0.29 | |
| A*02− | 3 | 8 | 63 | −41 | 90 | 0.15 | ||
—, not applicable.
Experimental validation of the A*02-KMQKEYALL epitope.
The T-cell-based sieve analysis identified differential HLA binding escape results in a predicted A*02-KMQKEYALL epitope. However, the HIV LANL epitope database (http://www.hiv.lanl.gov/content/immunology/) does not list any epitopes in the V2 region. To experimentally confirm the predicted A*02 CD8+ T-cell epitope, we first conducted HLA-binding experiments to test the binding of the KMQKEYALL peptide to HLA A*02. We found that the epitope binds strongly to A*0203 (IC50, 12 nM) and A*2402 (IC50, 21 nM) and less strongly to A*0201 (IC50, 719 nM) (see Table SA2 in the supplemental material). We then assayed PBMC from 18 HIV-1-infected individuals from a long-term nonprogressor cohort who were A*02+ (see the supplemental material for details). Using ELISpot and intracellular cytokine staining (ICS) assays, we identified one A*0201+ individual (A*02G1, A*03G1, B*08, B*15, Cw*04, Cw*07) that had a CD8+ T-cell response to the KMQKEYALL peptide (magnitude of 118 spot-forming cells [SFC] per million). Taken together, these data show that the KMQKEYALL peptide binds to HLA A*02 alleles and can elicit functional CD8+ responses in an HIV-infected individual.
HLA A*02 modifies immune correlates of risk of HIV infection.
The statistical association between the HLA A*02 allele and overall and K169-genotype-specific VE led us to question whether A*02 could also have a role in modifying the associations of vaccine-induced immune responses with the risk of infection. Therefore, we assessed A*02 as an effect modifier of the immune correlates of infection risk that were previously identified (15). Due to the small number of infected vaccine recipients who carry A*02 (19 individuals) and the large number of possible immune measurements to study, we preserved statistical power by restricting analyses to six immune measurements that were identified as correlates of risk in the previous analyses: (i) Env V1/V2 IgG antibody binding, (ii) IgA binding to a panel of Env isolates (M-B gD), (iii) IgA binding to consensus A Env gp140, (iv) IgA antibody binding to gp120-C1, (v) V2 hot spot peptide microarray, and (vi) PBMC Luminex cytokine score (see the supplemental material for details). For each of these measurements, we tested if the presence of A*02 significantly modified the previously reported correlation with the risk of infection.
The V2 hot spot immune variable, an average of the magnitude of antibody responses to overlapping V2 peptides from 6 HIV-1 subtypes in a peptide microarray, was previously found to correlate inversely with the risk of infection in vaccine recipients; we found no evidence that the presence of A*02 modified this effect. Following this, we performed a focused analysis that considered only responses to the two subtype B peptides that contained a variant of the predicted A*02-KMQKEYALL epitope. We found that responses to one peptide, TSIRDKVQKEYALFY (positions 163 to 182 [HXB2 numbering]), were directly correlated with the risk of infection in vaccine recipients lacking A*02 (OR of infection per standard deviation [SD] = 1.92; 95% CI, 1.07 to 3.45; P = 0.030) but were not correlated with that in vaccine recipients who expressed the A*02 allele (OR of infection per SD = 0.84; 95% CI, 0.42 to 1.70; P = 0.63) (A*02+/− interaction P value = 0.060) (Fig. 3A and Table 4). This analysis considered the peptide microarray measurement as a continuous quantitative variable. The finding was also reproduced in an analysis that categorized responses into high, medium, and low tertiles of vaccine recipient responses, similar to the categorical analysis by Haynes et al. (15). Medium responses compared to low responses were not significantly associated with the risk of infection in either of the A*02+/− subgroups (Table 4). However, high levels compared to low levels of antibodies were associated with increased risk of infection in the A*02− subgroup (OR for high versus low response = 6.04; 95% CI, 1.80 to 20.3; P = 0.0036) but not in the A*02+ subgroup (OR for high versus low response = 0.66; 95% CI, 0.15 to 2.96; P = 0.59) (Table 4). A P value for the test of the hypothesis that these categorized responses were significantly different in the A*02+/− vaccine recipients was not computed due to the low number of responders in each category.
FIG 3.
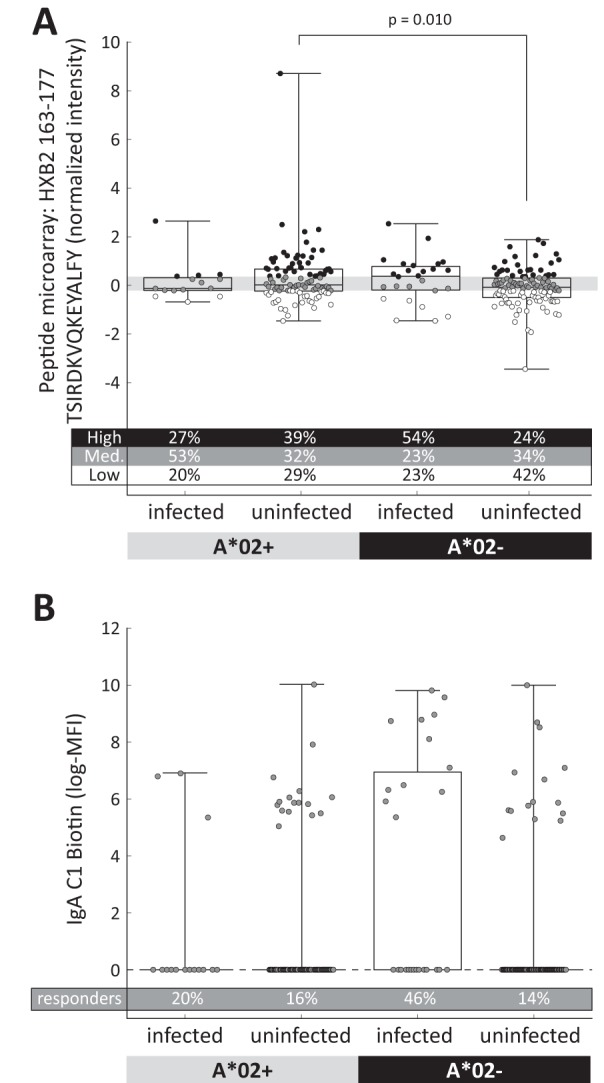
HLA A*02 modifies vaccine-induced immune responses and correlates of infection risk. Vaccine-induced immune responses were associated with the expression of HLA A*02. Assays were performed as part of the analysis of case-control immune correlates (15) using serum samples obtained 2 weeks after the fourth and final vaccine injection (week 26). Vaccine recipients were further divided into A*02+ (n = 15 infected, 97 uninfected) and A*02− (n = 26 infected, 108 uninfected) subgroups for analysis. (A) Normalized intensity representative of antibody responses to the TSIRDKVQKEYALFY peptide (Env V2 positions 163 to 177 [HXB2 numbering]) in the peptide microarray assay are shown using box plots. The extent of the box indicates the interquartile range, with a line indicating the median and whiskers indicating the full range. Responses from all case-control participants were divided into high, medium, and low tertiles, with the gray box showing the medium responses. Each dot represents a single response, its color indicating whether it is in the high (black), medium (gray), or low (white) tertile. The percentages of responses in each tertile are indicated below the box plot. One outlier response in the A*02+-uninfected subgroup (8.07) is not plotted. (B) The IgA-C1 biotin assay was analyzed as a binary variable, with positive responders having a measurement > 0 log(MFI [median fluorescence intensity]). The percentage of positive responders in each subgroup is indicated below the box plot.
TABLE 4.
HLA A*02 modifies immune correlates of risk of HIV-1 infectiona
| Immune variable | HLA type | Est. OR | 95% CI |
P value (H0: OR = 1) | A*02+/− interaction P value | A*02+/− interaction q value | |
|---|---|---|---|---|---|---|---|
| Lower | Upper | ||||||
| IgA C1 biotin* (positive vs negative response) | Overall | 3.15 | 1.48 | 6.71 | 0.003 | — | — |
| A*02+ | 1.26 | 0.31 | 5.15 | 0.75 | 0.13 | 0.44 | |
| A*02− | 6.01 | 2.19 | 16.5 | 0.0005 | |||
| Peptide microarray 163-TSIRDKVQKEYALFY (quantitative analysis**) | A*02+ | 0.84 | 0.42 | 1.70 | 0.63 | 0.060 | 0.41 |
| A*02− | 1.92 | 1.07 | 3.45 | 0.030 | |||
| Peptide microarray 163-TSIRDKVQKEYALFY (med vs low response***) | A*02+ | 1.71 | 0.40 | 7.37 | 0.47 | — | — |
| A*02− | 1.63 | 0.48 | 5.56 | 0.44 | — | — | |
| Peptide microarray 163-TSIRDKVQKEYALFY (high vs low response***) | A*02+ | 0.66 | 0.15 | 2.96 | 0.59 | — | — |
| A*02− | 6.04 | 1.80 | 20.3 | 0.0036 | — | — | |
*, OR is for vaccine recipients with a positive versus a negative response using positivity criteria determined previously (15); **, OR is per standard deviation change in the assay measurement; ***, OR is for vaccine recipients with a high (or medium) response versus a low response; —, not applicable.
We also found that a borderline-significant direct correlation between IgA-C1 binding and infection risk is evident only for the A*02− subjects (A*02+/− interaction P value = 0.13), with OR = 6.01 (P = 0.0005; 95% CI, 2.19 to 16.5) for A*02− subjects and OR = 1.26 (P = 0.75; 95% CI, 0.31 to 5.15) for A*02+ subjects (Fig. 3B and Table 4). These results suggest that the increased infection risk for vaccine recipients with positive IgA-C1 binding levels may have been present only in vaccine recipients who lacked the A*02 allele (53). In tests of the other four immune measurements that were previously identified as correlates of risk, we found no statistical evidence that either the correlations with the risk of infection or the immune responses were different in the A*02+ and A*02− vaccine recipient subgroups.
HLA A*02 modifies the vaccine-induced V2-specific immune response.
Whereas an assessment of A*02 as a possible modifier of immune CoRs involves comparing infected and uninfected vaccine recipients, it is also important to assess A*02 as a possible modifier of vaccine-induced immune responses, regardless of their impact on infection risk. To address this, we tested whether the immune responses in each of the assays tested above were significantly different in A*02+ versus A*02− uninfected vaccine recipients. We found that, in the peptide microarray, the antibody response to the subtype B peptide TSIRDKVQKEYALFY (positions 163 to 182 [HXB2 numbering]) was significantly greater in A*02+ (median = −0.039 normalized intensity) than in A*02− (median = −0.107 normalized intensity) uninfected vaccine recipients (P = 0.010, Wilcoxon rank sum test), indicating that the presence of A*02 may have modulated the vaccine-induced immune response, albeit the magnitude of the effect is small. There were no other significant differences found between the A*02+/− subgroups in the five additional assays that were tested (see the supplemental material).
DISCUSSION
We performed a novel sieve analysis of viruses isolated from infected RV144 participants focused on the V1/V2 region of envelope to assess whether the sieve effects observed in this region could be partially attributed to HLA binding escape under the selective pressure of vaccine-primed T cells. We found that the amino acid residues of the viruses differed from those of the MN strain rgp120 vaccine immunogen in a way that substantially decreased the predicted HLA binding affinity of three predicted CD8+ T-cell vaccine epitopes. These differences were significantly more common in viruses isolated from vaccine recipients than in viruses isolated from placebo recipients, providing evidence of a T-cell-based sieve effect. This effect implicated two HLA class I alleles, one of which was found to be associated with greater vaccine efficacy in the trial.
Previously, the effects of HLA—and, more generally, host genetics—on the efficacy of vaccines had not been well studied, in part due to the cost of HLA typing and to the difficulty in identifying associations within the highly diverse set of HLA haplotypes (estimated at >1013 unique alleles [4]). Despite these challenges, studies of influenza (36), measles/mumps/rubella (MMR) (38, 39), and hepatitis B (37) vaccination have identified indirect links between both class I and class II host-HLA alleles and putatively protective antibody responses to vaccination. While the mechanisms of these associations are not well understood, they may reflect a combination of known (54) and/or unknown links between class I and class II HLA alleles and humoral immunity.
Our analysis found evidence of T-cell-mediated escape in the V1/V2 region of the HIV envelope. However, CD8+ T-cell response rates of RV144 vaccine recipients were low overall; depending on the sample time point and the assay that was used, 12% to 63% of vaccine recipients had a T-cell response to Env peptides and 25% had a response to V2 peptides, but these were predominantly CD4+ T-cell-mediated responses (14, 19, 32). Furthermore, there are no CD8+ V1/V2 T-cell epitopes listed in the LANL HIV epitope database (http://www.hiv.lanl.gov/content/immunology/). Despite these facts, there are several findings that provide a rationale for studying T-cell epitopes in this region: (i) the variable loops of HIV envelope are epitope “hot spots” containing numerous CD4+ and CD8+ T-cell epitopes, likely due to the availability of the loops for antigen processing (55, 56); (ii) a cathepsin-D cleavage site was identified at L176 to Y177 (HXB2 numbering) that is highly conserved across subtypes (57); (iii) proteosomal degradation of the vaccine strain of envelope protein (92TH023) yielded several fragments, one of which, DKKQKVHALF (HXB2 167 to 176) (58), contained a homolog of the MN strain A*02-restricted epitope KMQKEYALL that was implicated in our analysis; and (iv) preliminary data suggest that the RV144 vaccine regimen elicited higher CD8+ T-cell response rates in the sigmoid mucosa than in those found in peripheral blood (Schuetz et al., unpublished). Finally, though the observation of robust T-cell responses would seem to be a necessary requirement for cytotoxic T lymphocyte (CTL)-mediated escape, in Step trial participants, despite high response rates overall (12), none of the observed T-cell responses correlated with the genetic signatures identified in the sieve analysis (25, 59).
In preliminary experiments to validate the predicted A*02 epitope KMQKEYALL, we showed that the peptide binds two A*02 alleles that were expressed by RV144 trial participants and that the peptide can elicit a CD8+ T-cell response in an HIV-1-infected A*0201+ individual. We note that all of the T-cell stimulation assays conducted with RV144 samples used CRF01_AE-derived peptides (92TH023) which matched the vaccine insertion but not the subtype B (MN) protein boost. It may therefore be important to study CD8+ T-cell responses to the V1/V2 region using subtype B (MN) peptides and/or mucosal samples in assays that are sensitive enough to detect potentially low-magnitude responses.
The T-cell-based sieve analysis investigated the hypothesis that T cells induced viral escape after acquisition, and yet we also found that both overall and genotype-specific vaccine efficacies were significantly increased in participants who carried the HLA A*02 allele implicated in our analysis. While these statistical associations do not imply that A*02 was a direct cause of greater vaccine efficacy in the trial, since it could be a marker of an unknown causal factor, they generate the hypothesis that A*02 contributed to greater vaccine efficacy. To consider this hypothesis, we entertain two possibilities: (i) that A*02-restricted CTLs offered some degree of protection in the trial and/or (ii) that the A*02 allele acted via a possibly undescribed CTL-independent mechanism. Recent studies of nonhuman primates have demonstrated prolonged CD8+ T-cell-mediated control of HIV replication (60), have identified lymph node T-cell responses as an immune correlate of risk in a trial of a live attenuated simian immunodeficiency virus (SIV) vaccine (61), and have shown that a SIV cytomegalovirus vector vaccine can induce T-cell responses that correlate with control and clearance of a SIV challenge infection (62, 63). Though it was antibody and not T-cell responses that correlated most significantly with infection risk in the RV144 trial (15), it is possible that lymph node or mucosal T-cell responses, previously thought to be important in SIV vaccines (61, 64), were also correlated. Therefore, it may be that A*02-restricted CD8+ T cells played a direct role in increasing vaccine efficacy.
Alternatively, we considered the possibility that HLA A*02 played a role in the efficacy of the vaccine that was independent of CD8+ T cells. To date, there have been numerous documented associations of HLA alleles with disease outcomes, many whose mechanisms have not yet been fully described. For example, in HIV-1 infection, specific HLA alleles have been implicated in the control of viral load and disease progression (47, 65–72). In dengue virus-related disease (73) and human T-cell leukemia virus type 1 (HTLV-1) infection (74), HLA has been associated with disease severity and progression. As noted above, there are also several documented examples of HLA alleles impacting vaccine-induced immune responses to influenza, hepatitis B, and MMR vaccination (36, 37, 39, 75). Though there are no canonical pathways allowing for a MHC class I allele to influence antibody production, a recent report describes one possible mechanism. The report proposes a model for MHC class I antigen cross-presentation operating in activated immune cells through MHC-I epitope recognition of exogenous polypeptides (76). The model predicts that MHC-I recognition of epitopes within an extended polypeptide chain facilitates exogenous antigen uptake and processing through a lysosomal, TAP-independent, antigen-presenting pathway. It is intriguing to posit that class II epitopes within the same polypeptide chain could be presented after class I-directed antigen acquisition, thereby facilitating CD4+ T-cell responses to the region and also potentially modulating humoral responses. On the basis of this model, we hypothesize that the MN protein immunogen may have been internalized via an HLA-F-dependent A*02-specific mechanism and subsequently presented by MHC-I and/or MHC-II. This provides a CTL-independent mechanism for the modulation of the immune response in A*02+ participants. Further studies must be completed to determine if the mechanism identified by Goodridge et al. was involved in the immune response to the RV144 vaccine.
The possibility that HLA A*02 modulated the antibody response to vaccination is consistent with our finding that the level of antibodies specific to the subtype B V2 peptide TSIRDKVQKEYALFY was significantly greater in vaccine recipients expressing A*02. Furthermore, two borderline-significant findings support the hypothesis that A*02 may have modified immune correlates of risk. First, we found that IgA-C1 antibodies, which were previously found to correlate directly with risk in the trial overall, were a significant correlate in participants without A*02. This suggests that the IgA antibodies, which were associated with greater risk of infection overall, may not have had the same association in A*02+ participants. Second, we found that the binding of vaccine-induced antibodies to a subtype B peptide that encompasses site 169 and the A*02-KMQKEYALL epitope was significantly greater in A*02+ than in A*02− vaccine recipients and that there was a direct correlation with risk only in participants lacking A*02. It was previously shown that high levels of V2-specific antibodies were associated with decreased risk of infection in the cohort overall (15); therefore, it seems inconsistent that low levels of V2-specific antibodies were associated with decreased risk in A*02− participants. One hypothesis to account for this inconsistency is that the antibodies specific to this subtype B peptide are different from those that correlate indirectly with risk overall and that their effects may therefore also differ. It is also possible that higher levels of antibodies binding this subtype B peptide are associated with decreased risk of infection within the A*02+ subgroup but that the low number of A*02+-infected participants reduced the statistical power needed to detect such an effect. Both possibilities implicate the A*02 allele in an immune modulatory role, offering a potential link between one of the genetic signatures found in the V2-region of breakthrough viruses, the V2-specific antibodies elicited by the vaccine, and the overall efficacy of the vaccine.
Understanding the interactions between the cellular and humoral immune responses upon vaccination is especially important in the RV144 trial since the vaccine regimen combined the ALVAC-HIV vector as a prime with the AIDSVAX B/E rgp120 as a boost. Though the ALVAC-HIV vector expressing a CRF01_AE strain of envelope and a clade B strain of gag and pol had previously been shown to induce T-cell responses (32) and the subtype B and CRF01_AE AIDSVAX proteins had been shown to induce antibody responses (77), negative results from a phase II trial of combined ALVAC and AIDSVAX B/B failed to trigger an efficacy trial (78) and trials of AIDSVAX B/B and AIDSVAX B/E products alone did not show efficacy (77, 79). Some researchers have speculated about the importance of the subtype-mismatched MN strain rgp120 included in the boost (16). Several recent findings seem to suggest that the V2 antibodies that correlated inversely with risk may have been cross-reactive: (i) the IgG V1/V2 CoR reported in RV144 by Haynes et al. (15) was based on an assay using a subtype B V1/V2 scaffold antigen mismatched to the predominant circulating HIV-1 subtype, (ii) the IgG V1/V2 CoR has been recapitulated using V1/V2 scaffolds from multiple different subtypes (16, 23), and (iii) the V2 hot spot immune variable, another CoR in RV144, was the average antibody response to peptides from 6 HIV-1 group M subtypes (15). One surprising aspect of our T-cell-based sieve analysis is that the sieve effect was driven by epitopes predicted in the MN sequence (subtype B) and not the 92TH023 (subtype E) or the CM244 (subtype E) sequences. Since 89% of the viruses isolated in the trial were CRF01_AE, the vaccine-induced T-cell responses driving the sieve effect would have needed to be cross-reactive, despite the fact that the MN vaccine sequence differed from each viral isolate sequence at 3, 4, or 5 amino acid residues within the KMQKEYALL epitope. The T-cell-based sieve method described here considered all residues within each potential epitope when predicting changes in the HLA binding affinity, effectively estimating the cross-reactivity of each potential viral epitope with the predicted vaccine epitope. Though often a single mutation can mediate viral escape, it was recently demonstrated that Pichinde virus (PV) and lymphocytic choriomeningitis virus (LCMV) encode two epitopes sharing 6 of 8 amino acids and induce distinct cross-reactive T-cell repertoires (80). Another study showed that an Epstein-Barr virus epitope cross-reacts with an influenza epitope despite sharing only 3 of 9 amino acid residues (81). In the V2 region of envelope, though the extreme sequence variability would seem to make cross-reactivity unlikely, there is evidence of structural conservation (82). Given these previous reports and our results, we hypothesize that the MN strain protein boost may have played a role in developing cross-reactive immune responses.
In considering the role of the HLA A*02 allele in the RV144 trial, the results could be attributed to underlying associations with other genes or other HLA alleles that are genetically linked to the A*02 allele. To partially address this possibility, we performed a genetic analysis to identify HLA alleles in linkage disequilibrium with the A*02 allele (44) (see Table SA4 in the supplemental material). The analysis revealed an HLA A*0207:B*4601:Cw*0102 haplotype that is common in the RV144 cohort (estimated prevalence of 14%) and has been previously documented as one of the most common haplotypes in ethnic northeast Thais (83). We estimated VE for each of several subgroups of participants who carried one or all of the alleles within this haplotype (see Table SA5). We found no conclusive evidence that VE was restricted to this haplotype or to any other HLA allele. However, because this haplotype spans the HIV-1-A, -B, and -C loci and is common in RV144 participants it remains possible that the A*02 associations we describe could be related to other unobserved genes that are included in the haplotype. In addition, given that the finding of differential VE in A*02+ versus A*02− individuals is of borderline significance (P = 0.050), it is possible that this finding is a false-positive result. Moreover, a comprehensive scan assessing all HLA alleles as modifiers of vaccine efficacy yielded no significant findings, due to the need to correct for the multiple hypothesis tests. Nevertheless, focusing the main analyses on A*02 and A*11 was justified by the fact that the vaccine-induced T-cell pressure was predicted only in these HLA-defined subgroups.
Follow-up analyses and ongoing experiments using samples from the RV144 study are providing in-depth information on the immune responses induced by RV144 vaccination. Both experimental and exploratory statistical analyses, such as this one, can generate hypotheses that will inform the design of future vaccine trials. Our findings emphasize the importance of considering HLA genotypes and, more generally, host genetics in vaccine clinical trials and their potential role in the complex web of humoral and cellular immune responses in vaccine-induced viral immunity.
Supplementary Material
ACKNOWLEDGMENTS
We thank the study participants, investigators, and sponsors of the RV144 Thai trial, including the U.S. Military HIV Research Program (MHRP); U.S. Army Medical Research and Materiel Command; NIAID; U.S. and Thai Components, Armed Forces Research Institute of Medical Science; Ministry of Public Health, Thailand; Mahidol University; SanofiPasteur; and Global Solutions for Infectious Diseases.
The study was funded by NIH NIAID R37 AI054165-11 (P.B.G.), NIH/NIAID UM1 AI06861 (M.J.M.) and NIH K25 AI087397-01 career award (T.H.).
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 14 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01164-14.
REFERENCES
- 1.Plotkin S, Orenstein W, Offit P. 2008. Vaccines, 5th ed. Elsevier Inc., Philadelphia, PA [Google Scholar]
- 2.Osterholm MT, Kelley NS, Sommer A, Belongia EA. 2012. Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect. Dis. 12:36–44. 10.1016/S1473-3099(11)70295-X [DOI] [PubMed] [Google Scholar]
- 3.Fine PE. 1995. Variation in protection by BCG: implications of and for heterologous immunity. Lancet 346:1339–1345. 10.1016/S0140-6736(95)92348-9 [DOI] [PubMed] [Google Scholar]
- 4.Poland GA, Kennedy RB, Ovsyannikova IG. 2011. Vaccinomics and personalized vaccinology: is science leading us toward a new path of directed vaccine development and discovery? PLoS Pathog. 7:e1002344. 10.1371/journal.ppat.1002344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plotkin SA. 2010. Correlates of protection induced by vaccination. Clin. Vaccine Immunol. 17:1055–1065. 10.1128/CVI.00131-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haralambieva IH, Ovsyannikova IG, Kennedy RB, Larrabee BR, Shane Pankratz V, Poland GA. 2013. Race and sex-based differences in cytokine immune responses to smallpox vaccine in healthy individuals. Hum. Immunol. 74:1263–1266. 10.1016/j.humimm.2013.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haralambieva IH, Ovsyannikova IG, Umlauf BJ, Vierkant RA, Shane Pankratz V, Jacobson RM, Poland GA. 2011. Genetic polymorphisms in host antiviral genes: associations with humoral and cellular immunity to measles vaccine. Vaccine 29:8988–8997. 10.1016/j.vaccine.2011.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haralambieva IH, Oberg AL, Dhiman N, Ovsyannikova IG, Kennedy RB, Grill DE, Jacobson RM, Poland GA. 2012. High-dimensional gene expression profiling studies in high and low responders to primary smallpox vaccination. J. Infect. Dis. 206:1512–1520. 10.1093/infdis/jis546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kennedy RB, Ovsyannikova IG, Pankratz VS, Haralambieva IH, Vierkant RA, Jacobson RM, Poland GA. 2012. Genome-wide genetic associations with IFNγ response to smallpox vaccine. Hum. Genet. 131:1433–1451. 10.1007/s00439-012-1179-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frahm N, DeCamp AC, Friedrich DP, Carter DK, Defawe OD, Kublin JG, Casimiro DR, Duerr A, Robertson MN, Buchbinder SP, Huang Y, Spies GA, De Rosa SC, McElrath MJ. 2012. Human adenovirus-specific T cells modulate HIV-specific T cell responses to an Ad5-vectored HIV-1 vaccine. J. Clin. Invest. 122:359–367. 10.1172/JCI60202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zak DE, Andersen-Nissen E, Peterson ER, Sato A, Hamilton MK, Borgerding J, Krishnamurty AT, Chang JT, Adams DJ, Hensley TR, Salter AI, Morgan CA, Duerr AC, De Rosa SC, Aderem A, McElrath MJ. 14 November 2012. Merck Ad5/HIV induces broad innate immune activation that predicts CD8+ T-cell responses but is attenuated by preexisting Ad5 immunity. Proc. Natl. Acad. Sci. U. S. A. 10.1073/pnas.1208972109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McElrath MJ, De Rosa SC, Moodie Z, Dubey S, Kierstead L, Janes H, Defawe OD, Carter DK, Hural J, Akondy R, Buchbinder SP, Robertson MN, Mehrotra DV, Self SG, Corey L, Shiver JW, Casimiro DR. 2008. HIV-1 vaccine-induced immunity in the test-of-concept Step Study: a case-cohort analysis. Lancet 372:1894–1905. 10.1016/S0140-6736(08)61592-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montefiori DC, Metch B, McElrath MJ, Self S, Weinhold KJ, Corey L. 2004. Demographic factors that influence the neutralizing antibody response in recipients of recombinant HIV-1 gp120 vaccines. J. Infect. Dis. 190:1962–1969. 10.1086/425518 [DOI] [PubMed] [Google Scholar]
- 14.Rerks-Ngarm S, Pitisuttithum P. 2009. Vaccination with ALVAC and ADISVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 361:2209–2220. 10.1056/NEJMoa0908492 [DOI] [PubMed] [Google Scholar]
- 15.Haynes BF, Gilbert PB, McElrath M, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, Liao H-X, DeVico AL, Lewis GK, Williams C, Pinter A, Fong Y, Janes H, DeCamp A, Huang Y, Rao M, Billings E, Karasavvas N, Robb ML, Ngauy V, de Souza MS, Paris R, Ferrari G, Bailer RT, Soderberg KA, Andrews C, Berman PW, Frahm N, De Rosa SC, Alpert MD, Yates NL, Shen X, Koup RA, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Michael NL, Kim JH, Alam M. 2012. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 366:1275–1286. 10.1056/NEJMoa1113425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zolla-Pazner S, Decamp AC, Cardozo T, Karasavvas N, Gottardo R, Williams C, Morris DE, Tomaras G, Rao M, Billings E, Berman P, Shen X, Andrews C, O'Connell RJ, Ngauy V, Nitayaphan S, de Souza M, Korber B, Koup R, Bailer RT, Mascola JR, Pinter A, Montefiori D, Haynes BF, Robb ML, Rerks-Ngarm S, Michael NL, Gilbert PB, Kim JH. 2013. Analysis of V2 antibody responses induced in vaccinees in the ALVAC/AIDSVAX HIV-1 Vaccine Efficacy Trial. PLoS One 8:e53629. 10.1371/journal.pone.0053629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rolland M, Edlefsen PT, Larsen BB, Tovanabutra S, Sanders-Buell E, Hertz T, deCamp AC, Carrico C, Menis S, Magaret CA, Ahmed H, Juraska M, Chen L, Konopa P, Nariya S, Stoddard JN, Wong K, Zhao H, Deng W, Maust BS, Bose M, Howell S, Bates A, Lazzaro M, O'Sullivan A, Lei E, Bradfield A, Ibitamuno G, Assawadarachai V, O'Connell RJ, deSouza MS, Nitayaphan S, Rerks-Ngarm S, Robb ML, McLellan JS, Georgiev I, Kwong PD, Carlson JM, Michael NL, Schief WR, Gilbert PB, Mullins JI, Kim JH. 10 September 2012. Increased HIV-1 vaccine efficacy against viruses with genetic signatures in Env V2. Nature 10.1038/nature11519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karasavvas N, Billings E, Rao M, Williams C, Zolla-Pazner S, Bailer RT, Koup RA, Madnote S, Arworn D, Shen X, Tomaras GD, Currier JR, Jiang M, Magaret C, Andrews C, Gottardo R, Gilbert P, Cardozo TJ, Rerks-Ngarm S, Nitayaphan S, Pitisuttithum P, Kaewkungwal J, Paris R, Greene K, Gao H, Gurunathan S, Tartaglia J, Sinangil F, Korber BT, Montefiori DC, Mascola JR, Robb ML, Haynes BF, Ngauy V, Michael NL, Kim JH, de Souza MS; MOPH TAVEG Collaboration. 2012. The Thai Phase III HIV Type 1 Vaccine Trial (RV144) regimen induces antibodies that target conserved regions within the V2 loop of gp120. AIDS Res. Hum. Retroviruses 28:1444–1457. 10.1089/aid.2012.0103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Souza MS, Ratto-Kim S, Chuenarom W, Schuetz A, Chantakulkij S, Nuntapinit B, Valencia-Micolta A, Thelian D, Nitayaphan S, Pitisuttithum P, Paris RM, Kaewkungwal J, Michael NL, Rerks-Ngarm S, Mathieson B, Marovich M, Currier JR, Kim JH. 2012. The Thai phase III trial (RV144) vaccine regimen induces T cell responses that preferentially target epitopes within the V2 region of HIV-1 envelope. J. Immunol. 188:5166–5176. 10.4049/jimmunol.1102756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montefiori DDC, Karnasuta C, Huang Y, Ahmed H, Gilbert P, de Souza MS, McLinden R, Tovanabutra S, Laurence-Chenine A, Sanders-Buell E, Moody MA, Bonsignori M, Ochsenbauer C, Kappes J, Tang H, Greene K, Gao H, LaBranche CC, Andrews C, Polonis VR, Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Self SG, Berman PW, Francis D, Sinangil F, Lee C, Tartaglia J, Robb ML, Haynes BF, Michael NL, Kim JH. 2012. Magnitude and breadth of the neutralizing antibody response in the RV144 and Vax003 HIV-1 vaccine efficacy trials. J. Infect. Dis. 206:431–441. 10.1093/infdis/jis367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu P, Yates NL, Shen X, Bonsignori M, Moody MA, Liao H-X, Fong Y, Alam SM, Overman RG, Denny T, Ferrari G, Ochsenbauer C, Kappes JC, Polonis V, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Montefiori DC, Gilbert P, Michael NL, Kim JH, Haynes BF, Tomaras GD. 8 May 2013. Infectious virion capture by HIV-1 gp120-specific IgG from RV144 vaccinees. J. Virol. 10.1128/JVI.02737-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao H-X, Bonsignori M, Alam SM, McLellan JS, Tomaras GD, Moody MA, Kozink DM, Hwang K-K, Chen X, Tsao C-Y, Liu P, Lu X, Parks RJ, Montefiori DC, Ferrari G, Pollara J, Rao M, Peachman KK, Santra S, Letvin NL, Karasavvas N, Yang Z-Y, Dai K, Pancera M, Gorman J, Wiehe K, Nicely NI, Rerks-Ngarm S, Nitayaphan S, Kaewkungwal J, Pitisuttithum P, Tartaglia J, Sinangil F, Kim JH, Michael NL, Kepler TB, Kwong PD, Mascola JR, Nabel GJ, Pinter A, Zolla-Pazner S, Haynes BF. 11 January 2013. Vaccine induction of antibodies against a structurally heterogeneous site of immune pressure within HIV-1 envelope protein variable regions 1 and 2. Immunity 10.1016/j.immuni.2012.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zolla-Pazner S, Decamp A, Gilbert PB, Williams C, Yates NL, Williams WT, Howington R, Fong Y, Morris DE, Soderberg KA, Irene C, Reichman C, Pinter A, Parks R, Pitisuttithum P, Kaewkungwal J, Rerks-Ngarm S, Nitayaphan S, Andrews C, O'Connell RJ, Yang Z-Y, Nabel GJ, Kim JH, Michael NL, Montefiori DC, Liao H-X, Haynes BF, Tomaras GD. 2014. Vaccine-induced IgG antibodies to V1V2 regions of multiple HIV-1 subtypes correlate with decreased risk of HIV-1 infection. PLoS One 9:e87572. 10.1371/journal.pone.0087572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yates NL, Liao H-X, Fong Y, deCamp A, Vandergrift NA, Williams WT, Alam SM, Ferrari G, Yang Z-Y, Seaton KE, Berman PW, Alpert MD, Evans DT, O'Connell RJ, Francis D, Sinangil F, Lee C, Nitayaphan S, Rerks-Ngarm S, Kaewkungwal J, Pitisuttithum P, Tartaglia J, Pinter A, Zolla-Pazner S, Gilbert PB, Nabel GJ, Michael NL, Kim JH, Montefiori DC, Haynes BF, Tomaras GD. 2014. Vaccine-induced Env V1–V2 IgG3 correlates with lower HIV-1 infection risk and declines soon after vaccination. Sci. Transl. Med. 6:228ra39. 10.1126/scitranslmed.3007730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rolland M, Tovanabutra S, DeCamp AC, Frahm N, Gilbert PB, Sanders-Buell E, Heath L, Magaret CA, Bose M, Bradfield A, O'Sullivan A, Crossler J, Jones T, Nau M, Wong K, Zhao H, Raugi DN, Sorensen S, Stoddard JN, Maust BS, Deng W, Hural J, Dubey S, Michael NL, Shiver J, Corey L, Li F, Self SG, Kim J, Buchbinder S, Casimiro DR, Robertson MN, Duerr A, McElrath MJ, McCutchan FE, Mullins JI. 2011. Genetic impact of vaccination on breakthrough HIV-1 sequences from the STEP trial. Nat. Med. 17:366–371. 10.1038/nm.2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Y, Gilbert PB. 2012. Estimation of stratified mark-specific proportional hazards models with missing marks. Scand. Stat. Theory Appl. 39:34–52. 10.1111/j.1467-9469.2011.00746.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, Macalalad AR, Berlin AM, Malboeuf CM, Ryan EM, Gnerre S, Zody MC, Erlich RL, Green LM, Berical A, Wang Y, Casali M, Streeck H, Bloom AK, Dudek T, Tully D, Newman R, Axten KL, Gladden AD, Battis L, Kemper M, Zeng Q, Shea TP, Gujja S, Zedlack C, Gasser O, Brander C, Hess C, Günthard HF, Brumme ZL, Brumme CJ, Bazner S, Rychert J, Tinsley JP, Mayer KH, Rosenberg E, Pereyra F, Levin JZ, Young SK, Jessen H, Altfeld M, Birren BW, Walker BD, Allen TM. 2012. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 8:e1002529. 10.1371/journal.ppat.1002529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goonetilleke N, Liu MKP, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, Keele BF, Learn GH, Turnbull EL, Salazar MG, Weinhold KJ, Moore S, Letvin N, Haynes BF, Cohen MS, Hraber P, Bhattacharya T, Borrow P, Perelson AS, Hahn BH, Shaw GM, Korber BT, McMichael AJ. 2009. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J. Exp. Med. 206:1253–1272. 10.1084/jem.20090365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferrari G, Korber B, Goonetilleke N, Liu MKP, Turnbull EL, Salazar-Gonzalez JF, Hawkins N, Self S, Watson S, Betts MR, Gay C, McGhee K, Pellegrino P, Williams I, Tomaras GD, Haynes BF, Gray CM, Borrow P, Roederer M, McMichael AJ, Weinhold KJ. 2011. Relationship between functional profile of HIV-1 specific CD8 T cells and epitope variability with the selection of escape mutants in acute HIV-1 infection. PLoS Pathog. 7:e1001273. 10.1371/journal.ppat.1001273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soghoian DZ, Jessen H, Flanders M, Sierra-Davidson K, Cutler S, Pertel T, Ranasinghe S, Lindqvist M, Davis I, Lane K, Rychert J, Rosenberg ES, Piechocka-Trocha A, Brass AL, Brenchley JM, Walker BD, Streeck H. 2012. HIV-specific cytolytic CD4 T cell responses during acute HIV infection predict disease outcome. Sci. Transl. Med. 4:123ra25. 10.1126/scitranslmed.3003165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herbeck JT, Rolland M, Liu Y, McLaughlin S, McNevin J, Zhao H, Wong K, Stoddard JN, Raugi D, Sorensen S, Genowati I, Birditt B, McKay A, Diem K, Maust BS, Deng W, Collier AC, Stekler JD, McElrath MJ, Mullins JI. 2011. Demographic processes affect HIV-1 evolution in primary infection before the onset of selective processes. J. Virol. 85:7523–7534. 10.1128/JVI.02697-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nitayaphan S, Pitisuttithum P, Karnasuta C, Eamsila C, de Souza M, Morgan P, Polonis V, Benenson M, VanCott T, Ratto-Kim S, Kim J, Thapinta D, Garner R, Bussaratid V, Singharaj P, El-Habib R, Gurunathan S, Heyward W, Birx D, McNeil J, Brown AE. 2004. Safety and immunogenicity of an HIV subtype B and E prime-boost vaccine combination in HIV-negative Thai adults. J. Infect. Dis. 190:702–706. 10.1086/422258 [DOI] [PubMed] [Google Scholar]
- 33.Jojic N, Reyes-Gomez M, Heckerman D, Kadie C, Schueler-Furman O. 2006. Learning MHC I–peptide binding. Bioinformatics 22:e227–e235. 10.1093/bioinformatics/btl255 [DOI] [PubMed] [Google Scholar]
- 34.Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Røder G, Peters B, Sette A, Lund O, Buus S. 2007. NetMHCpan, a method for quantitative predictions of peptide binding to any HIV-1-A and -B locus protein of known sequence. PLoS One 2:e796. 10.1371/journal.pone.0000796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nielsen M, Justesen S, Lund O, Lundegaard C, Buus S. 2010. NetMHCIIpan-2.0 - improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome Res. 6:9. 10.1186/1745-7580-6-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poland GA, Ovsyannikova I, Jacobson R. 12 September 2008. Immunogenetics of seasonal influenza vaccine response. Vaccine 10.1016/j.vaccine.2008.07.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alper CA, Kruskall MS, Marcus-Bagley D, Craven DE, Katz AJ, Brink SJ, Dienstag JL, Awdeh Z, Yunis EJ. 1989. Genetic prediction of nonresponse to hepatitis B vaccine. N. Engl. J. Med. 321:708–712. 10.1056/NEJM198909143211103 [DOI] [PubMed] [Google Scholar]
- 38.Ovsyannikova IG, Jacobson RM, Poland GA. 2004. Variation in vaccine response in normal populations. Pharmacogenomics 5:417–427. 10.1517/14622416.5.4.417 [DOI] [PubMed] [Google Scholar]
- 39.Ovsyannikova IG, Pankratz VS, Vierkant RA, Jacobson RM, Poland GA. 2012. Consistency of HLA associations between two independent measles vaccine cohorts: a replication study. Vaccine 30:2146–2152. 10.1016/j.vaccine.2012.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ovsyannikova IG, Jacobson RM, Vierkant RA, O'Byrne MM, Poland GA. 2009. Replication of rubella vaccine population genetic studies: validation of HLA genotype and humoral response associations. Vaccine 27:6926–6931. 10.1016/j.vaccine.2009.08.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edlefsen PT, Gilbert PB, Rolland M. 2013. Sieve analysis in HIV-1 vaccine efficacy trials. Curr. Opin. HIV AIDS 8:432–436. 10.1097/COH.0b013e328362db2b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schueler-Furman O, Altuvia Y, Sette A, Margalit H. 2000. Structure-based prediction of binding peptides to MHC class I molecules: application to a broad range of MHC alleles. Protein Sci. 9:1838–1846. 10.1110/ps.9.9.1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hurley CK, Mack SJ, Mickelson E, Marsh S, Tilanus MGJ, Gorodezky C, Wade JA, Müller C, Hartzman RJ, Baxter-Lowe LA, Raffoux C. 2006. HLA typing and informatics, in immunobiology of the human MHC, p 179–352 In Hansen J. A. (ed), 13th International Histocompatibility Workshop protocols. IHWG Press, Seattle, WA [Google Scholar]
- 44.Lancaster AK, Single RM, Solberg OD, Nelson MP, Thomson G. 2007. PyPop update–a software pipeline for large-scale multilocus population genomics. Tissue Antigens 69(Suppl 1):S192–S197. 10.1111/j.1399-0039.2006.00769.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dai JY, Li SS, Gilbert PB. 27 June 2013. Case-only method for cause-specific hazards models with application to assessing differential vaccine efficacy by viral and host genetics. Biostatistics 10.1093/biostatistics/kxt018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zelen M. 1971. The analysis of several 2 × 2 contingency tables. Biometrika 58:129–137. 10.1093/biomet/58.1.129 [DOI] [Google Scholar]
- 47.Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PIW, Walker BD, Ripke S, Brumme CJ, Pulit SL, Carrington M, Kadie CM, Carlson JM, Heckerman D, Graham RR, Plenge RM, Deeks SG, Gianniny L, Crawford G, Sullivan J, Gonzalez E, Davies L, Camargo A, Moore JM, Beattie N, Gupta S, Crenshaw A, Burtt NP, Guiducci C, Gupta N, Gao X, Qi Y, Yuki Y, Piechocka-Trocha A, Cutrell E, Rosenberg R, Moss KL, Lemay P, O'Leary J, Schaefer T, Verma P, Toth I, Block B, Baker B, Rothchild A, Lian J, Proudfoot J, Alvino DML, Vine S, Addo MM, et al. 2010. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330:1551–1557. 10.1126/science.1195271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sidney J, Southwood S, Oseroff C, del Guercio MF, Sette A, Grey HM. 2001. Measurement of MHC/peptide interactions by gel filtration. Curr. Protoc. Immunol. 31:18.3.1–18.3.19. 10.1002/0471142735.im1803s31 [DOI] [PubMed] [Google Scholar]
- 49.Sidney J, Assarsson E, Moore C, Ngo S, Pinilla C, Sette A, Peters B. 2008. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 4:2. 10.1186/1745-7580-4-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sidney J, Southwood S, Mann DL, Fernandez-Vina MA, Newman MJ, Sette A. 2001. Majority of peptides binding HIV-1 A*0201 with high affinity crossreact with other A2-supertype molecules. Hum. Immunol. 62:1200–1216. 10.1016/S0198-8859(01)00319-6 [DOI] [PubMed] [Google Scholar]
- 51.Cheng Y, Prusoff WH. 1973. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22:3099–3108. 10.1016/0006-2952(73)90196-2 [DOI] [PubMed] [Google Scholar]
- 52.Gulukota K, Sidney J, Sette A, DeLisi C. 1997. Two complementary methods for predicting peptides binding major histocompatibility complex molecules. J. Mol. Biol. 267:1258–1267. 10.1006/jmbi.1997.0937 [DOI] [PubMed] [Google Scholar]
- 53.Tomaras GD, Ferrari G, Shen X, Alam SM, Liao H-X, Pollara J, Bonsignori M, Moody MA, Fong Y, Chen X, Poling B, Nicholson CO, Zhang R, Lu X, Parks R, Kaewkungwal J, Nitayaphan S, Pitisuttithum P, Rerks-Ngarm S, Gilbert PB, Kim JH, Michael NL, Montefiori DC, Haynes BF. 9 May 2013. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc. Natl. Acad. Sci. U. S. A. 10.1073/pnas.1301456110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crotty S. 2011. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29:621–663. 10.1146/annurev-immunol-031210-101400 [DOI] [PubMed] [Google Scholar]
- 55.Surman S, Lockey TD, Slobod KS, Jones B, Riberdy JM, White SW, Doherty PC, Hurwitz JL. 2001. Localization of CD4+ T cell epitope hotspots to exposed strands of HIV envelope glycoprotein suggests structural influences on antigen processing. Proc. Natl. Acad. Sci. U. S. A. 98:4587–4592. 10.1073/pnas.071063898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhan X, Hurwitz JL, Brown SA, Slobod KS. 2007. HIV-1 envelope T cell epitope “hotspots” among mice and humans and among CD4+ and CD8+ T cell subpopulations. AIDS Res. Hum. Retroviruses 23:471–476. 10.1089/aid.2006.0241 [DOI] [PubMed] [Google Scholar]
- 57.Yu B, Fonseca DPA, O'Rourke JSM, Berman PW. 2010. Protease cleavage sites in HIV-1 gp120 recognized by antigen processing enzymes are conserved and located at receptor binding sites. J. Virol. 84:1513–1526. 10.1128/JVI.01765-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steers NJ, Ratto-Kim S, de Souza MS, Currier JR, Kim JH, Michael NL, Alving CR, Rao M. 2012. HIV-1 envelope resistance to proteasomal cleavage: implications for vaccine induced immune responses. PLoS One 7:e42579. 10.1371/journal.pone.0042579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Janes H, Frahm N, DeCamp A, Rolland M, Gabriel E, Wolfson J, Hertz T, Kallas E, Goepfert P, Friedrich DP, Corey L, Mullins JI, McElrath MJ, Gilbert P. 2012. MRKAd5 HIV-1 Gag/Pol/Nef vaccine-induced T-cell responses inadequately predict distance of breakthrough HIV-1 sequences to the vaccine or viral load. PLoS One 7:e43396. 10.1371/journal.pone.0043396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mudd PA, Martins MA, Ericsen AJ, Tully DC, Power KA, Bean AT, Piaskowski SM, Duan L, Seese A, Gladden AD, Weisgrau KL, Furlott JR, Kim YI, Veloso de Santana MG, Rakasz E, Capuano S, III, Wilson NA, Bonaldo MC, Galler R, Allison DB, Piatak M, Jr, Haase AT, Lifson JD, Allen TM, Watkins DI. 30 September 2012. Vaccine-induced CD8(+) T cells control AIDS virus replication. Nature 10.1038/nature11443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fukazawa Y, Park H, Cameron MJ, Lefebvre F, Lum R, Coombes N, Mahyari E, Hagen SI, Bae JY, Reyes MD, III, Swanson T, Legasse AW, Sylwester A, Hansen SG, Smith AT, Stafova P, Shoemaker R, Li Y, Oswald K, Axthelm MK, McDermott A, Ferrari G, Montefiori DC, Edlefsen PT, Piatak M, Jr, Lifson JD, Sékaly RP, Picker LJ. 9 September 2012. Lymph node T cell responses predict the efficacy of live attenuated SIV vaccines. Nat. Med. 10.1038/nm.2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M, Jr, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527. 10.1038/nature10003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hansen SG, Piatak M, Jr, Ventura AB, Hughes CM, Gilbride RM, Ford JC, Oswald K, Shoemaker R, Li Y, Lewis MS, Gilliam AN, Xu G, Whizin N, Burwitz BJ, Planer SL, Turner JM, Legasse AW, Axthelm MK, Nelson JA, Früh K, Sacha JB, Estes JD, Keele BF, Edlefsen PT, Lifson JD, Picker LJ. 11 September 2013. Immune clearance of highly pathogenic SIV infection. Nature 10.1038/nature12519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fuller DH, Rajakumar P, Che JW, Narendran A, Nyaundi J, Michael H, Yager EJ, Stagnar C, Wahlberg B, Taber R, Haynes JR, Cook FC, Ertl P, Tite J, Amedee AM, Murphey-Corb M. 2012. Therapeutic DNA vaccine induces broad T cell responses in the gut and sustained protection from viral rebound and AIDS in SIV-infected rhesus macaques. PLoS One 7:e33715. 10.1371/journal.pone.0033715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Honeyborne I, Prendergast A, Pereyra F, Leslie A, Crawford H, Payne R, Reddy S, Bishop K, Moodley E, Nair K, van der Stok M, McCarthy N, Rousseau CM, Addo M, Mullins JI, Brander C, Kiepiela P, Walker BD, Goulder PJR. 2007. Control of human immunodeficiency virus type 1 is associated with HLA-B*13 and targeting of multiple gag-specific CD8+ T-cell epitopes. J. Virol. 81:3667–3672. 10.1128/JVI.02689-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Duda A, Lee-Turner L, Fox J, Robinson N, Dustan S, Kaye S, Fryer H, Carrington M, McClure M, McLean A, Fidler RS, Weber J, Phillips RE, Frater AJ. 2009. HIV-1-infected ssociated clinical progression correlates with epitope reversion rates in early human immunodeficiency virus infection. J. Virol. 83:1228–1239. 10.1128/JVI.01545-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen H, Ndhlovu ZM, Liu D, Porter LC, Fang JW, Darko S, Brockman MA, Miura T, Brumme ZL, Schneidewind A, Piechocka-Trocha A, Cesa KT, Sela J, Cung TD, Toth I, Pereyra F, Yu XG, Douek DC, Kaufmann DE, Allen TM, Walker BD. 10 June 2012. TCR clonotypes modulate the protective effect of HLA class I molecules in HIV-1 infection. Nat. Immunol. 10.1038/ni.2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shea PR, Shianna KV, Carrington M, Goldstein DB. 27 September 2012. Host genetics of HIV acquisition and viral control. Annu. Rev. Med. 10.1146/annurev-med-052511-135400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Song H, Pavlicek JW, Cai F, Bhattacharya T, Li H, Iyer SS, Bar KJ, Decker JM, Goonetilleke N, Liu MKP, Berg A, Hora B, Drinker MS, Eudailey J, Pickeral J, Moody MA, Ferrari G, McMichael A, Perelson AS, Shaw GM, Hahn BH, Haynes BF, Gao F. 2012. Impact of immune escape mutations on HIV-1 fitness in the context of the cognate transmitted/founder genome. Retrovirology 9:89. 10.1186/1742-4690-9-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carrington M, Nelson GW, Martin MP, Kissner T, Vlahov D, Goedert JJ, Kaslow R, Buchbinder S, Hoots K, O'Brien SJ. 1999. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science 283:1748–1752. 10.1126/science.283.5408.1748 [DOI] [PubMed] [Google Scholar]
- 71.Kaslow RA, Carrington M, Apple R, Park L, Muñoz A, Saah AJ, Goedert JJ, Winkler C, O'Brien SJ, Rinaldo C, Detels R, Blattner W, Phair J, Erlich H, Mann DL. 1996. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med. 2:405–411. 10.1038/nm0496-405 [DOI] [PubMed] [Google Scholar]
- 72.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46–53. 10.1038/nm1520 [DOI] [PubMed] [Google Scholar]
- 73.Chaturvedi U, Nagar R, Shrivastava R. 2006. Dengue and dengue haemorrhagic fever: implications of host genetics. FEMS Immunol. Med. Microbiol. 47:155–166. 10.1111/j.1574-695X.2006.00058.x [DOI] [PubMed] [Google Scholar]
- 74.Macnamara A, Rowan A, Hilburn S, Kadolsky U, Fujiwara H, Suemori K, Yasukawa M, Taylor G, Bangham CRM, Asquith B. 2010. HLA class I binding of HBZ determines outcome in HTLV-1 infection. PLoS Pathog. 6:e1001117. 10.1371/journal.ppat.1001117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ovsyannikova IG, Jacobson RM, Vierkant RA, Jacobsen SJ, Pankratz VS, Poland GA. 2004. The contribution of HLA class I antigens in immune status following two doses of rubella vaccination. Hum. Immunol. 65:1506–1515. 10.1016/j.humimm.2004.07.001 [DOI] [PubMed] [Google Scholar]
- 76.Goodridge JP, Lee N, Burian A, Pyo C-W, Tykodi SS, Warren EH, Yee C, Riddell SR, Geraghty DE. 2013. HLA-F and MHC-I open conformers cooperate in a MHC-I antigen cross-presentation pathway. J. Immunol. 191:1567–1577. 10.4049/jimmunol.1300080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Flynn NM, Forthal DN, Harro CD, Judson FN, Mayer KH, Para MF. 2005. Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J. Infect. Dis. 191:654–665. 10.1086/428404 [DOI] [PubMed] [Google Scholar]
- 78.Russell ND, Graham BS, Keefer MC, McElrath MJ, Self SG, Weinhold KJ, Montefiori DC, Ferrari G, Horton H, Tomaras GD, Gurunathan S, Baglyos L, Frey SE, Mulligan MJ, Harro CD, Buchbinder SP, Baden LR, Blattner WA, Koblin BA, Corey L; National Institute of Allergy and Infectious Diseases HIV Vaccine Trials Network. 2007. Phase 2 study of an HIV-1 canarypox vaccine (vCP1452) alone and in combination with rgp120: negative results fail to trigger a phase 3 correlates trial. J. Acquir. Immune Defic. Syndr. 44:203–212. 10.1097/01.qai.0000248356.48501.ff [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pitisuttithum P, Gilbert P, Gurwith M, Heyward W, Martin M, van Griensven F, Hu D, Tappero JW, Choopanya K; Bangkok Vaccine Evaluation Group. 3 November 2006. Efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. Vaccine 10.1086/508748 [DOI] [PubMed] [Google Scholar]
- 80.Chen AT, Cornberg M, Gras S, Guillonneau C, Rossjohn J, Trees A, Emonet S, de la Torre JC, Welsh RM, Selin LK. 2012. Loss of anti-viral immunity by infection with a virus encoding a cross-reactive pathogenic epitope. PLoS Pathog. 8:e1002633. 10.1371/journal.ppat.1002633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Clute SC, Watkin LB, Cornberg M, Naumov YN, Sullivan JL, Luzuriaga K, Welsh RM, Selin LK. 23 November 2005. Cross-reactive influenza virus-specific CD8+ T cells contribute to lymphoproliferation in Epstein-Barr virus-associated infectious mononucleosis. J. Clin. Invest. 10.1172/JCI25078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zolla-Pazner S, Cardozo T. 2010. Structure-function relationships of HIV-1 envelope sequence-variable regions refocus vaccine design. Nat. Rev. Immunol. 10:527–535. 10.1038/nri2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Romphruk AV, Kongmaroeng C, Klumkrathok K, Paupairoj C, Leelayuwat C. 2010. HLA class I and II alleles and haplotypes in ethnic Northeast Thais. Tissue Antigens 75:701–711. 10.1111/j.1399-0039.2010.01448.x [DOI] [PubMed] [Google Scholar]
- 84.Schuetz A, Huangngern Y, de Souza M, Sukhumvittaya S, Jongrakthaitae S, Rerknimitr R, Saengtawan P, Ananworanich J, Vasan S, Ratto-Kim S, Pitisuttithum P, Michael N, O'Connell R, Ngauy V, Rerks-Ngarm S, Kim J. 2013. Evaluation of peripheral and mucosal cellular immune responses induced by late boost strategies in HIV-negative participants prior enrolled in RV144. AIDS Vaccine 2013, P12.56 LB. http://epostersonline.com/aidsvax2013/?q=node/4295 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.