

ABSTRACT
Cowpox viruses (CPXV) cause hemorrhagic lesions (“red pocks”) on infected chorioallantoic membranes (CAM) of embryonated chicken eggs, while most other members of the genus Orthopoxvirus produce nonhemorrhagic lesions (“white pocks”). Cytokine response modifier A (CrmA) of CPXV strain Brighton Red (BR) is necessary but not sufficient for the induction of red pocks. To identify additional viral proteins involved in the induction of hemorrhagic lesions, a library of single-gene CPXV knockout mutants was screened. We identified 10 proteins that are required for the formation of hemorrhagic lesions, which are encoded by CPXV060, CPXV064, CPXV068, CPXV069, CPXV074, CPXV136, CPXV168, CPXV169, CPXV172, and CPXV199. The genes are the homologues of F12L, F15L, E2L, E3L, E8R, A4L, A33R, A34R, A36R, and B5R of vaccinia virus (VACV). Mutants with deletions in CPXV060, CPXV168, CPXV169, CPXV172, or CPXV199 induced white pocks with a comet-like shape on the CAM. The homologues of these five genes in VACV encode proteins that are involved in the production of extracellular enveloped viruses (EEV) and the repulsion of superinfecting virions by actin tails. The homologue of CPXV068 in VACV is also involved in EEV production but is not related to actin tail induction. The other genes encode immunomodulatory proteins (CPXV069 and crmA) and viral core proteins (CPXV074 and CPXV136), and the function of the product of CPXV064 is unknown.
IMPORTANCE It has been known for a long time that cowpox virus induces hemorrhagic lesions on chicken CAM, while most of the other orthopoxviruses produce nonhemorrhagic lesions. Although cowpox virus CrmA has been proved to be responsible for the hemorrhagic phenotype, other proteins causing this phenotype remain unknown. Recently, we generated a complete single-gene knockout bacterial artificial chromosome (BAC) library of cowpox virus Brighton strain. Out of 183 knockout BAC clones, 109 knockout viruses were reconstituted. The knockout library makes possible high-throughput screening for studying poxvirus replication and pathogenesis. In this study, we screened all 109 single-gene knockout viruses and identified 10 proteins necessary for inducing hemorrhagic lesions. The identification of these genes gives a new perspective for studying the hemorrhagic phenotype and may give a better understanding of poxvirus virulence.
INTRODUCTION
Poxviruses comprise a family of complex DNA viruses that infect a wide spectrum of vertebrate and invertebrate animals. Among the eight genera of vertebrate poxviruses, orthopoxviruses (OPVs) have been studied most extensively. The best-known OPV is variola virus (VARV), the causative agent of smallpox, which was declared eradicated in 1980 (1). The prototype OPV is vaccinia virus (VACV), which was used as a vaccine against VARV. Cowpox virus (CPXV) has the largest and likely most complete genome among all known members of the OPV genus (2, 3) and has therefore become a popular model to study poxvirus biology and pathogenesis.
Although all OPVs can infect chicken embryos and cause distinctive visible lesions (pocks) on the chorioallantoic membrane (CAM) of embryonated eggs at 2 to 4 days postinfection (dpi), only CPXV and rabbitpox virus (RPXV) induce hemorrhagic (“red”) pocks on CAM (4). The first protein identified to be involved in inducing the hemorrhagic phenotype was CrmA (cytokine response modifier A) of CPXV (5). Mutant CPXV lacking the crmA gene is less virulent than wild-type virus in a mouse model (6). In a recent study, the CPXV crmA gene was introduced into the genome of modified vaccinia virus strain Ankara (MVA), but heterologous expression of CrmA did not confer on recombinant MVA the ability to produce hemorrhagic lesions on chicken CAM (7). The results clearly showed that CrmA is necessary but not sufficient for the hemorrhagic red-pock phenotype of poxviruses.
In RPXV, serine protease inhibitor 1 (serpin 1), serpin 2 (CrmA), and the product of the ps/hr gene (B5R homologue of VACV strain Copenhagen [VACV-COP]) are responsible for the induction of hemorrhagic pocks on CAM (4, 8). However, CPXV serpin 1 is not necessary for formation of red pocks, and nothing is known about the involvement of its B5 homologue in the process. Also, even though both CPXV and RPXV produce hemorrhagic lesions on CAM, the pocks as well as the hemorrhage induced by CPXV tend to be more pronounced than those produced by RPXV (4).
Besides CrmA, kelch-like proteins have an impact on the induction of red pocks on infected CAM. Deletion of four of the six kelch-like genes in CPXV strain GRI resulted in smaller, white pocks (9), suggesting that kelch-like proteins of CPXV might be involved in the induction of hemorrhages.
The present study aimed to identify CPXV proteins involved in the induction of hemorrhagic pocks on the CAM of infected chicken eggs. Recently, we generated a library of targeted knockout CPXV strain Brighton (CPXV-BR) mutants for each single viral open reading frame (ORF) (10) as well as a construct with a deletion of all six genes encoding kelch-like proteins. Reconstitution of infectious virus was successful for 109 of the 183 single-gene deletion mutants as well as for the mutant devoid of kelch-like genes. We screened all deletion mutant viruses that were able to replicate and identified 10 that produced white pocks on the chicken CAM. The results were verified by generating and testing repair viruses of each of the individual deletion mutants, which restored the red-pock phenotype in all cases.
MATERIALS AND METHODS
Cell lines.
African green monkey cells Vero 76 (Collection of Cell Lines in Veterinary Medicine, Friedrich-Loeffler-Institut, Greifswald-Insel Riems, Germany) were maintained in Eagle's minimal essential medium (MEM; Biochrom, Berlin, Germany) supplemented with 5% fetal bovine serum (FBS; Biochrom), 100 U/ml penicillin (Fisher Scientific, Schwerte, Germany), and 0.1 mg/ml streptomycin (AppliChem GmbH, Darmstadt, Germany) and incubated at 37°C with 5% CO2. Primary chicken embryo cells (CEC) were prepared from 11-day-old embryonated specific-pathogen-free (SPF) White Leghorn eggs (Valo BioMedia GmbH, Osterholz-Scharmbeck, Germany) according to standard procedures (11). CEC were cultured at 37°C in MEM containing 10% FBS (Biochrom) and antibiotics as described above.
Viruses.
Single-gene deletion mutants of CPXV-BR were reconstituted from mutant bacterial artificial chromosomes (BACs) previously (10). All CPXVs used in this study were propagated and titrated on Vero cells. Fowlpox virus (FWPV) (Nobilis-PD, strain WP [Intervet, Boxmeer, Netherlands]; kindly provided by D. Lüschow, Freie Universität Berlin, Germany) was grown on CEC and used for initial reconstitution of viruses. For preparation of virus stocks, Vero cells were grown in 10-cm cell culture dishes and infected with CPXV at a multiplicity of infection (MOI) of 0.1. After 48 h, infected cells and supernatants were frozen (−70°C) and thawed (37°C) three times. Cells were scraped and collected in 50-ml conical centrifuge tubes (Sarstedt, Nümbrecht, Germany). Glass beads with a diameter of 0.75 to 1 mm (Karl Roth, Karlsruhe, Germany) were added into the tube and vortexed for 90 s before the cell suspension was centrifuged for 15 min at 300 rpm. The supernatant was collected and stored at −70°C, and titers were determined by plaque assay of 10-fold serial dilutions on Vero cells (10).
BAC clones and BAC mutagenesis.
A BAC clone of CPXV-BR containing monomeric red fluorescent protein (mRFP) and enhanced green fluorescent protein (eGFP), driven by early and late poxviral promoters, respectively, in the mini-F region, was generated previously and termed pBRFseR (10). Virus reconstituted from pBRFseR was used as the wild-type control in this study and termed vBRFseR. Clones of a library of targeted knockout CPXV-BR BAC mutants for each single viral ORF (10) were maintained in Escherichia coli strain GS1783 (12). BAC clones were modified by two-step Red-mediated recombination (12, 13) using marker constructs containing kanamycin, tetracycline, or ampicillin resistance genes. Primers used for deletion of kelch-like genes and CPXV207 (crmA) using Red recombination are listed in Table 1. Primers used for repairing the mutant BAC constructs are given in Table 2.
TABLE 1.
Primers used for deletion of kelch-like genes and CPXV207 (crmA) and primers used for sequencing of kelch-like gene deletion mutants and the crmA deletion mutant
| Target ORF | Namea | Sequence (5′–3′) |
|---|---|---|
| CPXV013 | d13F | TATTAGTTTTATGGTTATATACATCAACATCATGTGTTGCACGCATGATAGCTATCTAATTAGGGATAACAGGGTAATCGATTT |
| d13R | GTGCGCTACCAGAACCATTAATTAGATAGCTATCATGCGTGCAACACATGATGTTGATGTGCCAGTGTTACAACCAATTAACC | |
| seq13F | TGAAAGTATCCATGTTCCATCG | |
| seq13R | CCAAGATGTGTACGCTGAATATAG | |
| CPXV035 | d35F | TGTCCAATAATAAAAAGTCATGCTATTGTAGGAATTGTTTTTATAAAAATCATTTCGACATAGGGATAACAGGGTAATCGATTT |
| d35R | GGAAATTTAAAAAGGAATTAATGTCGAAATGATTTTTATAAAAACAATTCCTACAATAGCAGCCAGTGTTACAACCAATTAACC | |
| seq35F | TGACGGAATAGTACAGCATGATAG | |
| seq35R | CTAGACAAGATGACTGCGGATAT | |
| CPXV050 | d50F | CAATAGCAGTTTATTATCCACTATGATCAATTCTGGATTATATTGGCATTTATGTTTCTTTAGGGATAACAGGGTAATCGATTT |
| d50R | TCTCTAGATGTTGACTTTTAAAGAAACATAAATGCCAATATAATCCAGAATTGATCATAGGCCAGTGTTACAACCAATTAACC | |
| seq50F | ATTCTACCATAGCAGAACTTAGGC | |
| seq50R | CAGATGCGGTATCCATTGAAC | |
| CPXV193 | d193F | TTTTTCCAATGGATATTTATGTTTAATAGGTTCGCATTTAGTTATGCATGATGACGCTGTTAGGGATAACAGGGTAATCGATTT |
| d193R | ATATAATTTATAATACACTTACAGCGTCATCATGCATAACTAAATGCGAACCTATTAAACGCCAGTGTTACAACCAATTAACC | |
| seq193F | TGAATATTGCGACGACATACG | |
| seq193R | GGCTCCTTATACCAAGCACTC | |
| CPXV204 | d204F | CCATGCACCATCATACTTTTCCACATATCCGATGTCCTTAAGAATCCATTCTAGAATTCATAGGGATAACAGGGTAATCGATTT |
| d204R | TAGTTCTTACAATTAGAGATTGAATTCTAGAATGGATTCTTAAGGACATCGGATATGTGGGCCAGTGTTACAACCAATTAACC | |
| seq204F | AACTGATACTCTAACCTGTGGAGC | |
| seq204R | ATCCGTATCCGTGTATTCGTC | |
| CPXV207 | d207F | AGACTATCTCTATCGTCACACAACAAAATCGATTGCCATGGCCGCGTGACGATTTATTCA |
| d207R | ATGATTCTTTTACAGATGCGTGATTAAATGCCTCGCCGTAGCGTAATGCTCTGCCAGTGT | |
| seq207F | AGTATGATGGTGCATGGAAGTTAG | |
| seq207R | CCGTTGAATATGGCTCATAACAC | |
| CPXV215b | d215F | TCATACTCATTTATTCTATTATATTTAGTAGATGGGTAGTTTCAATATTATAATCTTGATTAGGGATAACAGGGTAATCGATTT |
| d215R | TCTTATATAACACTAATTACATCAAGATTATAATATTGAAACTACCCATCTACTAAATATGCCAGTGTTACAACCAATTAACC | |
| seq215F | CCTGTACATCGTACAAATGACAAAC | |
| seq215R | CTTGACAAATTGGTATTCCGTACAC |
Primers with names starting with “d” are primers used for deletion of kelch-like genes. Primers with names starting with “seq” are primers used for sequencing of deletion mutants.
A pseudogene, CPXV214, was deleted together with CPXV215.
TABLE 2.
Primers used for generation of repaired BAC clones with Red recombination
| Target ORF | Name | Sequence (5′–3′) |
|---|---|---|
| CPXV060 | d60ampF | CGTGATAAACTACGGGAATATGGTCGTTAGTAGGTACGGTTGATCTTTTCTACGGGGTCTGAC |
| BR060R | GACGACGAATATGTTCATATCACTTC | |
| d60kanF | CGTGATAAACTACGGGAATATGGTCGTTAGTAGGTACGGTGACTTTACACAACGCGATATTAGGGATAACAGGGTAATCGATTT | |
| d60kanR | TGGTACAAAAGGAAAGTTATATATCGCGTTGTGTAAAGTCACCGTACCTACTAACGACCAGCCAGTGTTACAACCAATTAACC | |
| CPXV064 | d64ampF | TCATCACCACGATTAGAGATACAATACTTACATTCTTTTTTGATCTTTTCTACGGGGTCTGAC |
| BR064R | GCAGGGGTATTAATATCAGGGTATC | |
| d64kanF | TCATCACCACGATTAGAGATACAATACTTACATTCTTTTTGCTGTTTCGAAACTTTATCATAGGGATAACAGGGTAATCGATTT | |
| d64kanR | GGGTTTGTATTAACGTGTATTGATAAAGTTTCGAAACAGCAAAAAGAATGTAAGTATTGTGCCAGTGTTACAACCAATTAACC | |
| CPXV068 | d68ampF | TCTAGTTTCGTAATATCTATAGCATCCTCAAAAAATATATTGATCTTTTCTACGGGGTCTGAC |
| B68R | GCTGTAGATACACTTCTTGGTTACG | |
| d68kanF | TCTAGTTTCGTAATATCTATAGCATCCTCAAAAAATATATTCGCATATATTCCCAAGTCTTAGGGATAACAGGGTAATCGATTT | |
| d68kanR | TTTTTAGAAGATAGAACTGAAGACTTGGGAATATATGCGAATATATTTTTTGAGGATGCTGCCAGTGTTACAACCAATTAACC | |
| CPXV069 | BR069F | CGGAATAACATCATCGAAAGAC |
| BR069R | GGGTTAATCAGAGCCACATTC | |
| CPXV074 | BR074F | ACATAGTTGATAAAAAGCGGTAGG |
| BR074R | TACCGGAGAAAGATCCATTAGC | |
| CPXV136 | BR136F | GTTGTGCTGTTGTACATACTGTACC |
| BR136R | AATGGACTTCTTTAACAAGTTCTCAC | |
| CPXV168 | BR168F | CTAAGGTCGTTAGTAGGGAGGC |
| BR168R | GCATCCGAGAATGACTTGTAGTC | |
| CPXV169 | BR169F | CGATGTCTTGACTACCTGGCTC |
| BR169R | GAATCCGTCGTACTGTTTAGTTG | |
| CPXV172 | BR172F | CTTCATTCTGTATATCAGACGGC |
| BR172R | CCACAACATTGGATTCGTTATC | |
| CPXV199 | BR199F | GAGATAAATGGTCGTGTTTTCC |
| d199ampR | TCCGTGTTCTAATCGAAGAGGTTGGCATTCCGCATTAGGATGATCTTTTCTACGGGGTCTGAC | |
| d199kanF | AAAAAAATGGAAATACTTCTTGGAATGATACTGTTACGTGTCCTAATGCGGAATGCCAACTAGGGATAACAGGGTAATCGATTT | |
| d199kanR | TCCGTGTTCTAATCGAAGAGGTTGGCATTCCGCATTAGGACACGTAACAGTATCATTCCAGCCAGTGTTACAACCAATTAACC | |
| CPXV207 | BR207F | TGAGTGGTGGTAGTTACGGATATC |
| BR207R | AGTATCTCCAACATATGGCAGTTC |
Preparation of plasmid and BAC DNA and verification of BAC mutagenesis.
Plasmid and BAC DNAs were extracted by alkaline lysis (14) and verified by restriction fragment length polymorphism (RFLP) analysis. Briefly, BAC DNA was cleaved with selected restriction enzymes and separated by 0.8% agarose gel electrophoresis for 16 h at 75 V in TAE buffer (40 mM Tris, 20 mM acetic acid, 1 mM EDTA, pH 8.4). To confirm integrity of the respective gene in the repaired BAC clones, PCR was performed using the original cloning primers (Table 3). PCR products were checked by agarose gel electrophoresis, purified using a GF-1 AmbiClean (PCR & Gel) nucleic acid extraction kit (Vivantis Technologies, Subang Jaya, Malaysia), and finally sequenced (LGC Genomics GmbH, Berlin, Germany).
TABLE 3.
Primers used for cloning
| Name | Sequence (5′–3′) | Description of amplified sequence |
|---|---|---|
| I-SceIF | AAACTAGGGATAACAGGGTAATCTCATGTTTGACAGCTTATCATC | Tetracycline cassette, adding I-SceI restriction site |
| I-SceIR | AAGAATTGATTGGCTCCAATTC | |
| BR060F | GACTTTACACAACGCGATATATAACTTTC | Part of ORF CPXV060 |
| BR060R | GACGACGAATATGTTCATATCACTTC | |
| BR064F | GCTGTTTCGAAACTTTATCAATACAC | Part of ORF CPXV064 |
| BR064R | GCAGGGGTATTAATATCAGGGTATC | |
| BR068F | TCGCATATATTCCCAAGTCTTCAGT | Part of ORF CPXV068 |
| BR068R | GCTGTAGATACACTTCTTGGTTACG | |
| BR069F | CGGAATAACATCATCGAAAGAC | Part of ORF CPXV069 |
| BR069R | GGGTTAATCAGAGCCACATTC | |
| BR074F | ACATAGTTGATAAAAAGCGGTAGG | Part of ORF CPXV074 |
| BR074R | TACCGGAGAAAGATCCATTAGC | |
| BR136F | GTTGTGCTGTTGTACATACTGTACC | Part of ORF CPXV136 |
| BR136R | AATGGACTTCTTTAACAAGTTCTCAC | |
| BR168F | CTAAGGTCGTTAGTAGGGAGGC | Part of ORF CPXV168 |
| BR168R | GCATCCGAGAATGACTTGTAGTC | |
| BR169F | CGATGTCTTGACTACCTGGCTC | Part of ORF CPXV169 |
| BR169R | GAATCCGTCGTACTGTTTAGTTG | |
| BR172F | CTTCATTCTGTATATCAGACGGC | Part of ORF CPXV172 |
| BR172R | CCACAACATTGGATTCGTTATC | |
| BR199F | GAGATAAATGGTCGTGTTTTCC | Part of ORF CPXV199 |
| BR199R | CACGTAACAGTATCATTCCAAGAAG | |
| BR207F | TGAGTGGTGGTAGTTACGGATATC | Part of ORF CPXV207 |
| BR207R | AGTATCTCCAACATATGGCAGTTC |
Plasmid construction.
The tetracycline resistance gene (Tetr) was amplified from plasmid pACYC184 (NEB, Frankfurt, Germany) and cloned into the HincII site of pUC19 (NEB). An I-SceI restriction recognition site was added to the Tetr cassette by additional sequences in the primer and PCR amplification (Table 3). To generate transfer plasmids for the repair of the individual CPXV deletion mutants (12, 13), deleted sequences including sequences up- and downstream of the respective ORF were PCR amplified from pBRFseR (10) and cloned into the HincII site of pUC19 (primers are listed in Table 3). The Tetr and an adjoining I-SceI site were also amplified by PCR and inserted into unique restriction sites of the corresponding cloned genes (primers and restriction enzymes used are listed in Table 2). Duplicated sequences for removal of the Tetr cassette were added through 5′ extensions of the primers (Table 4).
TABLE 4.
Primers used for insertion of tetracycline cassette
| Target ORF | Name | Sequence (5′–3′) | Restriction site |
|---|---|---|---|
| CPXV069 | d069tetF | AATCTAGATCTTAGACATTTTTAGAGTAGGGATAACAGGGTAATCTCATG | BglII |
| d069tetR | AATCTAGATCTATATCGACGAACGTTCTGAAAGAATTGATTGGCTCCAATTC | ||
| CPXV074 | d074tetF | AACATACGCGTATGGATGGATACCAGTAGGGATAACAGGGTAATCTCATG | MluI |
| d074tetR | AACATACGCGTAATTATCAAATAGATATGTAAGAATTGATTGGCTCCAATTC | ||
| CPXV136 | d136tetF | AACGGTTTAAGGAGTGTACTTAGGGATAACAGGGTAATCTCATG | HincII |
| d136tetR | GACACCGAGCAATTCTATTCAAGAATTGATTGGCTCCAATTC | ||
| CPXV168 | d168tetF | AACATACGCGTGATTGGTCTATGTATTAGGGATAACAGGGTAATCTCATG | MluI |
| d168tetR | AACATACGCGTTTGCGTTTATTCTTTCCCTAAGAATTGATTGGCTCCAATTC | ||
| CPXV169 | d169tetF | TAAAGATTATTGGGTAAGTTTAGGGATAACAGGGTAATCTCATG | PsiI |
| d169tetR | TAAAAAATACTAAACAATACAAGAATTGATTGGCTCCAATTC | ||
| CPXV172 | d172tetF | TAAACCATGGAACAAAATAATGACGTAGGGATAACAGGGTAATCTCATG | NcoI |
| d172tetR | TAAACCATGGCGCTACAGTGATCCTCCCAAAGAATTGATTGGCTCCAATTC | ||
| CPXV207 | d207tetF | GACTTCACTGATTGTCGCACTAGGGATAACAGGGTAATCTCATG | HincII |
| d207tetR | AACAGTTTGGAAATTATCTCAAGAATTGATTGGCTCCAATTC |
Reconstitution of infectious virus from BAC DNA.
For virus reconstitution, 1 × 105 Vero cells were seeded in one well of a 24-well plate. Cells were transfected with approximately 2 μg of purified BAC DNA using 1 μl of FuGENE HD transfection reagent (Promega, Mannheim, Germany) according to the manufacturer's protocol and then infected with 20 to 500 PFU of FWPV at 2 h after transfection. Plaques formed by reconstituted viruses were monitored using an Axiovert S100 fluorescence microscope (Carl Zeiss, Jena, Germany). Viruses were passaged twice on Vero cells in order to remove helper virus.
CAM infections and analysis of size and shape of pocks.
Embryonated SPF White Leghorn eggs were incubated at 37°C and a relative humidity of 50% for 11 days. A small hole was made in the eggshell with an electric drill, and the CAM was infected with 100 PFU of virus in 200 μl of phosphate-buffered saline (PBS) or with the same volume of freeze-thawed Vero cells as a control. Eggs were sealed with wax and incubated at 37°C for 4 days without being moved. Finally, CAMs were harvested, washed three times with PBS, and photographed. Pock sizes were determined using ImageJ software (http://rsb.info.nih.gov/ij/). Statistical analysis was performed with Prism software (version 6; GraphPad Software, Inc.). CAMs were examined for pock shape using an Axiovert S100 fluorescence microscope to identify red and green fluorescence. Images were taken using an AxioCam MRm charge-coupled-device (CCD) camera (Carl Zeiss). Image processing was performed with the AxioVision, version 4.8.2, software package (Carl Zeiss). CAMs were finally fixed in neutral-buffered 4% formaldehyde and embedded in paraffin. Two-micrometer sections were cut and mounted on adhesive glass slides and stained with hematoxylin and eosin for histological examination.
Plaque size assays and correlation between plaque and pock sizes.
To determine virus plaque sizes, Vero cells seeded in six-well plates were infected with 100 PFU per well. After 90 min of incubation, medium was removed and replaced by 0.8% carboxymethyl cellulose (CMC; Sigma-Aldrich) in MEM with 3.5% FBS. Plaques were monitored using an Axiovert 100 fluorescence microscope, and fluorescent plaque images were taken before processing with the Axiovision, version 4.8.2, software. Plaque and pock sizes were determined using ImageJ software. Statistical analysis was performed with GraphPad Prism software.
Bioinformatics analysis.
Bioinformatics analyses were performed using NCBI BLAST, and comparisons were based on OPV sequences available at the Poxvirus Bioinformatics Resource Center (PBRC; www.poxvirus.org) and GenBank (15).
RESULTS
Generation of CPXV mutants with deletions in kelch-like genes or CPXV207 (crmA).
CPXV-BR contains six genes predicted to encode kelch-like proteins, CPXV013, CPXV035, CPXV050, CPXV193, CPXV204, and CPXV215 (www.poxvirus.org) (Fig. 1). First, kelch-like genes were deleted from BAC clone pBRFseR individually (Table 5). A complete kelch-like gene deletion mutant was generated by sequential deletion of all six kelch-like genes from CPXV013 to CPXV215 (Table 5). All BAC clones were verified by RFLP, and all patterns exactly matched in silico predictions (data not shown). Sequences covering deleted ORFs were amplified by PCR. The PCR products were analyzed by agarose gel electrophoresis (data not shown) and sequenced. The sequencing results confirmed the deletion of the kelch-like gene as intended (data not shown). Mutant viruses with deletions of a single kelch-like gene or of two to six kelch-like genes were reconstituted successfully from modified BAC clones. CPXV207 (crmA) was deleted from pBRFseR using a strategy described previously (10). The resulting mutant, pBRFseRd207, was verified by RFLP, and the pattern matched the in silico prediction (data not shown). Sequences covering the insertion site were amplified by PCR and analyzed by sequencing, which confirmed the deletion of CPXV207 (crmA) (data not shown). Eventually, the crmA deletion mutant vBRFseRd207 (crmA deletion) was reconstituted on Vero cells from the modified BAC clone pBRFseRd207.
FIG 1.
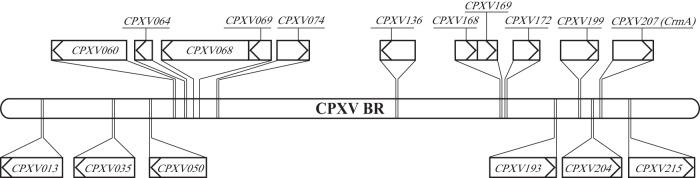
Schematic of genes necessary for induction of red pocks in the CPXV-BR genome and genes (boxes) encoding kelch-like proteins. The schematic illustrates the location, size, and direction of transcription of the respective gene (indicated by the arrow). The bar represents the genome of CPXV-BR (224 kbp). Genes above the bar are necessary for induction of red pocks. Genes below the bar are genes encoding kelch-like proteins.
TABLE 5.
Kelch-like deletion mutants
| Name of mutant |
kelch-like gene profilea |
|||||
|---|---|---|---|---|---|---|
| CPXV013 | CPXV035 (C2L) | CPXV050 (F3L) | CPXV193 (A55R) | CPXV204 | CPXV215 | |
| pBRFseRd035 | X | |||||
| pBRFseRd050 | X | |||||
| pBRFseRd193 | X | |||||
| pBRFseRd204 | X | |||||
| pBRFseRd215 | X | |||||
| pBRFseRdα | X | |||||
| pBRFseRdβ | X | X | ||||
| pBRFseRdγ | X | X | X | |||
| pBRFseRdδ | X | X | X | X | ||
| pBRFseRdε | X | X | X | X | X | |
| pBRFseRdζ | X | X | X | X | X | X |
Deletions are indicated by “X.” Homologous genes in the VACV Copenhagen strain are shown in parentheses.
Identification of mutant CPXV inducing nonhemorrhagic pocks on CAM.
Individual deletion mutant viruses were examined for their pock phenotypes on chicken CAM. The crmA deletion mutant vBRFseRd207 was used to infect CAMs, and nonhemorrhagic pocks were observed on CAMs infected with vBRFseR207, consistent with earlier results (7). All kelch-like deletion mutants, i.e., all mutants devoid of individual genes and the mutant missing all six kelch-like genes produced hemorrhagic pocks similar to those produced by wild-type virus. From the 109 viable knockout mutants, 10 produced nonhemorrhagic (white) pocks on infected CAM (Fig. 1 and 2A; also Table 6), while one, vBRFseRd061 (F13L deletion), failed to produce any visible lesions on the CAMs. All other single-gene deletion mutants induced formation of hemorrhagic (red) pocks upon CAM inoculation (data not shown). To confirm the results, reconstitutions from the 10 mutant BAC constructs were repeated twice, and CAMs were infected with newly reconstituted viruses. Consistently, mutant viruses reconstituted from the 10 BACs produced white pocks on infected CAMs (data not shown).
FIG 2.

Macroscopic photographs of pock lesions on chicken CAM. (A) White pocks caused by deletion mutants compared with red pocks produced by wild-type virus. (B) Red pocks produced by repaired viruses and wild-type virus. Scale bar, 0.5 cm. wt, wild type.
TABLE 6.
CPXV genes responsible for hemorrhagic phenotype, their functions, and homologue similarity
| Group and CPXV-BR ORF | Protein length (aa)f | VACV-COP homologue(s) | Function(s)a | Pock type of mutantb | Actin tail induction | EEV productionc | Conservationd | Similarity (%) of protein homologues in different OPVS |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CPXV-GRI | VACV-Lister | VACV-COP | VACV-MVA | VARV | MPXV | RPXV | ||||||||
| Immune suppressor | ||||||||||||||
| CPXV069 | 190 | E3L | Interferon resistance and PKR inhibitor | II | No | No | O | 94.74 | 96.32 | 96.32 | 96.32 | 96.84 | 90.85 | 95.79 |
| CPXV207 (crmA) | 341 | B13R/B14Re | Serine Protease Inhibitor-2 | I | No | No | O | 94.48 | 96.33 | 96.79 | 95.45 | 94.77 | 94.77 | 95.06 |
| Core protein | ||||||||||||||
| CPXV074 | 273 | E8R | Endoplasmic reticulum localized membrane protein | I | No | No | C | 99.63 | 99.27 | 99.27 | 99.27 | 98.90 | 99.27 | 99.27 |
| CPXV136 | 295 | A4L | Core protein | I | No | No | C | 92.2 | 90.85 | 91.19 | 88.81 | 87.50 | 89.49 | 91.86 |
| EEV-related protein | ||||||||||||||
| CPXV068 | 737 | E2L | IEV- associated protein | II | No | Yes* | C | 99.32 | 99.32 | 99.32 | 99.32 | 99.05 | 98.91 | 99.46 |
| CPXV060 | 634 | F12L | IEV- associated protein | III | Yes | Yes* | C | 98.42 | 98.11 | 98.11 | 98.27 | 97.01 | 98.43 | 98.27 |
| CPXV168 | 187 | A33R | EEV glycoprotein | III | Yes | Yes† | O | 98.40 | 97.33 | 97.33 | 97.33 | 93.58 | 97.25 | 97.33 |
| CPXV169 | 168 | A34R | EEV C-type lectin-like protein | III | Yes | Yes† | O | 100.00 | 100.00 | 99.40 | 99.40 | 100.00 | 100.00 | 100.00 |
| CPXV172 | 224 | A36R | IEV specific | III | Yes | Yes* | O | 97.78 | 96.89 | 96.89 | 93.18 | 93.78 | 95.11 | 96.89 |
| CPXV199 | 317 | B5R | EEV complement control protein | III | Yes | Yes* | O | 97.48 | 97.74 | 96.85 | 97.16 | 97.47 | 97.79 | 96.85 |
| Unknown | ||||||||||||||
| CPXV064 | 477 | F15L | Unknown | II | ? | ? | C | 98.10 | 98.10 | 97.47 | 97.47 | 97.39 | 97.47 | 97.47 |
Information about functions were based on studies of homologues in vaccinia virus and obtained from www.poxvirus.org.
Types of pocks were determined according to results of fluorescence microscopy (Fig. 3).
No, not involved in the production of EEV; yes, involved in the production of EEV; *, deletion mutant produces less EEV than the wild type; †, deletion mutant produces more EEV than the wild type (23).
C, gene conserved in chordopoxviruses (16); O, gene conserved only in orthopoxviruses (www.poxvirus.org).
B13R/B14R, a homologue of the CPXV207 (crmA) gene in VACV, is not functional due to formation of a premature stop codon.
aa, amino acids.
The shape of pocks induced by mutant CPXV on CAM.
Red and green fluorescent protein expressed by BAC-derived CPXV can readily be detected by fluorescence microscopy (Fig. 3). All pocks on the examined CAMs showed red and green fluorescence and allowed analysis of their respective shapes. Wild-type CPXV produced round pocks with dark areas in the middle, as did all red pock-producing deletion mutants (data not shown). In contrast, three different groups of nonhemorrhagic pocks were observed and were classified according to their morphologies as type I, type II, and type III (Fig. 3). Type I pocks produced by vBRFseRd074 (E8R deletion), vBRFseRd136 (A4L deletion), and vBRFseRd207 (crmA deletion) were similar in shape to those induced by wild-type virus. Type II pocks produced by vBRFseRd064 (F15L deletion), vBRFseRd068 (E2L deletion), and vBRFseRd069 (E3L deletion) were round and solid, whereas type III pocks produced by vBRFseRd060 (F12L deletion), vBRFseRd168 (A33R deletion), vBRFseRd169 (A36R deletion), vBRFseRd172 (A36R deletion), and vBRFseRd199 (B5R deletion) exhibited a comet-like shape (Fig. 3).
FIG 3.

Fluorescence microscopy of single pocks. The shapes of pocks can be classified into three types: type I, type II, and type III, as indicated on the figure. Type I pocks are round, with a dark area in the middle; type II pocks are round but without a dark area; type III pocks are comet-like and have no dark area. Scale bar, 500 μm.
Histological examination of pocks from infected CAMs.
Sections of pocks on CAMs infected with the white pock-inducing mutant viruses or with wild-type virus were stained with hematoxylin and eosin (Fig. 4). CPXV infection led to a number of microscopic lesions, including epithelial necrosis as well as epithelial and stromal proliferation. Consistent with the macroscopic data, hemorrhages were found in pocks induced by parental virus (Fig. 4) and in red pocks caused by single-deletion mutants (data not shown). In contrast, no microscopically evident hemorrhage was found in any of the white pocks examined. The white-pock phenotype was also associated with less necrosis and inflammation than the red-pock phenotype as well as with decreased proliferation of epithelial and stromal cells (Fig. 4).
FIG 4.

Histological examination of pocks from infected CAMs. Magnification, ×200.
Generation of repaired viruses and CAM infection.
Repaired viruses were reconstituted from BAC clones in which the deleted genes had been restored by two-step Red-mediated recombination (12, 13). Since all knockout BACs already harbored the kanamycin resistance gene, different positive selection markers were used to achieve the repair (12, 13). A tetracycline or ampicillin cassette was inserted into previously deleted sequences that had been cloned in pUC19. The entire insert was PCR amplified from the respective transfer plasmids (primers are given in Tables 2 and 3) and inserted into the target loci of each of the mutant BACs by Red recombination. The antibiotic cassette was removed by a second step of Red recombination with the expression of the I-SceI restriction enzyme within E. coli (Fig. 5) (12, 13).
FIG 5.
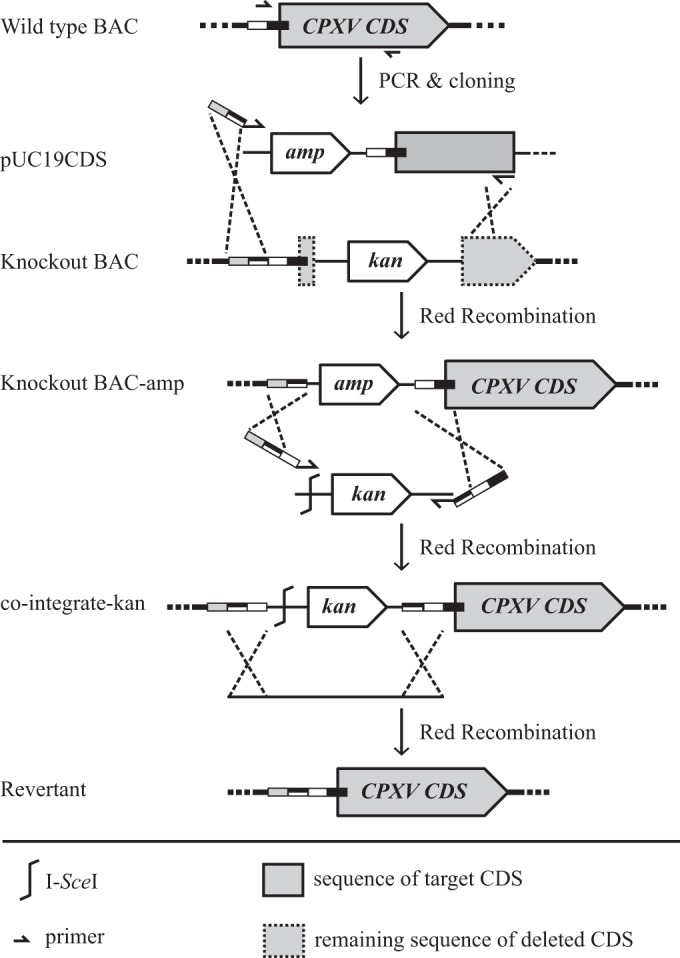
Schematic of generation of repaired BAC clones. Sequence flanking the deleted sequence of the target ORF was amplified from wild-type BAC DNA and cloned into the pUC19 plasmid. The cloned fragment together with the ampicillin resistance gene was PCR amplified and inserted into the knockout BAC by Red recombination, with primers containing additional sequences homologous to the target locus. The ampicillin resistance cassette was removed by two-step Red recombination. CDS, coding sequence.
All BAC clones were verified by RFLP, and all the patterns exactly matched in silico predictions (data not shown). Repaired sequences were amplified by PCR and analyzed by agarose gel electrophoresis. As intended, all repaired viruses contained the previously removed fragments, yet point mutations were present in repaired BAC clones pBRFseRd060rev (P237E and N448I) and pBRFseRd068rev (D13G and Y189N), likely as a result of PCR amplification. Repaired viruses were reconstituted and titrated on Vero cells. The ability to produce red pocks on chicken CAM was restored for all repaired viruses, including the two in which point mutations were present after repair (Fig. 2B).
Plaque and pock sizes induced by mutant viruses.
Plaque sizes of vBRFseR (wild-type virus), of all deletion mutants that produced white pocks, and of repaired viruses were determined on Vero cells. Compared with vBRFseR, all mutant viruses except vBRFseRd136 (A4L deletion) produced significantly smaller plaques (Fig. 6A). However, plaques produced by the respective repaired viruses and wild-type viruses did not significantly differ from each other (data not shown). Sizes of pocks produced by wild-type viruses and all deletion mutants that produced white pocks were determined. Compared with vBRFseR, all mutant viruses except vBRFseRd136 (A4L deletion) produced significantly smaller pocks (Fig. 6B). The shape of pocks and the correlation between plaque size and pock size of deletion mutants that produced white pocks were determined (Fig. 6C). Overall, there was a significant correlation between plaque and pock sizes. However, the reduction in pock size did not always correlate with the reduction in plaque size (as in the case for vBRFseRd064 and vBRFseRd068) (Fig. 6C).
FIG 6.
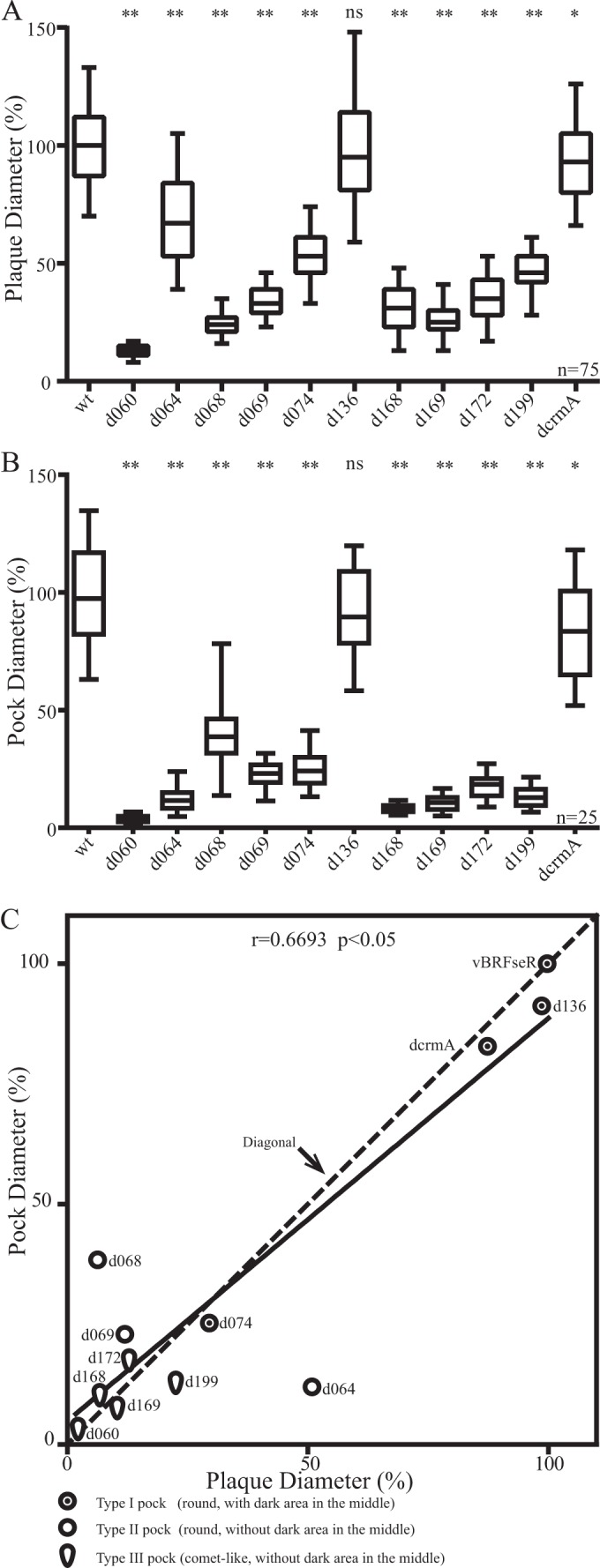
Analysis of plaque and pock sizes of mutant viruses. (A) Relative plaque sizes. Diameters of 75 plaques of each construct were measured in three independent experiments. Plaque diameters of all repaired viruses were not different from those of vBRFseR (data not shown). (B) Relative pock sizes. Diameters of 25 pocks of each of the viruses were measured. Plaque sizes and pock sizes were analyzed by analysis of variance for multiple comparisons with Bonferroni corrections. Significance is indicated as follows: *, significantly different from vBRFseR with an alpha of 5%; **, highly significant difference from the wild type with an alpha of 1%; ns, not significantly different from vBRFseR. (C) Correlation between plaque and pock sizes. The average relative sizes of plaques and pocks were analyzed by a nonparametric correlation (Spearman) test. Mutants with the same reductions in plaque and pock sizes are located on the diagonal. Mutants with greater reductions in pock sizes are above the diagonal. Mutants below the diagonal have greater reductions in plaque sizes. Three different symbols were used to represent different types of pocks. Abbreviated names of viruses are used in the figure (wt for parental vBRFseR, dcrmA for deletion of crmA, and d060 for vBRFseRd060, where d060 indicates the deletion of CPXV060, etc.).
CPXV ORFs necessary for hemorrhagic pocks and bioinformatics analysis.
Ten deletion mutants induced white pocks on the CAMs of infected chicken eggs, and the respective ORFs were therefore classified as necessary for the hemorrhagic pock phenotype (Table 6). All identified genes are conserved in either all chordopoxviruses (16) or the OPV (according to annotations of different OPV genomes in the database present on www.poxvirus.org) (Table 6). Protein function was determined based on protein homologues in other OPVs (www.poxvirus.org), as listed in Table 6, and the proteins were classified into four groups, as follows.
(i) Immune modulation.
CPXV CrmA and the CPXV069 product act as immune suppressors during infection. CrmA is involved in the modulation of inflammatory cytokines including interleukin-1β (IL-1β) and IL-18 (17). The homologue of CPXV069 in VACV is E3L, which encodes a protein inhibiting the expression of beta interferon (IFN-β) and tumor necrosis factor alpha (TNF-α) (18, 19).
(ii) Core proteins.
The homologues of CPXV074 and CPXV136 in VACV are E8R and A4L, respectively, and both the E8 and A4 proteins are involved in virion assembly and are also packaged in the viral core (20–22).
(iii) EEV production.
The homologues of CPXV060, CPXV068, CPXV168, CPXV169, CPXV172, and CPXV199 in VACV are F12, E2, A33, A34, A36, and B5, respectively. Products of all of these ORFs are involved in the process of the formation of extracellular enveloped viruses (EEV) (23, 24). Deletion of A36, B5, E2, or F12 in VACV resulted in decrease of EEV production (19, 23–26), whereas VACV lacking A33 or A34 produced more EEV (23, 27, 28).
(iv) Unknown.
The function of the VACV-COP protein encoded by F15L (corresponding to CPXV064) is not known.
The 10 genes can also be classified according to their role in actin tail formation. According to the functions of their VACV homologues, the products of CPXV060, CPXV168, CPXV169, CPXV172, and CPXV199 may be responsible for EEV production and actin tail induction (23). In VACV, the homologues of the products of CPXV168 and CPXV172, A33 and A36, were shown to induce actin tails in infected cells, a property that was shown to repulse progeny virions and enhance virus spread in VACV (29). NCBI BLAST results identified high similarities between the CPXV proteins and their homologues in other OPVs (Table 6). For example, alignment of CPXV-BR protein CPXV199 with its homologues in other OPVs showed that the amino acid sequences are identical between RPXV, VACV-COP, and CPXV for this protein.
DISCUSSION
The full-length BAC clone of CPXV-BR (30) and the generation of a complete single-gene knockout BAC library (10) have allowed us to perform high-throughput screening to study poxvirus gene functions. Here, we used the established CPXV knockout library to study the hemorrhagic phenotype of CPXV on CAM. Previous work had shown that CPXV CrmA is necessary but not sufficient for causing hemorrhagic pocks on chicken CAM (5, 7). In another study, CPXV GRI-90 with a deletion of four of the six kelch-like proteins produced smaller and less hemorrhagic pocks on CAM (9). Kelch proteins form a superfamily of proteins containing kelch repeats involved in diverse cellular functions (31). Previous studies revealed that CPXV encodes six kelch-like proteins, while VACV and monkeypox virus (MPXV) encode only three and one kelch-like protein, respectively (32–36). In VARV, no functional kelch-like protein is encoded (32, 33). In a mouse model of infection, VACV lacking each of the three kelch-like proteins and CPXV lacking four of the six kelch-like proteins were shown to be less virulent (9, 34–36). However, the function of orthopoxvirus kelch-like proteins remains poorly understood (32, 33).
Considering the fact that OPVs encoding fewer kelch-like proteins produce nonhemorrhagic pocks and that the absence of four kelch-like proteins in CPXV GRI-90 caused a change in pock phenotype (9), we hypothesized that all kelch-like proteins might be involved in causing hemorrhagic pocks on CAM. Therefore, we generated mutants of CPXV-BR with single, multiple, and complete deletions of the kelch-like proteins to study the role of kelch-like proteins in causing the red pocks. Surprisingly, we did not observe different forms of pocks produced by any of the kelch-like protein deletion mutants compared to those produced by wild-type viruses. The discrepancy between our findings and those reported by Kochenva et al. could result from the difference between CPXV-BR and CPXV GRI-90. It is also possible that spurious or compensatory mutations caused by standard homologous recombination used in the CPXV GRI-90 study arose in the process of generating the mutant with four kelch-like genes absent (9). Because of the low efficiency of homologous recombination in eukaryotic cells, progeny viruses containing the desired, modified genome are in the minority. Therefore, multiple rounds of plaque purification are necessary, which can increase the probability for compensatory mutations (37). In contrast, the BAC mutagenesis system used in this study is a suitable tool to propagate and modify large genomic DNA fragments (38) and also avoids the problem of selection. A prominent example is the stepwise introduction of six major deletions into the chorioallantois vaccinia virus Ankara (CVA) BAC (39). Sequencing of mutant viruses revealed that no mutations occurred during recombination, virus reconstitution, and subsequent passaging even though large portions of the viral genome had been rearranged (39). Likewise, we did not observe any spurious mutations at the modified loci with our own poxvirus BAC clones, and we therefore prefer them over traditional methods for mutant virus generation. To avoid mutations occurring during reconstitution and passage that could cause the virus to produce white pocks, all 10 mutant viruses that produced white pocks were reconstituted twice from the respective knockout BAC clones. All newly reconstituted viruses behaved identically and produced white pocks on CAMs, which we interpret as confirming the robustness of the methodology.
To identify proteins involved in the hemorrhagic phenotype, we screened 109 single-deletion mutants by infecting chicken CAMs. Most of the mutant viruses produced hemorrhagic pocks, while vBRFseRd061 (F13L deletion) failed to produce visible pocks and also to produce plaques on CEC (data not shown). A VACV mutant lacking the F13L gene is unable to form EEV and produce plaques but makes normal amounts of intracellular mature viruses (IMV) (40). Therefore, the failure of vBRFseRd061 (F13L deletion) to produce plaques on CEC and to induce pocks on CAM may be caused by small amounts of EEV produced and a concomitant reduction of virus particles that can spread directly from cell to cell. Ten deletion mutants were found to produce nonhemorrhagic white pocks, suggesting that the 10 corresponding proteins are essential for the formation of hemorrhagic pocks. All 10 proteins are highly conserved among poxviruses (Table 6). Homologues in CPXV GRI, VACV-Lister, VACV-COP, VACV-MVA, MPXV, VARV, and RPXV were chosen for comparative analysis. BLAST results revealed a high degree of similarity of the 10 CPXV-BR proteins and their homologues in different OPVs (Table 6). Alignment of homologues of the CPXV199 (B5R deletion) protein showed that RPXV ps/hr and VACV-COP B5 have exactly the same amino acid sequences. This analysis suggests that the functions of the identified proteins in CPXV are comparable to those of their counterparts in VACV. As most studies on poxvirus genes were performed using VACV, information on gene function was obtained from studies on the respective protein in VACV (Table 6).
Although proteins responsible for the hemorrhagic phenotype have a variety of functions, the proteins and their possible roles in causing hemorrhagic lesions fall into several categories (Table 6). One category contains proteins with immunosuppressive functions. It is known that CPXV CrmA is an inhibitor of granzyme B (41) and caspase-1 (42). CrmA also blocks the maturation of the proinflammatory cytokines IL-1β and IL-18 (17). Yet the role of CrmA in the formation of hemorrhagic pocks on CAM is still poorly understood. In pocks induced by CPXV BR lacking the crmA gene, inflammatory cells were absent, and increased levels of virus antigen were observed (43). CPXV-BR expressing myxoma virus serpin 2 or baculovirus P35 instead of CrmA still produced white pocks on CAM, while the inhibition of the terminal caspase and the antiapoptotic effects were restored (44). Moreover, CPXV-BR produced white pocks when the serpin function of CrmA was abolished by a point mutation (44). These results suggest that CrmA is involved in regulation of the inflammatory response, and the inhibition of protease function but not direct inhibition of caspase 3 is essential for the function mediated by CrmA with respect to the hemorrhagic pock phenotype.
Serpin 1 and serpin 2 were proven to be responsible for the red-pock phenotype of RPXV, while only serpin 2 (CrmA) but not serpin 1 is required in the case of CPXV (8). In the present study, we included vBRFseRd207 (crmA deletion mutant) and vBRFseRd217 (serpin 1 deletion mutant) in the screen. Pocks produced by vBRFseRd207 (crmA deletion) were white, while those produced by vBRFseRd217 (serpin 1 deletion) were hemorrhagic (data not shown). Our results are, therefore, consistent with those of the previous studies (8).
The VACV homologue of CPXV069, E3, encodes a function determining the host range of VACV (45). The E3 product is also involved in the inhibition of host immune responses (46). E3 is a double-stranded RNA binding protein that inhibits the activation of protein kinase R (PKR) (18), a multifunctional antiviral protein (47). PKR inhibits viral replication by blocking both host and viral protein synthesis and regulates cytokine expression (47–49). Therefore, the ability of E3 to inhibit PKR activation is crucial for maintaining the cellular translation machinery (50). Moreover, E3 is able to inhibit the expression of several cytokines, including IFN-β and TNF-α, through suppression of the PKR, NF-κB, and interferon regulatory factor 3 (IRF3) pathways (46). Considering the function of CrmA and E3, we hypothesize that the suppression of host immune responses by CrmA and CPXV069 is necessary for the hemorrhagic phenotype. Both CrmA and E3 evidently inhibit cell signaling pathways, which may be of vital importance for the induction of hemorrhagic pocks.
The VACV B5 protein (homologue of CPXV199) might also play a role in the inhibition of host immune responses as B5 shows sequence similarity to complement control proteins (51). However, whether B5 is involved in complement control is unknown (51). The B5 protein is known to be involved in EEV production and actin tail formation (19, 52, 53). Therefore, we included protein B5 in the second category, which is related to EEV production and virus spread. During VACV infection, intracellular mature viruses (IMV), which are released after cell lysis (23), are the majority fraction. Some IMV become wrapped by a double layer of intracellular membrane to form intracellular enveloped virus (IEV) (23). Subsequently, IEV moves to the cell surface and forms cell-associated enveloped viruses (CEV), which can be released to form EEV (23). However, the spread of VACV progeny can be accelerated by EEV being pushed by virus-induced actin tails or by the production of EEV before cell lysis (23, 54). The VACV homologues of CPXV060, CPXV068, CPXV168, CPXV169, CPXV172, and CPXV199 encode proteins that are all involved in IEV formation, IEV transport to the cell surface, actin tail formation, or EEV formation (23, 29). According to their different functions, the six proteins can be divided into three subgroups. The first subgroup contains proteins CPXV060, CPXV172 and CPXV199, whose VACV homologues (F12, A36, and B5) are involved in actin tail induction. Deletion of F12, A36, or B5 in VACV resulted in a decrease in EEV production (19, 25, 26). The second subgroup is comprised of A33 and A34 (VACV homologues of CPXV168 and CPXV169), which are also involved in the induction of actin tails. However, VACV lacking A33 or A34 produces more EEV than wild-type virus (23, 27, 28). The third subgroup consists of protein E2. VACV E2 protein is not involved in inducing actin tails, but the null mutant of E2 produces less EEV (24).
The ps/hr gene of RPXV (B5R homologue of VACV-COP) determines pock morphology on CAM (4), and it is also responsible for the hemorrhagic phenotype on CAM. However, the roles of the CPXV B5 homologue and other EEV proteins involved in the pock phenotype on CAM have not been determined. A possible explanation for a link between EEV production and induction of hemorrhagic pocks is that EEV determine cell entry. Compared to IMV, EEV possess an additional membrane and incorporate into virions at least six more proteins (55). Therefore, IMV and EEV are considered different forms of infectious particles, which likely utilize different entry pathways (56, 57). IMV are thought to mediate transmission between hosts, whereas EEV are specialized for spread within a host. The change in the number of EEV or the change in the ratio of EEV to IMV could possibly affect the efficiency and spread of infection in vivo.
While producing more EEV, A33 or A34 deletion mutants are much slower in virus spread in vitro, a phenomenon caused by their failure to induce actin tails (23). Although the release of EEV before cell lysis can benefit virus transmission, it is difficult for the EEV to cross polarized epithelial and endothelial cells and tight junctions. Considering that actin tails induced by poxvirus infection can push progeny virions to neighboring cells (29), this means of virus spread might help the virus to cross polarized epithelia and the tight-junction barrier. Although E2 is not involved in inducing actin tails, the decreased amount of EEV produced by an E2 null mutant also slows virus spread. Therefore, we speculate that rapid spread mediated by EEV production or actin tail formation of CPXV is of vital importance for formation of hemorrhagic pocks on CAM as effective transmission of viruses within tissues will damage endothelial cells of blood vessels and cause hemorrhage.
Another category of proteins apparently governing the red-pock phenotype are those involved in viral morphogenesis, more specifically, constituents of the viral core. VACV E8 is the homologue of CPXV74. It is synthesized early in infection but packaged into nascent virions in the late stages of infection (20). During virus assembly, E8 localizes to the endoplasmic reticulum (ER) and might mediate interaction between ER cisternae and developing viral factories (20, 21). VACV A4 is homologous to CPXV136 and is also a virus core protein required for efficient VACV morphogenesis (58) although the roles of E8 and A4 in VACV infection are not fully understood.
The function of VACV-COP F15 (corresponding to CPXV064) is not known. Homologues of F15 are conserved in chordopoxviruses (16) but are not essential for CPXV replication on Vero cells (10). BLAST analysis shows high similarity between CPXV064 and its OPV homologues (Table 6), which may suggest that F15 is highly conserved and that it may play an important role in producing red pocks on CAM. Revealing the function of F15 will help us better understand the mechanism of hemorrhage of CPXV on CAM.
Earlier findings on the induction of hemorrhage upon poxvirus infection of CAM identified only modulators of host immune responses (7, 44). We here add two categories of structural proteins (core and EEV) as being necessary for hemorrhaging, possibly shifting the focus from immunomodulation more toward efficient virus production, spread, and likely cellular, as well as endothelial, damage.
Although all 10 deletion mutants we describe here produce white pocks, the shapes of the lesions differ from virus to virus (Fig. 3). Pocks produced by vBRFseR (wild-type virus) are referred to as type I pocks (Fig. 3). The dark areas in the middle of red pocks are caused by necrotic cells in the center of the pocks that lost fluorescence. Pocks caused by vBRFseRd074 (E8R deletion), vBRFseRd136 (A4L deletion), and vBRFseRd207 (crmA deletion) also have dark areas in the middle, whereas other white pocks show solid fluorescence over the entire lesion (Fig. 3). This might be caused by the fact that vBRFseR, vBRFseRd074 (E8R deletion), vBRFseRd136 (A4L deletion), and vBRFseRd207 (crmA deletion) replicate better or cause more damage to the tissue than other nonhemorrhagic mutants. Similar to type I pocks, type II pocks produced by vBRFseRd064 (F15L deletion), vBRFseRd068 (E2L deletion), and vBRFseRd069 (E3L deletion) are also round but lack the dark areas in the middle (Fig. 3), which is caused by lower numbers of necrotic cells in the pocks.
Deletion of CPXV060, CPXV168, CPXV169, CPXV172, or CPXV199 resulted in the production of comet-like pocks (type III pocks) rather than round pocks like the type I or type II pocks produced by the other six deletion mutants. RPXV lacking the ps/hr gene (homologous to CPXV199 and VACV B5R) also produced white pocks with a similar shape (4). Microscopic examination of CAM showed that the comet-like shape is not caused by blood vessel architecture because the pocks did not localize to arteries or veins (data not shown). However, CPXV060, CPXV168, CPXV169, CPXV172, or CPXV199 deletion mutants produced small and round plaques (data not shown). RPXV lacking the ps/hr gene also produced smaller but round plaques (4), indicating that viruses can indeed behave differently in cell culture and live tissue and that the shape of pocks cannot be predicted from the shape of plaques. VACV lacking the E2L gene (homologue of CVXV068) produces less EEV (24), but vBRFseRd068 (E2L deletion) produced round pocks (Fig. 3). In contrast, VACV lacking protein F12, A36, or B5 produced less EEV (19, 25, 26), while VACV lacking protein A33 or A34 produced more EEV than wild-type virus (23, 27, 28). However, vBRFseRd060 (F12L deletion), vBRFseRd168 (A33R deletion), vBRFseRd169 (A36R deletion), vBRFseRd172 (A36R deletion), and vBRFseRd199 (B5R deletion) produced comet-like pocks (Fig. 3). We concluded from the data that the increase or decrease of EEV production is not related to the comet-like shape of the pocks. Considering that the A33, A34, A36, B5, and F12 proteins in VACV are all involved in actin tail formation (23), we hypothesize that deletion of actin tail-related genes in CPXV altered virus transmission, which might have caused the different pock shape.
Besides their shapes, the sizes of the pocks produced by different mutant viruses also varied. Although white pocks were usually smaller than red pocks produced by vBRFseR (wild-type virus), the sizes of pocks produced by vBRFseRd136 (A4L deletion) were similar to those produced by vBRFseR (wild-type virus) (Fig. 6B). We found that smaller lesion size is not necessarily a characteristic of white pocks as many mutants produced hemorrhagic pocks that were smaller than those induced by wild-type virus. Similarly, plaque size is also not related to hemorrhage of the lesions. Compared with wild-type virus, most mutant viruses producing white pocks on CAMs also produced significantly smaller plaques (Fig. 6A). However, vBRFseRd136 (A4L deletion) produced plaques as big as those produced by vBRFseR (wild type). We concluded, therefore, that plaque and pock sizes are not related to hemorrhage in lesions.
However, there is a positive correlation between plaque and pock sizes (Fig. 6C). Mutant viruses that produced smaller plaques in cell culture also caused smaller pocks on CAMs than vBRFseR (wild-type virus). Our results seem to be consistent with an earlier study showing that RPXV lacking the ps/hr gene (B5R homologue) yielded smaller plaques in cell culture and smaller white pocks on CAMs than parental wild-type RPXV (4). Likely, the sizes of plaques and lesions are influenced by factors determining virus entry, replication, spread, and interference with host control.
Nevertheless, the reduction in relative pock size does not always correlate with the reduction in relative plaque size. To compare relative plaque and relative pock sizes, we performed a regression analysis (Fig. 6C). Compared with vBRFseR (wild-type virus), mutant virus vBRFseRd068 (E2L deletion) and vBRFseRd069 (E3L deletion) that produced small plaques caused relatively large pocks, while mutant virus vBRFseRd064 (F15L deletion), vBRFseRd136 (A4L deletion), and vBRFseRd199 (B5R deletion) that produced large plaques caused relatively small pocks (Fig. 6C). It is known that VACV lacking E2 or B5 produces less EEV. We report here that vBRFseRd068 (E2L deletion) produced relatively large pocks while vBRFseRd199 (B5R deletion) produced relatively small pocks. Other deletion mutants that are not related to EEV production also exhibit differences with respect to the reduction of pock and plaque sizes. We concluded from our analysis, therefore, that EEV production seems not to be a determining factor for either pock or plaque size. Although protein E3 and CrmA are all immunomodulating molecules, vBRFseRd069 (E3L deletion) produced relative large pocks, whereas vBRFseRd207 (crmA deletion) showed a similar reduction in pock and plaque sizes. It seems that the difference in reduction of pock and plaque sizes could be due to different mechanisms. Besides, plaque sizes were analyzed in Vero cells covered with semisolid medium, while pocks on CAMs formed without restriction of virus movement.
Histological examination confirmed that pocks caused by wild-type virus are hemorrhagic while no hemorrhage was found in white pocks. Pocks induced by CPXV infection are characterized by epithelial necrosis and epithelial as well as stromal proliferation. Compared to red pocks, white pocks show decreased necrosis and inflammation and increased proliferation of epithelial and stromal cells.
In summary, we have identified 10 CPXV genes that are involved in the formation of hemorrhagic lesions on chicken CAM. The function of their homologues in VACV hints toward possible mechanisms for the formation of hemorrhages on infected CAM. Induction of actin tail and EEV could play an important role in causing the hemorrhagic phenotype.
ACKNOWLEDGMENTS
This study was supported by the Nationale Forschungsplattform für Zoonosen and the German Federal Ministry of Education and Research (BMBF 01KI1102) and by DFG grant (SPP 1596: Ecology and Species Barriers in Emerging Viral Diseases, Orthopoxvirus Working Group). Z.X. was supported by a scholarship from the Chinese Scholarship Council and the Dahlem Research School at Freie Universität Berlin. D.Z. received a scholarship from the International Max Planck Research School and is a member of the Center for Infection Biology and Immunology Graduate School, Berlin, Germany.
Footnotes
Published ahead of print 21 May 2014
REFERENCES
- 1.Fenner F, Anderson DA, Arita I, Jezek Z, Ladnyi ID. 1988. Smallpox and its eradication. World Health Organization, Geneva, Switzerland [Google Scholar]
- 2.Gubser C, Hue S, Kellam P, Smith GL. 2004. Poxvirus genomes: a phylogenetic analysis. J. Gen. Virol. 85:105–117. 10.1099/vir.0.19565-0 [DOI] [PubMed] [Google Scholar]
- 3.Shchelkunov SN, Safronov PF, Totmenin AV, Petrov NA, Ryazankina OI, Gutorov VV, Kotwal GJ. 1998. The genomic sequence analysis of the left and right species-specific terminal region of a cowpox virus strain reveals unique sequences and a cluster of intact ORFs for immunomodulatory and host range proteins. Virology 243:432–460. 10.1006/viro.1998.9039 [DOI] [PubMed] [Google Scholar]
- 4.Martinez-Pomares L, Stern RJ, Moyer RW. 1993. The ps/hr gene (B5R open reading frame homolog) of rabbitpox virus controls pock color, is a component of extracellular enveloped virus, and is secreted into the medium. J. Virol. 67:5450–5462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pickup DJ, Ink BS, Hu W, Ray CA, Joklik WK. 1986. Hemorrhage in lesions caused by cowpox virus is induced by a viral protein that is related to plasma protein inhibitors of serine proteases. Proc. Natl. Acad. Sci. U. S. A. 83:7698–7702. 10.1073/pnas.83.20.7698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacNeill AL, Moldawer LL, Moyer RW. 2009. The role of the cowpox virus crmA gene during intratracheal and intradermal infection of C57BL/6 mice. Virology 384:151–160. 10.1016/j.virol.2008.10.041 [DOI] [PubMed] [Google Scholar]
- 7.Roth SJ, Klopfleisch R, Osterrieder N, Tischer BK. 2012. Cowpox virus serpin CrmA is necessary but not sufficient for the red pock phenotype on chicken chorioallantoic membranes. Virus Res. 163:254–261. 10.1016/j.virusres.2011.10.002 [DOI] [PubMed] [Google Scholar]
- 8.Ali AN, Turner PC, Brooks MA, Moyer RW. 1994. The SPI-1 gene of rabbitpox virus determines host range and is required for hemorrhagic pock formation. Virology 202:305–314. 10.1006/viro.1994.1347 [DOI] [PubMed] [Google Scholar]
- 9.Kochneva G, Kolosova I, Maksyutova T, Ryabchikova E, Shchelkunov S. 2005. Effects of deletions of kelch-like genes on cowpox virus biological properties. Arch. Virol. 150:1857–1870. 10.1007/s00705-005-0530-0 [DOI] [PubMed] [Google Scholar]
- 10.Xu Z, Zikos D, Osterrieder N, Tischer BK. 2014. Generation of a complete single-gene knockout bacterial artificial chromosome library of cowpox virus and identification of its essential genes. J. Virol. 88:490–502. 10.1128/JVI.02385-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osterrieder N. 1999. Sequence and initial characterization of the U(L)10 (glycoprotein M) and U(L)11 homologous genes of serotype 1 Marek's disease virus. Arch. Virol. 144:1853–1863. 10.1007/s007050050710 [DOI] [PubMed] [Google Scholar]
- 12.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634:421–430. 10.1007/978-1-60761-652-8_30 [DOI] [PubMed] [Google Scholar]
- 13.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. 10.2144/000112096 [DOI] [PubMed] [Google Scholar]
- 14.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 15.Lefkowitz EJ, Upton C, Changayil SS, Buck C, Traktman P, Buller RM. 2005. Poxvirus Bioinformatics Resource Center: a comprehensive Poxviridae informational and analytical resource. Nucleic Acids Res. 33:D311–D316. 10.1093/nar/gki110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Upton C, Slack S, Hunter AL, Ehlers A, Roper RL. 2003. Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J. Virol. 77:7590–7600. 10.1128/JVI.77.13.7590-7600.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turner S, Kenshole B, Ruby J. 1999. Viral modulation of the host response via crmA/SPI-2 expression. Immunol. Cell Biol. 77:236–241. 10.1046/j.1440-1711.1999.00820.x [DOI] [PubMed] [Google Scholar]
- 18.Chang HW, Watson JC, Jacobs BL. 1992. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. U. S. A. 89:4825–4829. 10.1073/pnas.89.11.4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolffe EJ, Isaacs SN, Moss B. 1993. Deletion of the vaccinia virus B5R gene encoding a 42-kilodalton membrane glycoprotein inhibits extracellular virus envelope formation and dissemination. J. Virol. 67:4732–4741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doglio L, De Marco A, Schleich S, Roos N, Krijnse Locker J. 2002. The vaccinia virus E8R gene product: a viral membrane protein that is made early in infection and packaged into the virions' core. J. Virol. 76:9773–9786. 10.1128/JVI.76.19.9773-9786.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tolonen N, Doglio L, Schleich S, Krijnse Locker J. 2001. Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol. Biol. Cell 12:2031–2046. 10.1091/mbc.12.7.2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Risco C, Rodriguez JR, Demkowicz W, Heljasvaara R, Carrascosa JL, Esteban M, Rodriguez D. 1999. The vaccinia virus 39-kDa protein forms a stable complex with the p4a/4a major core protein early in morphogenesis. Virology 265:375–386. 10.1006/viro.1999.0046 [DOI] [PubMed] [Google Scholar]
- 23.Smith GL, Vanderplasschen A, Law M. 2002. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 83:2915–2931 [DOI] [PubMed] [Google Scholar]
- 24.Domi A, Weisberg AS, Moss B. 2008. Vaccinia virus E2L null mutants exhibit a major reduction in extracellular virion formation and virus spread. J. Virol. 82:4215–4226. 10.1128/JVI.00037-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parkinson JE, Smith GL. 1994. Vaccinia virus gene A36R encodes a M(r) 43–50 K protein on the surface of extracellular enveloped virus. Virology 204:376–390. 10.1006/viro.1994.1542 [DOI] [PubMed] [Google Scholar]
- 26.Ogawa R, Calvert JG, Yanagida N, Nazerian K. 1993. Insertional inactivation of a fowlpox virus homologue of the vaccinia virus F12L gene inhibits the release of enveloped virions. J. Gen. Virol. 74:55–64. 10.1099/0022-1317-74-1-55 [DOI] [PubMed] [Google Scholar]
- 27.Roper RL, Wolffe EJ, Weisberg A, Moss B. 1998. The envelope protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to-cell spread of vaccinia virus. J. Virol. 72:4192–4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McIntosh AA, Smith GL. 1996. Vaccinia virus glycoprotein A34R is required for infectivity of extracellular enveloped virus. J. Virol. 70:272–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doceul V, Hollinshead M, van der Linden L, Smith GL. 2010. Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science 327:873–876. 10.1126/science.1183173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roth S, Hoper D, Beer M, Feineis S, Tischer BK, Osterrieder N. 2011. Recovery of infectious virus from full-length cowpox virus (CPXV) DNA cloned as a bacterial artificial chromosome (BAC). Vet. Res. 42:3. 10.1186/1297-9716-42-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams J, Kelso R, Cooley L. 2000. The kelch repeat superfamily of proteins: propellers of cell function. Trends Cell Biol. 10:17–24. 10.1016/S0962-8924(99)01673-6 [DOI] [PubMed] [Google Scholar]
- 32.Shchelkunov SN, Totmenin AV, Kolosova IV, Sandakhchiev LS. 2002. Species-specific differences in the organization of genes encoding kelch-like proteins of orthopoxviruses pathogenic for humans. Dokl. Biochem. Biophys. 383:96–100. 10.1023/A:1015327500261 [DOI] [PubMed] [Google Scholar]
- 33.Shchelkunov S, Totmenin A, Kolosova I. 2002. Species-specific differences in organization of orthopoxvirus kelch-like proteins. Virus Genes 24:157–162. 10.1023/A:1014524717271 [DOI] [PubMed] [Google Scholar]
- 34.Froggatt GC, Smith GL, Beard PM. 2007. Vaccinia virus gene F3L encodes an intracellular protein that affects the innate immune response. J. Gen. Virol. 88:1917–1921. 10.1099/vir.0.82815-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beard PM, Froggatt GC, Smith GL. 2006. Vaccinia virus kelch protein A55 is a 64 kDa intracellular factor that affects virus-induced cytopathic effect and the outcome of infection in a murine intradermal model. J. Gen. Virol. 87:1521–1529. 10.1099/vir.0.81854-0 [DOI] [PubMed] [Google Scholar]
- 36.Pires de Miranda M, Reading PC, Tscharke DC, Murphy BJ, Smith GL. 2003. The vaccinia virus kelch-like protein C2L affects calcium-independent adhesion to the extracellular matrix and inflammation in a murine intradermal model. J. Gen. Virol. 84:2459–2471. 10.1099/vir.0.19292-0 [DOI] [PubMed] [Google Scholar]
- 37.Earl PL, Moss B, Wyatt LS, Carroll MW. 2001. Generation of recombinant vaccinia viruses. Curr. Protoc. Mol. Biol. Chapter 16:Unit 16.17. 10.1002/0471142727.mb1617s43 [DOI] [PubMed] [Google Scholar]
- 38.Tischer BK, BB Kaufer. 2012. Viral bacterial artificial chromosomes: generation, mutagenesis, and removal of mini-F sequences. J. Biomed. Biotechnol. 2012:472537. 10.1155/2012/472537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meisinger-Henschel C, Spath M, Lukassen S, Wolferstatter M, Kachelriess H, Baur K, Dirmeier U, Wagner M, Chaplin P, Suter M, Hausmann J. 2010. Introduction of the six major genomic deletions of modified vaccinia virus Ankara (MVA) into the parental vaccinia virus is not sufficient to reproduce an MVA-like phenotype in cell culture and in mice. J. Virol. 84:9907–9919. 10.1128/JVI.00756-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blasco R, Moss B. 1991. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-Dalton outer envelope protein. J. Virol. 65:5910–5920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quan LT, Caputo A, Bleackley RC, Pickup DJ, Salvesen GS. 1995. Granzyme B is inhibited by the cowpox virus serpin cytokine response modifier A. J. Biol. Chem. 270:10377–10379. 10.1074/jbc.270.18.10377 [DOI] [PubMed] [Google Scholar]
- 42.Ray CA, Black RA, Kronheim SR, Greenstreet TA, Sleath PR, Salvesen GS, Pickup DJ. 1992. Viral inhibition of inflammation: cowpox virus encodes an inhibitor of the interleukin-1 beta converting enzyme. Cell 69:597–604. 10.1016/0092-8674(92)90223-Y [DOI] [PubMed] [Google Scholar]
- 43.Palumbo GJ, Pickup DJ, Fredrickson TN, McIntyre LJ, Buller RM. 1989. Inhibition of an inflammatory response is mediated by a 38-kDa protein of cowpox virus. Virology 172:262–273. 10.1016/0042-6822(89)90128-1 [DOI] [PubMed] [Google Scholar]
- 44.Nathaniel R, MacNeill AL, Wang YX, Turner PC, Moyer RW. 2004. Cowpox virus CrmA, Myxoma virus SERP2 and baculovirus P35 are not functionally interchangeable caspase inhibitors in poxvirus infections. J. Gen. Virol. 85:1267–1278. 10.1099/vir.0.79905-0 [DOI] [PubMed] [Google Scholar]
- 45.Beattie E, Kauffman EB, Martinez H, Perkus ME, Jacobs BL, Paoletti E, Tartaglia J. 1996. Host-range restriction of vaccinia virus E3L-specific deletion mutants. Virus Genes 12:89–94. 10.1007/BF00370005 [DOI] [PubMed] [Google Scholar]
- 46.Myskiw C, Arsenio J, van Bruggen R, Deschambault Y, Cao J. 2009. Vaccinia virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-κB, and IRF3 pathways. J. Virol. 83:6757–6768. 10.1128/JVI.02570-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 70:1032–1060. 10.1128/MMBR.00027-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chakrabarti A, Sadler AJ, Kar N, Young HA, Silverman RH, Williams BR. 2008. Protein kinase R-dependent regulation of interleukin-10 in response to double-stranded RNA. J. Biol. Chem. 283:25132–25139. 10.1074/jbc.M804770200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meusel TR, Kehoe KE, Imani F. 2002. Protein kinase R regulates double-stranded RNA induction of TNF-alpha but not IL-1 beta mRNA in human epithelial cells. J. Immunol. 168:6429–6435. 10.4049/jimmunol.168.12.6429 [DOI] [PubMed] [Google Scholar]
- 50.Guerra S, Abaitua F, Martinez-Sobrido L, Esteban M, Garcia-Sastre A, Rodriguez D. 2011. Host-range restriction of vaccinia virus E3L deletion mutant can be overcome in vitro, but not in vivo, by expression of the influenza virus NS1 protein. PLoS One 6:e28677. 10.1371/journal.pone.0028677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Engelstad M, Howard ST, Smith GL. 1992. A constitutively expressed vaccinia gene encodes a 42-kDa glycoprotein related to complement control factors that forms part of the extracellular virus envelope. Virology 188:801–810. 10.1016/0042-6822(92)90535-W [DOI] [PubMed] [Google Scholar]
- 52.Engelstad M, Smith GL. 1993. The vaccinia virus 42-kDa envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology 194:627–637. 10.1006/viro.1993.1302 [DOI] [PubMed] [Google Scholar]
- 53.Sanderson CM, Frischknecht F, Way M, Hollinshead M, Smith GL. 1998. Roles of vaccinia virus EEV-specific proteins in intracellular actin tail formation and low pH-induced cell-cell fusion. J. Gen. Virol. 79:1415–1425 [DOI] [PubMed] [Google Scholar]
- 54.Condit RC. 2010. Surf. and turf: mechanism of enhanced virus spread during poxvirus infection. Viruses 2:1050–1054. 10.3390/v2051050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith GL, Vanderplasschen A. 1998. Extracellular enveloped vaccinia virus. Entry, egress, and evasion. Advances in experimental medicine and biology. 440:395–414 [PubMed] [Google Scholar]
- 56.Schmidt FI, Bleck CK, Mercer J. 2012. Poxvirus host cell entry. Curr. Opin. Virol. 2:20–27. 10.1016/j.coviro.2011.11.007 [DOI] [PubMed] [Google Scholar]
- 57.Moss B. 2012. Poxvirus cell entry: how many proteins does it take? Viruses 4:688–707. 10.3390/v4050688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williams O, Wolffe EJ, Weisberg AS, Merchlinsky M. 1999. Vaccinia virus WR gene A5L is required for morphogenesis of mature virions. J. Virol. 73:4590–4599 [DOI] [PMC free article] [PubMed] [Google Scholar]