

ABSTRACT
Natural transmission of prion diseases depends upon the spread of prions from the nervous system to excretory or secretory tissues, but the mechanism of prion transport in axons and into peripheral tissue is unresolved. Here, we examined the temporal and spatial movement of prions from the brain stem along cranial nerves into skeletal muscle as a model of axonal transport and transynaptic spread. The disease-specific isoform of the prion protein, PrPSc, was observed in nerve fibers of the tongue approximately 2 weeks prior to PrPSc deposition in skeletal muscle. Initially, PrPSc deposits had a small punctate pattern on the edge of muscle cells that colocalized with synaptophysin, a marker for the neuromuscular junction (NMJ), in >50% of the cells. At later time points PrPSc was widely distributed in muscle cells, but <10% of prion-infected cells exhibited PrPSc deposition at the NMJ, suggesting additional prion replication and dissemination within muscle cells. In contrast to the NMJ, PrPSc was not associated with synaptophysin in nerve fibers but was found to colocalize with LAMP-1 and cathepsin D during early stages of axonal spread. We propose that PrPSc-bound endosomes can lead to membrane recycling in which PrPSc is directed to the synapse, where it either moves across the NMJ into the postsynaptic muscle cell or induces PrPSc formation on muscle cells across the NMJ.
IMPORTANCE Prion diseases are transmissible and fatal neurodegenerative diseases in which prion dissemination to excretory or secretory tissues is necessary for natural disease transmission. Despite the importance of this pathway, the cellular mechanism of prion transport in axons and into peripheral tissue is unresolved. This study demonstrates anterograde spread of prions within nerve fibers prior to infection of peripheral synapses (i.e., neuromuscular junction) and infection of peripheral tissues (i.e., muscle cells). Within nerve fibers prions were associated with the endosomal-lysosomal pathway prior to entry into muscle cells. Since early prion spread is anterograde and endosome-lysosomal movement within axons is primarily retrograde, these findings suggest that endosome-bound prions may have an alternate fate that directs prions to the peripheral synapse.
INTRODUCTION
Dissemination of prions following entry into the nervous system is primarily along the neural circuitry. This is evident in the natural prion diseases of ruminants in which oral transmission leads to infection of the lymphoreticular and nervous systems of the gastrointestinal tract (1–7). After entry into the peripheral nervous system, prions spread along nerve tracts and between synaptically connected neurons into the central nervous system (CNS). In experimental models, targeted inoculation of prions into the peripheral nervous system such as the eye (8, 9), sciatic nerve (10, 11), or tongue (12) results in axonal and transynaptic spread to first-order and higher-order neurons. This property of prions is similar to viral transneuronal tracers such as rabies virus (13) and pseudorabies virus (14, 15), which have been useful for anatomical mapping of neural circuitry. These viral transneuronal tracers spread to higher-order neurons within hours to a few days, while prion spread along similar neural circuits can take several weeks. This delay in the spread of prions can be partially explained by a lower rate of prion agent replication, which is defined by the misfolding of the cellular prion protein, PrPC, into to the disease-specific isoform, PrPSc, and/or the lower rate of prion spread within neural circuits.
The centripetal spread of prions toward the central nervous system (CNS) following infection is necessary for induction of neurodegeneration. Less is understood regarding centrifugal spread of prions after they reach the CNS. Dissemination of prions to peripheral tissues is necessary to infect secretory and/or excretory tissues, which presumably leads to the release of prions into bodily fluids and secretions. Prion spread to these sites can lead to prion transmission to offspring, horizontal spread to other animals, and contamination of the environment. Prion dissemination within the lymphoreticular system is likely via the lymph and blood, but the mechanism of centrifugal prion spread to peripheral tissues by nonhematogenous routes, especially those linked to prion shedding, has not been elucidated. Specifically, it has not been determined how prions spread from the CNS into bodily fluids and excreta that contain low levels of prion infectivity (e.g., milk, saliva, urine, or feces) (16–21). Other studies investigating prion agent infection of the tongue mucosa illustrated prion infection of taste bud cells in fungiform papillae that are innervated but not in taste cells that do not receive neural input (22). These studies suggest that prions can spread from the brain along the chorda tympani branch of the facial nerve and across the neural synapse into the neuroepithelial taste cells. A more direct route of prion spread to the peripheral mucosa occurs in the olfactory system. Here, infection in the olfactory bulb leads to transynaptic spread of prions within glomeruli to olfactory sensory neurons (OSNs), and retrograde spread along the olfactory nerve results in PrPSc deposition in OSN somata and dendrites in the olfactory sensory epithelium (23–25). Subsequent release of prions into the nasal airway can explain the detection of moderate levels of prion infectivity and PrPSc in nasal secretions (24, 25). Whether prions can spread among nonneuronal cells in peripheral tissue or whether infection is limited to cells with direct neural input is not known outside the lymphoreticular system.
The molecular and cellular mechanisms of prion spread within axons and across synapses have not been defined. Prion infection of genetically modified mice that have altered axonal transport (e.g., overexpression of the human tau protein in neurons) or are partially deficient in molecular motors (e.g., dynein) do not exhibit alteration in the prion incubation period following peripheral inoculation of mouse-adapted scrapie (26, 27). These studies are limited in that these mice often exhibit neurological symptoms related to the genetic alteration of these essential cellular pathways. Furthermore, axonal transport of prions may not be the rate-limiting step that leads to a lengthening of the incubation period when these pathways are compromised. Synaptic prion spread is even less understood, but it could have parallels to observations in cell culture in which prion-infected neuronal cell lines could transfer infection to adjacent cells through direct contact of surface PrPSc on infected cells, with PrPC located on the cell surface of susceptible cells (28, 29). This pathway is consistent with the observation of PrPSc accumulation on neuronal membranes of dendrites and cell bodies in natural prion diseases (30). Release of prions from infected cells may also contribute to prion infection of cells in culture or adjacent cells in vivo such as glial cells. Despite these in vitro studies, the precise mechanism of cell-to-cell transfer across neural synapses is not known.
In the current study, we investigated the spatial and temporal pattern of centrifugal prion spread from the CNS to the periphery in order to define the sequence of events necessary to establish prion infection in peripheral tissues. To accomplish this goal, we monitored prion infection of skeletal muscle in the tongue since prior studies demonstrated that muscle cells of the tongue are susceptible to prion infection (12, 22, 31–33) and since each muscle cell has a single neural synapse, or neuromuscular junction. We demonstrate centrifugal spread of prions from the brain stem to nerve fibers of the tongue, followed by localization to the neuromuscular junction prior to entry into, and replication in, muscle cells. We then attempted to identify the axonal compartment in which prions are located in order to determine the pathway of spread along axons. Our findings indicate that prions are found within the endosomal-lysosomal compartment in nerve bundles, and they suggest that PrPSc formation or trafficking of prions could be related to intra-axonal transport of these organelles. There is also evidence that prions are associated with myelin produced by Schwann cells that surround axons in the nerve bundle.
MATERIALS AND METHODS
Animal inoculations and tissue collection.
Three- to four-month-old Syrian golden hamsters (Simonsen Laboratories, Gilroy, CA) were inoculated in the hypoglossal nerve, or cranial nerve XII (CN XII). Hamsters under general anesthesia (i.e., ketamine-xylazine mixture) were shaved in the neck area to remove fur, and the skin was cleansed with a Betadine scrub; a single incision of the skin was made on the left ventral aspect of the neck, and the hypoglossal nerve (CN XII) was exposed by dorsolaterally retracting the digastric muscle. Upon identification of CN XII, the nerve was bluntly dissected free of connective tissue and lifted out of the cavity using a spinal cord hook. Using a 5-μl Hamilton syringe equipped with a 30-gauge 2-point needle, 2 μl of a brain homogenate from either a normal hamster (i.e., mock infected) or a hamster containing 108.5 intracerebral median lethal doses (LD50) per ml of the hypertransmissible mink encephalopathy (HY TME) agent was slowly injected into the left CN XII. Using a serrated forceps, the nerve was crushed by gently applying pressure for 10 s. Surgical staples were used to close the incision. Hamsters were also inoculated into the cerebral cortex and tongue with 50 μl and 20 μl, respectively, of HY TME brain homogenate as previously described (12). Following inoculation, hamsters were observed three times per week for the onset of clinical symptoms, which included hyperesthesia, tremors of the head and trunk, and ataxia. Animals were euthanized at selected time points postinoculation or in the early stages of clinical disease.
For collection of tissues for immunohistochemical and immunofluorescence analyses, hamsters were intracardially perfused with periodate-lysine-paraformaldehyde (PLP) fixative, and tissues were dissected and processed for embedding in paraffin wax as previously described (12, 33).
Tissue culture analysis.
The CAD cell line was a generous gift of Dana M. Chikaraishi (Duke University, Durham, NC) (34) and the D5 scrapie-infected CAD cell line was a generous gift of Charles Weissmann (The Scripps Research Institute, Jupiter, FL) (35). CAD cells were grown on poly-l-lysine-coated coverslips in a 24-well tissue culture plate at 7,500 cells per well in HAM F-12/Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum for 24 h before serum-based growth medium was removed and replaced with HAM F-12/DMEM. This results in an arrest in cell division and morphological changes including extension of cellular processes or neurites and conversion to a phase-bright cell body. After 96 h in serum-free medium, cells were fixed with 4% paraformaldehyde in 4% sucrose and phosphate-buffered saline, and following several washes, the cells were treated with 0.1% digitonin in 100 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] buffer. Following treatment with 3 M guanidine hydrochloride for 10 min, cells were washed and prepared for immunofluorescence by blocking in 10% goat serum and 10% horse serum in 100 mM PIPES buffer in conjunction with separate streptavidin and biotin blocking steps (Vector Laboratories, Burlingame, CA, USA). Cells were incubated with anti-PrP monoclonal antibody ICSM 33 and either anti-LAMP-1 (lysosome-associated membrane protein 1) rabbit polyclonal antibody or anti-cathepsin D rabbit polyclonal antibody in 5% horse serum or goat serum in PIPES buffer, respectively (Table 1). Detection of primary antibodies was performed by incubation with either (i) horse anti-mouse IgG conjugated to biotin (Vector Laboratories, Burlingame, CA, USA), followed by an Alexa Fluor 488 streptavidin conjugate (Life Technologies, Grand Island, NY) at a 1:400 dilution for detection of PrPSc, or (ii) goat anti-rabbit antibody conjugated to Alexa Fluor 568 or Alexa Fluor 594 (1:400; Life Technologies, Grand Island, NY). The nuclear counterstain ToPro-3 (Life Technologies, Grand Island, NY) was applied at a 1:2,000 dilution for 10 min. Mowiol mounting medium was used to coverslip tissue sections.
TABLE 1.
Primary antibodies used in dual immunofluorescence
| Antigen and/or sample type | Host species | Source (location) | Concn | Name or catalog no. |
|---|---|---|---|---|
| Prion protein | ||||
| Tongue | Mouse | Rocky Mountain Laboratories, (Hamilton, MT) | 1:6,000 | 3F4a |
| dCAD cells | Mouse | D-Gen (London, UK) | 1 μg/ml | ICSM 33 |
| Synaptophysin | Rabbit | Epitomics (Burlingame, CA) | 1 μg/ml | 1485-S |
| βIII-Tubulin | Rabbit | Covance Research Products (USA) | 5 μg/ml | PRB-435P |
| LAMP-1 | Rabbit | Abcam (Cambridge, MA) | 2.25 μg/ml | ab24170 |
| Cathepsin D | ||||
| Tongue | Rabbit | Dako (Carpinteria, CA) | 5.8 μg/ml | A0561 |
| dCAD cells | Rabbit | Santa Cruz Biotechnology (Santa Cruz, CA) | 0.5 μg/ml | sc-10725 |
| Myelin P2 | Goat | Santa Cruz Biotechnology (Santa Cruz, CA) | 1 μg/ml | sc-49304 |
See reference 60.
PrPSc immunohistochemistry.
For PrPSc analysis, brain and tongue were collected, and PrPSc immunohistochemistry (IHC) was performed as previously described (12, 22, 33). Briefly, animals were intracardially perfused with PLP fixative, followed by immersion fixation in PLP for an additional 5 to 7 h. Prior to tissue processing and embedding in paraffin wax, the tongue was cut along the midline to generate two sagittal tongue tissue blocks that were either ipsilateral or contralateral to the site of inoculation in CN XII. Tissue sections were subjected to antigen retrieval by treatment with formic acid (99%, wt/vol) for 10 min, followed by successive incubation with anti-PrP monoclonal 3F4 antibody overnight at 4°C, horse anti-mouse IgG conjugated to biotin (1:400) at room temperature for 30 min, and streptavidin-horseradish peroxidase (HRP) at room temperature for 20 min. PrPSc was visualized by localization of HRP activity with either DAB+ (where DAB is diaminobenzidine) (Dako, Carpinteria, CA) or 3-amino-9-ethycarbazole in 50 mM sodium acetate (pH 5.0)–0.03% H2O2 as previously described (12, 33). Tissue sections were counterstained with hematoxylin and coverslip mounted with Aquamount (Lerner Laboratories, Pittsburgh, PA) for viewing with a Nikon Eclipse E600 microscope. Controls for PrPSc IHC included the use of mock-infected tissues and substitution of a similar concentration of murine IgG isotype control for the anti-PrP 3F4 monoclonal antibody. The specificity of PrPSc immunostaining is achieved through a combination of mild fixation, antigen retrieval using formic acid, and anti-PrP 3F4 monoclonal antibody. In hamster tissue this method results in negligible immunostaining signal in uninfected tissue and a punctate PrPSc signal in HY TME-infected tissues.
PrPSc dual immunofluorescence.
PrPSc immunofluorescence staining was combined with immunofluorescence for either synaptophysin, type III β-tubulin (βIII-tubulin), lysosome-associated membrane protein 1 (LAMP-1), cathepsin D, or myelin P2, as described in Table 1. Detection of primary antibodies was performed by incubation with either (i) horse anti-mouse IgG conjugated to biotin followed by an Alexa Fluor 488 streptavidin conjugate (1:400) for detection of PrPSc or (ii) donkey anti-goat antibody conjugated to Alexa Fluor 594 antibody (1:800; Life Technologies, Grand Island, NY) or goat anti-rabbit antibody conjugated to Alexa Fluor 568 antibody (1:800). The nuclear counterstain ToPro-3 was applied to some tissue sections at a 1:2,000 dilution for 10 min. Mowiol mounting medium was used to coverslip tissue sections. Tissues from a minimum of three HY TME-infected animals and three mock-infected animals were examined for each staining procedure at each time point. A minimum of 16 slides from tongue, both ipsilateral and contralateral to the injection site, was examined at each time point for each dual immunofluorescence combination for each tongue sample. For tongue sections used for quantification of PrPSc in axons and muscle cells, a distance of at least 50 μm was established between each tissue section used for analysis.
Laser scanning confocal microscopy (LSCM).
Images were visualized using a Zeiss LSM 510 Meta confocal system equipped with either a Zeiss Plan-Apochromat 63×/numerical aperture (NA) 1.4 or 100×/NA 1.0 oil objective. Double immunofluorescence was imaged after excitation of Alexa Fluor 488 with an argon laser at a wavelength of 488 nm and excitation of Alexa Fluor 568 or Alexa Fluor 594 with a helium/neon laser at a wavelength of 543 nm as previously described (22, 33). Images were scanned sequentially to minimize cross talk between channels, and the pinhole aperture was adjusted to <1.0 airy units for both channels while controlling for identical pinhole diameters and subsequent optical slice thickness. Individual images for stacks were at 0.1 μm per optical slice.
Deconvolution of confocal images.
Deconvolution was performed using Huygens Essential software (version 3.7; Scientific Volume Imaging, Hilversum, The Netherlands). Using the crop tool, the region of interest (e.g., nerve bundle, cell body, etc.,) within the image was chosen for deconvolution. A similarly sized region was analyzed for each comparison. A maximum-likelihood estimation algorithm was applied for deconvolution of the confocal images. All images for a particular experimental group were subjected to deconvolution using identical estimations of relative background and signal-to-noise ratio. Deconvolution was performed on entire image stacks, with a minimum of 27 stacks for each antibody combination per morphological area in each hamster group. Three-dimensional (3D) reconstructions of image stacks were generated using the 3D View Projection feature of Zeiss AIM software.
Colocalization analysis of confocal images.
After deconvolution digital images were evaluated for colocalization using the colocalization analyzer tool of Huygens Essential software, which provides information about the amount of spatial overlap between structures in different data channels, as previously described (22). Colocalization coefficients were generated by the analyzer module and included the Manders' overlap coefficient (MOC). For calculation of coefficients, a minimum of n = 27 for each region of interest was used for each comparison of mock- and HY TME-infected hamsters.
Statistical analysis.
The average MOC of the confocal images generated from HY TME-infected tissues was compared to the average MOC of images generated from mock-infected tissues to evaluate the probability (P value) that the colocalization observed in the experimental samples was greater than would be expected by chance. Statistical comparisons of MOCs were performed using a t test in Prism software (GraphPad, San Diego, CA).
Ethics statement.
All procedures involving animals were approved by the Montana State University IACUC and were in compliance with the Guide for the Care and Use of Laboratory Animals (36); these guidelines were established by the Institute of Laboratory Animal Resources and approved by the governing board of the U.S. National Research Council.
RESULTS
Prion spread into brain and nerve bundles of the tongue following inoculation of the hypoglossal nerve.
Prion agent dissemination within a host occurs by spread within the lymphoreticular and nervous systems. To investigate the pathway of prion agent entry into peripheral tissues from the nervous system, we monitored prion entry into the nervous system and skeletal muscle cells following inoculation of the HY TME agent into the left hypoglossal nerve (cranial nerve XII or CN XII) of a Syrian golden hamster. The incubation period following intra-CN XII inoculation was 94 ± 2.9 days compared to 62 ± 1 days and 80 ± 2 days following intracerebral and intratongue inoculations, respectively. At 4 weeks after intra-CN XII inoculation, immunohistochemistry in the brain stem revealed moderate to heavy levels of PrPSc deposition that were largely confined to the hypoglossal nucleus ipsilateral to the site of HY TME inoculation (Fig. 1A and B). Much lower levels of PrPSc were observed in the contralateral hypoglossal nucleus at this time. In the hypoglossal nucleus, PrPSc deposition was primarily localized at the periphery of the soma of motor neurons although small PrPSc deposits were observed within the soma at a lower frequency (Fig. 1C and D). Larger aggregates of PrPSc appeared to be associated with the adjacent glia cells in CN XII. At 7 weeks postinoculation strong PrPSc immunostaining was observed in the ipsilateral hypoglossal nucleus, and moderate levels of PrPSc were now observed in the contralateral hypoglossal nucleus as well as in several other regions of the brain stem, cerebellum, midbrain, and spinal cord (data not shown). This initial pattern of PrPSc deposition in the brain stem was consistent with transport of the HY TME agent along CN XII into the ipsilateral hypoglossal nucleus and subsequent spread to the contralateral portion of the hypoglossal nucleus.
FIG 1.
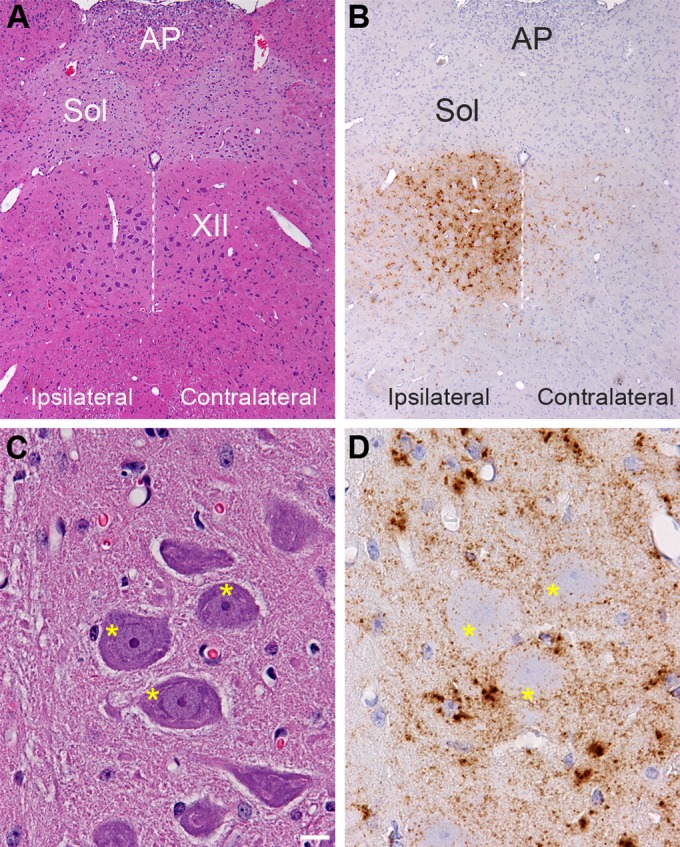
Distribution of PrPSc in the hypoglossal nucleus following HY TME infection. Hematoxylin and eosin staining of brain stem (A and C) and PrPSc immunohistochemistry (B and D) in adjacent tissue sections revealed a strong PrPSc pattern (brown) in the hypoglossal nucleus (XII) at 28 days postinoculation. PrPSc deposition was more intense ipsilateral to the inoculation site in the hypoglossal nerve than in the contralateral CN XII. Panels in C and D are higher magnifications of the ipsilateral CN XII illustrated in panels A and B. The dotted white line is located along the midline below the third ventricle, and it bisects CN XII. Higher magnification of ipsilateral CN XII is annotated with asterisks, which are located on the somata of hypoglossal motor neurons. This illustrates PrPSc deposition primarily at the edge of the neuronal soma and weakly within the soma. The PrPSc deposition pattern is also consistent with deposition in the neuropil or dendrites around the somata of motor neurons, and the dense clusters of PrPSc are likely to be associated with glial cells adjacent to motor neurons in CN XII. PrPSc immunohistochemistry was counterstained with hematoxylin. AP, area postremus; Sol, nucleus of the solitary tract. Scale bar, 10 μm.
A temporal and spatial analysis of PrPSc in the tongue was performed in order to investigate the route(s) of HY TME entry into skeletal muscle. Following intra-CN XII inoculation, hamsters were sacrificed on a weekly basis, and the ipsilateral and contralateral halves of the tongue relative to the inoculation site were examined for PrPSc deposition in nerve bundles and skeletal muscle cells. Nerve bundles were considered PrPSc positive based on the presence of at least one single PrPSc deposit, but often nerve bundles contained multiple PrPSc deposits (Fig. 2A). In nerve bundles PrPSc was primarily associated with axons and could sometimes be observed in the lumen of the axon. PrPSc deposition was not observed in nerve bundles prior to 7 weeks after intra-CN XII inoculation of the HY TME agent even though it was observed at earlier time points in CN XII in the brain stem. At 7 weeks postinfection, all hamsters (n = 3) had evidence of PrPSc deposition in nerve bundles in the ipsilateral tongue, and two of three had evidence for PrPSc deposition in the contralateral tongue (Fig. 3A). In hamsters 1 and 2, there were fewer than five PrPSc-positive nerve bundles per tongue histological section, but in hamster tongue 3 an average of 77 and 5 PrPSc-positive nerve bundles were observed ipsilateral and contralateral, respectively, to the site of inoculation. At 8 weeks postinfection hamsters 1 and 2 had fewer than five PrPSc-positive nerve bundles on each side of the tongue, while hamster 3 had no evidence of PrPSc deposition in nerve bundles (Fig. 3A). By 9 weeks postinfection, two of three hamsters had greater than 30 PrPSc-positive nerve bundles in tongue ipsilateral to the site of inoculation, while one hamster tongue (animal 1) had fewer than five PrPSc-positive nerve bundles. Contralateral to the site of inoculation, fewer than five PrPSc-positive nerve bundles per tongue section were observed in all three hamsters at this time (Fig. 3A). At later time points postinfection, higher numbers of PrPSc-positive nerve bundles per tongue section were observed, and by 11 and 13 (at clinical onset of HY TME) weeks postinfection, there were greater than 100 PrPSc-positive nerve bundles per tongue section (Fig. 3A). In the tongue of an individual animal the number of PrPSc-positive nerve bundles was always greater ipsilateral to the site of inoculation than contralateral to the site. These findings indicate that by 7 weeks after CN XII inoculation of the HY TME agent, PrPSc was consistently present in nerve bundles in the tongue, and there was a progressive increase in the level of HY TME infection in nerve bundles after 7 weeks postinoculation.
FIG 2.

PrPSc distribution in nerve bundles and skeletal muscle cells of the tongue following HY TME infection. Following intranerve inoculation of the hypoglossal nerve with HY TME, PrPSc immunohistochemistry was performed on tongue in order to identify the prion-infected cellular structures. Punctate PrPSc deposits were first observed within nerve bundles (NB) as early as 7 weeks postinfection (A). Two patterns of PrPSc deposition were found within skeletal muscle cells (m): a diffuse pattern with punctate PrPSc deposits spread throughout the muscle cell (B and C) and a focal pattern characterized by a few larger clusters of PrPSc at the cell periphery (D and E). Ganglia (G) in the tongue were found adjacent to muscle spindles, and PrPSc deposits were associated with neuronal somata within these structures (F). Tissue sections were counterstained with hematoxylin. Scale bar, 20 μm.
FIG 3.

Quantification of PrPSc deposition in the nerve bundle and muscle cells of the tongue. Following intranerve inoculation of the hypoglossal nerve with HY TME, PrPSc immunofluorescence was performed on tongue at weekly intervals, and the numbers of PrPSc-positive nerve bundles (A) and muscle cells (B) were counted both ipsilateral (ipsi; solid bars) and contralateral (contra; open bars) to the site of inoculation. At 6 weeks postinoculation and at earlier time points, PrPSc deposition was not observed in either nerve bundles or muscles cells. PrPSc deposition was initially observed in nerve bundles at 7 weeks postinoculation and, with the exception of animal 3 at 7 weeks, in muscle cells at 9 weeks postinoculation. The dashed line indicates the limit beyond which there were too many PrPSc-positive nerve bundles or muscle cells to count.
Prion spread into skeletal muscle cells via the neuromuscular junction.
A temporal and spatial analysis of PrPSc in skeletal muscle cells of the tongue was similar to that observed for PrPSc deposition in nerve bundles except for a delay in the onset of appearance by 1 to 2 weeks. The pattern of PrPSc deposition in skeletal muscle cells at clinical disease was characterized by intracellular deposition and often prominent localization to the outer periphery of the cell (Fig. 2B and C). Prior to 9 weeks postinoculation of CN XII, only a single hamster at 7 weeks, hamster 3, had evidence of PrPSc deposition in muscle cells, and the amount of deposition was high (>80 muscle cells per tongue section) ipsilateral to the site of inoculation (Fig. 3B). As previously noted, this hamster also had very high levels of PrPSc deposition in nerve bundles at 7 week postinfection (Fig. 3A). At 9 weeks postinfection the three hamster tongues had low levels of PrPSc deposition in muscle cells (<5 cells per tongue) both ipsilateral and contralateral to the site of inoculation (Fig. 3B). The number of PrPSc-positive muscle cells in the tongue continued to increase at 10 and 11 weeks postinoculation, and at the onset of clinical disease (i.e., 13 weeks postinoculation), the number of PrPSc-positive muscle cells on each side of the tongue was >100. Similar to our findings with PrPSc-positive nerve bundles, for each hamster a greater number of PrPSc-positive muscle cells were observed on the ipsilateral side than on the contralateral side of the tongue. These findings suggest that HY TME infection of muscle cells in the tongue likely occurred after infection of the nerve fibers in the tongue and that it was consistently found at 9 weeks postinoculation or 2 weeks after infection of nerve bundles.
At 9 weeks postinoculation, when initial PrPSc deposits were consistently associated with skeletal muscle cells of the tongue, two patterns of PrPSc deposition were observed. One was termed the focal pattern since one to three aggregated deposits or clusters of PrPSc were typically observed at or near the edge of the muscle cell (Fig. 2D and E and 4B). The second, or diffuse, pattern was characterized by a large number of PrPSc deposits that were located throughout the muscle cell (Fig. 2B and C and 4E and H). To investigate the spatial relationship of these two PrPSc patterns to the neuromuscular junction (NMJ; each muscle cell in the tongue will have a single NMJ that is innervated by CN XII), we performed laser scanning confocal microscopy (LSCM) for PrPSc and synaptophysin (a marker present in axons and presynaptic nerve terminals in muscle, blood vessels, and spindle fibers in the tongue). When the distribution of PrPSc was analyzed in skeletal muscle cells in the tongue, there was a strong correlation in the overlap between the focal PrPSc pattern and synaptophysin immunofluorescence (Fig. 4A to C; see also Movie S1 in the supplemental material). Deconvolution and colocalization analysis indicated that the Manders' overlap coefficients for the PrPSc focal pattern and synaptophysin at the edge of muscle cells were different for mock-infected and HY TME-infected animals (P < 0.05). We conclude that the focal PrPSc pattern represents PrPSc localization to the NMJ. In most cases, synaptophysin was not associated with the PrPSc deposits observed in the diffuse PrPSc pattern in muscle cells (Fig. 4D to F; see also below). However, in some cases we observed PrPSc deposits within the diffuse PrPSc pattern in muscle cells that were also associated with a cluster of synaptophysin at the periphery of the cell (Fig. 4G to I). PrPSc deposition was not observed in muscle cells of the tongue in hamsters inoculated in the CN XII with normal brain material (Fig. 4J to L).
FIG 4.

Laser scanning confocal microscopy for PrPSc and synaptophysin in skeletal muscle. Laser scanning confocal microscopy of synaptophysin (A, D, G, and J), PrPSc (B, E, H, and K), and both synaptophysin and PrPSc (C, F, I, and L) in tongue from HY TME-infected (A to I) and mock-infected (J to L) hamsters was performed. Panels A through C, D through F, G through I, and J through L are the same fields of view within their respective groups. The PrPSc deposition pattern in muscle cells was assigned to either a focal pattern (B), which was a small cluster of staining at the edge of the cell, or a diffuse pattern (E and H) that was characterized by many punctate deposits at the cell surface and throughout the cytoplasm. Synaptophysin immunofluorescence in muscle cells was also observed as a punctate pattern on the edge of muscle cells (arrowhead) and was consistent with localization to the neuromuscular junction. In the majority of examples of the PrPSc focal pattern, there were similar, but not identical, patterns of synaptophysin immunofluorescence in HY TME animals (A to C). In most muscle cells with the diffuse PrPSc deposition pattern, there was no evidence of synaptophysin immunofluorescence (D to F), but on occasion synaptophysin immunofluorescence was found in association with PrPSc deposition in muscle cells with the diffuse PrPSc pattern (G to H). Asterisks denote individual muscle cells, and a yellow asterisk denotes a muscle cell with PrPSc deposition. Scale bars, 10 (A) and 20 (D, G, and J) μm.
To quantify the spatial relationship between PrPSc deposition and the NMJ, we measured the percentage of muscle cells in the tongue with the focal and diffuse PrPSc patterns at 9 and 13 weeks following CN XII inoculation of HY TME agent. The 9-week time point represents the earliest time point that PrPSc was consistently found in muscle cells, while 4 weeks later hamsters began to exhibit clinical symptoms of HY TME. At least 100 PrPSc-positive muscle cells in the tongue were counted from multiple histological sections per hamster (n = 3) at the 9-week time point, while at clinical disease up to 260 PrPSc-positive muscle cells were counted since they were more frequent at this stage of infection. At 9 weeks postinfection, between 47% and 90% of the HY TME-infected muscle cells exhibited the focal PrPSc deposition pattern (Table 2). In all of the PrPSc-positive muscle cells, synaptophysin colocalized with PrPSc between 31% and 89% of the time. When analysis was limited to muscle cells with the focal pattern of PrPSc deposition, then 82% to 98% of cells exhibited colocalization of PrPSc with synaptophysin. We interpret these findings to indicate that at the initial time of PrPSc detection in muscle cells, it is localized to the neuromuscular junction in a high percentage of the cells. In contrast, at 13 weeks postinfection the focal PrPSc pattern was observed in only 2% to 7% of muscle cells, suggesting that the HY TME agent had entered muscle cells, replicated, and spread within muscle cells beyond the initial site of agent entry (Table 2). Even though the PrPSc focal pattern in muscle cells was less frequent at this time point, between 33% and 80% of cells with PrPSc and synaptophysin colocalization had the focal pattern (the wide range of values was due to the paucity of this event at clinical disease). However, overall the percentage of colocalization in all muscle cells was less than 10%. These findings suggest that PrPSc entry via the NMJ continues to occur at 13 weeks postinfection, but these cells represent a minor population of the HY TME-infected muscle cells. These data indicate that at the time of PrPSc entry into muscle cells, HY TME agent is present at the NMJ. These findings strongly suggest that prion infection is mediated via transynaptic spread or intercellular infection from CN XII to skeletal muscle cells.
TABLE 2.
Temporal pattern of PrPSc deposition and synaptophysin colocalization in muscle cells of the tongue
| Time postinfection and animala | No. of PrPSc-positive muscle cells | PrPSc deposition profile (no. of cells [%])b |
||
|---|---|---|---|---|
| PrPSc-positive muscle cells in a focal patternc | PrPSc and synaptophysin colocalization in muscle cells | PrPSc and synaptophysin colocalization in a focal pattern | ||
| Week 7 | ||||
| Animal 3 | ||||
| Ipsilaterald | 216 | 126 (58) | 117 (54) | 112 (96) |
| Contralaterald | 28 | 24 (86) | 23 (82) | 23 (100) |
| Week 9 | ||||
| Animal 1 | 101 | 47 (47) | 31 (31) | 27 (82) |
| Animal 2 | 100 | 60 (60) | 47 (47) | 46 (98) |
| Animal 3 | 100 | 90 (90) | 89 (89) | 83 (93) |
| Week 13 | ||||
| Animal 1 | 133 | 2 (2) | 3 (2) | 1 (33) |
| Animal 2 | 152 | 6 (4) | 5 (3) | 4 (80) |
| Animal 3 | 261 | 18 (7) | 26 (10) | 17 (65) |
Animal numbering corresponds to Fig. 3.
The percentage is calculated in terms of the counted PrPSc-positive cells except for percentages in the last column, which are percentages of the number of cells in the previous column.
Focal PrPSc pattern deposition is described in the legend of Fig. 4B.
Ipsilateral and contralateral to the site of inoculation in the hypoglossal nerve. Data for week 9 and 13 tongue are from an area ipsilateral to the site of inoculation.
There was a progression of PrPSc deposition in nerve bundles and muscle cells of the tongue beginning at 7 and 9 weeks postinoculation, respectively, but an accelerated pattern of prion infection of these structures was found in hamster 3 at the 7-week postinoculation group. This animal had high levels of PrPSc deposition in nerve bundles (>75 PrPSc-positive cells per tongue section) and in skeletal muscle cells (>80 PrPSc-positive cells per tongue section) in the ipsilateral tongue following intra-CN XII inoculation, suggesting that HY TME agent entry into the tongue in this animal occurred several weeks earlier (Fig. 3A and B). Spatial analysis of PrPSc deposition in both the ipsilateral and contralateral tongue revealed a moderate link to the NMJ in the ipsilateral tongue and a strong link to the NMJ in the contralateral tongue. In the ipsilateral tongue 216 PrPSc-positive skeletal muscle cells were counted, and 58% of these exhibited the PrPSc focal pattern of deposition compared to 24 of 28 (86%) PrPSc-positive skeletal muscle cells in the contralateral tongue (Table 2) (there were approximately 10-fold fewer PrPSc-positive skeletal muscle cells in the contralateral tongue). These findings indicate that within this animal entry into the contralateral tongue was slightly delayed compared to entry into the ipsilateral tongue and suggests that earlier events in prion infection of muscle cells were more prominent in the contralateral tongue. In both cases, there was a strong correlation between PrPSc and synaptophysin within the same muscle cell in the ipsilateral and contralateral tongue (54% and 82%, respectively) and a stronger correlation among PrPSc and synaptophysin colocalization in muscle cells with the PrPSc focal pattern (96% and 100%, respectively) (Table 2). These findings also support a role for the NMJ in the early stage of prion infection of muscle cells.
Association of prions with vesicles and organelles in nerve bundles and neuronal somata.
The transport of prions in axons is central to pathogenesis and agent dissemination in the nervous system, but the mechanism of prion transport within axons has not been established. This is partially due to the low level of PrPSc found in axons in the central nervous system. The high level of PrPSc deposition in nerve bundles of the tongue enabled us to investigate the spatial relationship between PrPSc and cellular structures associated with transport within the axon. We examined the relationship of prions to widely distributed macromolecular assemblies associated with axons, such as the microtubule network and the myelin sheath, and also with discrete structures, such as organelles that have previously been linked with PrPSc in cell bodies. The association of PrPSc with synaptophysin was also analyzed in nerve bundles since in axons synaptophysin is associated with discrete vesicles containing synaptic vesicle precursor proteins, which is in contrast to the NMJ, where there is a high concentration of synaptophysin and other synaptic proteins. As a control we examined the relationship of PrPSc to similar cellular structures in the soma of neurons in ganglia of the tongue (Fig. 2F), which are part of the autonomic nervous system and not innervated by CN XII, and in CAD neuronal cells in vitro. The rationale for using the CAD cell line is that following serum deprivation and differentiation (dCAD), the morphology of these cells changes, and they appear to have phase-bright cell bodies with long neurites, which were used as in vitro correlates for neuronal somata and axons, respectively.
Initially, we examined the association of PrPSc with synaptophysin in nerve fibers since we were able to colocalize these proteins at the NMJ in the tongue (Fig. 4A to C). Two approaches were used to assess the spatial relationship of PrPSc and synaptophysin in nerve bundles using LSCM: (i) PrPSc immunofluorescence was identified in nerve bundles, and then we determined whether synaptophysin immunofluorescence colocalized with PrPSc; and (ii) nerve bundles that contained both PrPSc and synaptophysin immunofluorescence were identified, and then colocalization analysis was performed. Using the first approach we rarely observed synaptophysin in nerve bundles that were prescreened for PrPSc deposition (data not shown), which led us to conclude that the majority of PrPSc does not specifically associate with synaptic vesicle precursors containing synaptophysin in axons. This was confirmed using the second approach, in which using LSCM, deconvolution image analysis, and colocalization analysis revealed that the Manders' overlap coefficient (MOC) was not different for PrPSc and synaptophysin in mock-infected tongue and HY TME-infected tongue (Table 3 and Fig. 5A to 5C; see also Movie S2 in the supplemental material). To investigate whether prions were associated with the microtubule network, we repeated the LSCM analysis for PrPSc and βIII-tubulin in axons in the tongue at 8 and 9 weeks postinoculation of HY TME into CN XII. In this experiment, we also did not observe different MOCs for mock-infected tongue and HY TME-infected tongue (Table 3 and Fig. 5D to F; see also and Movie S3). These findings suggested that PrPSc was not transported along axons in synaptophysin-containing vesicles and that PrPSc did not colocalize with the microtubule network within axons.
TABLE 3.
Colocalization analysis of PrPSc and cellular markers in nerve bundles and neuron soma in the tongue following HY TME inoculation
| Cellular maker and/or cell type | Sampling date (no. of wks p.i.)a | Nerve bundle MOCb | Neuronal soma (ganglion) MOCb | Cellular structure |
|---|---|---|---|---|
| βIII-Tubulin | 8–9 | NS | ND | Microtubule network in axons |
| Synaptophysin | 8–9 | NS | ND | SVPs in axonsd |
| LAMP-1 | 8–9 | ** | * | Late endosomes, lysosomes |
| 13 | *** | *** | ||
| dCAD cellsc | *** | *** | ||
| Cathepsin D | 13 | * | * | Mature lysosomes |
| Myelin P2 | 8–9 | * | ND | Myelin |
p.i., postinfection.
A t test (unpaired, two-tailed) comparing the Manders' overlap coefficient (MOC) of PrPSc and cellular markers in mock-infected and HY TME-infected tongue or between mock-infected and 22L scrapie-infected differentiated CAD cells in vitro was performed. ND, not done; NS, not statistically significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
dCAD, CAD cell line was switched to serum-free medium to induce differentiation, which results in cessation of cell division, extension of neurites, and formation of phase-bright cell bodies. For dCAD cells, LSCM analysis was performed in neurites and cell bodies and could be morphologically related to axons in nerve bundles and somata of ganglia cells, respectively, in the tongue.
SVPs, synaptic vesicle precursors.
FIG 5.

Laser scanning confocal microscopy for PrPSc and either synaptophysin or βIII-tubulin in nerve bundles of the tongue. Laser scanning confocal microscopy for synaptophysin (A), PrPSc (B), and both synaptophysin and PrPSc (C and G) or for βIII-tubulin (D), PrPSc (E), and both βIII-tubulin and PrPSc (F and H) in nerve bundles in tongue from HY TME-infected (A to F) or mock-infected (G and H) hamsters was performed. Panels A through C and D through F are the same fields of view within their respective groups. Synaptophysin immunofluorescence had a limited distribution and was observed in a discrete pattern within the nerve bundle and often in association with axons. The βIII-tubulin pattern was also observed within nerve bundles and had a widespread and bright immunofluorescence pattern within axons. Each group of panels represents a single image that was taken from a three-dimensional reconstruction of a LSCM stack assembled from 64 individual images. Colocalization analysis on 27 separate 3D reconstructions for each PrPSc and cell marker combination revealed that there was not an overlap of PrPSc with either synaptophysin or βIII-tubulin in nerve bundles (Table 3). 3D reconstructions in movie format of nerve bundles stained for PrPSc and either synaptophysin or βIII-tubulin illustrated in this figure can be observed in Movies S2 and S3 in the supplemental material. Scale bar, 10 μm.
Cell-associated PrPSc has been localized to the plasma membrane, primarily in dendrites, and within the endosomal-lysosomal pathway by immunoelectron microscopy in prion-infected brain and scrapie-infected cell lines (30). To examine whether prions were present within endosomes and lysosomes in axons, we investigated PrPSc colocalization with LAMP-1, which is found in late endosomes and lysosomes. In nerve bundles at 8 to 9 weeks postinoculation, there was a difference in the MOCs between mock-infected and HY TME-infected samples (P < 0.01), indicating a cooccurrence of PrPSc with LAMP-1, but the majority of PrPSc was not localized with LAMP-1 (Table 3 and Fig. 6; see also Movie S4 in the supplemental material). A similar cooccurrence of PrPSc with LAMP-1 was observed in nerve bundles at 13 weeks postinfection in mock- and HY TME-infected animals (MOC, P < 0.001). In the soma of neurons in the autonomic ganglia in tongue at both the preclinical and clinical time points, the MOC (P < 0.05 and P < 0.001, respectively) was consistent with findings in the nerve bundle and indicated colocalization of PrPSc with LAMP-1 (Table 3 and Fig. 7A to F; see also Movie S6 in the supplemental material). In addition, there were many PrPSc deposits that did not appear to colocalize with LAMP-1 in neuronal somata. As a positive control we compared the MOCs for PrPSc and LAMP-1 in the neurites and cell bodies of dCAD cells in vitro. There was a significant difference in the MOCs when colocalizations in the neurites (P < 0.001) and cell bodies (P < 0.01) from mock-infected and 22L scrapie-infected dCAD cells were compared (Table 3). These findings were consistent with PrPSc transport in a subset of LAMP-1-positive vesicles along the axons in nerve bundles of the tongue.
FIG 6.

Laser scanning confocal microscopy for PrPSc and LAMP-1 in nerve bundles of the tongue. Laser scanning confocal microscopy for LAMP-1 (A, D, and G), PrPSc (B, E, and H), and both LAMP-1 and PrPSc (C, F, and I) in nerve bundles in tongue from HY TME-infected hamsters was performed. Panels A through C, D through F, and G through I are the same fields of view within their respective groups. In nerve bundles, LAMP-1 immunofluorescence was observed in a discrete pattern within the nerve bundle and in association with axons. A cluster of LAMP-1 immunofluorescence was also observed in areas outside axons that were consistent with localization to the cell body of Schwann cells based on the proximity of immunofluorescence to nuclei (stained with ToPro-3 and shown in blue). A white arrow within a panel series indicates the area of colocalization of PrPSc and LAMP-1. Colocalization analysis on 27 separate three-dimensional (3D) reconstructions for PrPSc and LAMP-1 revealed that there was an overlap of PrPSc with LAMP-1 in nerve bundles (Table 3). Panels A to C represent a single image that was taken from a 3D reconstruction of an LSCM stack assembled from 64 individual images. This 3D reconstruction of nerve bundles stained for PrPSc and LAMP-1 is illustrated in Movie S4 in the supplemental material. Scale bars, 5 μm (A and D) and 1 μm (G).
FIG 7.

Laser scanning confocal microscopy for PrPSc and either LAMP-1 or cathepsin D in ganglia of the tongue. Laser scanning confocal microscopy for LAMP-1 (A and D), PrPSc (B and E), and both LAMP-1 and PrPSc (C, F, and M) or for cathepsin D (G and J), PrPSc (H and K), and both cathepsin D and PrPSc (I, L, and N) in ganglia of the tongue from HY TME-infected (A to L) and mock-infected (M and N) hamsters was performed. Panels A through C and G through I are the same fields of view within their respective groups. The boxed area in panel C is enlarged in panels D through F, and the boxed area in panel I is enlarged in panels J through L. Both LAMP-1 and cathepsin D immunofluorescence had a punctate pattern that was widespread within the somata of neurons. Nuclei (blue) were stained with ToPro-3. A white arrow within a panel series indicates the area of colocalization of PrPSc and the organelle marker. Each group of panels represents a single image that was taken from a three-dimensional (3D) reconstruction of an LSCM stack assembled from 64 individual images. Colocalization analysis on 27 separate 3D reconstructions for each PrPSc and cell marker combination revealed that there was an overlap of PrPSc with either LAMP-1 or cathepsin D in somata of ganglia (Table 3). 3D reconstructions of ganglia stained for PrPSc and the cellular markers in this figure are illustrated in Movies S6 and S7 in the supplemental material. Scale bar, 10 μm.
To investigate prion distribution in mature lysosomes in nerve bundles and the somata of neurons, we examined the spatial distribution of PrPSc in relation to cathepsin D. At 13 weeks postinoculation, there was a difference in the MOCs (P < 0.05) between mock- and HY TME-infected samples in nerve bundles and in the somata of ganglion cells (Table 3 and Fig. 7G to L and Fig. 8; see also Movie S7 in the supplemental material). These findings suggest that PrPSc also is localized to mature lysosomes in both the neuronal cell body and nerve bundles. In addition, there were many PrPSc deposits that did not appear to colocalize with cathepsin D in neuronal somata. We also investigated PrPSc in relation to myelin produced by Schwann cells that ensheath individual axons in nerve bundles. A comparison of PrPSc and myelin P2 immunofluorescence revealed a difference in the MOCs between mock-infected and HY TME-infected samples in nerve bundles (P < 0.05) (Table 3 and Fig. 9). Discrete PrPSc deposits were often found within the lumen of axons but were also observed in association with myelin P2 immunofluorescence at the border of the axon lumen and myelin sheath (Fig. 9D to I).
FIG 8.

Laser scanning confocal microscopy for PrPSc and cathepsin D in nerve bundles of the tongue. Laser scanning confocal microscopy for cathepsin D (A, D, and G), PrPSc (B, E, and H), and both cathepsin D and PrPSc (C, F, and I) in nerve bundles in tongue from HY TME-infected hamsters was performed. Panels A through C, D through F, and G through I are the same fields of view within their respective groups. In nerve bundles, cathepsin D immunofluorescence was observed in a discrete pattern within the nerve bundle and in association with axons, which exhibit low-level autofluorescence. A cluster of cathepsin D immunofluorescence was also observed in areas outside axons that were consistent with localization to the cell body of Schwann cells based on the proximity of immunofluorescence to nuclei (stained with ToPro-3 and shown in blue). A white arrow within a panel series indicates the area of colocalization of PrPSc and cathepsin D. Colocalization analysis on 27 separate three-dimensional (3D) reconstructions for each PrPSc and cathepsin D combination revealed that there was an overlap of PrPSc with cathepsin D in nerve bundles (Table 3). Panels A to C represent a single image that was taken from a 3D reconstruction of an LSCM stack assembled from 64 individual images. This 3D reconstruction of nerve bundles stained for PrPSc and cathepsin D is illustrated in Movie S5 in the supplemental material. Scale bars, 10 (A and D) and 1 (G) μm.
FIG 9.

Laser scanning confocal microscopy for PrPSc and myelin P2 in nerve bundles of the tongue. Laser scanning confocal microscopy for myelin P2 (A, D, and G), PrPSc (B, E, and H), and both myelin P2 and PrPSc (C, F, and I) in nerve bundles in tongue from HY TME-infected hamsters was performed. Panels A through C, D through F, and G through I are the same fields of view within their respective groups. Panels D to F are enlargements of the white boxed area in panel C. In nerve bundles, PrPSc immunofluorescence was observed most frequently within the lumen of the nerve bundle (arrowhead) and also in association with myelin P2 immunofluorescence (arrow) on the lumen wall of the axon. Panels A to C represent a single image that was taken from a three-dimensional (3D) reconstruction of an LSCM stack assembled from 64 individual images. Colocalization analysis on 27 separate 3D reconstructions for each PrPSc and cell marker combination revealed that there was an overlap of PrPSc with myelin P2 in nerve bundles (Table 3). Scale bar, 5 μm (A).
DISCUSSION
Prion spread along neural circuits and across synapses in the nervous system is well documented, but the mechanism(s) remains undefined (8–12). The current study established that prion infection of the brain stem was followed by PrPSc deposition in the axons and nerve fibers of the tongue at 1 to 2 weeks prior to infection of skeletal muscle cells. These findings indicate that axonal prion spread to the periphery is anterograde prior to infection of skeletal muscle. At the initial time of prion infection in muscle cells, the majority of the infected cells have a cluster of PrPSc deposits at the cell periphery in a focal pattern, and >80% of muscle cells with the focal PrPSc pattern also had evidence of synaptophysin colocalization within the PrPSc cluster. Since it has been estimated that only ∼0.1% of the surface of a muscle cell is occupied by the NMJ (37), these findings provide convincing evidence for localization of PrPSc to the NMJ during the earliest stage of muscle cell infection. They also strongly suggest that prion entry into muscle cells is via the NMJ. As infection progressed in muscle cells, the focal PrPSc pattern was reduced to <10% of all infected muscle cells, but of the cells that exhibited PrPSc and synaptophysin colocalization, 33% to 80% had the PrPSc focal pattern, suggesting that new prion infection of muscle cells via the NMJ is also occurring during the terminal stage of disease. It is also possible that infection of muscle cells can lead to subsequent spread into the NMJ or that prion replication in muscle cells could also occur at the NMJ. The PrPC substrate for PrPSc formation has been localized to the NMJ, specifically, to the subsynaptic region of postsynaptic muscle cells (38, 39).
Our findings also argue against a major role for muscle-to-muscle spread of prion infection. This conclusion is partially based on findings from hamster 3 at week 7 postinoculation in which there is a large number of PrPSc-positive muscle cells ipsilateral to the site of prion inoculation, while contralateral to the site of inoculation there are ∼90% fewer muscle cells with PrPSc deposits. If prion infection of muscle cells in the contralateral tongue were independent of neural input, then we would predict that spread from an area of high infection (i.e., ipsilateral tongue) to an area of low infection (i.e., contralateral tongue) should occur by an intercellular mechanism among adjacent muscle cells. In this scenario we would predict that in the contralateral tongue the focal PrPSc pattern should not predominate in muscle cells, and PrPSc and synaptophysin colocalization would be an infrequent event. However, the opposite results were found. Eighty-six percent of infected muscle cells in the contralateral tongue had the focal PrPSc pattern, and 82% of infected cells exhibited colocalization of PrPSc and synaptophysin. Of the latter group, 100% of the muscle cells had the focal PrPSc pattern. These results indicate that early infection of muscle cells is highly associated with the NMJ and strongly argue against muscle-to-muscle spread of prions although a low level of spread by this pathway would not likely be detected with the current experimental design. These findings suggest that innervation of muscle is necessary in order to establish prion infection. The mechanism of nerve-to-muscle spread of prions is unknown, despite demonstration of cell-to-cell transfer of prion infection in cell lines in vitro (28, 29). Perhaps, in vivo the NMJ or synapses provide a closely apposed cell membrane containing a high concentration of prions that can efficiently transmit infection to adjacent cells.
Our findings that PrPSc in axons was associated with markers of the endosomal and lysosomal compartments are consistent with the intracellular PrPSc distribution in the somata of neurons and glia in prion-infected brain (30, 40, 41) and the subcellular PrPSc distribution in prion-infected cell lines in vitro (42, 43). In the current study, we independently confirmed this finding in the somata of neurons in ganglia in the tongue and in dCAD cell lines. Studies in scrapie-infected cell lines indicate that PrPSc formation occurs after glycosylphosphatidylinositol (GPI)-linked PrPC traffics to the plasma membrane and/or enters the endosomal pathway or endosome recycling compartment, depending on the culture model (42–49). A recent study indicates that PrPSc destined for cellular degradation is directed to the Golgi apparatus and then to lysosomes (50). Other in vitro and in vivo models implicate autophagy as an important process for regulating PrPSc formation and clearance (51). In the current study, we speculate that intra-axonal PrPSc localization to endosomes and lysosomes could represent a site for new PrPSc formation within the axon or trafficking of these organelles after acquisition of PrPSc in the somatodendritic compartment.
It should be noted that the majority of PrPSc deposits did not colocalize with any single cellular marker, indicating that PrPSc was likely within multiple compartments within the axon. This raises the distinct possibility that some PrPSc deposits do not associate with the endosomal-lysosomal system. Dual immunofluorescence comparing myelin P2 and PrPSc clearly demonstrates myelin P2 surrounding PrPSc within the lumen of individual axons as well as colocalization of these markers within myelin. The latter finding may indicate that PrPSc also localizes to the axolemma, which is consistent with its known distribution to the plasma membrane of neuronal soma. This could represent a site of new PrPSc formation although PrPC has not been localized to the axolemma, or it may indicate trafficking of PrPSc to the plasma membrane through endosomes.
It is more difficult to interpret our findings with respect to the mechanism of prion trafficking in axons since endosome trafficking in axons is not as well characterized as in nonneuronal cells and is also distinct from organelle trafficking in neuronal somata and dendrites (52, 53). Endosomes in axons can move bidirectionally but primarily they display a net retrograde motion, and they become degradative lysosomes in the proximal region of axons (54–56). Although the axonal distribution of PrPSc is consistent with its localization to endosomes and lysosomes in neuronal somata, our findings suggest that between 7 and 9 weeks postinoculation, PrPSc displays anterograde movement within axons toward the nerve termini of the hypoglossal nerve and the NMJ. Therefore, if early PrPSc trafficking is within endosomes and toward the NMJ, then it is not likely to involve retrograde movement and fusion with degradative lysosomes. Axonal endosomes also function in cellular trafficking and membrane cycling, and one role of axonal endosomes may be to recycle membrane-bound PrPSc into the nerve terminal, where PrPSc can move across the NMJ or induce new PrPSc formation across the synapse in the closely apposed postsynaptic cell. Somatodendritic endosomes can also undergo anterograde transport by transcytosis, in which receptor-ligand complexes are transported the entire length of the axon in order to mediate cell signaling, and in this case, they are protected from degradation in lysosomes (53, 57). Although the best described examples are characterized by retrograde movement (e.g., neurotrophin trafficking), there are also examples of anterograde transcytosis within endosomes (e.g., L1/NgCAM, a neuron-glia cell adhesion molecule). Two extracellular receptors have been described for PrPSc, the 37-kDa/67-kDa laminin receptor and neuronal low-density lipoprotein receptor-related protein 1 (LRP1), but neither has been implicated in transcytosis (58, 59). Binding of PrPSc to LRP leads to endocytosis and trafficking of PrPSc to lysosomes. In the current study, localization of intra-axonal PrPSc to lysosomes is more consistent with retrograde spread than anterograde movement. Late in the disease course prion movement in axons may occur in both directions. Another possibility is that intra-axonal PrPSc trafficking occurs in a yet unidentified compartment, and PrPSc localization to endosome-lysosome compartments occurs after PrPSc formation at the nerve termini or NMJ.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant R01 AI055043 and USDA CSREES grant 2006-35201-16626.
We thank Scott Martinka and Crista DeJoia for technical assistance and the staff at the Animal Resource Center, especially Renee Arens, for excellent animal care.
Footnotes
Published ahead of print 21 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00378-14.
REFERENCES
- 1.van Keulen LJ, Schreuder BE, Vromans ME, Langeveld JP, Smits MA. 1999. Scrapie-associated prion protein in the gastrointestinal tract of sheep with natural scrapie. J. Comp. Pathol. 121:55–63. 10.1053/jcpa.1998.0300 [DOI] [PubMed] [Google Scholar]
- 2.Sigurdson CJ, Williams ES, Miller MW, Spraker TR, O'Rourke KI, Hoover EA. 1999. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J. Gen. Virol. 80:2757–2764 [DOI] [PubMed] [Google Scholar]
- 3.Andreoletti O, Berthon P, Marc D, Sarradin P, Grosclaude J, van Keulen L, Schelcher F, Elsen JM, Lantier F. 2000. Early accumulation of PrP(Sc) in gut-associated lymphoid and nervous tissues of susceptible sheep from a Romanov flock with natural scrapie. J. Gen. Virol. 81:3115–3126 [DOI] [PubMed] [Google Scholar]
- 4.Heggebo R, Press CM, Gunnes G, Lie KI, Tranulis MA, Ulvund M, Groschup MH, Landsverk T. 2000. Distribution of prion protein in the ileal Peyer's patch of scrapie-free lambs and lambs naturally and experimentally exposed to the scrapie agent. J. Gen. Virol. 81:2327–2337 [DOI] [PubMed] [Google Scholar]
- 5.Sigurdson CJ, Spraker TR, Miller MW, Oesch B, Hoover EA. 2001. PrP(CWD) in the myenteric plexus, vagosympathetic trunk and endocrine glands of deer with chronic wasting disease. J. Gen. Virol. 82:2327–2334 [DOI] [PubMed] [Google Scholar]
- 6.Heggebo R, Press CM, Gunnes G, Gonzalez L, Jeffrey M. 2002. Distribution and accumulation of PrP in gut-associated and peripheral lymphoid tissue of scrapie-affected Suffolk sheep. J. Gen. Virol. 83:479–489 [DOI] [PubMed] [Google Scholar]
- 7.van Keulen LJ, Vromans ME, van Zijderveld FG. 2002. Early and late pathogenesis of natural scrapie infection in sheep. APMIS 110:23–32. 10.1034/j.1600-0463.2002.100104.x [DOI] [PubMed] [Google Scholar]
- 8.Fraser H. 1982. Neuronal spread of scrapie agent and targeting of lesions within the retino-tectal pathway. Nature 295:149–150. 10.1038/295149a0 [DOI] [PubMed] [Google Scholar]
- 9.Fraser H, Dickinson AG. 1985. Targeting of scrapie lesions and spread of agent via the retino-tectal projection. Brain Res. 346:32–41. 10.1016/0006-8993(85)91091-1 [DOI] [PubMed] [Google Scholar]
- 10.Kimberlin RH, Hall SM, Walker CA. 1983. Pathogenesis of mouse scrapie. Evidence for direct neural spread of infection to the CNS after injection of sciatic nerve. J. Neurol. Sci. 61:315–325 [DOI] [PubMed] [Google Scholar]
- 11.Bartz JC, Kincaid AE, Bessen RA. 2002. Retrograde transport of transmissible mink encephalopathy within descending motor tracts. J. Virol. 76:5759–5768. 10.1128/JVI.76.11.5759-5768.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartz JC, Kincaid AE, Bessen RA. 2003. Rapid prion neuroinvasion following tongue infection. J. Virol. 77:583–591. 10.1128/JVI.77.1.583-591.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ugolini G. 1995. Specificity of rabies of virus as a transneuronal tracer of motor networks: transfer from hypoglossal motorneurons to connected second-order and higher-order central nervous system cell groups. J. Comp. Neurol. 356:457–480. 10.1002/cne.903560312 [DOI] [PubMed] [Google Scholar]
- 14.Kim E-S, Li H, McCulloch PF, Morrison LA, Yoon K-W, Xu XM. 2000. Spatial and temporal patterns of transneuronal labeling in CNS neurons after injection of pseudorabies virus into the sciatic nerve of adult rats. Brain Res. 857:41–55. 10.1016/S0006-8993(99)02332-X [DOI] [PubMed] [Google Scholar]
- 15.Rotto-Percelay DM, Wheeler JG, Osorio FA, Platt KB, Loewy AD. 1992. Transneuronal labeling of spinal interneurons and sympathetic preganglionic neurons after pseudorabies virus injections in the rat medial gastrocnemius muscle. Brain Res. 574:291–306. 10.1016/0006-8993(92)90829-X [DOI] [PubMed] [Google Scholar]
- 16.Konold T, Moore SJ, Bellworthy SJ, Simmons HA. 2008. Evidence of scrapie transmission via milk. BMC Vet. Res. 4:14. 10.1186/1746-6148-4-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lacroux C, Simon S, Benestad SL, Maillet S, Mathey J, Lugan S, Corbiere F, Cassard H, Costes P, Bergonier D, Weisbecker JL, Moldal T, Simmons H, Lantier F, Feraudet-Tarisse C, Morel N, Schelcher F, Grassi J, Andreoletti O. 2008. Prions in milk from ewes incubating natural scrapie. PLoS Pathog. 4:e1000238. 10.1371/journal.ppat.1000238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, Hays SA, Hayes-Klug J, Seelig DM, Wild MA, Wolfe LL, Spraker TR, Miller MW, Sigurdson CJ, Telling GC, Hoover EA. 2006. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science 314:133–136. 10.1126/science.1132661 [DOI] [PubMed] [Google Scholar]
- 19.Haley NJ, Seelig DM, Zabel MD, Telling GC, Hoover EA. 2009. Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. PLoS One 4:e4848. 10.1371/journal.pone.0004848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kruger D, Thomzig A, Lenz G, Kampf K, McBride P, Beekes M. 2009. Faecal shedding, alimentary clearance and intestinal spread of prions in hamsters fed with scrapie. Vet. Res. 40:4. 10.1051/vetres:2008042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamguney G, Miller MW, Wolfe LL, Sirochman TM, Glidden DV, Palmer C, Lemus A, DeArmond SJ, Prusiner SB. 2009. Asymptomatic deer excrete infectious prions in faeces. Nature 461:529–532. 10.1038/nature08289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeJoia C, Moreaux B, O'Connell K, Bessen RA. 2006. Prion infection of oral and nasal mucosa. J. Virol. 80:4546–4556. 10.1128/JVI.80.9.4546-4556.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zanusso G, Ferrari S, Cardone F, Zampieri P, Gelati M, Fiorini M, Farinazzo A, Gardiman M, Cavallaro T, Bentivoglio M, Righetti PG, Pocchiari M, Rizzuto N, Monaco S. 2003. Detection of pathologic prion protein in the olfactory epithelium in sporadic Creutzfeldt-Jakob disease. N. Engl. J. Med. 348:711–719. 10.1056/NEJMoa022043 [DOI] [PubMed] [Google Scholar]
- 24.Bessen RA, Shearin H, Martinka S, Boharski R, Lowe D, Wilham JM, Caughey B, Wiley JA. 2010. Prion shedding from olfactory neurons into nasal secretions. PLoS Pathog. 6:e1000837. 10.1371/journal.ppat.1000837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bessen RA, Wilham JM, Lowe D, Watschke CP, Shearin H, Martinka S, Caughey B, Wiley JA. 2012. Accelerated shedding of prions following damage to the olfactory epithelium. J. Virol. 86:1777–1788. 10.1128/JVI.06626-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kunzi V, Glatzel M, Nakano MY, Greber UF, Van Leuven F, Aguzzi A. 2002. Unhampered prion neuroinvasion despite impaired fast axonal transport in transgenic mice overexpressing four-repeat tau. J. Neurosci. 22:7471–7477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hafezparast M, Brandner S, Linehan J, Martin JE, Collinge J, Fisher EM. 2005. Prion disease incubation time is not affected in mice heterozygous for a dynein mutation. Biochem. Biophys. Res. Commun. 326:18–22. 10.1016/j.bbrc.2004.10.206 [DOI] [PubMed] [Google Scholar]
- 28.Kanu N, Imokawa Y, Drechsel DN, Williamson RA, Birkett CR, Bostock CJ, Brockes JP. 2002. Transfer of scrapie prion infectivity by cell contact in culture. Curr. Biol. 12:523–530. 10.1016/S0960-9822(02)00722-4 [DOI] [PubMed] [Google Scholar]
- 29.Paquet S, Langevin C, Chapuis J, Jackson GS, Laude H, Vilette D. 2007. Efficient dissemination of prions through preferential transmission to nearby cells. J. Gen. Virol. 88:706–713. 10.1099/vir.0.82336-0 [DOI] [PubMed] [Google Scholar]
- 30.Jeffrey M, McGovern G, Siso S, Gonzalez L. 2011. Cellular and sub-cellular pathology of animal prion diseases: relationship between morphological changes, accumulation of abnormal prion protein and clinical disease. Acta Neuropathol. 121:113–134. 10.1007/s00401-010-0700-3 [DOI] [PubMed] [Google Scholar]
- 31.Thomzig A, Schulz-Schaeffer W, Kratzel C, Mai J, Beekes M. 2004. Preclinical deposition of pathological prion protein PrPSc in muscles of hamsters orally exposed to scrapie. J. Clin. Invest. 113:1465–1472. 10.1172/JCI21083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomzig A, Cardone F, Kruger D, Pocchiari M, Brown P, Beekes M. 2006. Pathological prion protein in muscles of hamsters and mice infected with rodent-adapted BSE or vCJD. J. Gen. Virol. 87:251–254. 10.1099/vir.0.81277-0 [DOI] [PubMed] [Google Scholar]
- 33.Bessen RA, Robinson CJ, Seelig DM, Watschke CP, Lowe D, Shearin H, Martinka S, Babcock AM. 2011. Transmission of chronic wasting disease identifies a prion strain causing cachexia and heart infection in hamsters. PLoS One 6:e28026. 10.1371/journal.pone.0028026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi Y, Wang JK, McMillian M, Chikaraishi DM. 1997. Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J. Neurosci. 17:1217–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. 2007. Prion strain discrimination in cell culture: the cell panel assay. Proc. Natl. Acad. Sci. U. S. A. 104:20908–20913. 10.1073/pnas.0710054104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.National Research Council. 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC [Google Scholar]
- 37.Burden SJ. 1998. The formation of neuromuscular synapses. Genes Dev. 12:133–148. 10.1101/gad.12.2.133 [DOI] [PubMed] [Google Scholar]
- 38.Askanas V, Bilak M, Engel WK, Leclerc A, Tome F. 1993. Prion protein is strongly immunolocalized at the postsynaptic domain of human normal neuromuscular junctions. Neurosci. Lett. 159:111–114. 10.1016/0304-3940(93)90811-X [DOI] [PubMed] [Google Scholar]
- 39.Gohel C, Grigoriev V, Escaig-Haye F, Lasmezas CI, Deslys JP, Langeveld J, Akaaboune M, Hantai D, Fournier JG. 1999. Ultrastructural localization of cellular prion protein (PrPc) at the neuromuscular junction. J. Neurosci. Res. 55:261–267. [DOI] [PubMed] [Google Scholar]
- 40.Laszlo L, Lowe J, Self T, Kenward N, Landon M, McBride T, Farquhar C, McConnell I, Brown J, Hope J, et al. 1992. Lysosomes as key organelles in the pathogenesis of prion encephalopathies. J. Pathol. 166:333–341. 10.1002/path.1711660404 [DOI] [PubMed] [Google Scholar]
- 41.Lowe J, Fergusson J, Kenward N, Laszlo L, Landon M, Farquhar C, Brown J, Hope J, Mayer RJ. 1992. Immunoreactivity to ubiquitin-protein conjugates is present early in the disease process in the brains of scrapie-infected mice. J. Pathol. 168:169–177. 10.1002/path.1711680204 [DOI] [PubMed] [Google Scholar]
- 42.Caughey B, Raymond GJ, Ernst D, Race RE. 1991. N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J. Virol. 65:6597–6603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKinley MP, Taraboulos A, Kenaga L, Serban D, Stieber A, DeArmond SJ, Prusiner SB, Gonatas N. 1991. Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Lab. Invest. 65:622–630 [PubMed] [Google Scholar]
- 44.Borchelt DR, Taraboulos A, Prusiner SB. 1992. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 267:16188–16199 [PubMed] [Google Scholar]
- 45.Taraboulos A, Serban D, Prusiner SB. 1990. Scrapie prion proteins accumulate in the cytoplasm of persistently infected cultured cells. J. Cell Biol. 110:2117–2132. 10.1083/jcb.110.6.2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taraboulos A, Raeber AJ, Borchelt DR, Serban D, Prusiner SB. 1992. Synthesis and trafficking of prion proteins in cultured cells. Mol. Biol. Cell 3:851–863. 10.1091/mbc.3.8.851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marijanovic Z, Caputo A, Campana V, Zurzolo C. 2009. Identification of an intracellular site of prion conversion. PLoS Pathog. 5:e1000426. 10.1371/journal.ppat.1000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caughey B, Raymond GJ. 1991. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J. Biol. Chem. 266:18217–18223 [PubMed] [Google Scholar]
- 49.Goold R, Rabbanian S, Sutton L, Andre R, Arora P, Moonga J, Clarke AR, Schiavo G, Jat P, Collinge J, Tabrizi SJ. 2011. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat. Commun. 2:281. 10.1038/ncomms1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goold R, McKinnon C, Rabbanian S, Collinge J, Schiavo G, Tabrizi SJ. 2013. Alternative fates of newly formed PrPSc upon prion conversion on the plasma membrane. J. Cell Sci. 126:3552–3562. 10.1242/jcs.120477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heiseke A, Aguib Y, Schatzl HM. 2010. Autophagy, prion infection and their mutual interactions. Curr. Issues Mol. Biol. 12:87–97 http://www.horizonpress.com/cimb/v/v12/87.pdf [PubMed] [Google Scholar]
- 52.Winckler B, Yap CC. 2011. Endocytosis and endosomes at the crossroads of regulating trafficking of axon outgrowth-modifying receptors. Traffic 12:1099–1108. 10.1111/j.1600-0854.2011.01213.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Winckler B, Mellman I. 2010. Trafficking guidance receptors. Cold Spring Harb. Perspect. Biol. 2:a001826. 10.1101/cshperspect.a001826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hollenbeck PJ. 1993. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J. Cell Biol. 121:305–315. 10.1083/jcb.121.2.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Overly CC, Lee KD, Berthiaume E, Hollenbeck PJ. 1995. Quantitative measurement of intraorganelle pH in the endosomal-lysosomal pathway in neurons by using ratiometric imaging with pyranine. Proc. Natl. Acad. Sci. U. S. A. 92:3156–3160. 10.1073/pnas.92.8.3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Overly CC, Hollenbeck PJ. 1996. Dynamic organization of endocytic pathways in axons of cultured sympathetic neurons. J. Neurosci. 16:6056–6064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harrington AW, Ginty DD. 2013. Long-distance retrograde neurotrophic factor signalling in neurons. Nat. Rev. Neurosci. 14:177–187. 10.1038/nrn3253 [DOI] [PubMed] [Google Scholar]
- 58.Gauczynski S, Nikles D, El-Gogo S, Papy-Garcia D, Rey C, Alban S, Barritault D, Lasmezas CI, Weiss S. 2006. The 37-kDa/67-kDa laminin receptor acts as a receptor for infectious prions and is inhibited by polysulfated glycanes. J. Infect. Dis. 194:702–709. 10.1086/505914 [DOI] [PubMed] [Google Scholar]
- 59.Jen A, Parkyn CJ, Mootoosamy RC, Ford MJ, Warley A, Liu Q, Bu G, Baskakov IV, Moestrup S, McGuinness L, Emptage N, Morris RJ. 2010. Neuronal low-density lipoprotein receptor-related protein 1 binds and endocytoses prion fibrils via receptor cluster 4. J. Cell Sci. 123:246–255. 10.1242/jcs.058099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H. 1987. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J. Virol. 61:3688–3693 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.