

Abstract
Simeprevir (SMV), asunaprevir (ASV), daclatasvir (DCV), and sofosbuvir (SFV), which are newly developed direct-acting antiviral agents (DAAs) against hepatitis C virus (HCV) infection, are among the key components of anti-HCV regimens. Preclinical studies have identified inhibitory properties for some of these DAAs against organic anion transporting polypeptide 1B (OATP1B) functions. However, their details remain mostly uncharacterized. Because OATP1B1 and OATP1B3 play determinant roles in the pharmacokinetics of various drugs via their uptake into human hepatocytes, it is plausible that the inhibition of these OATP1Bs by a DAA would create a potential risk of drug-drug interaction, which has been an emerging concern in anti-HCV therapy. Accordingly, in the present study, we intended to clarify the inhibitory characteristics of newly developed DAAs toward OATP1B1 and -1B3 functions. The results of our coincubation inhibition assays have shown that all tested DAAs could inhibit OATP1B1 functions and that SMV, ASV, and DCV (to a lesser extent), but not SFV, exhibited long-lasting preincubation inhibitory effects on OATP1B1 functions. It was also found that the preincubation inhibitory effects of SMV and ASV could augment their coincubation inhibition potency. Furthermore, significant, but differential, inhibitory effects of the DAAs on the OATP1B3 function were identified. To summarize, our results clearly show that the newly developed DAAs are newly identified OATP1B1 and OATP1B3 inhibitors with distinctive interaction properties. It is believed that these inhibition profiles will provide essential information to all concerned parties with respect to the clinical significance of DAA-mediated inhibition of OATP1Bs in anti-HCV therapy.
INTRODUCTION
Direct-acting antiviral agents (DAAs) against hepatitis C virus (HCV) proteins have dramatically improved clinical outcomes in chronic hepatitis C therapy. Recent clinical studies have shown that addition of telaprevir (TLV) or boceprevir (BOC), which are the first nonstructural 3/4A (NS3/4A) protease inhibitors, to the combination therapy of pegylated alpha interferon and ribavirin significantly enhances the rate of a sustained virological response in up to approximately 80% of patients carrying the HCV genotype 1 (1, 2). In addition, even higher treatment efficacy can be expected with the introduction of newly developed DAAs, including the NS3/4 protease inhibitors simeprevir (SMV) and asunaprevir (ASV), the NS5A inhibitor daclatasvir (DCV), and the NS5B inhibitor sofosbuvir (SFV) (1). The significantly reduced toxic properties of these new DAAs in comparison with those of TLV and BOC have also been highlighted in clinical studies, which adds further value to the use of these new agents in anti-HCV therapy.
The high efficacy of TLV and BOC aside, it has become increasingly evident that there are clinically significant risks of drug-drug interaction (DDI) when DAAs are coprescribed with various drugs (3, 4). For example, it has been reported that TLV increased the area under the curve of atorvastatin, cyclosporine (CsA), and tacrolimus by 7.9-fold, 4.6-fold, and 70-fold, respectively (5, 6), and, consequently, precautions related to the coadministration of these drugs with TLV have been noted (Incivek prescribing information, Vertex Pharmaceuticals Inc., Cambridge, MA). Likewise, the DDI properties of BOC with numerous drugs have been shown previously although BOC interactions have occurred apparently to a lesser extent (3, 4). TLV and BOC are inhibitors of cytochrome P450 3A4 (CYP3A4) as well as organic anion transporting polypeptides (OATPs) (7–9), which play determinant roles in the pharmacokinetics of various drugs. Therefore, inhibition of these functions is considered likely to contribute to the aforementioned DDIs. Because a detrimental DDI often results in unintentional toxic effects of the victim drug due to its increased systemic exposure, addressing DDIs caused by DAAs can be seen as a key issue in anti-HCV therapy.
OATP1B1 and OATP1B3 (OATP1B1/1B3), which are members of the SLCO gene family, are drug transporters that are primarily expressed at the plasma membrane of human hepatocytes. It has been established that both OATP1B1 and OATP1B3 play determinant roles in the pharmacokinetics of various anionic amphipathic molecules via their uptake from the circulatory system. Therefore, these OATP1Bs have been acknowledged as pivotal targets of DDI study in drug development and/or clinical settings (e.g., reference 10). Although they show a certain level of redundancy in their substrate spectrum, each OATP1B has its own substrate preferences. For example, it has been reported that estradiol-17β-glucuronide (E2G) and statins (such as pravastatin, atorvastatin, and rosuvastatin) are substrates of both OATPs, whereas estrone-3-sulfate and cholecystokinin octapeptide (CCK-8) are primarily transported by OATP1B1 and OATP1B3, respectively. Both OATPs are also known as conjugated or unconjugated bilirubin uptake transporters (11, 12).
OATP1B1 (and likely OATP1B3 as well) can be considered important targets for DDI research efforts, as exemplified by the reports showing the significant contribution of these OATPs to the DDI occurring between cerivastatin and CsA (13). Interestingly, Amundsen et al. (14) have shown that, among OATP1B inhibitors, preincubation of CsA enhances its direct (coincubation) inhibition potency against OATP1B1 in a cell-based assay, while Shitara et al. (15) have shown that the preincubation effect lasts for some time. Thus, long-lasting preincubation inhibitory effects have emerged as important characteristics in the functional inhibition mechanisms of OATP1Bs. On the other hand, the functional inhibition of OATP1Bs is also believed to play an important role in hyperbilirubinemia induced by OATP1B inhibitors, such as rifamycin SV, CsA, and atazanavir (11). Further information about the roles of OATPs in DDIs and hyperbilirubinemia can be found elsewhere (10, 16, 17).
Considering the clinically important roles played by OATP1Bs, a more precise understanding of the inhibitory characteristics of each DAA against the OATP1Bs is necessary for better clinical management in DAA-based anti-HCV therapy. However, detailed interaction profiles between the newly developed DAAs and OATP1Bs remain uncharacterized. Therefore, in the present study, we intended to clarify the inhibition characteristics of SMV, ASV, DCV, and SFV toward OATP1B1 and OATP1B3 functions, while simultaneously comparing the results with those obtained from TLV in order to evaluate their clinical significance.
MATERIALS AND METHODS
OATP1B expression plasmids.
The development procedure of the pcDNA3.1(−)Zeo plasmid (Life Technologies, Carlsbad, CA) carrying OATP1B1 cDNA (OATP1B1/pcDNA3.1) and the pcDNA3.1(−)Neo plasmid (Life Technologies) carrying OATP1B3 cDNA (OATP1B3/pcDNA3.1) has been described previously (18, 19).
Plasmid transfection into HEK293 cells.
Human embryonic kidney 293 (HEK293) cells were obtained from the Human Science Company (Tokyo, Japan) and cultured in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies) supplemented with 10% fetal bovine serum and antibiotics in 5% CO2 at 37°C.
The development procedure for HEK293 cells stably expressing OATP1B1 (1B1/HEK) and the cells carrying empty plasmid (mock/HEK) has been described previously (18). The cells were grown in the presence of 300 μg/ml phleomycin D1 (Zeocin; Invivogen, San Diego, CA).
OATP1B3/pcDNA3.1 was transfected into HEK293 cells, and then cells showing resistance to 400 μg/ml G418 disulfate (Sigma, St. Louis, MO) were collected. Among the various cell clones that resulted from the colony individualization method, the one with the highest OATP1B3 level was isolated and used in this study (here referred to as 1B3/HEK).
Total RNA extraction, cDNA synthesis, and RT-PCR.
Total RNA extraction and cDNA synthesis using the HEK293 cells were performed according to the conventional methods described previously (18). Reverse transcription-PCR (RT-PCR) was performed to detect expression of an OATP1B isoform in the corresponding cells with the primers CAACAGTATGGTCAGCCTTCATCTAAGG (sense) and AATTTGGCAATTCCAACGGTGTTC (antisense) for detection of OATP1B1, the primers AACTCTTTGTTCTCTGCAACAGGAGGT (sense) and CTATAGATAAGCCCAAGTAGACCCTTCCA (antisense) for detection of OATP1B3, and the primers AGCCACATCGCTCAGACAC (sense) and GCCCAATACGACCAAATCC (antisense) for detection of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Western blotting.
Western blotting was performed essentially using the methods described in our previous paper (20). Briefly, whole-cell lysate prepared from 1B1/HEK, 1B3/HEK, or mock/HEK cells was centrifuged at 1,000 × g for 10 min at 4°C, and the supernatant was then subjected to ultracentrifugation (100,000 × g for 40 min at 4°C). The pellet was solubilized with Tris-sucrose-EDTA buffer containing 0.8% NP-40, 0.4% deoxycholic acid, and 0.08% sodium dodecyl sulfate (SDS), followed by a second ultracentrifugation (100,000× g for 40 min at 4°C). The supernatant (soluble membrane fraction) was mixed with the lysis buffer and then incubated for 30 min at 37°C. The proteins were separated by SDS-polyacrylamide gel electrophoresis, followed by transblotting onto a polyvinylidene difluoride membrane. Bovine serum albumin (BSA; 5%) or skim milk (5%) was used for membrane blocking of OATP1B1 or OATP1B3, respectively.
The primary antibodies used were rabbit anti-LST-1 IgG (500-fold dilution; Alpha Diagnostic International, San Antonio, TX), rabbit anti-SLCO1B3 IgG (1,000-fold dilution; Sigma), and mouse anti-Na+/K+ ATPase IgG (1,000-fold dilution; Sigma). The secondary antibodies used were horseradish peroxidase-conjugated goat anti-rabbit IgG (5,000-fold dilution; Sigma) and horseradish peroxidase-conjugated goat anti-mouse IgG (5,000-fold dilution; Abcam, Cambridge, United Kingdom). Immunocomplex was detected using chemiluminescence.
Immunocytochemistry.
Immunocytochemistry was performed essentially using the methods described in our previous paper (18). Briefly, 1B1/HEK, 1B3/HEK, or mock/HEK cells were seeded on a collagen-coated coverslip. The cells were fixed and permeabilized with a BD Cytofix/Cytoperm kit (BD Bioscience, Franklin Lakes, NJ). BSA (3%) was used for blocking. The primary antibodies used were rabbit anti-LST-1 IgG (200-fold dilution) or rabbit anti-SLCO1B3 IgG (200-fold dilution). The secondary antibodies used were Alexa Fluor 488-conjugated donkey anti-rabbit IgG (200-fold dilution; Life Technologies). Immunofluorescence was analyzed using confocal laser scanning immunofluorescence microscopy (FluoView FV-500; Olympus, Tokyo, Japan).
Transporter inhibition assays (coincubation method).
OATP activity level was determined in 1B1/HEK and 1B3/HEK cells based essentially on previously described transport assay methods (18). Briefly, 1 day after the cells were seeded, they were exposed to sodium butyrate (10 mM; Sigma) for 24 h, after which the transport assay was performed using E2G (100 nM; Sigma) for OATP1B1 or CCK-8 (10 nM; Peptide Institute, Osaka, Japan) for OATP1B3. 3H-labeled E2G and CCK-8 were obtained from American Radiolabeled Chemicals (St. Louis, MO) and PerkinElmer Life Science (Boston, MA), respectively. The uptake period was set to 3 min for OATP1B1 and to 5 min for OATP1B3, based on the results of preliminary experiments examining the uptake level linearity. The OATP activity level was calculated by subtracting the value obtained from mock/HEK cells from the value obtained from 1B1/HEK or 1B3/HEK cells.
Inhibition assays for validation of OATP1B expression in each cell line were performed using well-known inhibitors, rifampin (RIF; Wako, Osaka, Japan) for OATP1B1 and bromosulfophthalein (BSP; Sigma) for OATP1B3. Transport assays were performed using each cell line with the specific substrate in the presence of RIF (10 μM), BSP (100 μM), or their vehicle (dimethyl sulfoxide [DMSO]).
TLV, SMV, ASV, DCV, and SFV were purchased from Shanghai Biochempartner (Shanghai, China), ChemScene, LLC (Monmouth Junction, NJ), AdooQ BioScience LLC (Irvine, CA), ChemScene, LLC, and Medchemexpress, LLC (Princeton, NJ), respectively, and dissolved in DMSO. Inhibition assays using these DAAs were performed based on the above-described method. The E2G concentration was set to 100 nM, and CCK-8 concentration was set to 10 nM, levels that were far below the Km values of E2G uptake by OATP1B1 and CCK-8 uptake by OATP1B3 (8.3 and 3.8 μM, respectively) (21). Inhibitor concentrations are indicated in the figure legends. A concentration that inhibited OATP activity level by 50% (IC50) was calculated using the following formula: control (%) = 100/(1 + I/IC50), where control (%) represents the transporter-mediated uptake in the presence of various inhibitor concentrations relative to that in the absence of inhibitor, and I represents the inhibitor concentration.
R value calculation of OATP1B inhibition.
The maximum potential of OATP1B-mediated DDI was estimated by calculating the R value (17, 22). The R value was obtained by the following formula: R = 1 + [(fu × Iin,max)/IC50], where fu represents the blood unbound fraction of the inhibitor, and Iin,max represents the estimated maximum inhibitor concentration at the inlet to the liver. Iin,max was estimated using the following formula: Iin,max = Imax + [(Fa × dose × Ka)/Qh], where Imax is the maximum plasma concentration of the inhibitor, Fa is the dose fraction of the inhibitor that is absorbed, Ka is the absorption rate constant of the inhibitor, and Qh is the hepatic blood flow rate (1,500 ml/min) in humans. As shown in the literature (7), Fa was set at 1, Ka was set at 0.03 min−1, and the blood-to-plasma concentration ratio was assumed to be 1 for Iin,max estimation. Information related to the pharmacokinetic parameters of the DAAs used in this study are summarized in Table 1, in which Cmax, Cmax,u, and Cin,max,u are equivalent to Imax, Imax,u and Iin,max,u (estimated maximum unbound inhibitor concentration at the inlet to the liver), respectively.
TABLE 1.
Pharmacokinetic parameters of DAAs in humans
| DAA | Dose (mg) | MW (g/mol) | fu | Cmaxa (μM [ng/ml]) | Cmax,u (μM)b | Cin,max,u (μM) |
|---|---|---|---|---|---|---|
| TLV | 750 | 679.8 | 0.37 | 5.49 (3,732) | 2.031 | 10.2 |
| SMV | 150 | 749.9 | 0.01 | 5.85 (4,390) | 0.059 | 0.10 |
| ASV | 200 | 748.3 | 0.01 | 0.85 (639) | 0.007 | 0.06 |
| DCV | 60 | 738.9 | 0.01 | 2.34 (1,726) | 0.023 | 0.04 |
| SFV | 400 | 529.5 | 0.37 | 1.14 (603) | 0.421 | 6.01 |
Cmax values were obtained from the following reports: Buti et al. (36) for TLV, Sovriad interview form (Janssen Pharmaceutical K. K., Tokyo, Japan) for SMV, Eley et al. (presented at the Seventh International Workshop on Clinical Pharmacology of Hepatitis Therapy, Cambridge, MA, 27 to 28 June 2012) for ASV, Herbst and Reddy (37) for DCV, and Lawitz et al. (38) for SFV.
Cmax,u = Cmax × fu.
Transporter inhibition assays (preincubation method).
Based on the method described in a previous report (15), the 1B1/HEK, 1B3/HEK, or mock/HEK cells were preincubated with a DAA for 30 min at 0.1, 1.0, and 10 μM, after which the cells were washed twice with inhibitor-free transport assay buffer (Krebs-Henseleint buffer [KHB]). Immediately, assays of E2G or CCK-8 uptake by the cells were performed in inhibitor-free KHB, as described above. CsA (Tokyo Kasei, Tokyo, Japan), which is known to have preincubation inhibition effects on the OATP1B1/1B3 function, was used as a control in any experiments relevant to the preincubation inhibition effect.
Transporter inhibition assays (long-lasting preincubation method).
The long-lasting preincubation inhibition effects of DAAs on OATP1Bs were examined using a similar method to that described above. The cells were preincubated with a DAA for 30 min at 1.0 μM, after which they were washed once with inhibitor-free DMEM. Immediately thereafter, the cells were washed with KHB and then subjected to E2G or CCK-8 uptake assays, as described above, or they were further incubated with inhibitor-free DMEM in 5% CO2 at 37°C. After 1 or 3 h of additional incubation, the cells were washed with KHB, and the OATP1B functions were assessed by the transport assay.
Transporter inhibition assays (pre- and coincubation combination method).
The cells were preincubated with DMSO (0.1%) or a DAA at concentrations of 0.1, 0.4, and 1.0 μM as described in the preincubation method, immediately after which the OATP1B functions were determined in the presence of a DAA at the same concentration used in preincubation.
Statistical analysis.
Statistical analysis (Student's t test) was performed using a statistical software package (Statcell; OMS, Saitama, Japan) to determine whether the differences between two values were significant.
RESULTS
Validation of functional expression of OATP1B1 and OATP1B3 in HEK293 cells.
Since it has been well established that the HEK293-based OATP1B expression system is useful for drug transport assessment and determining the potential for DDI, the experiments began by examining functional OATP1B expression in HEK293 cells. The results of RT-PCR and Western blotting showed that OATP1B1 mRNA and protein expression were detected exclusively in 1B1/HEK cells (Fig. 1A and B). Cell surface localization of OATP1B1 was also detected (Fig. 1C). Consistently, the results of transport assays showed that significant E2G uptake levels were observed in 1B1/HEK cells and that this uptake was completely inhibited by RIF (Fig. 1D). Similarly, OATP1B3 mRNA and protein expression, as well as OATP1B3 cell surface localization, were detected in 1B3/HEK cells (Fig. 1E to G). As expected, BSP-sensitive CCK-8 uptake was observed in 1B3/HEK cells (Fig. 1H).
FIG 1.

Functional expression of OATP1B1 or OATP1B3 in HEK293 cells. (A and E) RT-PCR was performed to examine OATP1B1 or OATP1B3 expression in 1B1/HEK or 1B3/HEK cells, respectively. GAPDH mRNA was used as an experimental control. The representative results obtained from three independent experiments are shown. (B and F) Western blotting was performed to examine OATP1B1 or OATP1B3 protein expression in 1B1/HEK or 1B3/HEK cells, respectively. (Asterisks indicate presumably nonglycosylated forms of each OATP1B isoform.) Na+/K+ ATPase was used as a loading control. The representative results that were obtained from three independent experiments are shown. (C and G) Immunocytochemistry was performed to examine OATP1B1 or OATP1B3 cell surface localization in 1B1/HEK or 1B3/HEK cells, respectively. The representative results that were obtained from three independent experiments are shown. (D and H) The OATP1B1- or OATP1B3-mediated substrate uptake activity was determined. The substrate and inhibitor for OATP1B1 experiments were E2G (100 nM) and RIF (10 μM), respectively, and those for OATP1B3 experiments were CCK-8 (10 nM) and BSP (100 μM), respectively. Background activity level was determined by mock/HEK cells. Data represent the means ± standard deviations of the values obtained from three separate experiments, each performed in duplicate.
Characterization of interaction properties between OATP1B and DAAs using a coincubation inhibition method.
The interaction profiles of SMV, ASV, DCV, and SFV with OATP1B1 and OATP1B3 were examined by a classical coincubation inhibition assay, where TLV was also used as a reference DAA. E2G and CCK-8 were used as OATP1B1 and OATP1B3 substrates, respectively, because they have come to be regarded as good surrogate probes for evaluation of OATP1B-mediated DDIs (9, 23). The results showed that all DAAs tested were able to inhibit OATP1B1 function (Fig. 2); the IC50 values are listed in Table 2. The IC50 value of TLV for the OATP1B1 function was comparable to that reported in the literature (9). Compared with TLV, SMV and ASV were found to be potent inhibitors, while DCV had a similar level of inhibition and SFV was found to be a relatively weak inhibitor. Similarly, the inhibition profile of DAAs against the OATP1B3 function was also determined (Fig. 2 and Table 2). Again, the IC50 value of TLV for OATP1B3 was comparable to that reported in the literature (9), and other IC50 values showed that SMV, ASV, and DCV were all strong OATP1B3 inhibitors, while SFV did not significantly affect OATP1B3 function.
FIG 2.

Interaction properties between OATP1B and DAAs determined by coincubation inhibition method. Coincubation inhibition experiments were performed in 1B1/HEK and 1B3/HEK cells using E2G (100 nM) and CCK-8 (10 nM) for OATP1B1 and OATP1B3 substrates, respectively. TLV, SMV, ASV, DCV, and SFV were used as test inhibitors at concentrations of 0.1 to 100 μM (TLV, DCV, and SFV) and of 0.01 to 10 μM (SMV and ASV). The OATP activity level was calculated by subtracting the value obtained from mock/HEK cells from the value obtained from 1B1/HEK or 1B3/HEK cells. Data represent the means ± standard deviations of relative percentages where OATP activity level in the absence of an inhibitor (DMSO alone) was set to 100%. The values were obtained from three separate experiments, each performed in duplicate. The IC50 values of TLV, SMV, ASV, DCV, and SFV are summarized in Table 2.
TABLE 2.
Inhibition properties of DAAs to OATP1B1/1B3 and in vitro evaluation of their DDI potentials through OATP1B inhibition
| DAAa | OATP1B1 |
OATP1B3 |
||||
|---|---|---|---|---|---|---|
| IC50 (μM) | Cmax/IC50 | R | IC50 (μM) | Cmax/IC50 | R | |
| TLV | 1.36 ± 0.58 | 4.04 | 8.50 | 9.69 ± 3.10 | 0.57 | 2.05 |
| SMV | 0.30 ± 0.06 | 19.5 | 1.33 | 0.22 ± 0.07 | 26.6 | 1.45 |
| ASV | 0.79 ± 0.21 | 0.85 | 1.08 | 0.65 ± 0.26 | 1.03 | 1.10 |
| DCV | 1.50 ± 0.33 | 1.55 | 1.03 | 3.27 ± 0.57 | 0.71 | 1.01 |
| SFV | 16.5 ± 7.60 | 0.07 | NAb | 61.9 ± 31.6 | 0.02 | NAb |
The International Transporter Consortium has proposed a decision tree for use in determining if a drug has the potential to cause OATP1B-mediated DDI (17). Using that tree, Cmax/IC50 values were calculated as the initial step (Table 2). All Cmax/IC50 values (except for SFV) were above the cutoff value (0.1), which suggested the need to proceed with an R value calculation for SMV, ASV, and DCV. It was also found that, even though they are less significant than those of TLV, the SMV R values for both OATP1B1 and OATP1B3 were over 1.25 (the suggested value according to the upper limit of equivalence range) (Table 2). In contrast, the R values of ASV and DCV were below 1.25.
Identification of preincubation inhibition effects of DAAs on OATP1B functions.
Although available literature is still limited, recent evidence suggests that the preincubation inhibition effect is one of the intrinsic characteristics of OATP1B inhibitors. Therefore, we sought to clarify whether the DAAs have the capability to exert such inhibitory effects by conducting preincubation inhibition assays using CsA as a reference inhibitor. As shown in Fig. 3, preincubation with SMV at 1.0 and 10 μM results in a substantial decrease in the OATP1B1 function level by 67.7 ± 13.4% and 88.4 ± 12.9%, respectively, and a decrease in the OATP1B3 function level by 95.1 ± 3.1% and 98.1 ± 1.1%, respectively. These effects were as potent as those of CsA. Unexpectedly, the preincubation inhibition profile of ASV on OATP1B1 function was somewhat different from that of SMV, and ASV preincubation affected OATP1B3 function only at 10 μM. DCV also exhibited significant preincubation inhibition effects on both OATP1Bs but only at 10 μM. In contrast, preincubation with TLV and SFV did not influence OATP1B functions at all.
FIG 3.

Preincubation inhibition effects of DAAs on OATP1B functions. E2G (100 nM) and CCK-8 (10 nM) uptake by OATP1B1 and OATP1B3 were examined, respectively, under inhibitor-free conditions immediately after a 30-min preincubation with TLV, SMV, ASV, DCV, SFV, or CsA at 0.1, 1.0, and 10 μM (preincubation method). The OATP activity level was calculated by subtracting the value obtained from mock/HEK cells from the value obtained from 1B1/HEK or 1B3/HEK cells. Data represent the means ± standard deviations of relative percentages where the OATP activity level preincubated with DMSO alone was set to 100%. The values were obtained from three separate experiments, each performed in duplicate.
Examination of the long-lasting effect of preincubation inhibition of DAAs on OATP1B functions.
It has been shown that preincubation inhibition effects of CsA on OATP1Bs can be maintained for several hours (15). Therefore, the long-lasting properties of the preincubation inhibition effects of SMV and ASV (1 μM) were investigated using CsA as a reference inhibitor (Fig. 4). The results showed that the residual OATP1B1 functional levels at 1 h after SMV and ASV exposure were found to be 65.1 ± 9.2% and 85.3 ± 6.1%, respectively, and that the complete recovery of OATP1B1 function was observed as early as 3 h after SMV or ASV exposure, while CsA imposed significantly prolonged inhibition on OATP1B1 function. On the other hand, SMV continuously repressed OATP1B3 function for as long as CsA did. The residual OATP1B3 activity level was 53.0 ± 12.0% at 3 h after SMV exposure. As expected, ASV lacked a preincubation inhibition effect at this concentration.
FIG 4.
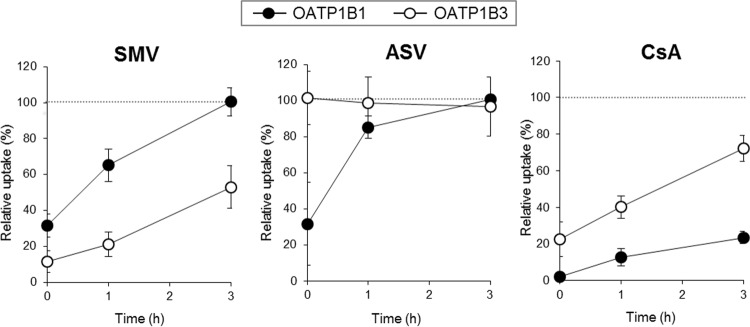
Long-lasting effect of preincubation inhibition of DAAs on OATP1B functions. E2G (100 nM) and CCK-8 (10 nM) uptake by OATP1B1 and OATP1B3 was examined, respectively, under inhibitor-free conditions at 1 h, 3 h, or after just 30 min of preincubation with SMV, ASV, and CsA at 1.0 μM (long-lasting preincubation inhibition method). The OATP activity level was calculated by subtracting the value obtained from mock/HEK cells from the value obtained from 1B1/HEK or 1B3/HEK cells. Data represent the means ± standard deviations of relative percentages where the OATP activity level preincubated with DMSO alone was set to 100%. The values were obtained from three separate experiments, each performed in duplicate.
Determination of enhancing effect of DAAs' long-lasting preincubation inhibition on their overall inhibition potency against OATP1B functions.
An examination was conducted to determine if the long-lasting preincubation inhibition effects of SMV and ASV augment their coincubation inhibitory level against OATP1B functions because such effects have been identified in the case of CsA (14). Our results showed that preincubation with SMV (0.4 μM) significantly strengthened the coincubation inhibition effect (0.4 μM) on OATP1B1 activity (from 46.4 ± 2.4% to 22.3 ± 10.3%; P < 0.05) (Fig. 5). Preincubation with ASV (0.1 and 0.4 μM) was also found to enhance the coincubation inhibition effect on OATP1B1 activity (from 78.0 ± 2.3% to 56.1 ± 7.5% and from 50.6 ± 1.3% to 29.4 ± 7.6%, respectively; P < 0.01). Similar tendencies were observed at other concentrations of SMV and ASV.
FIG 5.

Enhancing effect of DAAs' long-lasting preincubation inhibition on their overall inhibition potency against OATP1B functions. E2G (100 nM) and CCK-8 (10 nM) uptake by OATP1B1 and OATP1B3 were examined, respectively, in the presence of an inhibitor immediately after a 30-min preincubation with SMV or ASV (pre- and coincubation combination method). The inhibitor concentrations used for pre- and coincubation were equal and set at 0.1, 0.4, and 1.0 μM. The OATP activity level was calculated by subtracting the value obtained from mock/HEK cells from the value obtained from 1B1/HEK or 1B3/HEK cells. Data represent the means ± standard deviations of relative percentages where OATP activity level pre- and coincubated with DMSO alone was set to 100%. The values were obtained from three separate experiments, each performed in duplicate. Significance is indicated as follows: *, P < 0.05; **, P < 0.01.
Similarly, the coincubation inhibition effects of SMV at 0.1 μM on OATP1B3 function level was strengthened by preincubation with the same concentration of SMV (from 43.1 ± 6.4% to 26.5 ± 4.4%). Preincubation with ASV only marginally affected its coincubation inhibition effect on the OATP1B3 function, which was consistent with the results shown in Fig. 3 and 4.
DISCUSSION
Our results show that all of the new DAAs examined in this study can inhibit OATP1B functions in a classical manner (coincubation inhibition) and also that some of them have distinctive long-lasting preincubation inhibitory effects on OATP1B functions. These inhibition characteristics should be seen as essential information when the clinical significance of DAA-mediated OATP1B inhibition is considered, as we have discussed herein.
Based on the criteria of R value significance (≥1.25), the values of SMV indicate that SMV creates a mild DDI risk when it is administered with OATP1B substrates. In contrast, the DDI risk potential for ASV, DCV, and SFV was found to be from very modest to negligible. These predictions are roughly consistent with the lack of literature reporting “detrimental” DDIs in association with these new agents.
Nevertheless, it has been shown that SMV administration (150 mg) leads to apparently 2- to 3-fold increases in atorvastatin (40 mg) and rosuvastatin (10 mg) exposure, respectively (Sovriad interview form, Janssen Pharmaceutical K. K., Tokyo, Japan), and that ASV administration (200 mg) results in a 1.4-fold increase in rosuvastatin (10 mg) exposure (T. Eley, Y.-H. Han, S.-P. Huang, B. He, W. Li, W. Bedford, M. Stonier, D. Gardiner, K. Sims, P. Balimane, D. Rodrigues, and R. J. Bertz, presented at the Seventh International Workshop on Clinical Pharmacology of Hepatitis Therapy, Cambridge, MA, 27 to 28 June 2012). (Please note that the clinically used dosage amounts of SMV, rosuvastatin, and atorvastatin are 100 to 150 mg once a day [QD], 5 to 40 mg QD, and 10 to 80 mg QD, respectively, based on their prescribing information, and that 200 mg of ASV QD or twice a day [BID] has been used in clinical trials.) Based on the results of our preincubation inhibition experiments, it can be assumed that SMV or ASV imposes long-lasting inhibition effects on OATP1B functions, in addition to their coincubation effects, in such DDIs. Preclinical data have shown that SMV and ASV accumulate significantly in rat livers (32- to 65-fold for SMV and 315-fold for ASV) (24, 25), which implies that, to the extent that they sufficiently exert preincubation inhibitory effects on OATP1B functions, unbound liver SMV or ASV concentrations might become greater than their plasma unbound concentrations. Therefore, it is reasonable to presume that the long-lasting preincubation inhibition effects of SMV or ASV play a clinically significant role in the reduction of OATP1Bs' functional levels during SMV- or ASV-containing therapy. Despite this likelihood, in order to enhance the reliability of this concept, an integrated prediction method is likely to be necessary during further investigations aimed at determining the significance of OATP1B inhibition by drugs that possess both co- and preinhibition properties. Once established, such a prediction method can be expected to provide additional quantitative explanations as to why SMV and ASV affect systemic exposure of OATP1B substrates including statins.
In addition to the inhibition properties obtained in this study, it will be necessary to pay attention to the following viewpoints in order to estimate the DDI potential of DAAs in vivo. First, it is likely that the Cmax and the Cin,max,u of a DAA are highly variable among patients. For example, the Cmax of SMV ranged from 1.8 to 13.5 μM (n = 123, HPC3003 clinical trial in Japan). This range could be further expanded as the patient population increases, partially because of the presence of rare variants in the drug-metabolizing and transporter genes (26, 27). Second, although E2G and CCK-8 have been considered good surrogate probes for use in OATP1B inhibition studies (9, 23), the R value obtained from other OATP1B substrates may be different from the values obtained in this study, as exemplified by the report showing that the IC50 values of rifamycin SV toward OATP1B1-mediated E2G and rosuvastatin uptake are 0.34 ± 0.05 and 0.05 ± 0.02 (μM), respectively (23). Finally, because various enzymes and transporters, such as CYP3A4, cooperatively determine a drug pharmacokinetic profile together with OATP1Bs, multifactor evaluation of clinical DDI likelihood is necessary in order to minimize over- or underprediction, as depicted in the recent literature (28). Therefore, DAA R values related to other gene functions should be determined and consolidated with those obtained in this study in order to create a more advanced evaluation of DDI potential of DAAs.
Taking these results together, we can suggest that SMV and (to a lesser degree) ASV have a latent potential to cause DDIs at the level of OATP1B1/1B3 even though the clinical DDI likelihood of DAAs is still open to further investigation. The possibility of this potential for DCV cannot be ruled out due to its simultaneous possession of co- and preinhibition properties, while SFV seems to be harmless to OATP1Bs.
Because chronic hepatitis C patients may often take multiple medications in addition to anti-HCV drugs, these findings will call the physician's attention to the cautions related to the DDIs associated with these DAAs (especially SMV and ASV). In addition to statins, examples of potential victim OATP substrates are repaglinide (antidiabetic), fexofenadine (antihistaminic), olmesartan and telmisartan (antihypertensives), and torsemide (diuretic) based on the literature (10, 29). Furthermore, the number of such examples can be expected to increase as new OATP1B substrate drugs are developed in the future. Therefore, even though DDIs may not be critical factors in drug decision making, an understanding of the possible DDI risks for a DAA intended for use is nevertheless important for appropriate clinical management.
In addition, potential interactions between a DAA and an anti-human immunodeficiency virus (HIV) agent should be mentioned because ∼33% of HCV patients are estimated to be coinfected with HIV in Western countries (30). According to the Sovriad interview form (Janssen Pharmaceutical K. K., Tokyo, Japan), SMV (150 mg QD) does not significantly affect rilpivirine (25 mg QD), raltegravir (400 mg QD), tenofovir (300 mg QD), or efavirenz (600 mg QD) exposure. However, although it has not yet been reported, SMV could affect lopinavir exposure, based on a report showing that its plasma concentration has been affected by the OATP1B1 function level (31, 32). On the other hand, it has been shown that efavirenz (600 mg QD) significantly reduces SMV (150 mg QD) exposure, while ritonavir (100 mg BID) increases SMV (200 mg QD) exposure in preliminary DDI tests although these are believed to be caused by alteration of SMV metabolism rate (Sovriad interview form, Janssen Pharmaceutical K. K., Tokyo, Japan). Due to the distinctive pleiotropic effects of each anti-HIV drug (or DAA) on drug-metabolizing enzymes and transporters, the in vivo interaction profile between an anti-HIV drug and a DAA or between any antiretroviral/DAA and its coadministered drug(s) is considerably complex. Therefore, caution related to such interactions is advisable prior to the accumulation of experimental and clinical data. A similar suggestion has been made in a recent review (33), where additional DDI information can be obtained. Considering the level of complexity, it is assumed that if antivirus-specialized clinical pharmacists could be trained, they would contribute to safer and more effective treatment for HCV/HIV-coinfected patients.
Another important issue related to the inhibition of the OATP1B function is hyperbilirubinemia. OATP1Bs play pivotal roles in development of hyperbilirubinemia because, in addition to identification of OATP1B1 and OATP1B3 as conjugated and unconjugated bilirubin transporters (11, 12), it has been shown that patients who carry biallelic inactivating mutations in both genes show rotor syndrome, which is characterized by conjugated hyperbilirubinemia (34). Clinical studies have shown that transient and benign hyperbilirubinemia without association of concomitant elevation of liver enzymes is often observed in SMV-containing regimens (35) but not in TLV-containing regimens, despite the fact that either DAA can inhibit OATP1Bs. This observation may indicate that cooperative coincubation and long-lasting preincubation inhibitory effects of SMV on both OATP1B1/1B3 functions may play a crucial role in the development of hyperbilirubinemia in SMV-containing regimens. In addition, although not fully characterized, it has been shown that SMV inhibits the bilirubin glucuronidation enzyme (UDP-glucuronosyltransferase 1A1 [UGT1A1]) and conjugated bilirubin extrusion pump (multidrug resistance protein 2 [MRP2]), while TLV does not. Therefore, the involvement of UGT1A1 and/or MRP2 inhibition in the development of SMV-mediated hyperbilirubinemia should not be ruled out even if the IC50 values of SMV for the bilirubin glucuronidation and MRP2 functions are higher than those for OATP1Bs (119 μM and 6.4 to 19 μM, respectively). Collectively, it is considered likely that elevation of the blood bilirubin level could reflect a functional disturbance of OATP1B1/1B3 along with UGT1A1 and/or MRP2 by SMV. Therefore, it can be speculated that extensive hyperbilirubinemia could provide a warning for SMV-caused DDI with drugs that utilize those pathways for their elimination in such patients.
Finally, the differential long-lasting preincubation effects among our DAAs should be mentioned due to their important relevant aspects in OATP1B studies. It is intriguing that SMV, ASV, and (to a lesser extent) DCV are newly listed as members of the long-lasting dual OATP1B1/1B3 inhibitor lineup, in which only CsA has been identified to date (15). In addition, it was surprising to find that, despite the outstanding similarities of their physicochemical properties (data not shown) as well as their IC50 values against OATP1Bs, there are significant differences in the preincubation inhibition profiles of SMV and ASV. Although the mechanisms behind their long-lasting preincubation inhibition effects remain unknown, our findings suggest that their long-lasting inhibitions against OATP1B1 and OATP1B3 do not share common cellular mechanisms and that the physicochemical property of a particular drug is unlikely to play a decisive role in its inhibition effects. Since these results may provide important insights into the clarification of inhibitory mechanisms, further mechanistic exploration using SMV and ASV can be expected to identify a key factor or process underlying long-lasting preincubation effects.
In conclusion, our results not only show that all tested DAAs are capable of inhibiting OATP1B1 and 1B3 functions but also that SMV, ASV, and DCV are newly identified, distinctive long-lasting OATP1B inhibitors. Our results also suggest that the cooperative coincubation and long-lasting preincubation inhibitory effects of SMV on OATP1B functions at least partially account for the increased exposure of statins and transient hyperbilirubinemia in SMV-containing regimens. The inhibitory effects of ASV, but not SFV, on OATP1B functions may also be clinically important, while the possibility in relation to DCV remains elusive. We expect that although such DDIs may not be the sole determinants in treatment decision making, these inhibition profiles and estimations for OATP1B-mediated DDI potentials will provide useful information that will facilitate safer and more effective anti-HCV therapy. In addition, our results point toward the need for elucidation of the detailed characteristics underlying long-lasting preincubation effects of DAAs on OATP1Bs in order to facilitate the development of an improved quantitative DDI evaluation method that takes such effects, along with other relevant factors, into consideration.
ACKNOWLEDGMENTS
We thank Yuki Suzuki and Hanae Morio (Laboratory of Pharmacology and Toxicology, Chiba University) for their technical support.
This work is funded by a Ministry of Health, Labor and Welfare Grant-in-Aid for Scientific Research (Emergency Research Project to Conquer Hepatitis), Japan.
Footnotes
Published ahead of print 27 May 2014
REFERENCES
- 1.Shah N, Pierce T, Kowdley KV. 2013. Review of direct-acting antiviral agents for the treatment of chronic hepatitis C. Expert Opin. Investig. Drugs 22:1107–1121. 10.1517/13543784.2013.806482 [DOI] [PubMed] [Google Scholar]
- 2.Tsubota A, Furihata T, Matsumoto Y, Chiba K. 2013. Sustained and rapid virological responses in hepatitis C clinical trials. Clin. Investig. 3:1083–1093. 10.4155/cli.13.98 [DOI] [Google Scholar]
- 3.Burger D, Back D, Buggisch P, Buti M, Craxí A, Foster G, Klinker H, Larrey D, Nikitin I, Pol S, Puoti M, Romero-Gómez M, Wedemeyer H, Zeuzem S. 2013. Clinical management of drug-drug interactions in HCV therapy: challenges and solutions. J. Hepatol. 58:792–800. 10.1016/j.jhep.2012.10.027 [DOI] [PubMed] [Google Scholar]
- 4.Kiser JJ, Burton JR, Jr, Everson GT. 2013. Drug-drug interactions during antiviral therapy for chronic hepatitis C. Nat. Rev. Gastroenterol. Hepatol. 10:596–606. 10.1038/nrgastro.2013.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garg V, van Heeswijk R, Lee JE, Alves K, Nadkarni P, Luo X. 2011. Effect of telaprevir on the pharmacokinetics of cyclosporine and tacrolimus. Hepatology 54:20–27. 10.1002/hep.24443 [DOI] [PubMed] [Google Scholar]
- 6.Lee JE, van Heeswijk R, Alves K, Smith F, Garg V. 2011. Effect of the hepatitis C virus protease inhibitor telaprevir on the pharmacokinetics of amlodipine and atorvastatin. Antimicrob. Agents Chemother. 55:4569–4574. 10.1128/AAC.00653-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu X, Cai X, Cui D, Tang C, Ghosal A, Chan G, Green MD, Kuo Y, Liang Y, Maciolek CM, Palamanda J, Evers R, Prueksaritanont T. 2013. In vitro assessment of drug-drug interaction potential of boceprevir associated with drug metabolizing enzymes and transporters. Drug Metab. Dispos. 41:668–681. 10.1124/dmd.112.049668 [DOI] [PubMed] [Google Scholar]
- 8.Garg V, Chandorkar G, Farmer HF, Smith F, Alves K, van Heeswijk RP. 2012. Effect of telaprevir on the pharmacokinetics of midazolam and digoxin. J. Clin. Pharmacol. 52:1566–1573. 10.1177/0091270011419850 [DOI] [PubMed] [Google Scholar]
- 9.Kunze A, Huwyler J, Camenisch G, Gutmann H. 2012. Interaction of the antiviral drug telaprevir with renal and hepatic drug transporters. Biochem. Pharmacol. 84:1096–1102. 10.1016/j.bcp.2012.07.032 [DOI] [PubMed] [Google Scholar]
- 10.Shitara Y, Maeda K, Ikejiri K, Yoshida K, Horie T, Sugiyama Y. 2013. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 34:45–78. 10.1002/bdd.1823 [DOI] [PubMed] [Google Scholar]
- 11.Chiou WJ, de Morais SM, Kikuchi R, Voorman RL, Li X, Bow DA. 2014. In vitro OATP1B1 and OATP1B3 inhibition is associated with observations of benign clinical unconjugated hyperbilirubinemia. Xenobiotica 44:276–282. 10.3109/00498254.2013.820006 [DOI] [PubMed] [Google Scholar]
- 12.Cui Y, König J, Leier I, Buchholz U, Keppler D. 2001. Hepatic uptake of bilirubin and its conjugates by the human organic anion transporter SLC21A6. J. Biol. Chem. 276:9626–9630. 10.1074/jbc.M004968200 [DOI] [PubMed] [Google Scholar]
- 13.Shitara Y, Itoh T, Sato H, Li AP, Sugiyama Y. 2003. Inhibition of transporter-mediated hepatic uptake as a mechanism for drug-drug interaction between cerivastatin and cyclosporin A. J. Pharmacol. Exp. Ther. 304:610–616. 10.1124/jpet.102.041921 [DOI] [PubMed] [Google Scholar]
- 14.Amundsen R, Christensen H, Zabihyan B, Asberg A. 2010. Cyclosporine A, but not tacrolimus, shows relevant inhibition of organic anion-transporting protein 1B1-mediated transport of atorvastatin. Drug Metab. Dispos. 38:1499–1504. 10.1124/dmd.110.032268 [DOI] [PubMed] [Google Scholar]
- 15.Shitara Y, Takeuchi K, Nagamatsu Y, Wada S, Sugiyama Y, Horie T. 2012. Long-lasting inhibitory effects of cyclosporin A, but not tacrolimus, on OATP1B1- and OATP1B3-mediated uptake. Drug Metab. Pharmacokinet. 27:368–378. 10.2133/dmpk.DMPK-11-RG-096 [DOI] [PubMed] [Google Scholar]
- 16.Keppler D. 2014. The roles of MRP2, MRP3, OATP1B1, and OATP1B3 in conjugated hyperbilirubinemia. Drug Metab. Dispos. 42:561–565. 10.1124/dmd.113.055772 [DOI] [PubMed] [Google Scholar]
- 17.Tweedie D, Polli JW, Berglund EG, Huang SM, Zhang L, Poirier A, Chu X, Feng B, International Transporter Consortium 2013. Transporter studies in drug development: experience to date and follow-up on decision trees from the International Transporter Consortium. Clin. Pharmacol. Ther. 94:113–125. 10.1038/clpt.2013.77 [DOI] [PubMed] [Google Scholar]
- 18.Kameyama Y, Yamashita K, Kobayashi K, Hosokawa M, Chiba K. 2005. Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet. Genomics 15:513–522. 10.1097/01.fpc.0000170913.73780.5f [DOI] [PubMed] [Google Scholar]
- 19.Nagai M, Furihata T, Matsumoto S, Ishii S, Motohashi S, Yoshino I, Ugajin M, Miyajima A, Matsumoto S, Chiba K. 2012. Identification of a new organic anion transporting polypeptide 1B3 mRNA isoform primarily expressed in human cancerous tissues and cells. Biochem. Biophys. Res. Commun. 418:818–823. 10.1016/j.bbrc.2012.01.115 [DOI] [PubMed] [Google Scholar]
- 20.Furihata T, Satoh N, Ohishi T, Ugajin M, Kameyama Y, Morimoto K, Matsumoto S, Yamashita K, Kobayashi K, Chiba K. 2009. Functional analysis of a mutation in the SLCO1B1 gene (c.1628T>G) identified in a Japanese patient with pravastatin-induced myopathy. Pharmacogenomics J. 9:185–193. 10.1038/tpj.2009.3 [DOI] [PubMed] [Google Scholar]
- 21.Hirano M, Maeda K, Shitara Y, Sugiyama Y. 2004. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J. Pharmacol. Exp. Ther. 311:139–146. 10.1124/jpet.104.068056 [DOI] [PubMed] [Google Scholar]
- 22.Hirano M, Maeda K, Shitara Y, Sugiyama Y. 2006. Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab. Dispos. 34:1229–1236. 10.1124/dmd.106.009290 [DOI] [PubMed] [Google Scholar]
- 23.Sharma P, Butters CJ, Smith V, Elsby R, Surry D. 2012. Prediction of the in vivo OATP1B1-mediated drug-drug interaction potential of an investigational drug against a range of statins. Eur. J. Pharm. Sci. 47:244–255. 10.1016/j.ejps.2012.04.003 [DOI] [PubMed] [Google Scholar]
- 24.Lin TI, Lenz O, Fanning G, Verbinnen T, Delouvroy F, Scholliers A, Vermeiren K, Rosenquist A, Edlund M, Samuelsson B, Vrang L, de Kock H, Wigerinck P, Raboisson P, Simmen K. 2009. In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor. Antimicrob. Agents Chemother. 53:1377–1385. 10.1128/AAC.01058-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McPhee F, Sheaffer AK, Friborg J, Hernandez D, Falk P, Zhai G, Levine S, Chaniewski S, Yu F, Barry D, Chen C, Lee MS, Mosure K, Sun LQ, Sinz M, Meanwell NA, Colonno RJ, Knipe J, Scola P. 2012. Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS-650032). Antimicrob. Agents Chemother. 56:5387–5396. 10.1128/AAC.01186-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Brien TJ, Kidd RS, Richard CA, Ha NH, Witcher P, Tran LV, Barbour A, Tuck M, McIntosh SD, Douglas JN, Harralson AF. 2013. First report of warfarin dose requirements in patients possessing the CYP2C9*12 allele. Clin. Chim. Acta 424:73–75. 10.1016/j.cca.2013.05.008 [DOI] [PubMed] [Google Scholar]
- 27.Ramsey LB, Bruun GH, Yang W, Treviño LR, Vattathil S, Scheet P, Cheng C, Rosner GL, Giacomini KM, Fan Y, Sparreboom A, Mikkelsen TS, Corydon TJ, Pui CH, Evans WE, Relling MV. 2012. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 22:1–8. 10.1101/gr.129668.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Camenisch G, Umehara K. 2012. Predicting human hepatic clearance from in vitro drug metabolism and transport data: a scientific and pharmaceutical perspective for assessing drug-drug interactions. Biopharm. Drug Dispos. 33:179–194. 10.1002/bdd.1784 [DOI] [PubMed] [Google Scholar]
- 29.Gong IY, Kim RB. 2013. Impact of genetic variation in OATP transporters to drug disposition and response. Drug Metab. Pharmacokinet. 28:4–18. 10.2133/dmpk.DMPK-12-RV-099 [DOI] [PubMed] [Google Scholar]
- 30.Sulkowski MS. 2008. Viral hepatitis and HIV coinfection. J. Hepatol. 48:353–367. 10.1016/j.jhep.2007.11.009 [DOI] [PubMed] [Google Scholar]
- 31.Kohlrausch FB, de Cássia Estrela R, Barroso PF, Suarez-Kurtz G. 2010. The impact of SLCO1B1 polymorphisms on the plasma concentration of lopinavir and ritonavir in HIV-infected men. Br. J. Clin. Pharmacol. 69:95–98. 10.1111/j.1365-2125.2009.03551.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartkoorn RC, Kwan WS, Shallcross V, Chaikan A, Liptrott N, Egan D, Sora ES, James CE, Gibbons S, Bray PG, Back DJ, Khoo SH, Owen A. 2010. HIV protease inhibitors are substrates for OATP1A2, OATP1B1 and OATP1B3 and lopinavir plasma concentrations are influenced by SLCO1B1 polymorphisms. Pharmacogenet. Genomics 20:112–120. 10.1097/FPC.0b013e328335b02d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karageorgopoulos DE, El-Sherif O, Bhagani S, Khoo SH. 2014. Drug interactions between antiretrovirals and new or emerging direct-acting antivirals in HIV/hepatitis C virus coinfection. Curr. Opin. Infect. Dis. 27:36–45. 10.1097/QCO.0000000000000034 [DOI] [PubMed] [Google Scholar]
- 34.van de Steeg E, Stránecký V, Hartmannová H, Nosková L, Hřebíček M, Wagenaar E, van Esch A, de Waart DR, Oude Elferink RP, Kenworthy KE, Sticová E, al-Edreesi M, Knisely AS, Kmoch S, Jirsa M, Schinkel AH. 2012. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J. Clin. Invest. 122:519–528. 10.1172/JCI59526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeuzem S, Berg T, Gane E, Ferenci P, Foster GR, Fried MW, Hezode C, Hirschfield GM, Jacobson I, Nikitin I, Pockros PJ, Poordad F, Scott J, Lenz O, Peeters M, Sekar V, De Smedt G, Sinha R, Beumont-Mauviel M. 2014. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology 146:430–441.e6. 10.1053/j.gastro.2013.10.058 [DOI] [PubMed] [Google Scholar]
- 36.Buti M, Agarwal K, Horsmans Y, Sievert W, Janczewska E, Zeuzem S, Nyberg L, Brown RS, Jr, Hezode C, Rizzetto M, Parana R, De Meyer S, De Masi R, Luo D, Bertelsen K, Witek J. 2014. Telaprevir twice daily is noninferior to telaprevir every 8 hours for patients with chronic hepatitis C. Gastroenterology 146:744–753.e3. 10.1053/j.gastro.2013.11.047 [DOI] [PubMed] [Google Scholar]
- 37.Herbst DA, Reddy KR. 2013. NS5A inhibitor, daclatasvir, for the treatment of chronic hepatitis C virus infection. Expert Opin. Investig. Drugs 22:1337–1346. 10.1517/13543784.2013.826189 [DOI] [PubMed] [Google Scholar]
- 38.Lawitz EJ, Rodriguez-Torres M, Denning J, Mathias A, Mo H, Gao B, Cornpropst MT, Berrey MM, Symonds WT. 2013. All-oral therapy with nucleotide inhibitors sofosbuvir and GS-0938 for 14 days in treatment-naive genotype 1 hepatitis C (nuclear). J. Viral. Hepat. 20:699–707. 10.1111/jvh.12091 [DOI] [PubMed] [Google Scholar]
- 39.Perni RB, Almquist SJ, Byrn RA, Chandorkar G, Chaturvedi PR, Courtney LF, Decker CJ, Dinehart K, Gates CA, Harbeson SL, Heiser A, Kalkeri G, Kolaczkowski E, Lin K, Luong YP, Rao BG, Taylor WP, Thomson JA, Tung RD, Wei Y, Kwong AD, Lin C. 2006. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3-4A serine protease. Antimicrob. Agents Chemother. 50:899–909. 10.1128/AAC.50.3.899-909.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR, II, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100. 10.1038/nature08960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tong X, Le Pogam S, Li L, Haines K, Piso K, Baronas V, Yan JM, So SS, Klumpp K, Nájera I. 2014. In vivo emergence of a novel mutant L159F/L320F in the NS5B polymerase confers low-level resistance to the HCV polymerase inhibitors mericitabine and sofosbuvir. J. Infect. Dis. 209:668–675. 10.1093/infdis/jit562 [DOI] [PubMed] [Google Scholar]