

Abstract
While β-lactam antibiotics are a critical part of the antimicrobial arsenal, they are frequently compromised by various resistance mechanisms, including changes in penicillin binding proteins of the bacterial cell wall. Genetic deletion of the penicillin binding protein and serine/threonine kinase-associated protein (PASTA) kinase in methicillin-resistant Staphylococcus aureus (MRSA) has been shown to restore β-lactam susceptibility. However, the mechanism remains unclear, and whether pharmacologic inhibition would have the same effect is unknown. In this study, we found that deletion or pharmacologic inhibition of the PASTA kinase in Listeria monocytogenes by the nonselective kinase inhibitor staurosporine results in enhanced susceptibility to both aminopenicillin and cephalosporin antibiotics. Resistance to vancomycin, another class of cell wall synthesis inhibitors, or antibiotics that inhibit protein synthesis was unaffected by staurosporine treatment. Phosphorylation assays with purified kinases revealed that staurosporine selectively inhibited the PASTA kinase of L. monocytogenes (PrkA). Importantly, staurosporine did not inhibit a L. monocytogenes kinase without a PASTA domain (Lmo0618) or the PASTA kinase from MRSA (Stk1). Finally, inhibition of PrkA with a more selective kinase inhibitor, AZD5438, similarly led to sensitization of L. monocytogenes to β-lactam antibiotics. Overall, these results suggest that pharmacologic targeting of PASTA kinases can increase the efficacy of β-lactam antibiotics.
INTRODUCTION
Listeria monocytogenes is a common environmental Gram-positive bacterium that upon ingestion can cause the serious disease listeriosis (1). Listeriosis is normally contracted from ingestion of contaminated food by at-risk populations, which include the elderly, the immunocompromised, and pregnant women (2, 3). Disease symptoms can range from mild gastroenteritis to severe meningitis and spontaneous miscarriage (4). Current therapy calls for high-dose aminopenicillins combined with gentamicin (5). Although L. monocytogenes is highly susceptible to this treatment in vitro, the fatality rate from confirmed cases of listeriosis remains high, sometimes reaching ∼30%, suggesting an increased need for better therapeutic strategies (6, 7).
β-Lactam antibiotics have been a critical part of treatment for Gram-positive bacterial infections since they were discovered (8). Unfortunately, due to the increasing frequency of antibiotic resistance, β-lactams are no longer effective against many pathogens, including certain penicillin-resistant streptococci and enterococci and, most notoriously, methicillin-resistant Staphylococcus aureus (MRSA) (9). MRSA strains, including community-associated strains such as USA300, contain the mecA gene, which encodes penicillin binding protein 2A (PBP2A), a PBP that confers resistance to all approved β-lactams with the exception of ceftaroline (10, 11). This increase in the development of antibiotic resistance, particularly to β-lactams, has resulted in a need for new strategies for antimicrobial therapy.
S. aureus and many other important pathogens, including L. monocytogenes, Mycobacterium tuberculosis, and Enterococcus faecalis, express a bipartite membrane-associated eukaryote-like serine/threonine kinase (eSTK) that has one or more extracellular repeats of a homologous family of PBPs (12). This family of proteins is known as the penicillin binding protein and serine/threonine kinase-associated protein (PASTA) kinases (12). PASTA kinases have extracellular penicillin binding domains that were previously shown to bind fragments of peptidoglycan, likely generated by cell wall damage or remodeling, and an intracellular serine/threonine kinase domain similar to those found in eukaryotic cells (13). While the substrates and function of the PASTA kinases are incompletely defined, they appear to have various functions in different organisms, ranging from playing a role in biofilm formation (Streptococcus mutans) to being essential in some organisms (M. tuberculosis) (14, 15). As such, pharmacologic inhibition of PASTA kinases has been preliminarily pursued as a novel antimicrobial therapeutic strategy against the M. tuberculosis kinase PknB (16–18). Deletion of Stk1, the PASTA kinase in S. aureus, reverses the methicillin-resistant phenotype in MRSA (19, 20). In addition, deletion of the PASTA kinase in E. faecalis, PrkC, led to a >100-fold sensitization to certain β-lactam antibiotics (21).
Here, we test the hypothesis that pharmacologic inhibition of the PASTA kinase leads to increased β-lactam susceptibility in the Gram-positive pathogen L. monocytogenes. Using a conditional knockout strategy, we demonstrate that the PASTA kinase in L. monocytogenes (PrkA) is essential for resistance to β-lactam antibiotics. We demonstrate that L. monocytogenes is resistant to treatment with the nonspecific kinase inhibitor staurosporine but that combination therapy with β-lactam antibiotics and staurosporine leads to an ∼100-fold increase in susceptibility to the β-lactam antibiotic. Importantly, the synergistic effect was observed only with β-lactams and not with other cell-wall-acting antibiotics such as vancomycin or non-cell-wall-active antibiotics such as kanamycin. We furthermore show that staurosporine inhibits autophosphorylation of the L. monocytogenes PASTA kinase as well as substrate-level phosphorylation, while the S. aureus kinase is resistant to staurosporine treatment. Finally, we demonstrate that inhibition of PrkA using a more selective kinase inhibitor, AZD5438, similarly results in sensitization of L. monocytogenes to β-lactam antibiotics. Taken together, this work suggests that pharmacologic inhibition of PASTA kinases, in combination with β-lactam treatment, is a novel and viable antibiotic development strategy.
MATERIALS AND METHODS
Antibiotics.
Ampicillin (AMP), ceftriaxone (CRO), cephalexin (LEX), and vancomycin (VAN) were purchased from Sigma-Aldrich (St. Louis, MO) and resuspended according to the manufacturer's protocols. Kanamycin (KAN) was purchased from Fisher Scientific (Waltham, MA) and resuspended according to the manufacturer's protocols.
Bacterial strains and growth.
All L. monocytogenes strains used and generated in this study were derived from the 10403s background. Conditional deletion of prkA was achieved by first placing the gene under the control of a theophylline-controlled riboswitch (22). Briefly, promoterless prkA was amplified and fused to a T5 promoter and theophylline riboswitch E (22), using splice overlap extension (SOE) PCR (23) (MLR50 to MLR53) (Table 1). The SOE product was then ligated into an erythromycin-resistant derivative of the phage integration vector pPL2 (24), facilitating single-copy, theophylline-inducible expression from the chromosome in L. monocytogenes to create strain prkAtheo. Subsequently, clean deletion of prkA was achieved in this strain background in the presence of theophylline through pKSV7-mediated allelic exchange, as previously described (BK38 to BK41) (Table 1) (25). Staphylococcus aureus strain USA300 LAC was used both as a source of PASTA kinase DNA for cloning as well as in antibiotic treatment assays. Escherichia coli strains XL-1Blue and Rosetta BL21 were used for subcloning and protein expression, respectively. When needed, erythromycin (Sigma-Aldrich) was used at a final concentration of 2 μg/ml, chloramphenicol (Sigma-Aldrich) was used at 10 μg/ml, and kanamycin (Sigma-Aldrich) was used at 20 μg/ml.
TABLE 1.
Primers used in this study
| Primer | Sequence (5′–3′)a | Reference |
|---|---|---|
| MLR50 (prkAribo A) | GGCCGGGCCCGGAAATCATAAAAAATTTATTTGC | This study |
| MLR51 (prkAribo B) | CTTAATCGCTTACCAATCATCATCTTGTTGTTACCTCCTTAGCA | This study |
| MLR52 (prkAribo C) | TGCTAAGGAGGTAACAACAAGATGATGATTGGTAAGCGATTAAG | This study |
| MLR53 (prkAribo D) | GGCCCTCGAGTAATTTGGATAAGGGACTGTAC | This study |
| BK38 (prkA KO A) | ATATTATCTAGAGTACCATTGACAAGGAAGAAAATGAAACG | This study |
| BK39 (prkA KO B) | GCACATTTCCTCCGTTCTATTTTTAATTTGGAATCATCATGAAGCATCCCTCCCTTTCTG | This study |
| BK40 (prkA KO C) | CAGAAAGGGAGGGATGCTTCATGATGATTCCAAATTAAAAATAGAACGGAGGAAATGTGC | This study |
| BK41 (prkA KO D) | TATAATTCTAGAACGTCAATATGGATGTAATCTGCACCG | This study |
| JDS50 (PrkA KD F) | ATATTATGGATCCATGATGATTGGTAAGCGATTAAGCGATCGAT | This study |
| JDS54 (PrkA KD R) | ATTATACAATTGTTTCTTTTTCTTGCTCATTTTTTTCTTTTTCTTATCTTTTTTCTC | This study |
| JDS52 (Lmo0618 KD F) | ATATTATGGATCCATGGGAGAAATGACACTTGCTTTTATAGAAGAACA | This study |
| JDS55 (Lmo0618 KD R) | ATTATACAATTGGCCCTCTGTTGGTGGGCTGAAT | This study |
| SA-STPK-F (Stk1 KD F) | TAGGATCCATGATAGGTAAAATAATAAATGAAC | This study |
| SA-STPK-R (Stk1 KD R) | TATAGAATTCTTATCGTGTTGATTTCTTTTTAGGTTTTG | This study |
Boldface type indicates restriction sites.
Broth growth curves.
For in vitro growth experiments, L. monocytogenes strains were grown in brain heart infusion (BHI) medium at 30°C overnight to stationary phase without shaking. Complementation strains were grown in BHI medium supplemented with 2 mM theophylline at 30°C overnight without shaking to stationary phase. Methicillin-resistant S. aureus (strain USA300) was grown in tryptic soy broth (TSB) at 37°C overnight with gyratory shaking (250 rpm) to stationary phase. Stationary-phase cultures grown overnight were back-diluted 1:50 (L. monocytogenes) or 1:100 (S. aureus). Growth was measured at an optical density at 600 nm (OD600) at 15-min intervals over the course of 12 h in a 96-well plate format using an Eon microplate spectrophotometer or Synergy HT microplate spectrophotometer (BioTek Instruments, Inc., Winooski, VT). All growth experiments were repeated at least three times. For all in vitro growth assays, staurosporine (Calbiochem, Billerica, MA) was used at a final concentration of 10 μM, AZD5438 (Selleck Chemicals, Houston, TX) was used at 50 μM, and antibiotics were used at the concentrations specified in the figure legends.
For minimal medium experiments, cultures were grown in BHI medium at 30°C overnight to stationary phase without shaking. Cultures were washed three times in phosphate-buffered saline (PBS) and back-diluted 1:50 into improved minimal medium with glucose as the primary carbon source (26).
Lmo0618, PrkA, and SaStk1 protein expression and purification.
Using L. monocytogenes or S. aureus genomic DNA as the template, prkA, lmo0618, and S. aureus stk1 (Sastk1) kinase domains were amplified (JDS50–SA-STPK-R) (Table 1) and ligated into the expression vector pGEX-2T to construct a glutathione S-transferase (GST) fusion protein. The plasmids were transformed into E. coli Rosetta BL21 cells, and protein expression was analyzed by SDS-PAGE. The bacteria were pelleted by centrifugation, resuspended in 15 ml of lysis buffer (1× PBS, 1% Triton X-100, 2 μg/ml aprotinin, 1 μg/ml leupeptin, 25 μg/ml phenylmethylsulfonyl fluoride [PMSF]), and lysed with a cell disruptor (Branson, Danbury, CT). Cell debris was pelleted by centrifugation for 15 min. The supernatant was incubated with a slurry of glutathione-Sepharose 4B beads (GE Healthcare Life Sciences, Pittsburgh, PA) and 1× PBS (50:50, vol/vol) for 1 h at 4°C with gentle agitation. Following incubation, beads were pelleted by centrifugation, washed with 5 ml cold 1× PBS, and resuspended in 5 ml 1× PBS. The bound protein was eluted by using disposable chromatography columns (Thermo Scientific, Rockford, IL) and an elution buffer containing 50 mM Tris (pH 8.0) and 20 mM reduced glutathione. The fractions were assessed for purity via SDS-PAGE, fractions with an estimated >95% purity were concentrated via centrifugation, and glutathione was removed and exchanged with 1× PBS by using a buffer exchange unit (Amicon, Billerica, MA).
In vitro protein phosphorylation.
Phosphorylation assays were performed by mixing 1 μg of PrkA, Lmo0618, or SaStk1 in a 10-μl reaction mixture containing 50 mM Tris-HCl (pH 7.4), 1 mM dithiothreitol (DTT), 5 mM MnCl2, 250 μM ATP, and 1 μCi of [γ-32P]ATP, followed by incubation at room temperature overnight. To investigate substrate-level phosphorylation, ∼10 μg of myelin basic protein (MBP; Novatein Biosciences, Woburn, MA) was added to the reaction mixture described above and incubated at room temperature overnight. The reactions were terminated by the addition of 5× SDS loading buffer to the mixture. Samples were fractionated by SDS-PAGE, fixed, dried, and analyzed by autoradiography. Quantification was performed by using the Typhoon imager (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) and ImageQuant software using local average background subtraction.
Confocal microscopy.
Cultures of wild-type L. monocytogenes grown overnight were back-diluted and grown for 6 h in the presence or absence of antibiotic and/or staurosporine. Following 6 h of growth, bacteria were harvested, fixed in 4% paraformaldehyde, and washed three times in PBS, and wet mounts of each strain were imaged. Single z-stack fluorescence images were taken by laser scanning confocal microscopy (Fluoview FV-1000; Olympus, Center Valley, PA) using a PLAPO correction 1.45-numerical-aperture (NA)/60× lens with an additional 6× optical zoom. During image acquisition, fluorescent channel (488 nm) and differential interference contrast (DIC) images were acquired by one-way sequential line scanning using a 105-μm pinhole.
RESULTS
L. monocytogenes prkA mutants are hypersusceptible to β-lactam antibiotics.
In many Gram-positive pathogens, deletion of the PASTA kinase leads to hypersusceptibility to β-lactam antibiotics (19–21). Repeated attempts to delete the homologous protein in L. monocytogenes were unsuccessful (Daniel Portnoy and Michelle Reniere, personal communication). Therefore, we adapted, for the first time, a theophylline-inducible riboswitch system for use in L. monocytogenes for the purpose of creating a conditional prkA deletion mutant (22). Construction of the prkA deletion mutant resulted in the expected ratios of wild-type to mutant recombinant bacteria only when theophylline was present during the allelic exchange process. As predicted, compared to wild-type L. monocytogenes, ΔprkA mutants were hypersusceptible to a variety of β-lactam antibiotics while showing little to no increased susceptibility to cell-wall-acting non-β-lactams or non-cell-wall-acting antibiotics (Fig. 1, black versus red lines). Susceptibility to β-lactam antibiotics was rescued by the addition of 2 mM theophylline to induce translation of prkA expressed in trans (Fig. 1, blue lines). Taken together, these data suggest that, similar to what has been observed for other Gram-positive organisms, the L. monocytogenes ΔprkA mutants are selectively susceptible to β-lactam antibiotics.
FIG 1.

ΔprkA mutants are hypersusceptible to β-lactam antibiotics. Cultures of wild-type (wt), Δprk, or complemented ΔprkA L. monocytogenes bacteria grown overnight were back-diluted and treated with 10-fold serial dilutions of ampicillin (A), ceftriaxone (B), cephalexin (C) vancomycin (D), or kanamycin (E). Antibiotic concentrations are in μg/ml. Growth was analyzed for 12 h at 15-min intervals. Data are representative of at least 3 independent repeat experiments.
Staurosporine sensitizes L. monocytogenes to β-lactam antibiotics.
Deletion of the PASTA kinase in L. monocytogenes (Fig. 1), S. aureus, or E. faecalis leads to an increased susceptibility to β-lactam antibiotics (19–21). To test the hypothesis that pharmacologic inhibition of bacterial serine/threonine kinases could result in a synergistic sensitization to antibiotics, we incubated L. monocytogenes with or without the nonselective kinase inhibitor staurosporine in the presence of various antibiotics. Staurosporine treatment alone had a minimal effect on L. monocytogenes growth (Fig. 2A to E; see also Fig. S1 in the supplemental material). Similarly, subinhibitory concentrations of antibiotic had no effect on L. monocytogenes growth (Fig. 2A to E). However, treatment of L. monocytogenes with subinhibitory concentrations of β-lactam antibiotics (ampicillin, cephalexin, and ceftriaxone) in the presence of 10 μM staurosporine led to a 10- to 100-fold increase in susceptibility (Fig. 2A to C, black lines versus red lines) and significant chaining of combination-treated bacteria (see Fig. S1 in the supplemental material). Importantly, as was previously shown for S. aureus and E. faecalis kinase deletion mutants, susceptibility to other cell-wall-acting antibiotics such as vancomycin, or ribosome inhibitors such as kanamycin, was unaffected (Fig. 2D and E). Additionally, similar to the deletion of PrkC in E. faecalis, treatment of wild-type L. monocytogenes with staurosporine resulted in a minor growth defect in chemically defined, minimal media but not in rich media (see Fig. S2 in the supplemental material) (21). Finally, while staurosporine had potent effects on the susceptibility of L. monocytogenes to β-lactam antibiotics, S. aureus susceptibility to ceftriaxone or other β-lactam or non-β-lactam antibiotics was unaffected (Fig. 2F and data not shown). Taken together, these data suggest that pharmacologic kinase inhibition by staurosporine in L. monocytogenes specifically sensitizes bacteria to β-lactam antibiotics.
FIG 2.

Staurosporine sensitizes L. monocytogenes to β-lactam antibiotics. (A to E) Cultures of wild-type L. monocytogenes grown overnight were back-diluted and treated with 10-fold serial dilutions of ampicillin (A), ceftriaxone (B), cephalexin (C), vancomycin (D), or kanamycin (E) in the presence or absence of 10 μM staurosporine (str). (F) Cultures of S. aureus grown overnight were back-diluted and treated with ceftriaxone in the presence or absence of 10 μM staurosporine. Antibiotic concentrations are in μg/ml. Growth was analyzed for 12 h at 15-min intervals. Data are representative of at least 3 independent repeat experiments.
Staurosporine selectively prevents L. monocytogenes PrkA phosphorylation.
L. monocytogenes, like many Gram-positive pathogens, encodes two predicted serine/threonine kinases, one with an extracellular PASTA domain (PrkA) and one without (Lmo0618) (27). To determine if staurosporine, a broad-spectrum kinase inhibitor (28), can selectively inhibit either or both of these kinases, the catalytic domains of both kinases were cloned into a GST bacterial expression vector, purified, and assayed for activity in the presence or absence of increasing concentrations of staurosporine. Both PrkA as well as Lmo0618 have kinase activity, as evidenced by both autophosphorylation as well as phosphorylation of the nonspecific substrate myelin basic protein (MBP) (Fig. 3). However, only PrkA, and not Lmo0618, was inhibited by staurosporine at the levels of both autophosphorylation as well as substrate phosphorylation in a staurosporine concentration-dependent manner. As predicted by a staurosporine dose-dependent effect on PrkA, inhibition of growth at a fixed ceftriaxone concentration (1.25 μg/ml) was also staurosporine dose dependent, demonstrating at least minor growth inhibition at doses of staurosporine as low as 1.25 μM (see Fig. S3 in the supplemental material). Consistent with what we observed in antibiotic sensitization assays, the purified PASTA kinase from S. aureus was also resistant to staurosporine treatment. Taken together, these data suggest that staurosporine specifically inhibits PrkA, the PASTA kinase in L. monocytogenes, leading to sensitization to β-lactam antibiotics.
FIG 3.
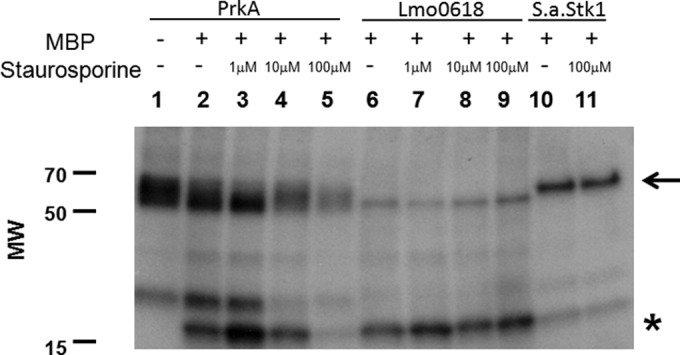
Staurosporine inhibits in vitro PrkA phosphorylation in a dose-dependent manner. Autophosphorylation (arrow) and myelin basic protein (MBP) phosphorylation (asterisk) activities were assayed for PrkA (lanes 1 to 5), Lmo0618 (lanes 6 to 9), and SaStk1 (lanes 10 and 11) in the presence or absence of 1 μM, 10 μM, or 100 μM staurosporine. MW, molecular weight (in thousands).
Staurosporine activity is dependent on the presence of PrkA.
Biochemical phosphorylation assays suggested that staurosporine acts on PrkA but not on Lmo0618, the non-PASTA-containing kinase. To test the hypothesis that the activity of staurosporine is dependent on PrkA inhibition, we tested whether or not ΔprkA mutants demonstrate increased sensitivity to β-lactam antibiotics in the presence of staurosporine. As previously demonstrated, ΔprkA mutants are hypersusceptible to β-lactam antibiotics (Fig. 4A, red lines); however, whereas wild-type L. monocytogenes became at least 10-fold more susceptible to ceftriaxone in the presence of 5 μM staurosporine, the sensitivity of ΔprkA mutants did not change appreciably in the presence of staurosporine (Fig. 4B, red lines). These data, taken together with the biochemical inhibition data, further suggest that staurosporine sensitizes wild-type L. monocytogenes to β-lactam antibiotics through the inhibition of the PASTA kinase PrkA.
FIG 4.
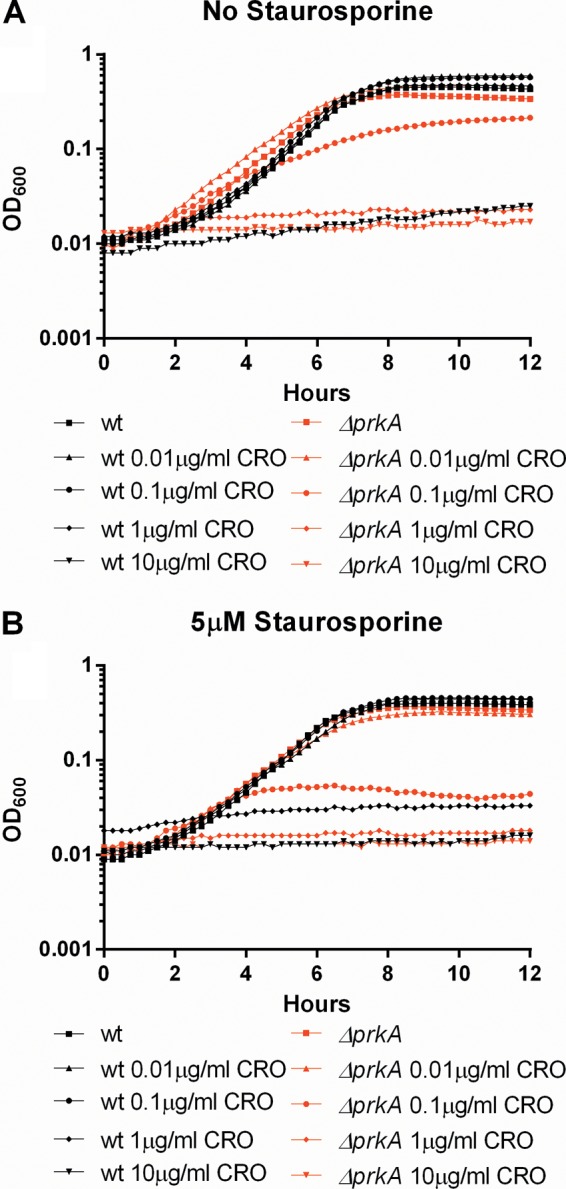
The activity of staurosporine is dependent on PrkA. (A) Cultures of wild-type or ΔprkA L. monocytogenes bacteria grown overnight were back-diluted and treated with 10-fold serial dilutions of ceftriaxone. (B) Cultures of wild-type or ΔprkA L. monocytogenes bacteria grown overnight were back-diluted and treated with 10-fold serial dilutions of ceftriaxone in the presence of 5 μM staurosporine. Antibiotic concentrations are in μg/ml. Growth was analyzed for 12 h at 15-min intervals. Data are representative of at least 3 independent repeat experiments.
AZD5438 sensitizes L. monocytogenes to β-lactam treatment via inhibition of PrkA.
Although staurosporine shows specificity for L. monocytogenes PrkA relative to Lmo0618 or SaStk1, it has no therapeutic potential due to its nonselective nature and its promiscuity as an inhibitor of eukaryotic kinases. To determine if more selective kinase inhibitors could function to sensitize L. monocytogenes to β-lactam antibiotics, we screened a small library of kinase inhibitors for β-lactam-dependent inhibition of L. monocytogenes growth. We identified one compound, AZD5438 (a cyclin-dependent kinase [CDK] inhibitor [29]), capable of inhibiting L. monocytogenes growth in a β-lactam-synergism-dependent manner (Fig. 5A). Furthermore, AZD5438 acted synergistically with β-lactam antibiotics against L. monocytogenes but not S. aureus (data not shown). To determine if the mechanism of action is similar to that of staurosporine, we analyzed the ability of AZD5438 to inhibit PrkA and Lmo0618 activities in a biochemical phosphorylation assay. Similarly to staurosporine, we observed that AZD5438 specifically inhibits PrkA while not inhibiting Lmo0618 (Fig. 5B). A higher-resolution dose-response curve shows both autophosphorylation and substrate phosphorylation being inhibited in a dose-dependent manner in parallel (Fig. 5C). Taken together, these data suggest that both nonselective (staurosporine) and specific (AZD5438) kinase inhibitors can synergistically act to sensitize L. monocytogenes but not MRSA to β-lactam antibiotic treatment in a manner dependent upon the bacterium's PASTA kinase.
FIG 5.

The CDK inhibitor AZD5438 also sensitizes L. monocytogenes to β-lactam treatment through inhibition of PrkA. (A) Cultures of wild-type L. monocytogenes grown overnight were back-diluted and treated with 10-fold serial dilutions of ceftriaxone in the presence or absence of 50 μM AZD5438. Antibiotic concentrations are in μg/ml. Growth was analyzed for 12 h at 15-min intervals. (B) Autophosphorylation (arrow) and myelin basic protein (MBP) phosphorylation (asterisk) activities were assayed for PrkA (lanes 1 to 5) and Lmo0618 (lanes 6 to 9) in the presence or absence of 1 μM, 10 μM, or 100 μM AZD5438. (C) Autophosphorylation (arrow) and MBP phosphorylation (asterisk) activities were assayed for PrkA in the presence or absence of 2-fold serial dilutions of AZD5438. Quantification was performed by using a Typhoon imager and ImageQuant software. Data are representative of at least 3 independent repeat experiments. A.U., arbitrary units.
DISCUSSION
The 518 human kinases share common structural features, with ∼30% similarity across the ∼250-amino-acid catalytic domain (30). Despite this structural similarity, pharmacologic selectivity has been achieved and resulted in the widespread use of kinase inhibitors for cancer as well as other conditions (31). Until recently, prokaryotic phosphorylation was thought to be mediated largely by kinases specific to histidine and aspartyl residues (so-called two-component regulators) (32, 33). These histidine kinases have very little sequence homology to eukaryotic kinases (34). Histidine kinases typically phosphorylate a single target, the second component of the two-component signaling system, which is usually a DNA binding response regulator (35). However, it is now clear that eukaryote-like serine/threonine kinases (eSTKs) frequently occur in both Gram-positive and -negative prokaryotes (13). The PASTA kinases, however, appear to be specific to Gram-positive bacteria (Firmicutes and Actinobacteria) (13). Similar to eukaryotic serine/threonine kinases, bacterial eSTKs have many targets, and in L. monocytogenes, PrkA has already been shown to phosphorylate or interact with 62 unique substrates (36). These substrates imply a role for PrkA in carbohydrate metabolism, protein synthesis, cell wall synthesis, and division. Indeed, pharmacologic inhibition of PrkA in L. monocytogenes leads to chaining and septation defects in the presence of subinhibitory concentrations of β-lactam antibiotics (see Fig. S1 in the supplemental material). While most bacteria have only a few eSTKs (four or less) Streptomycetes and mycobacterial genomes can have 10 or more (13). The PASTA kinase in M. tuberculosis, PknB, is essential; however, in other pathogens such as S. aureus, the PASTA kinase (Stk1) is not an essential gene, and deletion mutants have only minor defects in the absence of specific stresses such as β-lactam antibiotics. These phenotypes demonstrate that while there are clearly some shared functions, there are likely to be species-specific differences as well (14, 19, 37). The specific targets downstream of PrkA that mediate resistance to β-lactam antibiotics are a current area of research in the laboratory.
Staurosporine is a relatively nonselective kinase inhibitor (28). Despite the broad activity of staurosporine against human kinases, and the similarity of bacterial eSTKs to human kinases, we demonstrated selective inhibition of some (PrkA) but not other (Lmo0618 and Stk1) bacterial kinases. Others have reported that staurosporine can inhibit the PASTA kinases of Staphylococcus epidermidis (38) and E. faecalis (39) in biochemical assays at high concentrations. Importantly, although staurosporine is a nonselective kinase inhibitor, it does not increase the β-lactam susceptibility of MRSA (Fig. 2F) or directly inhibit Stk1 (Fig. 3), suggesting that selectivity of bacterial kinase inhibition is possible. Furthermore, we observed additional evidence of kinase-specific selectivity with a different inhibitor, AZD5438. While AZD5438 is not as potent of an inhibitor of PrkA as it is of the human kinases that it was optimized to inhibit (CDK-2) (29), AZD5438 does inhibit PrkA at concentrations at which no effect is seen on Lmo0618. The specific residues that confer inhibitor specificity to bacterial eSTKs remain to be defined. These differences are likely in the ATP binding pocket of the kinase domain and unrelated to the presence, absence, or number of repeats of the extracellular penicillin binding domain of the PASTA kinases. Understanding how inhibitor specificity is conferred is an important step in the rational design of inhibitors that will have specificity for bacterial PASTA kinases while avoiding nonspecific inhibition of host kinases.
The penicillin binding domain of the PASTA kinases likely acts as a receptor for peptidoglycan fragments generated through cell wall damage or remodeling. For M. tuberculosis, it was demonstrated that there was specificity for the second and third residues of the stem peptide as well as the presence of an N-acetylmuramic acid sugar moiety of peptidoglycan fragments to facilitate binding and signaling through the PASTA domain of PknB (40). Presumably, binding of cell wall fragments transmits a signal to the kinase through a conformational change that allows the regulation of substrates involved in cell wall remodeling and homeostasis. In the case of L. monocytogenes, these include cell shape-determining proteins (MreB) and the peptidoglycan synthesis proteins GlmU and MurG (36). For other organisms, the PASTA kinases have been suggested to regulate cell wall homeostasis through the phosphorylation of PBPs and autolysins (41, 42). An understanding of how cell wall damage is recognized by the PASTA domain and what specific responses this triggers will lead us to a mechanistic understanding of how the PASTA kinases work to maintain cell wall homeostasis and how this affects β-lactam susceptibility.
In summary, PrkA but not Lmo0618 is sensitive to staurosporine inhibition. The result of this selective inhibition is the sensitization of L. monocytogenes to cell wall stress, similar to the phenotype seen with PASTA kinase deletions in S. aureus and E. faecalis. While antibiotic resistance in the model pathogen L. monocytogenes is not a burgeoning issue, we believe that these data act as a proof of principle that specificity in the targeting of bacterial PASTA eSTKs is possible. Prior to our work, pharmacologic inhibitors that target the M. tuberculosis kinase PknB have also been identified, although their development as potential therapeutic agents is still a work in progress (16–18). Inhibitors of other bacterial PASTA kinases that work in a β-lactam-synergistic manner remain to be identified. However, our results suggest that the generation of kinase inhibitors with specificity for bacterial PASTA kinases is a potentially viable approach to the development of novel antimicrobials that will work in combination therapy with β-lactam antibiotics. Importantly, we propose that these inhibitors should work synergistically with β-lactam antibiotics independent of the resistance phenotype of the organisms that they target, as demonstrated by the resensitization of MRSA to β-lactams upon deletion of its homologous PASTA eSTK (19, 20). A full analysis of the PASTA kinase phosphorylation substrates will lead to a mechanistic understanding of how kinase inhibition leads to increased β-lactam susceptibility. In addition, a systematic understanding of the biochemical and biophysical interactions between kinase inhibitors and the PASTA kinases will facilitate the rational design of inhibitors with specificity for bacterial kinases and with limited cross-reactivity with host serine/threonine kinases.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by an individual biomedical research award from The Hartwell Foundation to R.S. and by an NIH grant (U54 AI57153) to J.-D.S.
Footnotes
Published ahead of print 27 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02396-14.
REFERENCES
- 1.Freitag NE, Port GC, Miner MD. 2009. Listeria monocytogenes—from saprophyte to intracellular pathogen. Nat. Rev. Microbiol. 7:623–628. 10.1038/nrmicro2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lecuit M. 2007. Human listeriosis and animal models. Microbes Infect. 9:1216–1225. 10.1016/j.micinf.2007.05.009 [DOI] [PubMed] [Google Scholar]
- 3.Schlech WF, III, Lavigne PM, Bortolussi RA, Allen AC, Haldane EV, Wort AJ, Hightower AW, Johnson SE, King SH, Nicholls ES, Broome CV. 1983. Epidemic listeriosis—evidence for transmission by food. N. Engl. J. Med. 308:203–206. 10.1056/NEJM198301273080407 [DOI] [PubMed] [Google Scholar]
- 4.Drevets DA, Bronze MS. 2008. Listeria monocytogenes: epidemiology, human disease, and mechanisms of brain invasion. FEMS Immunol. Med. Microbiol. 53:151–165. 10.1111/j.1574-695X.2008.00404.x [DOI] [PubMed] [Google Scholar]
- 5.Temple ME, Nahata MC. 2000. Treatment of listeriosis. Ann. Pharmacother. 34:656–661. 10.1345/aph.19315 [DOI] [PubMed] [Google Scholar]
- 6.McCollum JT, Cronquist AB, Silk BJ, Jackson KA, O'Connor KA, Cosgrove S, Gossack JP, Parachini SS, Jain NS, Ettestad P, Ibraheem M, Cantu V, Joshi M, DuVernoy T, Fogg NW, Jr, Gorny JR, Mogen KM, Spires C, Teitell P, Joseph LA, Tarr CL, Imanishi M, Neil KP, Tauxe RV, Mahon BE. 2013. Multistate outbreak of listeriosis associated with cantaloupe. N. Engl. J. Med. 369:944–953. 10.1056/NEJMoa1215837 [DOI] [PubMed] [Google Scholar]
- 7.Jones EM, MacGowan AP. 1995. Antimicrobial chemotherapy of human infection due to Listeria monocytogenes. Eur. J. Clin. Microbiol. Infect. Dis. 14:165–175. 10.1007/BF02310351 [DOI] [PubMed] [Google Scholar]
- 8.Lewis K. 2013. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 12:371–387. 10.1038/nrd3975 [DOI] [PubMed] [Google Scholar]
- 9.Sader HS, Jones RN. 2009. Antimicrobial susceptibility of Gram-positive bacteria isolated from US medical centers: results of the Daptomycin Surveillance Program (2007-2008). Diagn. Microbiol. Infect. Dis. 65:158–162. 10.1016/j.diagmicrobio.2009.06.016 [DOI] [PubMed] [Google Scholar]
- 10.Villegas-Estrada A, Lee M, Hesek D, Vakulenko SB, Mobashery S. 2008. Co-opting the cell wall in fighting methicillin-resistant Staphylococcus aureus: potent inhibition of PBP 2a by two anti-MRSA beta-lactam antibiotics. J. Am. Chem. Soc. 130:9212–9213. 10.1021/ja8029448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richter SS, Heilmann KP, Dohrn CL, Riahi F, Costello AJ, Kroeger JS, Biek D, Critchley IA, Diekema DJ, Doern GV. 2011. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob. Agents Chemother. 55:4154–4160. 10.1128/AAC.00315-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeats C, Finn RD, Bateman A. 2002. The PASTA domain: a beta-lactam-binding domain. Trends Biochem. Sci. 27:438. 10.1016/S0968-0004(02)02164-3 [DOI] [PubMed] [Google Scholar]
- 13.Pereira SF, Goss L, Dworkin J. 2011. Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol. Mol. Biol. Rev. 75:192–212. 10.1128/MMBR.00042-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandez P, Saint-Joanis B, Barilone N, Jackson M, Gicquel B, Cole ST, Alzari PM. 2006. The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J. Bacteriol. 188:7778–7784. 10.1128/JB.00963-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hussain H, Branny P, Allan E. 2006. A eukaryotic-type serine/threonine protein kinase is required for biofilm formation, genetic competence, and acid resistance in Streptococcus mutans. J. Bacteriol. 188:1628–1632. 10.1128/JB.188.4.1628-1632.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lougheed KE, Osborne SA, Saxty B, Whalley D, Chapman T, Bouloc N, Chugh J, Nott TJ, Patel D, Spivey VL, Kettleborough CA, Bryans JS, Taylor DL, Smerdon SJ, Buxton RS. 2011. Effective inhibitors of the essential kinase PknB and their potential as anti-mycobacterial agents. Tuberculosis (Edinb.) 91:277–286. 10.1016/j.tube.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seal A, Yogeeswari P, Sriram D, Consortium O, Wild DJ. 2013. Enhanced ranking of PknB inhibitors using data fusion methods. J. Cheminform. 5:2. 10.1186/1758-2946-5-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chapman TM, Bouloc N, Buxton RS, Chugh J, Lougheed KE, Osborne SA, Saxty B, Smerdon SJ, Taylor DL, Whalley D. 2012. Substituted aminopyrimidine protein kinase B (PknB) inhibitors show activity against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 22:3349–3353. 10.1016/j.bmcl.2012.02.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamber S, Schwartzman J, Cheung AL. 2010. Role of PknB kinase in antibiotic resistance and virulence in community-acquired methicillin-resistant Staphylococcus aureus strain USA300. Infect. Immun. 78:3637–3646. 10.1128/IAI.00296-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beltramini AM, Mukhopadhyay CD, Pancholi V. 2009. Modulation of cell wall structure and antimicrobial susceptibility by a Staphylococcus aureus eukaryote-like serine/threonine kinase and phosphatase. Infect. Immun. 77:1406–1416. 10.1128/IAI.01499-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kristich CJ, Wells CL, Dunny GM. 2007. A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. Proc. Natl. Acad. Sci. U. S. A. 104:3508–3513. 10.1073/pnas.0608742104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Topp S, Reynoso CM, Seeliger JC, Goldlust IS, Desai SK, Murat D, Shen A, Puri AW, Komeili A, Bertozzi CR, Scott JR, Gallivan JP. 2010. Synthetic riboswitches that induce gene expression in diverse bacterial species. Appl. Environ. Microbiol. 76:7881–7884. 10.1128/AEM.01537-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528–535 [PubMed] [Google Scholar]
- 24.Lauer P, Chow MY, Loessner MJ, Portnoy DA, Calendar R. 2002. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J. Bacteriol. 184:4177–4186. 10.1128/JB.184.15.4177-4186.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Camilli A, Tilney LG, Portnoy DA. 1993. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol. Microbiol. 8:143–157. 10.1111/j.1365-2958.1993.tb01211.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phan-Thanh L, Gormon T. 1997. A chemically defined minimal medium for the optimal culture of Listeria. Int. J. Food Microbiol. 35:91–95. 10.1016/S0168-1605(96)01205-6 [DOI] [PubMed] [Google Scholar]
- 27.Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, Baquero F, Berche P, Bloecker H, Brandt P, Chakraborty T, Charbit A, Chetouani F, Couve E, de Daruvar A, Dehoux P, Domann E, Dominguez-Bernal G, Duchaud E, Durant L, Dussurget O, Entian KD, Fsihi H, Garcia-del Portillo F, Garrido P, Gautier L, Goebel W, Gomez-Lopez N, Hain T, Hauf J, Jackson D, Jones LM, Kaerst U, Kreft J, Kuhn M, Kunst F, Kurapkat G, Madueno E, Maitournam A, Vicente JM, Ng E, Nedjari H, Nordsiek G, Novella S, de Pablos B, Perez-Diaz JC, Purcell R, Remmel B, Rose M, Schlueter T, Simoes N, Tierrez A, Vazquez-Boland JA, Voss H, Wehland J, Cossart P. 2001. Comparative genomics of Listeria species. Science 294:849–852. 10.1126/science.1063447 [DOI] [PubMed] [Google Scholar]
- 28.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. 2008. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26:127–132. 10.1038/nbt1358 [DOI] [PubMed] [Google Scholar]
- 29.Byth KF, Thomas A, Hughes G, Forder C, McGregor A, Geh C, Oakes S, Green C, Walker M, Newcombe N, Green S, Growcott J, Barker A, Wilkinson RW. 2009. AZD5438, a potent oral inhibitor of cyclin-dependent kinases 1, 2, and 9, leads to pharmacodynamic changes and potent antitumor effects in human tumor xenografts. Mol. Cancer Ther. 8:1856–1866. 10.1158/1535-7163.MCT-08-0836 [DOI] [PubMed] [Google Scholar]
- 30.Brooijmans N, Chang YW, Mobilio D, Denny RA, Humblet C. 2010. An enriched structural kinase database to enable kinome-wide structure-based analyses and drug discovery. Protein Sci. 19:763–774. 10.1002/pro.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chahrour O, Cairns D, Omran Z. 2012. Small molecule kinase inhibitors as anti-cancer therapeutics. Mini Rev. Med. Chem. 12:399–411. 10.2174/138955712800493915 [DOI] [PubMed] [Google Scholar]
- 32.Bakal CJ, Davies JE. 2000. No longer an exclusive club: eukaryotic signalling domains in bacteria. Trends Cell Biol. 10:32–38. 10.1016/S0962-8924(99)01681-5 [DOI] [PubMed] [Google Scholar]
- 33.Munoz-Dorado J, Inouye S, Inouye M. 1991. A gene encoding a protein serine/threonine kinase is required for normal development of M. xanthus, a gram-negative bacterium. Cell 67:995–1006. 10.1016/0092-8674(91)90372-6 [DOI] [PubMed] [Google Scholar]
- 34.Hanks SK, Hunter T. 1995. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 9:576–596 [PubMed] [Google Scholar]
- 35.Laub MT, Goulian M. 2007. Specificity in two-component signal transduction pathways. Annu. Rev. Genet. 41:121–145. 10.1146/annurev.genet.41.042007.170548 [DOI] [PubMed] [Google Scholar]
- 36.Lima A, Duran R, Schujman GE, Marchissio MJ, Portela MM, Obal G, Pritsch O, de Mendoza D, Cervenansky C. 2011. Serine/threonine protein kinase PrkA of the human pathogen Listeria monocytogenes: biochemical characterization and identification of interacting partners through proteomic approaches. J. Proteomics 74:1720–1734. 10.1016/j.jprot.2011.03.005 [DOI] [PubMed] [Google Scholar]
- 37.Debarbouille M, Dramsi S, Dussurget O, Nahori MA, Vaganay E, Jouvion G, Cozzone A, Msadek T, Duclos B. 2009. Characterization of a serine/threonine kinase involved in virulence of Staphylococcus aureus. J. Bacteriol. 191:4070–4081. 10.1128/JB.01813-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Q, Fan J, Niu C, Wang D, Wang J, Wang X, Villaruz AE, Li M, Otto M, Gao Q. 2011. The eukaryotic-type serine/threonine protein kinase Stk is required for biofilm formation and virulence in Staphylococcus epidermidis. PLoS One 6:e25380. 10.1371/journal.pone.0025380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hall CL, Tschannen M, Worthey EA, Kristich CJ. 2013. IreB, a Ser/Thr kinase substrate, influences antimicrobial resistance in Enterococcus faecalis. Antimicrob. Agents Chemother. 57:6179–6186. 10.1128/AAC.01472-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mir M, Asong J, Li X, Cardot J, Boons GJ, Husson RN. 2011. The extracytoplasmic domain of the Mycobacterium tuberculosis Ser/Thr kinase PknB binds specific muropeptides and is required for PknB localization. PLoS Pathog. 7:e1002182. 10.1371/journal.ppat.1002182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shah IM, Dworkin J. 2010. Induction and regulation of a secreted peptidoglycan hydrolase by a membrane Ser/Thr kinase that detects muropeptides. Mol. Microbiol. 75:1232–1243. 10.1111/j.1365-2958.2010.07046.x [DOI] [PubMed] [Google Scholar]
- 42.Dasgupta A, Datta P, Kundu M, Basu J. 2006. The serine/threonine kinase PknB of Mycobacterium tuberculosis phosphorylates PBPA, a penicillin-binding protein required for cell division. Microbiology 152:493–504. 10.1099/mic.0.28630-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.