

Abstract
The emergence of drug-resistant herpesviruses represents a significant problem in clinical practice, primarily in immunocompromised patients. Furthermore, effective antiviral therapies against gammaherpesvirus-associated diseases are lacking. Here, we present two thiothymidine derivatives, KAY-2-41 and KAH-39-149, with different spectra of antiviral activity from those of the reference antiherpetic drugs, showing inhibitory activities against herpes simplex virus, varicella-zoster virus (VZV), and particularly against Epstein-Barr virus, with high selectivity in vitro. While KAY-2-41- and KAH-39-149-resistant herpesviruses were found to harbor mutations in the viral thymidine kinase (TK), these mutations conferred only low levels of resistance to these drugs but high levels to other TK-dependent drugs. Also, antiviral assays in HeLa TK-deficient cells showed a lack of KAY-2-41 and KAH-39-149 activities against herpes simplex virus 1 (HSV-1) and HSV-2 TK-deficient mutants. Furthermore, enzymatic TK assays showed the ability of HSV-1 TK, VZV TK, and cellular TK1 and TK2 to recognize and phosphorylate KAY-2-41 and KAH-39-149. These results demonstrate that the compounds depend on both viral and host TKs to exert antiviral activity. Additionally, the antiviral efficacy of KAH-39-149 proved to be superior to that of KAY-2-41 in a mouse model of gammaherpesvirus infection, highlighting the potential of this class of antiviral agents for further development as selective therapeutics against Epstein-Barr virus.
INTRODUCTION
Among the diseases associated with Epstein-Barr virus (EBV), one of the more serious complications in transplant patients is posttransplant lymphoproliferative disorder (PTLD) (1). The current treatment modalities for PTLD and other EBV-associated malignancies include conventional cancer therapies, such as chemotherapy, but also involve anti-CD20 monoclonal antibody (rituximab) therapy and adoptive transfer of EBV-specific cytotoxic T lymphocytes (2). The administration of antiviral agents, mainly acyclovir (ACV) and ganciclovir (GCV), alone or in combination with these conventional cancer therapies has been used for prophylaxis (as they can reduce the risk of developing this disease) and treatment for PTLD (1, 3–6). Nevertheless, antiviral treatment has been shown to be moderately effective against EBV-associated infections in the lytic phase (7). Novel targeted therapies for EBV-related lymphomas are emerging and they are focused on (i) the activation of lytic viral genes to render the tumor cells susceptible to antiviral therapies, (ii) the inhibition of downstream cell cycle or antiapoptotic pathways that may be activated by latent EBV proteins, and (iii) the development of an EBV vaccine (8).
Mechanism-based inhibitors are currently not available for EBV (a gammaherpesvirus), in comparison with current antiviral options (i.e., inhibitors of viral DNA replication) for alpha- and betaherpesviruses. ACV is highly effective for the treatment of herpes simplex virus (HSV) infections, but selective pressure of the drug increases the frequency of ACV resistance, particularly in immunocompromised patients (9). In immunocompetent individuals with mucocutaneous disease, long-term therapy with ACV has not been associated with the development of drug-resistant HSV, but the high ACV resistance rates in patients with recurrent herpetic keratitis raise serious concerns (9–14). Other currently approved drugs (i.e., foscarnet and cidofovir) are active against most (cidofovir), but not all (foscarnet), ACV-resistant mutants, yet they have major limitations in terms of toxicity and pharmacokinetic liability (14). As a consequence, the emergence of drug-resistant HSV strains stresses the need for novel antiviral drugs.
Thionucleosides, such as 4′-thiothymidine, were previously explored for their potential in antiviral treatment, although their antiviral activities were masked by severe cytotoxicity (15, 16). Among the several derivatives of 4′-thiothymidine that were subsequently developed (15–19), 5-iodo-4′-thio-2′-deoxyuridine (4′-thioIDU) exhibited good activity against many of the herpesviruses (18). The mechanism of action of 4′-thioIDU was shown to be similar to that described for IDU in that it is preferentially phosphorylated by the thymidine kinase (TK) of vaccinia virus and HSV, and the triphosphate metabolite is a substrate of the DNA polymerases in both viruses (18, 20). Another 4′-thiothymidine analog is 1′-methyl-substituted 4′-thiothymidine or KAY-2-41, which was proven to be a strong inhibitor of orthopoxviruses (21) but also exerted activity against HSV-1 in vitro (17). The 4′-azido analog of 4′-thiothymidine, designated KAH-39-149, exhibited promising antiviral activity against human immunodeficiency virus, but its activity against herpesviruses was not evaluated (22).
In this study, we determined the sensitivities of alpha-, beta-, and gammaherpesviruses to KAY-2-41 and KAH-39-149. Other gammaherpesviruses, including the murine gammaherpesvirus 68 (MHV-68), herpesvirus saimiri (HVS), and rhesus rhadinovirus (RRV), were also included in these studies as surrogate viruses for EBV and Kaposi's sarcoma-associated herpesvirus (KSHV). With the aim of better understanding the modes of action of these two novel antivirals, drug-resistant viruses were selected in vitro, and enzymatic TK assays were performed. KAY-2-41 and KAH-39-149 treatment of mice infected with MHV-68 enabled us to assess their inhibitory effects in vivo.
MATERIALS AND METHODS
Cells and viruses.
Murine fibroblasts (NIH 3T3 cells; ATCC CRL-1685), owl monkey kidney cells (OMK) (ATCC CRL-1556), primary rhesus monkey fibroblasts (RF) (kindly provided by Scott Wong, Oregon Health and Science University, Beaverton, OR, USA), human embryonic lung (HEL) fibroblasts (ATCC CCL-137), BCBL-1 (NIH AIDS Research & Reference Reagent Program), and P3HR-1 cells (ATCC HTB-62) were grown as previously described (23). HeLa TK− cells (thymidine kinase-deficient human epithelial cell lines derived from a cervical carcinoma) were kindly provided by Y. C. Cheng, Yale University School of Medicine, New Haven, CT, USA, and grown in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Merelbeke, Belgium) containing 10% heat-inactivated fetal calf serum (FCS), 2 mM l-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, and 10 mM HEPES. MHV-68 (clone G2.4; kindly provided by A. A. Nash, Edinburgh, United Kingdom), HVS (strain C-488; ATCC VR-1414), and RRV (strain 17577; kindly provided by Scott Wong) were replicated in NIH 3T3, OMK, and RF cells, respectively. The following viruses were replicated in HEL cells: HSV-1 (KOS strain, ATCC VR-1493), HSV-2 (G strain, ATCC VR-734), varicella-zoster virus (VZV) strain Oka (ATCC VR-795), and two human cytomegalovirus (HCMV) strains, AD-169 and Davis (ATCC VR-538 and ATCC VR-807, respectively).
Viruses bearing mutations in the viral TK and that were previously selected in our laboratory were also used for antiviral testing: HSV-1 TK-deficient (TK−) KOS strain bearing a G insertion at nucleotides 430 to 436 (24), VZV TK− strain 07-1, the MHV-68 TK T364P mutant (G2.4 clone) (23), and the HVS TK R45stop mutant (C488 strain) (23).
Compounds.
KAY-2-41 and KAH-39-149 were synthesized and characterized at Showa University, Tokyo, Japan. Their chemical structures are shown in Fig. 1. The sources of the compounds used in the antiviral assays were as follows: HDVD (1-[(2S,4S-2-(hydroxymethyl)-1,3-dioxolan-4-yl]5-vinylpyrimidine-2,4(1H,3H)-dione), synthesized at the University of Georgia College of Pharmacy, Athens, GA, USA; ACV (9-(2-hydroxyethoxymethyl)guanine), GlaxoSmithKline, Stevenage, United Kingdom; brivudin (BVDU) [(E)-5-(2-bromovinyl)-1-β-d-2′-deoxyribofuranos-1-yl-uracil], Searle, United Kingdom; GCV [9-(1,3-dihydroxy-2-propoxymethyl)guanine], Roche, Basel, Switzerland; and PFA (foscarnet, phosphonoformate sodium salt), Sigma Chemicals, St. Louis, MO; HPMPC [(S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine], and adefovir (PMEA) (9-[2-(phosphonylmethoxyethyl)adenine]), Gilead Sciences, Foster City, CA; and HPMP-5-azaC (1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine), HPMPA ((S)-9-[3-hydroxy-2-(phosphonomethoxy)propyl]adenine), HPMPO-DAPy [(R)-(2,4-diamino-3-hydroxy-6-[2-(phosphonomethoxy)propoxy])pyrimidine], PMEDAP (9-[2-(phosphonomethoxy)ethyl]-2,6-diaminopurine), and PMEO-DAPy (2,4-diamino-6-[2-(phosphonomethoxy)ethoxy]pyrimidine), Marcela Krecmerova, Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Prague, Czech Republic.
FIG 1.
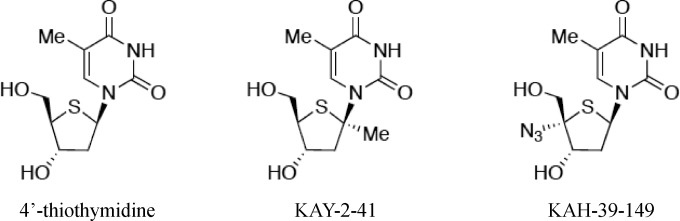
Chemical structures of 4′-thiothymidine, KAY-2-41, and KAH-39-149.
Antiviral assays.
The drug susceptibilities of all viruses were evaluated in their corresponding cell lines, as previously described (23). Briefly, for HSV-1, HSV-2, HCMV, MHV-68, HVS, and RRV, a viral cytopathic effect (CPE) reduction assay was used, and plaque reduction assays were performed for VZV. Drug susceptibility assays on HeLa TK− mutants were performed under similar conditions as those carried out with the HEL cells. The antiviral assays for KSHV and EBV were performed in BCBL-1 and P3HR-1 cell cultures, respectively, induced to the viral lytic cycle by adding 20 ng/ml 12-O-tetradecanoylphorbol 13-acetate (TPA) (Sigma-Aldrich, Bornem, Belgium) to the growing cells. After 5 days, intracellular DNA was quantified by real-time quantitative PCR (qPCR), targeting ORF73 of KSHV and BNRF1 of EBV (25). The 50% effective concentrations (EC50s) were further calculated.
Cytotoxicity assays.
The cytostatic effect of the drugs was based on the inhibition of cell growth (HEL, NIH 3T3, OMK, and RF cells), as previously described (23). Briefly, HEL cells were seeded at a density of 5 ×103 cells/well into 96-well microtiter plates. NIH 3T3, OMK, and RF cells were seeded at a density of 3 × 103 cells/well, and uninduced BCBL-1 and P3HR-1 cells were seeded at densities of 3 × 105 cells/ml and 1 × 106 cells/ml, respectively. After 24 h of cell growth, medium containing different concentrations of the test compounds was added. After 3 days of incubation at 37°C, the cell counts were determined using a Coulter counter (Analis, Namur, Belgium). The 50% cytostatic concentration (CC50) is the concentration of the compound required to reduce cell growth by 50% relative to the number of cells in the untreated controls. Alternatively, cytotoxicity was expressed as the minimum cytotoxic concentration (MCC), which is the compound concentration that causes a microscopically detectable alteration in cell morphology.
Thymidine kinase assay.
The assay was performed as previously described (23), and the inhibitory activities of KAY-2-41 and KAH-39-149 were determined against [methyl-3H]thymidine (dThd) phosphorylation by purified cellular TK-1 and TK-2, HSV-1 TK, and VZV TK. Briefly, the enzymes were incubated at 37°C for 30 min in the presence of different concentrations of the test compounds in a 50-μl reaction mixture containing 50 mM Tris-HCl (pH 8.0), 2.5 mM MgCl2, 10 mM dithiothreitol, 0.5 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propane-sulfonic acid, 3 mg/ml bovine serum albumin, 2.5 mM ATP, 1 μM [methyl-3H]deoxythymidine ([methyl-3H]dThd), and enzyme. Forty-five-microliter aliquots of the reaction mixtures were spotted on Whatman DE-81 filter paper disks. The filters were washed three times for 5 min in 1 mM ammonium formate, once for 1 min in water, and once for 5 min in ethanol. The radioactivity was determined by scintillation counting.
To determine the monophosphorylation of KAY-2-41, KAH-39-149, and BVDU by HSV-1 TK, VZV TK, TK1, and TK2, 500 μM of the test compounds was added in the mixture as described above but without [methyl-3H]dThd. After 0, 30, and 120 min of incubation at 37°C, 50 μl of phosphate-buffered saline (PBS) and 67% cold methanol were added the mixture. After centrifugation, the supernatant was injected into the high-pressure liquid chromatography (HPLC) system (strong-anion exchange [SAX] column). The relative peak areas of the drugs and their monophosphate forms were used to determine the enzymatic activities of the TKs.
Selection and characterization of drug-resistant viruses.
Drug-resistant HSV-1, HSV-2, HVS, and MHV-68 isolates were obtained by serial passages (between 4 and 15 passages) in their respective cell lines in the presence of increasing concentrations of KAY-2-41 or KAH-39-149 (starting at a concentration equivalent to their EC50). After confirming the resistant phenotype of the parent stock, viruses were plaque purified by limiting dilution. These clones were used for sequencing of the viral TK and DNA polymerase genes, as well as the protein kinase genes of MHV-68 and HVS. Phenotyping of the different viral clones was assessed by the corresponding antiviral assays.
Animal studies.
All animal work was approved by the Katholieke Universiteit Leuven Ethics Committee for Animal Care and Use (permit P097-2010). Intranasal infection of 4-week-old BALB/c mice was performed with MHV-68 (104 PFU/animal) under anesthesia using ketamine-xylazine in saline. KAY-2-41 and KAH-39-149, diluted in PBS, were administered intraperitoneally at a dose of 50 mg/kg of body weight once per day for 5 consecutive days, starting the day of infection. At days 6 and 12 postinfection (p.i.), each mouse was euthanized with a lethal injection of pentobarbital. The lungs, mediastinal lymph nodes (MLNs), and spleen were stored at −20°C in PBS for DNA extraction, at −80°C in RNAlater (Ambion) for RNA extraction, and then were fixed in 10% buffered formalin. The fixed tissues were embedded in paraffin, and 5-μm sections were stained with hematoxylin and eosin stain for histopathological examination. Quantification of viral DNA load and relative open reading frame 73 (ORF73) (latent) and glycoprotein B (gB) (lytic) gene expression in the tissues was performed as previously described (23).
RESULTS
Antiherpetic activity.
The inhibitory effects of KAY-2-41 and KAH-39-149 against a broad variety of herpesviruses are expressed as the 50% effective concentration (EC50), and the results are shown in Table 1. Among the gammaherpesviruses, EBV DNA synthesis was strongly inhibited by KAY-2-41 (EC50, 0.7 μM) and KAH-39-149 (EC50, 0.3 μM), whereas none of the compounds were active against KSHV (EC50, ≥130 μM). MHV-68 was more sensitive to the inhibitory effects of KAH-39-149 than of KAY-2-41 (EC50s, 0.2 μM versus 13 μM, respectively). In contrast, HVS and RRV replication were weakly or not inhibited by KAY-2-41 and KAH-39-149.
TABLE 1.
Antiviral activities of KAY-2-41 and KAH-39-149 against wild-type and mutant herpesviruses
| Virus or TK mutant | EC50 (μM) (fold resistance) fora: |
||||
|---|---|---|---|---|---|
| KAY-2-41 | KAH-39-149 | ACV | GCV | BVDU | |
| Viruses (strains) | |||||
| HSV-1 (KOS) | 3.3 ± 2.1 | 1.4 ± 1.4 | 0.1 ± 0.06 | 0.04 ± 0.01 | 0.08 ± 0.03 |
| HSV-2 (G) | 3.3 ± 1.2 | 1.5 ± 1.3 | 0.3 ± 0.1 | 0.06 ± 0.03 | ≥30 |
| VZV (Oka) | 37 ± 14 | 0.4 ± 0.02 | 5.0 ± 1.2 | NDb | 0.01 ± 0.02 |
| HCMV (AD-169) | >20 | >4 | ND | 3.0 ± 1.0 | ND |
| HCMV (Davis) | >20 | >20 | ND | 5.6 ± 2.3 | ND |
| EBV (P3HR-1) | 0.7 ± 0.9 | 0.3 ± 0.07 | 3.8 ± 0.9 | 4.3 ± 3.6 | 6.9 ± 5.1 |
| KSHV (BCBL-1) | ≥130 | ≥200 | 137 ± 44 | 6.3 ± 2.4 | 0.09 ± 0.09 |
| MHV-68 (G2.4) | 13 ± 6 | 0.2 ± 0.05 | 6.2 ± 1.3 | 10 ± 5.5 | 0.1 ± 0.06 |
| HVS (C488) | 27 ± 14 | 42 ± 8 | 116 ± 49 | 121 ± 47 | 6.0 ± 5.4 |
| RRV (VR-1414) | 24 ± 5 | >50 | >200 | 32 ± 5 | 0.4 ± 0.03 |
| TK mutants | |||||
| HSV-1 TK− | 26 ± 4.7 (8) | 16 ± 3.5 (11) | 45 ± 1.5 (450) | 47 ± 0 (1,175) | 188 ± 40 (2,350) |
| VZV TK−(07/1) | ≥45 (≥1) | 1.4 ± 0.1 (4) | 74 ± 21 (15) | ND | >150 (>15,000) |
| MHV-68 TK T364P | 55 ± 11 (4) | 1.9 ± 0.4 (10) | 6.2 ± 1.8 (1) | 10 ± 3 (1) | 19 ± 21 (190) |
| HVS TK R45stop | >200 (>8) | >200 (>5) | 209 ± 156 (1) | 141 ± 25 (1) | 468 ± 192 (1,170) |
Data are the mean values of at least three independent experiments ± standard deviations (SD). EC50, 50% effective concentration, the drug concentration required to reduce virus plaque formation, CPE, or viral DNA copies by 50%.
ND, not determined.
Both compounds showed antiviral activities against HSV-1 and HSV-2 in the micromolar range (EC50s, 1.4 μM to 3.3 μM), but KAH-39-149 was about 2-fold-more potent than KAY-2-41. The inhibitory effect of KAY-2-41 was weak against the VZV Oka strain (EC50, 37 μM), whereas KAH-39-149 proved to be approximately 100-fold-more potent (EC50, 0.4 μM). No antiviral activity was found against the two HCMV strains evaluated. The activity of the parent compound, 4′-thiothymidine, was previously reported, and the molecule was found to be active against HSV-1 (EC50, 0.01 μM), HSV-2 (EC50, 0.04 μM), VZV (EC50, 0.09 μM), and HCMV (EC50, 1 μM), and it showed severe inhibition of fibroblast proliferation (CC50, 0.8 μM) (26).
The cytostatic activities of KAY-2-41 and KAH-39-149 on HEL cells, measured as CC50 values, were 12 μM and 10 μM, respectively, but both compounds were less cytostatic on P3HR-1 cells (CC50s, 157 μM and 109 μM, respectively) and BCBL-1 cells (CC50s, 75 μM and 25 μM, respectively) (Table 2). Also, these drugs did not alter HEL cell morphology at the highest concentration tested (MCC, >100 μM). Thus, the selectivity indices (SIs) (calculated by CC50/EC50) of KAH-39-149 against human herpesviruses in decreasing order were as follows: 363 for EBV, 25 for VZV, and 7 for HSV-1 and HSV-2. KAY-2-41 was highly selective against EBV (SI, 224), whereas only low SIs were found toward HSV-1 and HSV-2 (SI, 3), and the compound was not selective against VZV (SI, <1). Both KAY-2-41 and KAH-39-149 showed a cytostatic effect on growing NIH 3T3 cells, with CC50 values of 3 μM, but these effects were less pronounced on OMK or RF cells (CC50, ≥28 μM). For both compounds, the MCC values were 50 μM and >100 μM on NIH 3T3 and OMK cells, respectively.
TABLE 2.
Cytostatic concentrations and selectivity indices of KAY-2-41 and KAH-39-149a
| Virus (strain) | Cell type | KAY-2-41 |
KAH-39-149 |
||
|---|---|---|---|---|---|
| CC50b | SIc | CC50 | SI | ||
| HSV-1 (KOS) | HEL | 12 ± 8 | 4 | 10 ± 4.5 | 7 |
| HSV-2 (G) | HEL | 12 ± 8 | 4 | 10 ± 4.5 | 7 |
| VZV (Oka) | HEL | 12 ± 8 | <1 | 10 ± 4.5 | 25 |
| HCMV (AD-169) | HEL | 12 ± 8 | <1 | 10 ± 4.5 | <3 |
| HCMV (Davis) | HEL | 12 ± 8 | <1 | 10 ± 4.5 | <1 |
| EBV | P3HR-1 | 157 ± 57 | 224 | 109 ± 83 | 363 |
| KSHV | BCBL-1 | 75 ± 19 | <1 | 25 ± 7 | <1 |
| MHV-68 (G2.4) | NIH 3T3 | 3.2 ± 1.0 | 1 | 3.4 ± 2.3 | 15 |
| HVS (C488) | OMK | 79 ± 48 | 3 | >50 | >1 |
| RRV (VR-1414) | RF | ≥112 | ≥5 | ≥28 | ≤1 |
The values shown are the averages of three independent experiments ± SD.
CC50, 50% cytostatic concentration, the drug concentration required to reduce cell growth by 50% relative to the number of cells in the untreated controls.
SI, selectivity index (ratio of CC50 to EC50).
Since the viral TK was reported to be involved in the activation of various thionucleosides (18), we evaluated the inhibitory effects of KAY-2-41 and KAH-39-149 against TK mutants of HSV-1, VZV, MHV-68, and HVS (Table 1). A 4-fold to ≥8-fold reduction in the sensitivity to KAY-2-41 was observed for the HSV-1, MHV-68, and HVS TK mutants, while a decrease in the antiviral activity of KAH-39-149 (up to 11 times) was shown against HSV-1, VZV, MHV-68, and HVS TK mutants. For the reference antiherpesvirus TK-dependent drugs (i.e., ACV, GCV, and/or BVDU), the decrease in inhibitory activity was much more pronounced than those for KAY-2-41 and KAH-39-149. These results indicate that mutations in the herpesvirus TK that give resistance to reference antiherpesvirus TK-dependent drugs conferred only low levels of resistance to either KAY-2-41 or KAH-39-149.
Inhibition of TK-catalyzed dThd phosphorylation.
Since herpesvirus TK mutants showed a modest decrease in sensitivity to KAY-2-41 and KAH-39-149, we further investigated the role of the viral TK in the mode of action of these nucleoside analogs. Enzymatic assays with TKs of different origins demonstrated that KAY-2-41 and KAH-39-149 were able to inhibit the phosphorylation of dThd by HSV-1 and VZV TK (Table 3). KAH-39-149 appeared to be a much more potent inhibitor of the viral TK-catalyzed reaction than KAY-2-41 but exerted less inhibitory activity for HSV-1 TK (inhibitory concentration [IC50], 3.0 μM) than its parent compound, 4′-thiothymidine (IC50, 0.09 μM). The inhibitory activity of KAH-39-149 was comparable to that of BVDU, with IC50s of 1.5 μM for VZV TK and 2.6 μM for HSV-1 TK. Additionally, KAH-39-149 inhibited dThd phosphorylation catalyzed by cellular TK1 and TK2, with IC50s of 26 μM and 6.2 μM, respectively, while BVDU inhibited the reaction catalyzed by TK2 (IC50, 0.4 μM) but not by TK1 (IC50, >500 μM). dThd phosphorylation by cytosolic TK1 was most potently inhibited by 4′-thiothymidine, with an IC50 of 6.3 μM, as previously reported (26). On the contrary, KAY-2-41 was a poor inhibitor of TK1- and TK2-catalyzed reactions (IC50s, ≥500 μM and 321 μM, respectively). These results showed that both thionucleosides are inhibitors of viral and cellular TKs and that KAH-39-149 is a better inhibitor of dThd phosphorylation than is KAY-2-41.
TABLE 3.
Inhibitory activities of KAY-2-41, KAH-39-149, and BVDU on dThd phosphorylation and phosphorylation of these drugs by 2′-deoxynucleoside kinases from different origins
| Compound | IC50 (μM) fora: |
% of nucleoside monophosphate formb |
||||||
|---|---|---|---|---|---|---|---|---|
| HSV-1 TK | VZV TK | TK1 | TK2 | HSV-1 TK | VZV TK | TK1 | TK2 | |
| KAY-2-41 | 49 ± 1 | 272 ± 21 | ≥500 | 321 ± 16 | 0.6 ± 0.07 | 0.5 ± 0.2 | 0.9 ± 0.2 | 2.3 ± 0.4 |
| KAH-39-149 | 3.0 ± 0 | 1.4 ± 0.7 | 26 ± 11 | 6.2 ± 2.3 | 0.5 ± 0.04 | 0.7 ± 0.1 | 0.9 ± 0.3 | 4.8 ± 0.01 |
| BVDU | 2.6 ± 1.5 | 1.5 ± 0.4 | >500 | 0.4 ± 0.05 | 7.0 ± 0.04 | 5.8 ± 0.3 | 0 | 17 ± 1.3 |
| 4′-Thiothymidine | 0.09c | 6.3c | ||||||
The IC50 is the 50% inhibitory concentration of the test compounds, which was calculated as the compound concentration required to inhibit TK-catalyzed [methyl-3H]dThd (1μM) phosphorylation by 50%. The data are mean values from two independent experiments ± SD.
Values represent the percentage of the respective drug monophosphate form present in the reaction mixture after 120 min of incubation of the nucleoside with viral or cellular TK. The data are the mean values of two independent experiments ± SD.
Mean values of two experiments. The SD was never ±20% of each reported value (data from reference 26).
Phosphorylation of KAY-2-41 and KAH-39-149 by viral and cellular TKs.
In order to determine whether viral and cellular TKs are able to phosphorylate KAY-2-41 and KAH-39-149 to the monophosphate form, the products generated from the enzymatic reaction were resolved by HPLC (Table 3). Although KAY-2-41, KAH-39-149, and BVDU were phosphorylated by HSV-1 and VZV TK, both enzymes preferentially phosphorylated BVDU, as approximately 10-fold-higher levels of the monophosphate form were detected for BVDU compared to those of KAY-2-41 and KAH-39-149. Yet approximately 0.6% of the monophosphate forms of KAY-2-41 and KAH-39-149 was produced by HSV-1 TK and VZV TK after 120 min. At this time point, approximately 1% of the monophosphate forms were produced by cytosolic TK1 for KAY-2-41 and KAH-39-149. Mitochondrial TK2 did phosphorylate KAY-2-41 and KAH-39-149, but the highest levels of the monophosphate form were observed for BVDU (17% versus 2.3% for KAY-2-41 and 4.8% for KAH-39-149).
Inhibition of HSV-1 and HSV-2 wild-type and mutant strains by KAY-2-41 and KAH-39-149 in HeLa TK− cells.
We further examined the inhibitory effects of KAY-2-41 and KAH-39-149 against HSV-1 and HSV-2 wild-type (WT) and TK− strains in HeLa cells deficient in TK1 in order to further assess the roles of cellular and viral TKs in the mode of action of these thionucleosides. HSV-1 and HSV-2 WT strains were sensitive to the inhibitory effects of KAY-2-41 and KAH-39-149 in HeLa TK− cells, with EC50s in the range of 13 μM and 3 μM, respectively (Table 4). In contrast, these compounds lost their antiviral activities against HSV-1 and HSV-2 TK− strains (EC50s, >147 μM and ≥40 μM, respectively). As a control, we observed a marked reduction in the activities of ACV and GCV against TK− HSV-1 and HSV-2, while the antiviral effect of the TK-independent viral DNA polymerase inhibitor foscarnet (PFA) remained unaffected. These results demonstrated that KAY-2-41 and KAH-39-149 lost their activities when both HSV TK and cellular TK1 were absent, suggesting that both of these TKs are able to activate the compounds.
TABLE 4.
Antiviral activities of KAY-2-41 and KAH-39-149 against wild-type and mutant HSV-1 and HSV-2 in HeLa TK− cells
| Virus | EC50 (μM) fora: |
||||
|---|---|---|---|---|---|
| KAY-2-41 | KAH-39-149 | ACV | GCV | PFA | |
| HSV-1 WT | 13 ± 0.8 | 3.6 ± 0.4 | 0.3 ± 0.1 | 0.6 ± 0.05 | 286 ± 0 |
| HSV-1 TK− | >147 | ≥40 | 14 ± 15 | 13 ± 9 | 163 ± 95 |
| HSV-2 WT | 13 ± 1.1 | 2.7 ± 0.6 | 0.5 ± 0.2 | 0.1 ± 0.04 | 179 ± 38 |
| HSV-2 TK− | >147 | ≥40 | 28 ± 9 | 7.8 ± 2.8 | 252 ± 50 |
EC50 (50% effective concentration) is the drug concentration required to reduce virus-induced CPE by 50%. The data are the mean values of three independent experiments ± SD.
Genotypic analysis of in vitro-selected drug-resistant herpesvirus strains.
To further understand the mode of action of KAY-2-41 and KAH-39-149, drug-resistant viruses were selected under pressure of each thiothymidine derivative (Table 5). HSV-1, HSV-2, HVS, and MHV-68 were serially passaged in increasing concentrations of each compound. Different independent selection procedures were performed with the HSV-1 strains, indicated as KOS/A, KOS/B, KOS/C, and KOS/D, and HSV-2 strains, indicated as G/A, G/B, and G/C (Table 5). The different viral stocks were then tested for their drug susceptibility phenotypes in CPE reduction assays, and when a drug-resistant profile was observed, the clones of each drug-resistant virus were further isolated. These clones were then used for genotyping. Based on the proposed mode of action of the thionucleosides (18), the viral TK and DNA polymerase genes were sequenced, as well as the protein kinase genes from the HVS and MHV-68 clones. As shown in Fig. 2, several mutations were identified in the viral TK and they consisted of single nucleotide substitutions, insertions, or deletions. None of the clones carried mutations in the viral DNA polymerase or in the protein kinase genes of MHV-68 and HVS.
TABLE 5.
Genotypic characterization of drug-resistant herpesvirus TK mutants obtained in vitro under selective pressure of KAY-2-41 and KAH-39-149a
| Viruses by compound | Strain selection | No. of clones/no. total | Nucleotide change in viral TKb | Amino acid substitution or frameshift in viral TKb |
|---|---|---|---|---|
| KAY-2-41 | ||||
| HSV-1 | KOS/A | 1/6 | nt 430–436, G insertion | Frameshift |
| 5/6 | C151T | R51W | ||
| KOS/B | 6/6 | C664T | R222C | |
| KOS/C | 2/6 | C664T | R222C | |
| 4/6 | A703T | I235F | ||
| KOS/D | 4/4 | nt 430–436, G insertion | Frameshift | |
| HSV-2 | G/A | 2/2 | nt 433–439, G insertion | Frameshift |
| G/B | 4/5 | nt 433–439, G insertion | Frameshift | |
| 1/5 | C863T | T288M | ||
| G/C | 1/4 | A263G | Y88C | |
| 1/4 | T385C | M129T | ||
| 1/4 | G545A | S182N | ||
| 1/4 | nt 433–439, G insertion | Frameshift | ||
| MHV-68 | G2.4/A | 3/3 | T1116G | C372W |
| HVS | C488/A | 1/6 | G160T | E54stop |
| 1/6 | C976A | P326T | ||
| 2/6 | nt 440–445, A deletion | Frameshift | ||
| 2/6 | nt 1094–1095, A insertion | Frameshift | ||
| KAH-39-149 | ||||
| HSV-1 | KOS/A | 5/5 | G159A | V187M |
| KOS/C | 4/4 | G159A | V187M | |
| HSV-2 | G/B | 5/5 | A518G | Y173C |
| MHV-68 | G2.4/A | 2/2 | nt 661–665, C insertion | Frameshift |
| HVS | C488/A | 3/3 | nt 1094–1095, A insertion | Frameshift |
TK, thymidine kinase.
No mutations were found in the viral DNA polymerase or in the gammaherpesvirus protein kinase. nt, nucleotides.
FIG 2.

Mapping of amino acid changes and frameshift mutations in the herpesvirus TK selected under pressure of KAY-2-41 and KAH-39-149. Shown is a schematic representation of the TK of HSV-1, MHV-68, and HVS. The ATP-binding site, the nucleoside-binding site, and the regions conserved among herpesvirus TKs are indicated by black boxes. Amino acid changes and frameshift mutations in the herpesvirus TK that have not previously been reported in vitro or in patients are underlined. nts, nucleotides.
Under selective pressure of KAY-2-41, an insertion of a single guanosine (G) was identified between nucleotides 430 and 436 in a stretch of a homopolymer of seven Gs, resulting in a frameshift mutation in the HSV-1 TK. This mutation was observed in HSV-1 clones isolated from two independent selection procedures (KOS/A and KOS/D). KAY-2-41-resistant (KAY-2-41r) HSV-1 clones isolated from the two other selection procedures (KOS/B and KOS/C) showed single nucleotide substitutions leading to the following amino acid substitutions: R51W, I235F, and R222C. HSV-2 clones selected under pressure of KAY-2-41 carried a frameshift mutation resulting from an insertion of one G between nucleotides 433 and 439. Other HSV-2 mutants harbored single amino acid substitutions, including Y88C, M129T, S182N, and T288M.
All KAY-2-41r MHV-68 clones showed the C372W substitution in the viral TK. Among the HVS clones isolated under pressure of KAY-2-41, different amino acid substitutions and frameshift mutations were identified and consisted of E54stop, P326T, the deletion of one adenine (A) between nucleotides 440 and 445, and the insertion of one A between nucleotides 1094 and 1095.
Under KAH-39-149 pressure, all HSV-1 clones carried the V187M amino acid substitution, and HSV-2 clones showed the Y173C substitution. An insertion of A between nucleotides 1094 and 1095 was found in all KAH-39-149r HVS clones and, in two MHV-68 clones, an insertion of C occurred in a string of five Cs at nucleotide positions 661 to 665 in the viral TK.
Phenotypic analysis of the different mutants.
To assess the levels of drug resistance of the viral TK mutants isolated following KAY-2-41 and KAH-39-149 pressure, drug susceptibility profiles were determined (Fig. 3 and 4). We also calculated the ratio of EC50mutant to EC50WT, representing the fold resistance.
FIG 3.

Phenotyping of drug-resistant HSV isolates bearing mutations in the viral TK. The data are presented as a dot plot of the EC50s of drug-resistant HSV-1 (A) and HSV-2 (B) versus the EC50 for wild-type (WT) virus. The horizontal bars indicate the mean values of three independent experiments.
FIG 4.

Phenotyping of drug-resistant gammaherpesvirus isolates bearing mutations in the viral TK. The data are presented as a dot plot of the EC50s of drug-resistant HVS (A) and MHV-68 (B) versus the EC50 for wild-type (WT) virus. The horizontal bars indicate the mean values of three independent experiments.
For HSV-1, mutants with a frameshift (G-string insertion) or an amino acid substitution in the ATP-binding site (R51W) had the highest levels of resistance to KAY-2-41 and KAH-39-149 (ranging from 13- to 20-fold), as well as to ACV, GCV, and BVDU, compared to that of WT virus (Fig. 3A). In general, the I235F, R222C, and particularly V187M HSV-1 mutants showed lower levels of resistance against KAY-2-41 and KAH-39-149 (ranging from 2- to 7-fold) and also lower levels of resistance to ACV, GCV, and BVDU than the R51W and frameshift TK mutants.
The HSV-2 mutant viruses (G insertion, Y88C, M129T, S182N, and T288M) isolated under pressure of KAY-2-41 showed 2- to 5-fold resistance against KAY-2-41 and KAH-39-149, except for the S182N mutant, which remained sensitive to KAH-39-149. In contrast, the Y137C amino acid change conferred 2-fold resistance to KAH-39-149 but no resistance to KAY-2-41. Notably, the sensitivities of the HSV-2 mutants to ACV and GCV were markedly reduced, ranging from 10- to 600-fold compared to that of the WT virus (Fig. 3B). The HSV-2 TK mutant with a frameshift (G-string insertion) mutation showed markedly lower levels of resistance for the thiothymidine analogs than did the HSV-1 TK mutants with the same type of mutation (2- to 4-fold for HSV-2 versus 13- to 20-fold for HSV-1).
HVS clones bearing an insertion or a deletion of a single nucleotide in the viral TK gene, as well as the E54stop and P326T amino acid substitutions, conferred noticeable levels of resistance to KAY-2-41 and KAH-39-149 (Fig. 4A). Yet a much higher degree of resistance was found with other TK-dependent drugs, such as the pyrimidine derivatives BVDU and HDVD (23). However, for these compounds, the EC50s calculated for the HVS TK mutants were higher than the highest concentrations used in the phenotypic assays, which were 200 μM for KAY-2-41 and KAH-39-149, 600 μM for BVDU, and 700 μM for HDVD. The HVS TK mutants showed no altered sensitivities to ACV and GCV, which are known to be dependent on the virus protein kinase (ORF36) for their activation.
MHV-68 clones bearing the C372W TK amino acid substitution showed higher EC50s for KAY-2-41, KAH-39-149, BVDU, and HDVD than did the WT, and 4- and 5-fold levels of resistance were observed against KAY-2-41 and KAH-39-149, respectively (Fig. 4B). The MHV-68 TK mutant with a frameshift (C insertion, nucleotides [nt] 661 to 665) showed higher levels of resistance to KAY-2-41 and KAH-39-149 (3- and 15-fold, respectively), as well as BVDU and HDVD (26- and 115-fold), compared to the C372W TK mutant.
No changes in the sensitivity of HPMPC were observed between the WT virus and the various TK mutants. Other acyclic nucleoside phosphonates, i.e., HPMPA, HPMP-5-azaC, HPMPO-DAPy, PMEO-DAPy, and the pyrophosphate analog PFA, were also included in the phenotypic assays, showing no variations in the EC50s between the WT and the different TK mutant viruses (data not shown).
In vivo activity.
The in vitro antiviral activities and selectivities of KAY-2-41 and KAH-39-149 were the most promising against EBV replication, and therefore we aimed to assess their in vivo activities in a mouse model of gammaherpesvirus infection. Intranasal infection of BALB/c mice with MHV-68 results in an acute productive infection of alveolar lung epithelial cells (23, 27). Next, the virus enters the mediastinal lymph nodes (MLNs) and establishes a latent infection in the spleen (28). Previously, we determined the kinetics of gene expression of two genes, the glycoprotein B (gB) gene, mainly expressed during lytic replication, and the ORF73 gene, mainly expressed during latency, in this mouse model (23). We showed that following a rapid onset, both gB and ORF73 expression peaked in the lungs after 4 and 6 days postinfection (p.i.), whereas gene expression peaked at approximately 12 to 14 days p.i. in the spleen. BALB/c mice were infected intranasally and treated intraperitoneally with 50 mg/kg of KAY-2-41 or KAH-39-149 once a day for 5 days, starting 2 h after infection. To evaluate drug efficacy, we examined (i) the inhibition of acute MHV-68 replication in the lungs after 6 days p.i. and (ii) the effect of antiviral treatment on the establishment of viral latency in MLNs and the spleen after 12 days p.i. To this end, viral DNA copies, as well as the expression of lytic gB and the latent ORF73 gene, were determined (Fig. 5).
FIG 5.

Analysis of MHV-68 infection in different organs of untreated and treated mice at the acute and latent stages. (A) Viral DNA copies were quantified in the lungs, mediastinal lymph nodes (MLNs), and spleens of intranasally infected mice (104 PFU of MHV-68) and treated intraperitoneally with KAY-2-41 and KAH-39-149 for five consecutive days. Each group contained five mice. The values are given as the mean log viral copy number per mg of tissue ± standard deviation (SD). The dashed line represents the limit of detection set on 10 copies of viral DNA. (B) The upper panels represent the level of gB (white bars) and ORF73 (black bars) expression relative to those of the untreated control and normalized to the endogenous control glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The error bars display the calculated maximum (RQMax) and minimum (RQMin) expression levels that are equivalent to the standard error of the mean expression level (RQ value). The RQMax is represented as ΔΔCT + s and the RQMin as ΔΔCT − s, where s is the SD of the ΔΔCT value. The lower panels represent the 1/delta CT values obtained from each mouse in the untreated and treated groups, and on which the Mann-Whitney U tests were done to compare the untreated and treated groups: *, P < 0.05; **, P < 0.01; ***, P < 0.001. The dashed line represents the limit of detection.
At day 6 p.i., the viral DNA load in the lung tissue of the untreated infected control was as high as 106 copies/mg (Fig. 5A). In the drug-treated mice, viral DNA copies were significantly reduced by 1.5 log for KAY-2-41 and by 3 log for KAH-39-149. KAY-2-41 treatment also decreased the relative expression levels of a lytic gene (gB) and a latent gene (ORF73) by approximately one log compared to the infected control, whereas KAH-39-149 was more potent in reducing gB and ORF73 expression levels in the lungs of infected mice (by approximately 2 log) (Fig. 5B). In the MLNs, but particularly in the spleen, KAY-2-41-treated animals harbored reduced MHV-68 DNA copies compared with the control group. In contrast, viral DNA copies were not detected in these tissues after KAH-39-149 treatment (Fig. 5A).
At day 12 p.i., the untreated infected mice showed low numbers of viral DNA copies in the lungs (103 copies/mg). At this time point, viral DNA copies were still significantly reduced in the lungs of the KAY-2-41-treated mice (P = 0.004) but not of the KAH-39-149-treated mice (Fig. 5A). Yet gB and ORF73 expression was not significantly lower in the lungs of KAY-2-41-treated animals than that in those of the control group (Fig. 5B). In the MLNs, similar levels of viral DNA loads were observed between the control and KAY-2-41-treated groups, while three out of five KAH-39-149-treated mice showed the presence of MHV-68 DNA. Nevertheless, the five mice of the KAH-39-149-treated group did not harbor MHV-68 DNA copies in the spleen at day 12 p.i. (P = 0.0008) (Fig. 5A). In line with this, the KAH-39-149-treated animals did not show gB or ORF73 expression in this tissue. Thus, until 12 days p.i., KAH-39-149 treatment was able to inhibit the spread of MHV-68 to the spleen; while less active, KAY-2-41 still significantly diminished MHV-68 loads in this tissue.
At day 6 p.i., a histological examination of lung tissue in MHV-68-infected mice showed a diffusely increased interstitial cellularity, particularly at the perivascular and peribronchial sites, compared to that in the uninfected control mice. At day 12 p.i., this inflammatory reaction was dominated by mononuclear cells (Fig. 6). After treatment with KAY-2-41 and KAH-39-149, a similar inflammatory reaction was present in the lungs of the virus-infected treated mice. The white pulp of the spleens of the MHV-68-infected untreated and KAY-2-41-treated mice was composed of large poorly delineated lymphoid follicles and an unremarkable red pulp. In addition, tingible body macrophages were noted in the white pulp of the spleens of the untreated and KAY-2-41-treated infected mice but not in the KAH-39-149-treated mice.
FIG 6.

Histopathological analysis of lungs and spleen of uninfected, infected, and treated infected mice. Shown are hematoxylin and eosin staining of the lung tissues of uninfected mice, MHV-68-infected mice, KAY-2-41-treated mice, and KAH-39-149-treated mice at days 6 and/or 12 p.i. The histopathology of the spleen at day 12 p.i. is shown in the right panels, and the black arrows indicate the presence of tingible body macrophages in the spleens of the infected mice. Magnification, ×20 (inset, ×100).
DISCUSSION
In this report, we demonstrate the inhibitory activities of KAY-2-41 and KAH-39-149 against alpha- and gammaherpesviruses, which are dependent on phosphorylation by viral and cellular TKs. The antiviral activity of KAH-39-149 was superior to that of KAY-2-41 against HSV-1, HSV-2, VZV, EBV, and MHV-68. It was previously shown that the parent molecule 4′-thiothymidine was also an inhibitor of herpesvirus replication; however, this drug demonstrated considerably higher cellular toxicity than KAY-2-41 and KAH-39-149 (26). Among all herpesvirus evaluated here, both compounds were the most active against EBV, with 5- to 10-fold higher anti-EBV activity than that against ACV, GCV, and HPMPC. The guanosine analogs ACV and GCV are the antiviral drugs commonly administered off-label for the treatment of EBV-associated malignancies, and the acyclic nucleoside phosphonate HPMPC exhibits selective anti-EBV activity in vitro (3, 29). KAY-2-41 and KAH-39-149 were poorly active against KSHV and two genetically related rhadinoviruses, HVS and RRV. Since our findings showed the involvement of viral TK in the mode of action of the thionucleosides, future studies aimed at determining the affinity and phosphorylation capacity of EBV TK for KAY-2-41 and KAH-39-149 would be helpful for assessing the eventual contribution of EBV TK to the selective inhibition of EBV replication by these drugs.
In contrast to the study of Rahim et al. (19), which reported that thionucleosides may be poor substrates of HSV-2 TK, and despite the fact that HSV-2 TK does not exhibit thymidylate kinase activity, KAY-2-41 and KAH-39-149 showed pronounced antiviral activity against this virus. Still, HSV-2 TK mutants with a frameshift mutation exhibited approximately 5-fold-lower levels of resistance for KAY-2-41 and KAH-39-149 than the HSV-1 TK mutant with a similar mutation, but this was not seen for ACV and GCV. These results might indicate that the activities of the 4′-thiothymidine derivatives are more dependent on the phosphorylation by cellular kinases in HSV-2-infected cells than in HSV-1-infected cells. Furthermore, the enzymatic assays demonstrated that KAH-39-149 had a higher inhibitory effect on the phosphorylation of dThd catalyzed by HSV-1 and VZV TK than did KAY-2-41. These results are consistent with the higher antiviral activity of KAH-39-149 against HSV-1 and VZV, notwithstanding that the viral TK activity was similar for these drugs and that the viral DNA polymerase likely represents the final target responsible for virus inhibition.
As mentioned above, our data demonstrate that not only herpesvirus TKs but also the cellular TK1 are needed for the full antiviral effects of KAY-2-41 and KAH-39-149. Evidence supporting the involvement of these TKs was obtained by an evaluation of the sensitivities of TK− herpesviruses, e.g., HSV-1, VZV, MHV-68, and HVS, to KAY-2-41 and KAH-39-149. These mutants showed modest decreases in sensitivity (4- to 11-fold) to these drugs, while they were markedly resistant to other TK-dependent drugs (ACV, GCV, and/or BVDU), with levels of resistance in the range of 100- to 1,000-fold (Table 1); this might point toward the ability of cellular kinases to activate the thionucleosides. Similar observations were made with our KAY-2-41r and KAH-39-149r herpesvirus mutants, and our data are in agreement with those reported for another thionucleoside, 4′-thioIDU (18). In addition, in all the different herpesviruses replicated under pressure of KAY-2-41 and KAH-39-149, various mutations were identified in the viral TK and not in the DNA polymerase or protein kinase (of gammaherpesviruses). In agreement with this, a functional viral TK was necessary for HSV-1 and HSV-2 to remain sensitive to each of the thionucleoside derivatives in cells defective in TK1 (HeLa TK− cells). Additionally, KAH-39-149, and to a lesser extent KAY-2-41, inhibited HSV-1 and VZV TKs, as well as cytosolic TK1 and mitochondrial TK2 dThd phosphorylation. Finally, KAY-2-41 and KAH-39-149 were shown to be phosphorylated to their monophosphate forms by HSV-1 TK, VZV TK, TK1, and TK2. These results further support that the viral TK and cytosolic TK1 are the kinases involved in the activation of these 4′-thiothymidine derivatives. Although the thionucleoside derivatives were phosphorylated in vitro by TK2, the role of TK2 in the activation of the cell compounds appears to be minor, because no activity of KAY-2-41 or KAH-39-149 against TK− HSV mutants was seen in the HeLa cells that lacked TK1. However, to further work out the mechanism of action of KAY-2-41 and KAH-39-149, metabolic studies are required to assess the formation of the active metabolite (triphosphate form) and the incorporation of these compounds into DNA by DNA polymerases.
In line with our findings, Prichard et al. (18) provided further evidence supporting that besides viral kinases, cellular kinases may be implicated in the first phosphorylation step of thionucleosides. The anti-HSV-1 activity of 4′-thioIDU was reduced by 10-fold against an ACVr TK− mutant, while a >100-fold reduction in the efficacy of ACV was observed. Also, the compound was incorporated into the viral DNA of HCMV-infected cells, yet HCMV does not encode a TK homolog, and the UL97 (protein kinase) was not shown to contribute significantly to the phosphorylation of 4′-thioIDU. The triphosphate form of 4′-IDU was most probably produced by cellular kinases. Therefore, these data support a model for the mechanism of action of KAY-2-41 and KAH-39-149. As the drugs are converted to their monophosphate forms by the herpesvirus and cellular TKs, we hypothesize that they may further inhibit DNA synthesis by either incorporation into DNA or inhibition of the viral DNA polymerase after two subsequent phosphorylations by cellular nucleoside kinases.
Mitochondrial TK2 has an important role in the maintenance of deoxynucleoside triphosphate pools in the mitochondria and has been suggested to contribute to the mitochondrial toxicity of pyrimidine nucleoside analogues (30). It could be asked whether the antiviral activities seen against HSV-1, HSV-2, VZV, and MHV-68 might be related in part to cytostatic effects, as KAH-39-149 was particularly able to inhibit TK2 activity. Yet no inhibitory activity was observed against HCMV, which was evaluated in the same cell type as HSV-1, HSV-2, and VZV. In addition, the antiviral assays were performed on confluent monolayers, for which KAY-2-41 and KAH-39-149 had an MCC value of >100 μM, and not on growing cells, for which low CC50 values were obtained. Similarly, the anti-EBV and anti-KSHV activities of KAY-2-41 and KAH-39-149 were evaluated in dividing lymphocytes, and whereas KSHV was not inhibited by these drugs, they were highly selective against EBV, with minimal cytostatic effects. Therefore, we assumed that the antiherpesvirus activity is likely related to the inhibition of viral DNA synthesis rather than to mitochondrial toxicity, but this should still be evaluated in further studies. Nevertheless, immunofluorescence studies have suggested that 4′-thioIDU might be incorporated into the DNA of uninfected cells, indicating that the triphosphate form was a substrate for the cellular DNA polymerase (18). While this may have accounted for the inhibition of cell proliferation seen with 4′-thioIDU, one may speculate that it is a situation comparable to those of KAY-2-41 and KAH-39-149. Yet, no noticeable signs of toxicity (i.e., weight loss) were seen in mice repetitively treated with each of the drugs for 5 days, once per day.
Although cellular kinases might be involved in the phosphorylation of these thiothymidine derivatives, drug-resistant HSV-1, HSV-2, HVS, and MHV-68 clones that were selected under pressure of KAY-2-41 and KAH-39-149 all harbored mutations in the viral TK, indicating that these antiviral agents interact with the viral TK. In HSV-1 clinical isolates, the most common TK mutation described to date is a single G insertion in the run of seven guanosine nucleotides from nucleotide positions 433 to 439, which appears to be a hot spot for mutations (31). This frameshift mutation localized closed to the nucleoside-binding site, leading to the production of a truncated TK polypeptide (32, 33). In this study, we found this mutation in HSV-1 and HSV-2 clones replicated under pressure of KAY-2-41. Other previously known drug resistance mutations mapped in the TK of HSV-1 and HSV-2 of clinical samples have been also identified here, such as the amino acid substitutions R51W, R222C, V187M, S182N, T288M, and Y173C (13, 24, 34–41). Three amino acid changes in the HSV TK were never reported before, i.e., I235F, Y88C, and M129T. Strikingly, amino acid changes in the TK of HSV-1 (V187M, R222C, and I235F) that conferred the lowest levels of resistance against ACV, GCV, and BVDU were also associated with low levels of resistance to KAY-2-41 and KAH-39-149. Similar observations were made in HSV-1 clones harboring mutations that conferred the highest level of resistance against all TK-dependent drugs.
While drug resistance has not yet been studied in detail for gammaherpesviruses, we recently isolated a drug-resistant HVS mutant under pressure of HDVD, a pyrimidine nucleoside that depends on the viral TK for its activation, and this clone had a P326T amino acid substitution located in the nucleoside-binding site of the TK (23). Here, we selected the P326T HVS mutant under pressure of KAY-2-41, and in line with our previous study, we observed cross-resistance to other TK-dependent drugs, i.e., BVDU and HDVD. Besides the P326T amino acid substitution in HVS, the Y173C amino acid substitution in HSV-2 TK also occurred in the substrate binding site, and more precisely, both changes are located in a motif of three conserved amino acids, presented as CYP in HSV-2 TK (positions 172 to 174) and VFP in HVS (positions 324 to 326). Previously, Munir et al. (42) revealed that in the nucleoside-binding site encoded by HSV-1, a high degree of flexibility in accommodating different types of amino acid substitutions existed. However, no replacement of the proline residue was tolerated at position 173 of HSV-1 TK (corresponding to Pro174 in HSV-2 and Pro326 in HVS), whereas the tyrosine residue at position 172 (HSV-1) could be replaced by the structurally related phenylalanine only (42). Consistent with this study, Wu et al. (43) reported that in EBV, the full activity of the TK was retained only when a tyrosine residue replaced the phenylalanine at this position. Thus, the very limited substitution of proline and tyrosine/phenylalanine in the conserved site IV (i.e., nucleoside-binding site) suggests that these residues are essential for maintaining functional TK activity (42, 43).
Furthermore, the inhibitory effects of KAY-2-41 and KAH-39-149 were demonstrated in vivo in a mouse model of gammaherpesvirus infection, which proved that KAH-39-149 was superior to KAY-2-41 in inhibiting MHV-68 replication and both lytic and latent gene expression in the lungs (day 6 p.i.), as well as viral dissemination to other organs (MLNs and spleen). A similar course of infection was previously observed after treatment with HPMPC but not with the pyrimidine analog HDVD (23, 29). In addition, we evaluated KAY-2-41 and KAH-39-149 in athymic nude mice infected intracutaneously with HSV-1; however, both drugs did not show antiviral efficacy in this model (our unpublished data). Indeed, the treatment of HSV-1-infected mice with KAY-2-41 and KAH-39-149, administered topically at 1% and 0.3% twice daily, did not reduce the development of lesions and mortality among the infected animals. The failure of drug treatment in HSV-1-infected mice compared to that in MHV-68-infected mice might be related to differences in (i) the route of infection (intracutaneously versus intranasal) and/or (ii) pathogenicity, as HSV-1 led to severe disease development and further death of the infected animals. Additionally, as reported by Prichard et al. (18), the thionucleoside 4-thioIDU was not effective at reducing the mortality of mice infected with HSV-2 by the intranasal route and treated orally with the drug at a dose of up to 30 mg/kg.
The data presented here prove that KAH-39-149 is an active and selective inhibitor of EBV replication in cell culture and in a mouse model of gammaherpesvirus acute infections (44, 45). Although KAY-2-41 showed less efficacy in vivo, our study demonstrated that 1′-substituted- and 4′-substituted-4′-thiothymidine derivatives should be considered attractive classes of antiviral agents. The in vitro results presented here strongly suggest that the mechanism of action of KAY-2-41 and KAH-39-149 involves phosphorylation by viral and cellular TKs prior to inhibition of the viral DNA polymerase. Still, further investigations, such as metabolic studies and marker transfer experiments, are needed to support the proposed mode of action in order to determine the impact of specific mutations in the herpesvirus TK on the activity of the thionucleosides. Besides the promising anti-EBV activities of these drugs, the development of new thionucleoside derivatives, with a higher affinity for herpesvirus TKs, may offer enhanced selectivity against HSV-1, HSV-2, and VZV.
ACKNOWLEDGMENTS
We thank Anita Camps, Sarah Gillemot, Steven Carmans, Lies Van den Heurck, Pierre Fiten, Lizette van Berckelaer, and Ria Van Berwaer for their excellent technical assistance.
This work was supported by a grant from the Geconcerteerde Onderzoeksacties (GOA) (10/014) and the Fonds voor Wetenschappelijk Onderzoek (FWO) (G-0608-08) of the KU Leuven.
Footnotes
Published ahead of print 12 May 2014
REFERENCES
- 1.Höcker B, Böhm S, Fickenscher H, Küsters U, Schnitzler P, Pohl M, John U, Kemper MJ, Fehrenbach H, Wigger M, Holder M, Schröder M, Feneberg R, Köpf-Shakib S, Tönshoff B. 2012. (Val-)Ganciclovir prophylaxis reduces Epstein-Barr virus primary infection in pediatric renal transplantation. Transpl. Int. 25:723–731. 10.1111/j.1432-2277.2012.01485.x [DOI] [PubMed] [Google Scholar]
- 2.Davis JE, Moss DJ. 2004. Treatment options for post-transplant lymphoproliferative disorder and other Epstein-Barr virus-associated malignancies. Tissue Antigens 63:285–292. 10.1111/j.0001-2815.2004.00227.x [DOI] [PubMed] [Google Scholar]
- 3.Rafailidis PI, Mavros MN, Kapaskelis A, Falagas ME. 2010. Antiviral treatment for severe EBV infections in apparently immunocompetent patients. J. Clin. Virol. 49:151–157. 10.1016/j.jcv.2010.07.008 [DOI] [PubMed] [Google Scholar]
- 4.Davis CL, Harrison KL, McVicar JP, Forg PJ, Bronner MP, Marsh CL. 1995. Antiviral prophylaxis and the Epstein Barr virus-related post-transplant lymphoproliferative disorder. Clin. Transplant. 9:53–59 [PubMed] [Google Scholar]
- 5.Funch DP, Walker AM, Schneider G, Ziyadeh NJ, Pescovitz MD. 2005. Ganciclovir and acyclovir reduce the risk of post-transplant lymphoproliferative disorder in renal transplant recipients. Am. J. Transplant. 5:2894–2900. 10.1111/j.1600-6143.2005.01115.x [DOI] [PubMed] [Google Scholar]
- 6.Malouf MA, Chhajed PN, Hopkins P, Plit M, Turner J, Glanville AR. 2002. Anti-viral prophylaxis reduces the incidence of lymphoproliferative disease in lung transplant recipients. J. Heart Lung Transplant. 21:547–554. 10.1016/S1053-2498(01)00407-7 [DOI] [PubMed] [Google Scholar]
- 7.Mentzer SJ, Perrine SP, Faller DV. 2001. Epstein-Barr virus post-transplant lymphoproliferative disease and virus-specific therapy: pharmacological re-activation of viral target genes with arginine butyrate. Transpl. Infect. Dis. 3:177–185. 10.1034/j.1399-3062.2001.003003177.x [DOI] [PubMed] [Google Scholar]
- 8.Kanakry JA, Ambinder RF. 2013. EBV-related lymphomas: new approaches to treatment. Curr. Treat. Options Oncol. 14:224–236. 10.1007/s11864-013-0231-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Velzen M, van de Vijver DA, van Loenen FB, Osterhaus AD, Remeijer L, Verjans GM. 2013. Acyclovir prophylaxis predisposes to antiviral-resistant recurrent herpetic keratitis. J. Infect. Dis. 208:1359–1365. 10.1093/infdis/jit350 [DOI] [PubMed] [Google Scholar]
- 10.Fife KH, Crumpacker CS, Mertz GJ, Hill EL, Boone GS. 1994. Recurrence and resistance patterns of herpes simplex virus following cessation of > or = 6 years of chronic suppression with acyclovir. Acyclovir Study Group. J. Infect. Dis. 169:1338–1341. 10.1093/infdis/169.6.1338 [DOI] [PubMed] [Google Scholar]
- 11.James SH, Prichard MN. 2013. A possible pitfall in acyclovir prophylaxis for recurrent herpetic keratitis? J. Infect. Dis. 208:1353–1355. 10.1093/infdis/jit379 [DOI] [PubMed] [Google Scholar]
- 12.Burrel S, Boutolleau D, Azar G, Doan S, Deback C, Cochereau I, Agut H, Gabison EE. 2013. Phenotypic and genotypic characterization of acyclovir-resistant corneal HSV-1 isolates from immunocompetent patients with recurrent herpetic keratitis. J. Clin. Virol. 58:321–324. 10.1016/j.jcv.2013.05.001 [DOI] [PubMed] [Google Scholar]
- 13.Duan R, de Vries RD, Osterhaus AD, Remeijer L, Verjans GM. 2008. Acyclovir-resistant corneal HSV-1 isolates from patients with herpetic keratitis. J. Infect. Dis. 198:659–663. 10.1086/590668 [DOI] [PubMed] [Google Scholar]
- 14.Pan D, Kaye SB, Hopkins M, Kirwan R, Hart IJ, Coen DM. 2013. Common and new acyclovir resistant herpes simplex virus-1 mutants causing bilateral recurrent herpetic keratitis in an immunocompetent patient. J. Infect. Dis. 209:345–349. 10.1093/infdis/jit437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dyson MR, Coe PL, Walker RT. 1991. The synthesis and antiviral activity of some 4′-thio-2′-deoxy nucleoside analogues. J. Med. Chem. 34:2782–2786. 10.1021/jm00113a016 [DOI] [PubMed] [Google Scholar]
- 16.Secrist JA, III, Tiwari KN, Riordan JM, Montgomery JA. 1991. Synthesis and biological activity of 2′-deoxy-4′-thio pyrimidine nucleosides. J. Med. Chem. 34:2361–2366. 10.1021/jm00112a007 [DOI] [PubMed] [Google Scholar]
- 17.Haraguchi K, Takahashi H, Tanaka H, Hayakawa H, Ashida N, Nitanda T, Baba M. 2004. Synthesis and antiviral activities of 1′-carbon-substituted 4′-thiothymidines. Bioorg. Med. Chem. 12:5309–5316. 10.1016/j.bmc.2004.07.057 [DOI] [PubMed] [Google Scholar]
- 18.Prichard MN, Quenelle DC, Hartline CB, Harden EA, Jefferson G, Frederick SL, Daily SL, Whitley RJ, Tiwari KN, Maddry JA, Secrist JA, III, Kern ER. 2009. Inhibition of herpesvirus replication by 5-substituted 4′-thiopyrimidine nucleosides. Antimicrob. Agents Chemother. 53:5251–5258. 10.1128/AAC.00417-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rahim SG, Trivedi N, Bogunovic-Batchelor MV, Hardy GW, Mills G, Selway JW, Snowden W, Littler E, Coe PL, Basnak I, Whale RF, Walker RT. 1996. Synthesis and anti-herpes virus activity of 2′-deoxy-4′-thiopyrimidine nucleosides. J. Med. Chem. 39:789–795. 10.1021/jm950029r [DOI] [PubMed] [Google Scholar]
- 20.Kern ER, Prichard MN, Quenelle DC, Keith KA, Tiwari KN, Maddry JA, Secrist JA., III 2009. Activities of certain 5-substituted 4′-thiopyrimidine nucleosides against orthopoxvirus infections. Antimicrob. Agents Chemother. 53:572–579. 10.1128/AAC.01257-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duraffour S, Drillien R, Haraguchi K, Balzarini J, Topalis D, van den Oord JJ, Andrei G, Snoeck R. 2013. KAY-2-41, a novel nucleoside analogue inhibitor of orthopoxviruses in vitro and in vivo. Antimicrob. Agents Chemother. 58:27–37. 10.1128/AAC.01601-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haraguchi K, Shimada H, Tanaka H, Hamasaki T, Baba M, Gullen EA, Dutschman GE, Cheng YC. 2008. Synthesis and anti-HIV activity of 4′-substituted 4′-thiothymidines: a new entry based on nucleophilic substitution of the 4′-acetoxy group. J. Med. Chem. 51:1885–1893. 10.1021/jm070824s [DOI] [PubMed] [Google Scholar]
- 23.Coen N, Singh U, Vuyyuru V, Van den Oord JJ, Balzarini J, Duraffour S, Snoeck R, Cheng YC, Chu CK, Andrei G. 2013. Activity and mechanism of action of HDVD, a novel pyrimidine nucleoside derivative with high levels of selectivity and potency against gammaherpesviruses. J. Virol. 87:3839–3851. 10.1128/JVI.03338-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrei G, Balzarini J, Fiten P, De Clercq E, Opdenakker G, Snoeck R. 2005. Characterization of herpes simplex virus type 1 thymidine kinase mutants selected under a single round of high-dose brivudin. J. Virol. 79:5863–5869. 10.1128/JVI.79.9.5863-5869.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedrichs C, Neyts J, Gaspar G, De Clercq E, Wutzler P. 2004. Evaluation of antiviral activity against human herpesvirus 8 (HHV-8) and Epstein-Barr virus (EBV) by a quantitative real-time PCR assay. Antiviral Res. 62:121–123. 10.1016/j.antiviral.2003.12.005 [DOI] [PubMed] [Google Scholar]
- 26.Verri A, Focher F, Duncombe RJ, Basnak I, Walker RT, Coe PL, de Clercq E, Andrei G, Snoeck R, Balzarini J, Spadari S. 2000. Anti-(herpes simplex virus) activity of 4′-thio-2′-deoxyuridines: a biochemical investigation for viral and cellular target enzymes. Biochem. J. 351(Pt 2):319–326. 10.1042/0264-6021:3510319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nash AA, Dutia BM, Stewart JP, Davison AJ. 2001. Natural history of murine gamma-herpesvirus infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356:569–579. 10.1098/rstb.2000.0779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flano E, Husain SM, Sample JT, Woodland DL, Blackman MA. 2000. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J. Immunol. 165:1074–1081. 10.4049/jimmunol.165.2.1074 [DOI] [PubMed] [Google Scholar]
- 29.Coen N, Duraffour S, Naesens L, Krecmerová M, Van den Oord J, Snoeck R, Andrei G. 2013. Evaluation of novel acyclic nucleoside phosphonates against human and animal gammaherpesviruses revealed an altered metabolism of cyclic prodrugs upon Epstein-Barr virus reactivation in P3HR-1 cells. J. Virol. 87:12422–12432. 10.1128/JVI.02231-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pérez-Pérez MJ, Hernández AI, Priego EM, Rodríguez-Barrios F, Gago F, Camarasa MJ, Balzarini J. 2005. Mitochondrial thymidine kinase inhibitors. Curr. Top. Med. Chem. 5:1205–1219. 10.2174/156802605774463097 [DOI] [PubMed] [Google Scholar]
- 31.Sasadeusz JJ, Tufaro F, Safrin S, Schubert K, Hubinette MM, Cheung PK, Sacks SL. 1997. Homopolymer mutational hot spots mediate herpes simplex virus resistance to acyclovir. J. Virol. 71:3872–3878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown DG, Visse R, Sandhu G, Davies A, Rizkallah PJ, Melitz C, Summers WC, Sanderson MR. 1995. Crystal structures of the thymidine kinase from herpes simplex virus type-1 in complex with deoxythymidine and ganciclovir. Nat. Struct. Biol. 2:876–881. 10.1038/nsb1095-876 [DOI] [PubMed] [Google Scholar]
- 33.Graham D, Larder BA, Inglis MM. 1986. Evidence that the ‘active centre' of the herpes simplex virus thymidine kinase involves an interaction between three distinct regions of the polypeptide. J. Gen. Virol. 67(Pt 4):753–758 [DOI] [PubMed] [Google Scholar]
- 34.Gaudreau A, Hill E, Balfour HH, Jr, Erice A, Boivin G. 1998. Phenotypic and genotypic characterization of acyclovir-resistant herpes simplex viruses from immunocompromised patients. J. Infect. Dis. 178:297–303. 10.1086/515626 [DOI] [PubMed] [Google Scholar]
- 35.Harris W, Collins P, Fenton RJ, Snowden W, Sowa M, Darby G. 2003. Phenotypic and genotypic characterization of clinical isolates of herpes simplex virus resistant to aciclovir. J. Gen. Virol. 84:1393–1401. 10.1099/vir.0.18880-0 [DOI] [PubMed] [Google Scholar]
- 36.Morfin F, Souillet G, Bilger K, Ooka T, Aymard M, Thouvenot D. 2000. Genetic characterization of thymidine kinase from acyclovir-resistant and -susceptible herpes simplex virus type 1 isolated from bone marrow transplant recipients. J. Infect. Dis. 182:290–293. 10.1086/315696 [DOI] [PubMed] [Google Scholar]
- 37.Schmit I, Boivin G. 1999. Characterization of the DNA polymerase and thymidine kinase genes of herpes simplex virus isolates from AIDS patients in whom acyclovir and foscarnet therapy sequentially failed. J. Infect. Dis. 180:487–490. 10.1086/314900 [DOI] [PubMed] [Google Scholar]
- 38.Piret J, Boivin G. 2011. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob. Agents Chemother. 55:459–472. 10.1128/AAC.00615-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andrei G, Topalis D, Fiten P, McGuigan C, Balzarini J, Opdenakker G, Snoeck R. 2012. In vitro-selected drug-resistant varicella-zoster virus mutants in the thymidine kinase and DNA polymerase genes yield novel phenotype-genotype associations and highlight differences between antiherpesvirus drugs. J. Virol. 86:2641–2652. 10.1128/JVI.06620-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frobert E, Ooka T, Cortay JC, Lina B, Thouvenot D, Morfin F. 2005. Herpes simplex virus thymidine kinase mutations associated with resistance to acyclovir: a site-directed mutagenesis study. Antimicrob. Agents Chemother. 49:1055–1059. 10.1128/AAC.49.3.1055-1059.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horsburgh BC, Chen SH, Hu A, Mulamba GB, Burns WH, Coen DM. 1998. Recurrent acyclovir-resistant herpes simplex in an immunocompromised patient: can strain differences compensate for loss of thymidine kinase in pathogenesis? J. Infect. Dis. 178:618–625. 10.1086/515375 [DOI] [PubMed] [Google Scholar]
- 42.Munir KM, French DC, Dube DK, Loeb LA. 1992. Permissible amino acid substitutions within the putative nucleoside binding site of herpes simplex virus type 1 encoded thymidine kinase established by random sequence mutagenesis [corrected]. J. Biol. Chem. 267:6584–6589 [PubMed] [Google Scholar]
- 43.Wu CC, Chen MC, Chang YR, Hsu TY, Chen JY. 2004. Identification and characterization of the conserved nucleoside-binding sites in the Epstein-Barr virus thymidine kinase. Biochem. J. 379:795–803. 10.1042/BJ20031832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flaño E, Woodland DL, Blackman MA. 2002. A mouse model for infectious mononucleosis. Immunol. Res. 25:201–217. 10.1385/IR:25:3:201 [DOI] [PubMed] [Google Scholar]
- 45.Tripp RA, Hamilton-Easton AM, Cardin RD, Nguyen P, Behm FG, Woodland DL, Doherty PC, Blackman MA. 1997. Pathogenesis of an infectious mononucleosis-like disease induced by a murine gamma-herpesvirus: role for a viral superantigen? J. Exp. Med. 185:1641–1650. 10.1084/jem.185.9.1641 [DOI] [PMC free article] [PubMed] [Google Scholar]