

Abstract
This study was designed to verify the in vivo efficacy of sulfoxide and sulfone fexinidazole metabolites following oral administration in a murine model of Chagas disease. Female Swiss mice infected with the Y strain of Trypanosoma cruzi were treated orally once per day with each metabolite at doses of 10 to 100 mg/kg of body weight for a period of 20 days. Parasitemia was monitored throughout, and cures were detected by parasitological and PCR assays. The results were compared with those achieved with benznidazole treatment at the same doses. Fexinidazole metabolites were effective in reducing the numbers of circulating parasites and protecting mice against death, compared with untreated mice, but without providing cures at daily doses of 10 and 25 mg/kg. Both metabolites were effective in curing mice at 50 mg/kg/day (30% to 40%) and 100 mg/kg/day (100%). In the benznidazole-treated group, parasitological cure was detected only in animals treated with the higher dose of 100 mg/kg/day (80%). Single-dose pharmacokinetic parameters for each metabolite were obtained from a parallel group of uninfected mice and were used to estimate the profiles following repeated doses. Pharmacokinetic data suggested that biological efficacy most likely resides with the sulfone metabolite (or subsequent reactive metabolites formed following reduction of the nitro group) following administration of either the sulfoxide or the sulfone and that prolonged plasma exposure over the 24-h dosing window is required to achieve high cure rates. Fexinidazole metabolites were effective in treating T. cruzi in a mouse model of acute infection, with cure rates superior to those achieved with either fexinidazole itself or benznidazole.
INTRODUCTION
Chagas disease, or American trypanosomiasis, represents an important public health problem in Latin America. The prevalence of Chagas disease in the poorer parts of the world has meant that it has been largely neglected, with limited progress being made in the identification of new drugs for treatment. The nitroheterocyclic drugs nifurtimox (Lampit) and benznidazole (Rochagan or Radanil), which were introduced in the 1960s and 1970s, are available for the treatment of Chagas disease; however, these drugs are unsatisfactory due to toxicity, long treatment regimens, and lack of efficacy against the chronic forms of the disease (1).
There is a continuing need for safe and cost-effective drugs suitable for the control of Chagas disease. Nitroimidazoles are a well-known class of pharmacologically active compounds, several of which have shown good activity against trypanosomes (2). In the early 1980s, the nitroimidazole fexinidazole was shown to have activity against Trypanosoma cruzi, trichomonads, and Entamoeba histolytica (3). More recently, the molecule was “rediscovered” and selected for development by the Drugs for Neglected Diseases initiative (DNDi) in the exploration of new and old nitroimidazoles as drug leads for human African trypanosomiasis and Chagas disease. Fexinidazole has recently been identified as a promising new drug candidate for the treatment of African trypanosomiasis (4, 5). Fexinidazole is orally available and is rapidly converted by oxidative metabolism to two metabolites, the sulfoxide and the sulfone, which have equivalent in vitro activities against Trypanosoma brucei rhodesiense and T. brucei gambiense, compared with the parent compound, and contribute significantly to the in vivo efficacy in animal models of both stage 1 and stage 2 African trypanosomiasis (4). The DNDi carried out phase I clinical trials in 2010 and 2011, assessing the safety and pharmacokinetics of fexinidazole in human volunteers given a single dose or multiple doses, and a phase II/III study of clinical safety and efficacy in sleeping sickness patients was commenced in 2012 (6).
Fexinidazole was described previously as having superior efficacy, compared with the current standard of care, i.e., benznidazole or nifurtimox, in an acute murine infection model of Chagas disease using the T. cruzi Brazil 32 strain (3). More recently, studies have confirmed that fexinidazole is effective in curing experimental T. cruzi infections in models of both acute and chronic stages of the disease, including infections with benznidazole-resistant T. cruzi strains (7). At higher doses, fexinidazole treatment was shown to achieve better cure rates and prevention of cardiac inflammation than was benznidazole.
The aim of the present study was to characterize the anti-T. cruzi activity of the two principal oxidative metabolites of fexinidazole, namely, fexinidazole sulfoxide and fexinidazole sulfone, using an in vivo screening model of Chagas disease. In addition, the pharmacokinetic properties of the two metabolites following oral administration were evaluated in a parallel group of uninfected mice, to assess the time course of exposure for each compound and to relate this exposure to efficacy.
MATERIALS AND METHODS
Study drugs.
The fexinidazole sulfoxide and sulfone derivatives, i.e., 1-methyl-2-(4-methylsulfinyl phenoxymethyl)-5-nitroimidazole and 1-methyl-2-(4-methylsulfonyl phenoxymethyl)-5-nitroimidazole, respectively, were prepared by Axyntis/Centipharm (France) and were provided by the Drugs for Neglected Diseases initiative (DNDi). Fexinidazole metabolites were each formulated in an aqueous suspension medium containing methyl cellulose (0.5% [wt/vol]) and polysorbate 80 (5% [vol/vol]). Benznidazole (2-nitroimidazole-N-benzyl-2-nitro-1-imidazole acetamide) (Lafepe, Brazil) was used as the reference treatment and was administered orally, in an aqueous suspension containing 4% (wt/vol) methyl cellulose, for the efficacy studies.
Parasite strain.
The Trypanosoma cruzi Y (DTU II) strain (8) was used in this study. The Y strain is partially resistant to benznidazole and was used as the standard strain since, in Swiss mice, it induces high levels of parasitemia and 100% mortality rates, which is generally observed 10 to 19 days postinfection in the absence of treatment.
In vitro activity assessment.
For in vitro assays, stock solutions (100 mM for fexinidazole sulfoxide, fexinidazole sulfone, and benznidazole) were prepared in dimethyl sulfoxide (DMSO). All subsequent dilutions were prepared in fresh culture medium (RPMI 1640 medium) on the day of the assay. The final DMSO concentration never exceeded 0.05% and had no deleterious effect on parasite growth. For analysis of the effects against intracellular parasites, the maximum concentration was 120 μM for each of the fexinidazole metabolites and 60 μM for benznidazole, and a seven-point profile with 3-fold serial dilutions was used in a 72-h assay. The highly invasive trypomastigotes of the Y strain of T. cruzi, which were shown to infect >90% of exposed murine macrophages, were used for evaluation of the activities of the fexinidazole metabolites in cellular infection assays. Murine peritoneal macrophages were harvested 24 h after starch (Life Technologies) induction (2% aqueous starch solution, administered intraperitoneally), and 500-μl aliquots were dispensed into 24-well tissue culture plates at 4 × 104 cells/well. After 24 h, the cells were infected with trypomastigotes (obtained from Vero cells) at a ratio of 5 parasites to 1 macrophage. After 24 h of incubation, the cultures were exposed to the compounds for 3 days. All tissue culture plates were maintained at 37°C in a 5% CO2-air mixture. After 72 h, the cultures were fixed with methanol, stained with Giemsa stain, and examined microscopically to determine the percentages of infected cells in treated and untreated control cultures. IC50s were calculated using CalcuSyn software (Biosoft, United Kingdom).
In vivo efficacy studies.
All procedures and experimental protocols were conducted in accordance with Brazilian School of Animal Experimentation (COBEA) guidelines for the use of animals in research and were approved by the Ethics Committee in Animal Research at Ouro Preto Federal University (UFOP) (Brazil). Female Swiss mice from the animal facility at UFOP were used in this study. Mice were maintained on a diet of commercial food, and water was available ad libitum. Mice (20 to 24 g) were inoculated intraperitoneally with 5.0 × 103 blood-stage trypomastigotes. Groups (10 animals/group) were treated by oral gavage (200 μl) with doses of 10, 25, 50, and 100 mg/kg of body weight fexinidazole sulfoxide or fexinidazole sulfone daily for 20 days, beginning at the time of detection of parasitemia, which generally occurred 4 days postinfection. Parasitological cure was determined as described below, and the results were compared with those achieved with benznidazole at the same doses. A group of 10 animals that were infected with the parasite but received no treatment was used as a control group.
Determination of parasitological cure.
Parasitological cure was determined following the methodology described by Caldas et al. (9) and was based on two independent tests, namely, fresh blood examinations (FBEs) before and after cyclophosphamide immunosuppression and real-time PCR assays performed with blood samples from mice with negative parasitemia results. Animals with negative results for all tests were considered cured.
Fresh blood examination.
During all treatments and up to 30 days posttreatment, parasitemia was evaluated by FBEs to determine the potential for natural reactivation of infection. Animals that did not exhibit reactivation of parasitemia after treatment were subjected to immunosuppression consisting of three cycles of treatment with 50 mg/kg cyclophosphamide for 4 consecutive days, with intervals of 3 days between cycles. Parasitemia was evaluated during cyclophosphamide immunosuppression and for 10 days after immunosuppression.
Real-time PCR assay.
For quantitative PCR (qPCR) assays, animals were bled from the orbital venous sinus at 30 and 180 days posttreatment. Two hundred microliters of collected blood was mixed with 35 μl of sodium citrate solution (129 mM) and used for DNA extraction with the Wizard genomic DNA purification kit (Promega Corp., Madison, WI), with minor modifications (10). qPCR assays were performed using a SYBR green system (Roche Applied Science, Mannheim, Germany), according to the manufacturer's instructions, with the primers TCZ-F (5′-GCTCTTGCCCACAMGGGTGC-3′, where M indicates A or C) and TCZ-R (5′-CCAAGCAGCGGATAGTTCAGG-3′) (Invitrogen, Life Technologies, Grand Island, NY) for T. cruzi DNA amplification (11). The internal control (corresponding to a segment of the murine tumor necrosis factor alpha [TNF-α] gene) was amplified using the primers TNF-5241 (5′-TCCCTCTCATCAGTTCTATGGCCCA-3′) and TNF-5411 (5′-CAGCAAGCATCTATGCACTTAGACCCC-3′) (Invitrogen) (11). Amplification cycles were carried out in an ABI 7300 real-time PCR system (Applied Biosystems, Life Technologies). The cycles consisted of an initial denaturation step of 10 min at 95°C followed by 40 cycles of 15 s at 94°C and 1 min at 64.3°C, with fluorescence data acquisition. Amplification was immediately followed by a melting program with initial denaturation for 15 s at 95°C, cooling to 60°C for 1 min, and then a stepwise temperature increase from 60 to 95°C at 0.3°C/s. All samples were analyzed in duplicate, and negative samples and reagent controls were processed in parallel in each assay.
IgG levels.
Blood from treated mice was collected from the orbital venous sinus (500 μl) at 180 days after treatment completion. T. cruzi-specific antibodies were detected with the technique described by Voller et al. (12). Enzyme-linked immunosorbent assay (ELISA) plates were coated with T. cruzi antigen prepared by alkaline extraction from the Y strain during exponential growth in liver infusion tryptose (LIT) medium. Anti-mouse IgG peroxidase-conjugated antibody (Sigma Chemical Co., St. Louis, MO) was used. The mean values for absorbance at 490 nm for 10 negative-control samples were used to determine the reactivity index value, which was obtained by dividing the absorbance for each serum sample by the mean value for the control sample.
Myocardial tissue assessment.
For analysis of cellular inflammatory infiltration, mice were sacrificed 180 days posttreatment completion, and heart tissues were fixed with 10% formalin and embedded in paraffin. Blocks were cut into 4-μm sections and stained with hematoxylin and eosin for inflammation assessment. Twenty fields from hematoxylin/eosin-treated slides were randomly chosen at ×40 magnification, for a total area of analyzed myocardium of 1.49 by 106 μm2. Images were captured using a Leica DM 5000 B microcamera (Leica Application Suite, model 2.4.0R1; Leica Camera AG, Germany) and were processed with a Leica QWin V3 image analyzer. The inflammatory process was evaluated using the correlation index for the number of inflammatory cells observed in myocardial muscle from uninfected versus infected animals (13).
Statistical analysis.
Morphological and serological data were analyzed with Tukey's nonparametric multiple-comparison test, with significance set at an α value of 0.05.
Pharmacokinetic studies.
Pharmacokinetic studies were conducted using established procedures, in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, and the study protocols were reviewed and approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee. Fexinidazole sulfoxide and fexinidazole sulfone were formulated as described for the efficacy studies and were administered orally by gavage (10-ml/kg dose volume), at doses of 10 to 100 mg/kg, to uninfected female Swiss mice weighing 20 to 26 g. Benznidazole was formulated in 10% (vol/vol) polyethylene glycol (PEG) 400 with 0.4% (vol/vol) Tween 80 and was administered by gavage (10 ml/kg), at 100 mg/kg, to uninfected female Swiss mice weighing 23 to 26 g. While this formulation differs from that used in the efficacy studies, previous studies in our lab have shown that these formulations are likely to provide similar absorption and bioavailability for benznidazole. Mice had free access to food and water both prior to and following compound administration. Following administration, blood samples were collected for up to 30 h (3 samples per time point). No more than two samples were obtained from each mouse, with samples being taken via either submandibular bleeding (conscious sampling) or terminal cardiac puncture (under inhaled isoflurane anesthesia). Blood was collected directly into polypropylene Eppendorf tubes containing heparin as anticoagulant and a stabilization cocktail (containing Complete protease inhibitor cocktail [Roche, Switzerland], potassium fluoride, and EDTA), to minimize the potential for ex vivo degradation prior to analysis. Blood samples were centrifuged immediately after collection, and the plasma supernatant was removed and stored at −20°C until analysis by liquid chromatography-mass spectrometry (LC-MS).
Concentrations of fexinidazole, fexinidazole sulfoxide, and fexinidazole sulfone were determined by LC-MS. Plasma proteins were precipitated with the addition of acetonitrile (2:1, relative to the plasma volume), and the supernatant was removed following centrifugation. Chromatography was conducted using a Waters (Waters Corp., Milford, MA) Acquity ultraperformance liquid chromatography (UPLC) system coupled to either a Waters Micromass Xevo TQ (for fexinidazole and metabolites) or a Waters Micromass Quattro Premier (for benznidazole) mass spectrometer, a Supelco Ascentis Express RP-Amide column (50 mm by 2.1 mm, 2.7-μm particle size; Sigma-Aldrich, St. Louis, MO), and a mobile phase consisting of either acetonitrile (for fexinidazole and metabolites) or methanol (for benznidazole) and water, with both phases containing 0.5% formic acid. The mobile-phase solvents were delivered using a linear gradient from 0 to 100% acetonitrile or methanol, with reequilibration prior to the next injection. The injection volume was 3 μl, the flow rate was 0.4 ml/min, and the cycle time was approximately 4 min. Detection was achieved using positive electrospray ionization with multiple-reaction monitoring, a cone voltage of 30 to 35 V, and a collision energy of 15 V. Quantitation was achieved by comparison of the sample responses to calibration curves prepared by spiking compounds into blank mouse plasma and processing the samples in an identical manner. The accuracy of the assay for each analyte was within ±10% and precision was <10% across the calibration range of 0.001 to 100 μg/ml.
Plasma concentration-time data were analyzed by noncompartmental methods with sparse sampling, using WinNonlin 5.2.1 (Pharsight, Certara USA, Inc., St. Louis, MO). Parameters included the maximum plasma concentration (Cmax), the time to reach the maximum plasma concentration (Tmax), the area under the plasma concentration-time curve (AUC) extrapolated to infinity (AUCinf), and the terminal elimination half-life. Mean concentration-time profiles for benznidazole, fexinidazole sulfoxide, and fexinidazole sulfone at 100 mg/kg were fit to a one-compartment model with first-order absorption and elimination (WinNonlin), using a weighting factor of 1/y or 1/y2, where y is the concentration, and the estimated parameters were used to simulate the profiles following repeated dosing, with the assumption that there was no change in the kinetics across the 20-day dosing period.
RESULTS
In vitro activity of fexinidazole metabolites.
First, we investigated the activities of the fexinidazole metabolites with intracellular parasites by incubating T. cruzi-infected macrophages with selected nontoxic concentrations of each metabolite. Microscopic examination of the macrophages showed that the fexinidazole metabolites were not cytotoxic at concentrations of up to 120 μM. Direct quantification of the numbers of parasites in Giemsa-stained T. cruzi-infected cultures after 72 h of treatment showed a concentration-dependent reduction in the number of infected cells with both of the fexinidazole metabolites and benznidazole (Fig. 1 and 2). The fexinidazole metabolites exhibited similar IC50s in the low micromolar range, i.e., 1.8 μg/ml (5.8 μM) for the sulfone and 1.6 μg/ml (5.4 μM) for the sulfoxide, similar to that observed for benznidazole (IC50 of 1.8 μg/ml [6.9 μM]).
FIG 1.
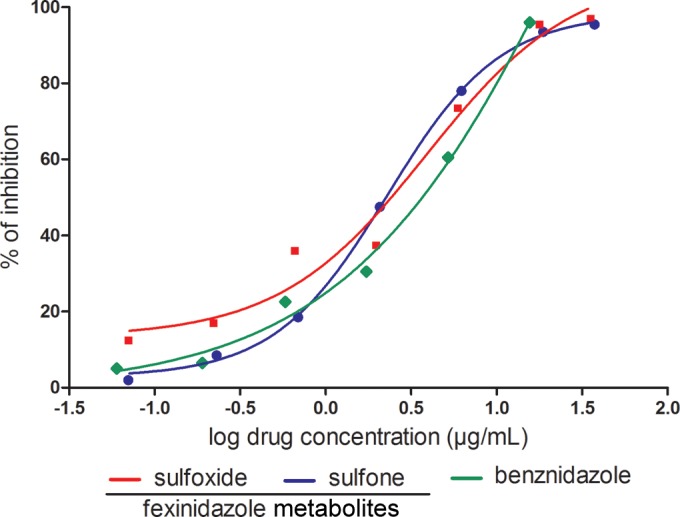
In vitro assay with Trypanosoma cruzi. The comparative concentration-dependent antiparasitic effects of fexinidazole metabolites and benznidazole were evaluated in murine macrophages infected with T. cruzi Y strain, in a 72-h assay. Data represent the mean values of percent inhibition, which were obtained by dividing the number of infected cells in treated cultures by the mean value for infected control cultures and multiplying the result by 100. The results represent the means of results from two independent experiments.
FIG 2.
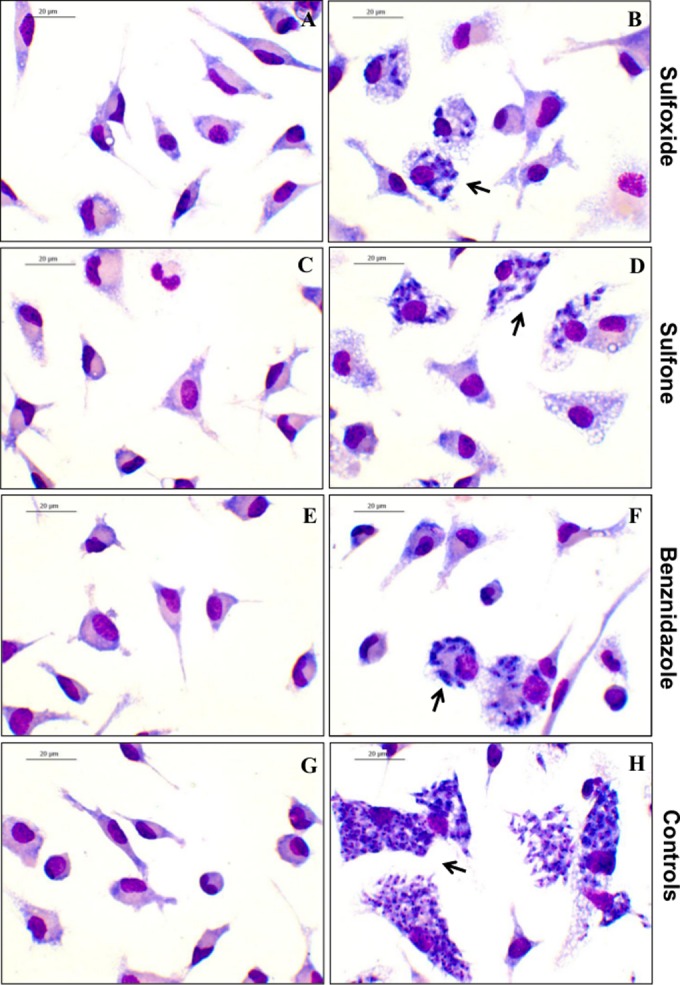
Activity of fexinidazole metabolites against intracellular amastigotes of Trypanosoma cruzi. Peritoneal murine macrophages were infected with T. cruzi Y strain and incubated with different concentrations of fexinidazole metabolites or benznidazole for 72 h. Methanol-fixed cells were stained with Giemsa. Arrows, intracellular parasites. (A, C, and E) Drug activity at the 90% inhibitory concentration (IC90) (note the absence of parasites); (B, D, and F) drug activity at the IC50; (G) uninfected cells; (H) infected and untreated cells. No cytotoxic effects were observed in host cells.
Tolerability of fexinidazole metabolites.
Both of the fexinidazole metabolites were well tolerated in the mouse model, and mortality due to high levels of parasitemia was observed only among animals treated with 10 or 25 mg/kg/day. No deaths were detected at the higher doses of 50 and 100 mg/kg/day (Table 1). Additionally, no significant differences in weight loss/gain were found among animals treated with 50 or 100 mg/kg/day and uninfected control animals (Fig. 3). Lower doses of fexinidazole sulfoxide (10 and 25 mg/kg/day) or fexinidazole sulfone (10 mg/kg/day) were also effective in preventing weight loss, relative to infected and untreated animals but not relative to uninfected animals. In contrast, similar weight losses were detected among infected and untreated animals and animals treated with benznidazole at 10 or 25 mg/kg/day.
TABLE 1.
Efficacy of fexinidazole sulfone, fexinidazole sulfoxide, and benznidazole in a murine model of acute Trypanosoma cruzi infectiona
| Group | Dose (mg/kg/day) | No. with parasitemia clearance/total no. | No. with parasitemia PT/total no.b | No. with positive PCR results 1 and/or 6 mo PT/total no. | No. surviving until 1 mo PT/total no. (%) | No. with positive test results/total no. (%) |
|---|---|---|---|---|---|---|
| Control uninfected | 0 | ND | ND | 0/10 | 6/6 (100) | 0/6 (0) |
| Control infected | 0 | ND | ND | ND | 0/6 (0) | 6/6 (100) |
| Benznidazole | 10 | 0/7 | 3/3c | ND | 3/7 (43) | 7/7 (100) |
| 25 | 0/7 | 7/7 | ND | 6/7 (86) | 7/7 (100) | |
| 50 | 7/7 | 5/7 | 2/2 | 7/7 (100) | 7/7 (100) | |
| 100 | 10/10 | 1/10 | 1/9 | 10/10 (100) | 2/10 (20) | |
| Sulfoxide metabolite | 10 | 0/10 | 3/3d | ND | 3/10 (30) | 10/10 (100) |
| 25 | 7/10 | 5/7 | 2/2 | 8/10 (80) | 10/10 (100) | |
| 50 | 10/10 | 7/10 | 0/3 | 10/10 (100) | 7/10 (70) | |
| 100 | 10/10 | 0/10 | 0/10 | 10/10 (100) | 0/10 (0) | |
| Sulfone metabolite | 10 | 0/10 | 3/3d | ND | 2/10 (20) | 10/10 (100) |
| 25 | 8/10 | 6/8 | 2/2 | 7/10 (70) | 10/10 (100) | |
| 50 | 10/10 | 6/10 | 0/4 | 10/10 (100) | 6/10 (60) | |
| 100 | 10/10 | 0/10 | 0/10 | 10/10 (100) | 0/10 (0) |
Swiss female mice (20 to 24 g) were inoculated with 5 × 103 trypomastigotes (Y strain). Treatment was initiated 4 days after inoculation, followed by 20 days of consecutive dosing; treatment was via the oral route.
PT, posttreatment; ND, not detected.
Four animals died before the end of treatment.
Seven animals died before the end of treatment.
FIG 3.
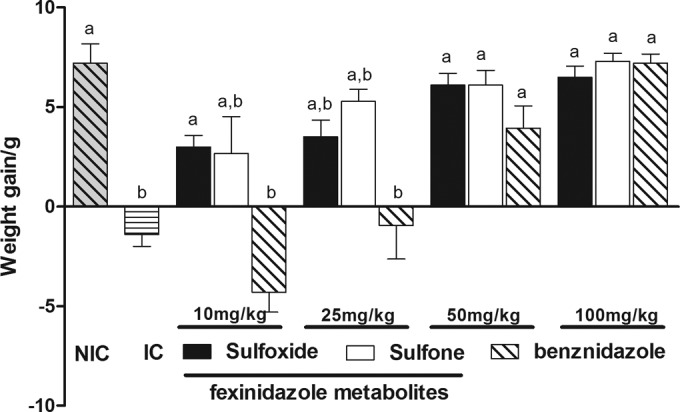
Weight loss or gain for animals infected with Trypanosoma cruzi Y strain and treated with fexinidazole sulfoxide or fexinidazole sulfone. Infected mice were treated once daily with 10, 25, 50, or 100 mg/kg of fexinidazole sulfoxide, fexinidazole sulfone, or benznidazole for 20 consecutive days, and results were compared with those for infected and untreated control (IC) and noninfected control (NIC) groups. The weight loss or gain was calculated as the difference between the weights of animals before and after treatment. The weights of animals in the uninfected control group were assessed over the same period. The weights of the animals in the infected and untreated control group were assessed 14 days after infection (all animals died before the end of the treatment). a, significant difference relative to infected control (P < 0.005); b, significant difference relative to uninfected control (P < 0.005).
In vivo efficacy of fexinidazole oxidative metabolites.
Both of the fexinidazole metabolites showed dose-related responses in the acute murine infection model with the T. cruzi Y strain, at oral doses of 10, 25, 50, and 100 mg/kg given once per day for 20 consecutive days. Parasitemia was suppressed in 70 to 100% of animals treated with 25 to 100 mg/kg/day (Table 1 and Fig. 4). This efficacy compares well with that of the currently used anti-T. cruzi drug benznidazole at doses of 50 and 100 mg/kg/day. At the lower doses (i.e., 10 mg/kg for the fexinidazole metabolites and 10 and 25 mg/kg for benznidazole), there was transient suppression of parasitemia, with subsequent relapse and increases in parasitemia.
FIG 4.
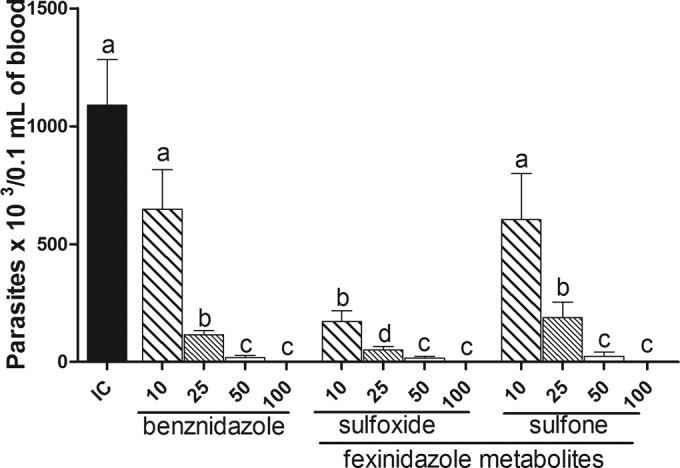
Parasitemia levels during and after oral administration of fexinidazole sulfoxide or fexinidazole sulfone. Bars represent the means of the maximum parasitemia values obtained in peripheral blood samples from mice (n = 10) infected with 5,000 trypomastigotes of Trypanosoma cruzi Y strain during and up to 30 days after 20 days of treatment with doses of 10, 25, 50, or 100 mg/kg/day of fexinidazole sulfoxide, fexinidazole sulfone, or benznidazole. IC, infected and untreated control. a, b, and c, bars with similar letters are not significantly different, and bars with different letters are significantly different (P < 0.05).
In assessments of the definitive clearance of parasitemia using a combination of parasitological and qPCR analyses, 100% of mice treated with 100 mg/kg/day of each of the fexinidazole metabolites were cured at the 6-month follow-up evaluation, versus 80% of mice treated with benznidazole at the same dose (Table 1). Lower doses of the fexinidazole metabolites were also effective in treating infections, with a dose of 50 mg/kg/day inducing a 30 to 40% cure rate, while the same dose of benznidazole cured none of the infected animals.
Analyses for T. cruzi-specific antibodies in blood samples collected from treated mice 6 months posttreatment were performed. For these analyses, animals were classified based on the results of parasitological and PCR evaluations (positive or negative test results). The results suggest that there was concordance among parasitological and serological test results, given that, 6 months after treatment of the acute infection, animals deemed to be cured by the parasitological tests also had reduced levels of specific anti-T. cruzi IgG antibodies compared with animals that were not cured (Fig. 5A).
FIG 5.

Anti-Trypanosoma cruzi activity of fexinidazole metabolites in mice infected with Y strain. Mice were inoculated intraperitoneally with 5,000 trypomastigotes of T. cruzi Y strain and treated with doses of 10, 25, 50, or 100 mg/kg fexinidazole sulfoxide or fexinidazole sulfone or 100 mg/kg benznidazole once daily for 20 consecutive days. (A) IgG antibodies in treated mice as detected by ELISA. The reactivity index was obtained by dividing the absorption value of each serum sample by the mean value of the uninfected control sample. (B) Myocardial inflammatory cell counts in mice infected with T. cruzi Y strain at 6 months posttreatment. (C) Hematoxylin/eosin-stained slides. Magnification, ×40. NIC, noninfected control; −50 and +50, mice treated with 50 mg/kg/day of fexinidazole sulfoxide or fexinidazole sulfone with negative (−50) or positive (+50) results for parasites in fresh blood examinations and qPCR assays. a and b, bars with similar letters are not significantly different, and bars with different letters are significantly different (P < 0.05).
The efficacy of the treatments in preventing chronic lesions in Y strain-infected mice was determined through the quantitative analysis of inflammatory cells present in the heart tissue of treated mice versus uninfected mice. The fexinidazole metabolites were shown to prevent or to lessen the typical inflammation of heart tissue associated with the chronic phase of experimental Chagas disease, showing similar levels of inflammatory cells in comparison with benznidazole at the effective dose (100 mg/kg). In contrast, animals treated with suboptimal doses demonstrated more inflammatory cells in the heart tissue than did either uninfected mice or infected mice that were treated and cured (Fig. 5B and C).
Pharmacokinetics of fexinidazole oxidative metabolites.
Following oral administration, fexinidazole sulfoxide was rapidly absorbed, with the time to reach the maximum plasma concentration (Tmax) being approximately 30 min (Table 2 and Fig. 6A). Plasma concentrations of fexinidazole sulfoxide declined with a half-life of approximately 1 to 2 h. For the three lower doses, concentrations could be detected only up to 8 h; for the highest dose, concentrations could be detected up to 24 h. Maximum plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC) extrapolated to infinity (AUCinf) values increased approximately in proportion to the increase in the fexinidazole sulfoxide dose over the range of 10 to 100 mg/kg (Table 2).
TABLE 2.
Pharmacokinetic parameters for fexinidazole, fexinidazole sulfoxide, and fexinidazole sulfone after single oral administration of fexinidazole sulfoxide or fexinidazole sulfone to uninfected mice
| Drug administered and dose (mg/kg) | Parameter | Fexinidazole | Fexinidazole sulfoxide | Fexinidazole sulfone |
|---|---|---|---|---|
| Fexinidazole sulfoxide | ||||
| 10 | Cmax (μg/ml) | 0.012 | 3.56 | 6.88 |
| Tmax (h) | 0.5 | 0.5 | 2.0 | |
| AUCinf (μg·h/ml) | CNCa | 7.68 | 43.4 | |
| Half-life (h) | CNC | 0.8 | 1.9 | |
| 25 | Cmax (μg/ml) | 0.053 | 13.1 | 14.8 |
| Tmax (h) | 0.5 | 0.5 | 2.0 | |
| AUCinf (μg·h/ml) | 0.070 | 27.1 | 117 | |
| Half-life (h) | 0.80 | 0.7 | 2.1 | |
| 50 | Cmax (μg/ml) | 0.130 | 21.6 | 35.4 |
| Tmax (h) | 0.5 | 0.5 | 2.0 | |
| AUCinf (μg·h/ml) | 0.180 | 54.4 | 308 | |
| Half-life (h) | 1.8 | 1.3 | 2.0 | |
| 100 | Cmax (μg/ml) | 0.330 | 32.9 | 58.4 |
| Tmax (h) | 0.25 | 0.5 | 4.0 | |
| AUCinf (μg·h/ml) | 0.480 | 160 | 806 | |
| Half-life (h) | 2.6 | 1.5 | 2.2 | |
| Fexinidazole sulfone | ||||
| 10 | Cmax (μg/ml) | ND | ND | 8.48 |
| Tmax (h) | 1.0 | |||
| AUCinf (μg·h/ml) | 46.6 | |||
| Half-life (h) | 2.0 | |||
| 25 | Cmax (μg/ml) | ND | ND | 21.0 |
| Tmax (h) | 1.0 | |||
| AUCinf (μg·h/ml) | 124 | |||
| Half-life (h) | 3.2 | |||
| 50 | Cmax (μg/ml) | ND | ND | 33.5 |
| Tmax (h) | 2.0 | |||
| AUCinf (μg·h/ml) | 304 | |||
| Half-life (h) | 2.3 | |||
| 100 | Cmax (μg/ml) | ND | ND | 67.6 |
| Tmax (h) | 2.0 | |||
| AUCinf (μg·h/ml) | 624 | |||
| Half-life (h) | 2.2 |
CNC, could not calculate; ND, not detected.
FIG 6.

Plasma concentration-time profiles following a single oral administration of fexinidazole sulfoxide or fexinidazole sulfone to mice. Mice were dosed orally with 10 (●), 25 (△), 50 (◼), or 100 (◇) mg/kg of fexinidazole sulfoxide or fexinidazole sulfone. (A) Fexinidazole sulfoxide concentrations after administration of fexinidazole sulfoxide; (B) fexinidazole concentrations after administration of fexinidazole sulfoxide; (C) fexinidazole sulfone concentrations after administration of fexinidazole sulfoxide; (D) fexinidazole sulfone concentrations after administration of fexinidazole sulfone.
Fexinidazole was detected as a metabolite following administration of fexinidazole sulfoxide; however, concentrations of fexinidazole were low and of short duration (Table 2). The sulfoxide was also extensively converted to fexinidazole sulfone, and Cmax and AUCinf values increased in a dose-proportional manner (Table 2 and Fig. 6C). Concentrations of the sulfone were measurable up to 24 h at the two lower doses of the sulfoxide and for the duration of the sampling period at the higher doses.
Following administration of fexinidazole sulfone, only the sulfone could be detected in plasma samples, and the half-life across the dose range was approximately 2 h (Fig. 6D). As with administration of the sulfoxide, Cmax and AUCinf values for the sulfone increased approximately in proportion to the increase in dose (Table 2). In comparisons of the sulfone profiles and parameters following administration of either fexinidazole sulfone or fexinidazole sulfoxide, Cmax and Tmax were marginally reduced and delayed, respectively, following dosing of the sulfoxide, while the half-life and AUCinf values were similar for the two dosing groups at each dose level. Oral administration of benznidazole at 100 mg/kg resulted in Cmax, AUCinf, and half-life values (34.8 μg/ml, 183 μg·h/ml, and 0.9 h, respectively) that were comparable to the data for fexinidazole sulfoxide, but AUC and half-life values were both lower than those seen for fexinidazole sulfone.
Figure 7 illustrates the simulated repeated-dose profiles for benznidazole, fexinidazole sulfoxide, and fexinidazole sulfone, each administered once daily at 100 mg/kg. Given the similarity in the profiles and pharmacokinetic parameters for the sulfone following administration of either the sulfoxide or the sulfone (Table 2 and Fig. 6C and D), simulations were conducted for the sulfone only following administration of the sulfone. The simulations suggest that accumulation with repeated administration is not likely for either the sulfoxide or the sulfone, consistent with previous reports on the metabolites following repeated administration of fexinidazole (4).
FIG 7.

Simulated plasma concentration-time profiles following repeated once-daily administration of 100 mg/kg benznidazole (A), fexinidazole sulfoxide (B), or fexinidazole sulfone (C) to mice. For ease of interpretation, only the first 3 days of the 20-day dosing regimen are shown. Symbols represent the experimental data, and lines represent the repeated-dose simulated profiles. Dashed lines indicate the in vitro IC50 for each compound.
DISCUSSION
Benznidazole has been the standard of care in the treatment of Chagas disease for many years, although its efficacy and tolerance profiles are suboptimal (14). Interest in fexinidazole, which also belongs to the nitroimidazole class of antiparasitic agents, has recently been rekindled following demonstration of its efficacy in the treatment of T. brucei gambiense and T. brucei rhodesiense (4, 5), Leishmania (15), and T. cruzi (7) in animal models of infection. Fexinidazole is known to undergo oxidative metabolism to form both the sulfoxide and the sulfone, and both of these metabolites are active against trypanosomes (4, 5). As in other nitroimidazole drugs, the nitro group present in fexinidazole and its oxidative metabolites would be expected to undergo reductive metabolism catalyzed by nitroreductases, to form the biologically active species (16).
The results of the present study suggest that higher cure rates can be achieved in animals infected with the T. cruzi Y strain following treatment with either fexinidazole sulfoxide or fexinidazole sulfone, compared with the same doses of benznidazole. Both the sulfoxide and the sulfone are also more efficacious than the parent drug, fexinidazole, for which a dose of 300 mg/kg provided an 80% cure rate (7). In addition, both of the fexinidazole metabolites were well tolerated in this animal model, corroborating the data of a number of studies that demonstrated that these drugs did not induce toxicity in different experimental models (2, 5, 7, 15, 17), on the basis of genotoxicity, cardiac, and neurobehavioral assessments.
For practical reasons, the pharmacokinetic properties of benznidazole, fexinidazole sulfoxide, and fexinidazole sulfone were assessed in parallel groups of uninfected mice, and parameter estimates for a one-compartment fit of the single-dose data were used to simulate the repeated-dose profiles at the 100-mg/kg dose level for each compound. This approach assumes that the pharmacokinetic profiles in uninfected mice are representative of those in infected mice and that there are no changes in the pharmacokinetic properties with repeated dosing. The latter assumption is likely valid, based on a previous report in which the pharmacokinetic properties of fexinidazole and the two oxidative metabolites were found to be similar following repeated oral administration of fexinidazole for 14 days to either rats or dogs (4).
Following administration of the sulfoxide, both fexinidazole and fexinidazole sulfone were detected as metabolites, with fexinidazole presumably being formed via metabolic reductases. Fexinidazole concentrations were relatively low and declined rapidly; therefore, this species is not likely to have contributed significantly to the efficacy profile. Interestingly, the AUC values for fexinidazole sulfone were comparable following dosing of either the sulfoxide or the sulfone itself (Table 2), suggesting that the sulfoxide is extensively converted to the sulfone. The results further suggest that there is no benefit in terms of efficacy in having both the sulfoxide and the sulfone present, compared to only the sulfone, given that the cure rates were similar for treatment with either fexinidazole sulfoxide or fexinidazole sulfone.
Simulations of the repeated-dose profiles for both benznidazole and fexinidazole sulfoxide suggested that concentrations were likely to be below the in vitro IC50s of 1.8 and 1.6 μg/ml, respectively, for much of the 24-hour dosing window even at the highest dose of 100 mg/kg (Fig. 7A and B). In comparison, concentrations of the sulfone (after dosing of either the sulfoxide or the sulfone) are likely to remain above the IC50 of 1.8 μg/ml for most of the 24-h dosing window (Fig. 7C). Collectively, these results suggest that high concentrations need to be maintained to ensure efficacy and that the sulfone (with its associated nitroreduced metabolites) is the species most likely associated with efficacy following administration of either fexinidazole sulfoxide or fexinidazole sulfone.
In summary, the results presented here suggest that both fexinidazole sulfoxide and fexinidazole sulfone are effective in treating T. cruzi in a mouse model of acute infection, with cure rates being superior to those obtained with either fexinidazole itself (7) or benznidazole. Furthermore, the data suggest that the primary efficacious species are likely to be the sulfone and its nitroreduced metabolites and extended exposure throughout the dosing interval is necessary to achieve cures.
ACKNOWLEDGMENTS
We thank Isabela Ribeiro for critical reading of the manuscript.
This study was funded by the Drugs for Neglected Diseases initiative (DNDi) the Fundação de Amparo a Pesquisa do Estado de Minas Gerais, the Universidade Federal de Ouro Preto, and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (research fellowships to M.T.B. and A.T.) and was partially supported by a grant from the Australian Research Council. For the work described in this paper, the DNDi received financial support from the Department for International Development (United Kingdom), the Reconstruction Credit Institution-Federal Ministry of Education and Research (Germany), the Spanish Agency for International Development Cooperation (Spain), the Directorate-General for International Cooperation (Netherlands), and Doctors without Borders.
We have no conflicts of interest to declare.
Footnotes
Published ahead of print 19 May 2014
REFERENCES
- 1.Rassi A, Rassi A, Jr, Marin-Neto JA. 2010. Chagas disease. Lancet 375:1388–1402. 10.1016/S0140-6736(10)60061-X [DOI] [PubMed] [Google Scholar]
- 2.Sokolova AY, Wyllie S, Patterson S, Oza SL, Read KD, Fairlamb AH. 2010. Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrob. Agents Chemother. 54:2893–2900. 10.1128/AAC.00332-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raether W, Seidenath H. 1983. The activity of fexinidazole (HOE 239) against experimental infections with Trypanosoma cruzi, Trichomonas and Entamoeba histolytica. Ann. Trop. Med. Parasitol. 77:13–26 [DOI] [PubMed] [Google Scholar]
- 4.Torreele E, Trunz BB, Tweats D, Kaiser M, Brun R, Mazué G, Bray MA, Pécoul B. 2010. Fexinidazole: a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl. Trop. Dis. 4:e923. 10.1371/journal.pntd.0000923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaiser M, Bray MA, Cal M, Bourdin Trunz B, Torreele E, Brun R. 2011. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 55:5602–5608. 10.1128/AAC.00246-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tarral A, Blesson S, Mordt OV, Torreele E, Sassella D, Bray MA, Hovsepian L, Evène E, Gualano V, Felices M, Strub-Wourgaft N. 2014. Determination of an optimal dosing regimen for fexinidazole, a novel oral drug for the treatment of human African trypanosomiasis: first-in-human studies. Clin. Pharmacokinet. 53:565–580. 10.1007/s40262-014-0136-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bahia MT, de Andrade IM, Martins TA, do Nascimento AF, Diniz LF, Caldas IS, Talvani A, Trunz BB, Torreele E, Ribeiro I. 2012. Fexinidazole: a potential new drug candidate for Chagas disease. PLoS Negl. Trop. Dis. 6:e1870. 10.1371/journal.pntd.0001870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moreno M, D'Avila DA, Silva MN, Galvão LM, Macedo AM, Chiari E, Gontijo ED, Zingales B. 2010. Trypanosoma cruzi benznidazole susceptibility in vitro does not predict the therapeutic outcome of human Chagas disease. Mem. Inst. Oswaldo Cruz 105:918–924. 10.1590/S0074-02762010000700014 [DOI] [PubMed] [Google Scholar]
- 9.Caldas S, Santos FM, Lana M, Diniz LF, Machado-Coelho GL, Veloso VM, Bahia MT. 2008. Trypanosoma cruzi: acute and long-term infection in the vertebrate host can modify the response to benznidazole. Exp. Parasitol. 118:315–323. 10.1016/j.exppara.2007.08.016 [DOI] [PubMed] [Google Scholar]
- 10.Caldas S, Caldas IS, Diniz Lde F, Lima WG, Oliveira Rde P, Cecílio AB, Ribeiro I, Talvani A, Bahia MT. 2012. Real-time PCR strategy for parasite quantification in blood and tissue samples of experimental Trypanosoma cruzi infection. Acta Trop. 123:170–177. 10.1016/j.actatropica.2012.05.002 [DOI] [PubMed] [Google Scholar]
- 11.Cummings KL, Tarleton RL. 2003. Rapid quantitation of Trypanosoma cruzi in host tissue by real-time PCR. Mol. Biochem. Parasitol. 129:53–59. 10.1016/S0166-6851(03)00093-8 [DOI] [PubMed] [Google Scholar]
- 12.Voller A, Bidwell DE, Bartlett A. 1976. Enzyme immunoassays in diagnostic medicine: theory and practice. Bull. World Health Organ. 53:55–65 [PMC free article] [PubMed] [Google Scholar]
- 13.Caldas IS, Talvani A, Caldas S, Carneiro CM, de Lana M, da Matta Guedes PM, Bahia MT. 2008. Benznidazole therapy during acute phase of Chagas disease reduces parasite load but does not prevent chronic cardiac lesions. Parasitol. Res. 103:413–421. 10.1007/s00436-008-0992-6 [DOI] [PubMed] [Google Scholar]
- 14.Guedes PMM, Fietto JLR, Lana M, Bahia MT. 2006. Advances in Chagas disease chemotherapy. Antiinfect. Agents Med. Chem. 5:175–186. 10.2174/187152106776359020 [DOI] [Google Scholar]
- 15.Wyllie S, Patterson S, Stojanovski L, Simeons FR, Norval S, Kime R, Read KD, Fairlamb AH. 2012. The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis. Sci. Transl. Med. 4:119re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilkinson SR, Bot C, Kelly JM, Hall BS. 2011. Trypanocidal activity of nitroaromatic prodrugs: current treatments and future perspectives. Curr. Top. Med. Chem. 11:2072–2084. 10.2174/156802611796575894 [DOI] [PubMed] [Google Scholar]
- 17.Tweats D, Bourdin Trunz B, Torreele E. 2012. Genotoxicity profile of fexinidazole: a drug candidate in clinical development for human African trypanomiasis (sleeping sickness). Mutagenesis 27:523–532. 10.1093/mutage/ges015 [DOI] [PubMed] [Google Scholar]