

Abstract
We investigated biofilms of two pathogens, Acinetobacter baumannii and Staphylococcus aureus, to characterize mechanisms by which the extracellular polymeric substance (EPS) found in biofilms can protect bacteria against tobramycin exposure. To do so, it is critical to study EPS-antibiotic interactions in a homogeneous environment without mass transfer limitations. Consequently, we developed a method to grow biofilms, harvest EPS, and then augment planktonic cultures with isolated EPS and tobramycin. We demonstrated that planktonic cultures respond differently to being treated with different types of EPS (A. baumannii versus S. aureus) in the presence of tobramycin. By harvesting EPS from the biofilms, we found that A. baumannii EPS acts as a “universal protector” by inhibiting tobramycin activity against bacterial cells regardless of species; S. aureus EPS did not show any protective ability in cell cultures. Adding Mg2+ or Ca2+ reduced the protective effect of A. baumannii EPS. Finally, when we selectively digested the proteins or DNA of the EPS, we found that the protective ability did not change, suggesting that neither has a significant role in protection. To the best of our knowledge, this is the first study that demonstrates how EPS protects pathogens against antibiotics in a homogeneous system without mass transfer limitations. Our results suggest that EPS protects biofilm communities, in part, by adsorbing antibiotics near the surface. This may limit antibiotic diffusion to the bottom of the biofilms but is not likely to be the only mechanism of protection.
INTRODUCTION
A bacterial biofilm represents a structurally complex system that protects the microbial community encompassed within the biofilm. Biofilms are known for their resistance to eradication and high tolerance toward antimicrobial treatments (1–3). Biofilm removal in a host or clinical setting is an ongoing challenge. Two opportunistic pathogens, Acinetobacter baumannii and Staphylococcus aureus, have been classified as a “serious” threat in hospitals by the Centers for Disease Control and Prevention (4). Many nosocomial and biofilm-related illnesses are associated with these particular pathogens, for example, pneumonia and bacteremia, along with common infections (5–8). Emerging drug-resistant strains of these bacteria have caused further treatment challenges (9).
One factor contributing to the success of these pathogens is the production of a biofilm-enveloping matrix of biomolecules, commonly called the extracellular polymeric substance (EPS). The EPS matrix is formed when biomolecules accumulate around the residing microorganisms, serving as a protective barrier (10, 11). Primarily composed of polysaccharides, proteins, and DNA, EPS accumulates through active cellular secretion, retention of cell lysis products, and adsorption of external molecules. EPS molecules use a diverse range of interactions for their cohesion, including electrostatic, ionic, and van der Waals interactions. These interactions, along with the molecular composition, give rise to many important properties, including electrical charge, three-dimensional (3D) structure, and porosity (3).
The type of polysaccharides present in EPS makes a significant contribution to the general properties of EPS, and these polysaccharides have been characterized in many cases. Furthermore, the genes responsible for the production of specific EPS polysaccharides have been identified, although expression of the polysaccharide is not guaranteed. For example, the pgaABCD gene cluster that is responsible for the production of poly-β-(1-6)-N-acetylglucosamine (PNAG) is present in A. baumannii, but PNAG production was not universal in all tested strains and levels of production varied throughout the producing strains. PNAG is also expressed by S. aureus (12), although it is a minor component of the EPS (13) and its production level is low under aerobic conditions (14). Both A. baumannii and S. aureus express capsular polysaccharides (referred to here as bound EPS [bEPS]) that may convey protective advantages for the cells (15–17).
The composition of EPS determines the type and extent of the protective advantages. For example, EPS that is rich in polysaccharides may bind significant quantities of water, staving off dehydration (18). EPS can act as a “backup” nutrient source, supplying the bacteria with energy under starvation conditions (19). It can also act as a barrier, protecting the hosts from attack by blocking, repelling, or trapping molecules and preventing them from reaching the bacteria (3).
Biofilms are known to have high resistance to antibiotics compared to their planktonic counterparts, although the causes for this are both biofilm and single cell related (20). Understanding the protective role of EPS would be beneficial to treating biofilm-related infections or diseases by offering clues to strategies that might be used to counteract or bypass biofilm protection (6). With respect to antibiotics, EPS slows or delays the penetration of aminoglycoside antibiotics into a biofilm in vivo. This observation can be linked to the presence of the polysaccharide alginate, which was thought to represent an aminoglycoside-alginate interaction (21, 22), although this interaction was later dismissed as an insignificant factor in overall resistance (10, 23). More recently, the EPS interaction with antibiotics has been attributed to binding by extracellular DNA (eDNA) (24). The majority of studies have investigated these interactions inside the biofilm, where many different factors may confound the observations. Changes in hydrodynamic conditions, diffusion processes, cell density, ionic strength, or other environmental factors could affect the structure and function of EPS. These factors, combined with the heterogeneous nature of EPS, make it difficult to determine exactly how EPS protects against antibiotics.
When EPS is studied separately from the biofilm community, however, it is possible to work in a more replicable and homogeneous environment. This allows us to determine the protective properties of EPS regardless of the biofilm's growth environment. Furthermore, allowing EPS and an antibiotic to interact in solution eliminates diffusion limitations and allows a more direct study of antibiotic interactions. To the best of our knowledge, the protective mechanism of EPS has never been evaluated in this type of model system.
In this study, we targeted biofilms of pathogens A. baumannii and S. aureus. Overall, our work was designed to address the following questions in a homogeneous system without a mass transfer limitation: (i) does biofilm EPS from these species protect against tobramycin, (ii) what component of EPS is protective, and (iii) can we reduce the protective ability of EPS? We first harvested EPS from mature biofilms in two steps, one to collect loosely associated EPS (laEPS) and the second to collect bound EPS (bEPS). Cell growth in planktonic cultures of A. baumannii and S. aureus was then studied in the presence of EPS and tobramycin. For selected control experiments, we also cultured Escherichia coli K-12 with EPS and tobramycin. Last, we investigated the role of divalent cations, extracellular proteins, and extracellular DNA as potential mechanisms by which EPS protects planktonic cells.
MATERIALS AND METHODS
Culture growth.
Acinetobacter baumannii ATCC strain BAA-1605, Staphylococcus aureus ATCC strain BAA-1747, and Escherichia coli K-12 were used for this study. Cultures were grown in tryptic soy broth (TSB; BD catalog no. 211825) at a concentration of 10 g/liter in deionized water. Cell cultures were grown for 16 h at 37°C at an agitation speed of 100 rpm. While all three bacteria were used for planktonic test cultures, only A. baumannii and S. aureus biofilms were grown and used for EPS harvesting.
Biofilm growth.
Biofilms were grown using a custom-built membrane bioreactor (MBR) (Fig. 1). The reactor was built using a Sterifil filter funnel from Millipore (catalog no. XX11J4750). The polyvinylidene fluoride membrane filter had a pore size of 0.1 μm (Millipore; catalog no. VVLP04700). The reactor and tubing were sterilized by autoclaving, and the filter membrane was sterilized by 15 min of exposure to UV radiation (27 kJ).
FIG 1.
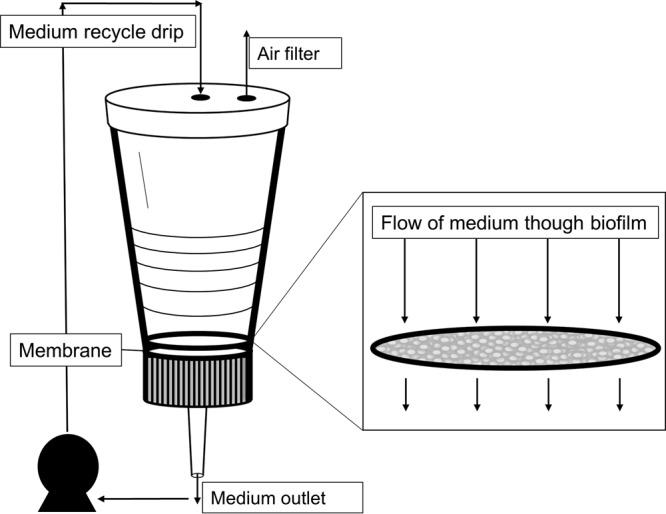
Biofilm membrane reactor. A pump is used to pull medium through the membrane. The medium exits the bottom of the funnel. It is recycled using a drip inlet through the top of the reactor. An air filter is used to maintain constant pressure. The closeup of the membrane shows the biofilm forming on the surface.
After sterilization, the membrane was placed in the funnel, and 25 ml of sterilized deionized water with 100 μg/ml carbenicillin (Sigma; catalog no. 1389) was passed through the membrane filter. Next, 50 ml of 0.33× TSB (10 g/liter) was poured into the filter funnel and allowed to circulate for 15 min. The reactor funnel was then inoculated with 2 to 3 ml of overnight A. baumannii culture. Biofilms were grown for 3 days at room temperature with a circulation rate of 2.5 ml/min.
EPS extraction from A. baumannii biofilms.
EPS extraction methods were adapted from the work of Cao et al. (25, 26). EPS was extracted as loosely associated EPS (laEPS) or bound EPS (bEPS) (see Fig. S1 in the supplemental material). The biofilm was first removed from the reactor by keeping the pump pressure constant while the medium was very gently poured off into a waste flask. Sterile water (2 ml) was used to wash the top of the biofilm, and the pump was then restarted to pull off any excess medium from the biofilm. Next, the membrane was removed from the reactor and placed into a 50-ml conical tube along with 3 ml of sterile 0.9% NaCl. This was vortexed to remove the biofilm from the membrane. The biofilm suspension was poured into another conical tube, and the membrane was rinsed twice more with 3 ml of 0.9% NaCl. Once the biofilm suspension was collected, the mixture was centrifuged (4,180 × g) for 8 min. Supernatant was removed and kept as laEPS. Pellets were washed by resuspension in 1 ml 0.9% NaCl in a 1.5-ml microcentrifuge tube and centrifuged at 5,000 × g for 5 min. Supernatant was added to laEPS, which was then filtered twice through a 0.45-μm filter to remove any remaining bacteria. Cell pellets were retained for bEPS extraction.
For bEPS extractions, two methods were tested: extraction by EDTA and extraction by heat. For the EDTA method, the cell suspension (in 0.9% NaCl) was mixed with an equal volume of 0.9% NaCl plus 2% EDTA. This was stored at 4°C for 3 h and then centrifuged for 10 min at 5,000 × g. The supernatant (bEPS fraction) was filtered twice through a 0.45-μm filter to remove any remaining bacteria, and the sample was then dialyzed (molecular mass cutoff, 3.5 to 5.0 kDa) in 1 liter of 0.9% NaCl overnight.
The initial step in the heating method requires the cell suspension (cell pellet resuspended in 0.9% NaCl) to be incubated at 80°C for 10 min and then centrifuged for 15 min at 15,000 × g and 4°C. The supernatant was filtered twice through a 0.45-μm filter to remove bacteria and kept as bEPS. Remaining pellets were weighed and then discarded. EPS was stored at −20°C; SDS-PAGE showed no noticeable protein degradation after months of storage at 4°C (data not shown). Carbohydrate, protein, and DNA concentrations were quantified for laEPS and bEPS using the phenol-sulfuric acid method (27), the bicinchoninic acid (BCA) assay (Sigma; catalog no. BCA1), and PicoGreen dye (Invitrogen; catalog no. P7581), respectively.
Planktonic well plate cultures.
Growth curves were taken at room temperature in 48-well plates at a volume of 0.6 ml. A composite sample consisting of laEPS (9 ml) and bEPS (1 ml) was used for these experiments and will be referred to as EPS here. Each well contained 0.33× TSB (200 μl), EPS or 0.9% NaCl (300 μl), a cell suspension (variable volume), tobramycin (variable volume), and 0.9% NaCl to a complete volume of 600 μl. An overnight cell culture was incubated for 16 h at 37°C under constant shaking. Cells were centrifuged at 4,000 × g for 5 min and washed with 0.9% NaCl before resuspension in 0.9% NaCl. An appropriate volume of cell suspension was added to the assay for an approximate optical density at 595 nm (OD595) of ∼0.12 at a volume of 0.6 ml. OD measurements were taken using a BioAssay Reader (PerkinElmer HTS 7000 Plus). Cell growth rates were determined as OD/h using linear regression where OD changed linearly over time. Tobramycin was previously tested at various concentrations and cell concentrations over a 9-h time frame to optimize sensitivity. Assays were monitored over 9 to 14 h (or until a stationary phase was reached) by taking the OD595.
Protein digest and SDS-PAGE.
Proteinase K was purchased from Invitrogen (catalog no. EO0491). Protease experiments were performed with degradation at 55°C with constant shaking overnight and a protease-to-protein ratio of 1:8 (wt/wt). Digests were then added to cultures. For SDS-PAGE analysis, samples were extracted with a 1:4 trichloroacetic acid-to-sample ratio (vol/vol) and incubated at 4°C for 15 min. Samples were then centrifuged at 14,000 × g for 10 min at 4°C. The supernatant was carefully decanted, and 200 μl of acetone was added to the remaining EPS pellet. This was centrifuged again and then dried under heat and resuspended in Laemmli buffer with 50 mM dithiothreitol (DTT). Bio-Rad 4 to 20% gradient mini-Protean TGX stain-free gels were used, and imaging was accomplished using a Bio-Rad MP imaging system.
DNA digest and agarose gels.
DNase I was purchased from New England BioLabs (catalog no. M0303S), and conditions were optimized for use with EPS. In particular, the Mg2+ and Ca2+ concentrations in solution were increased to compensate for possible ion binding to the matrix. DNase digestion of EPS was carried out overnight at 37°C in the presence of 10 to 20 units of DNase I, 1.0 mM Ca2+, and 5.0 mM Mg2+. Positive controls contained amounts of λ DNA (Invitrogen; catalog no. 25250-010) equal to those of the DNA present in the EPS; these were digested under the same conditions. Digested EPS was dialyzed using SpectraPor 3.5-kDa-molecular-mass-cutoff dialysis tubing (catalog no. 132724) or Amicon Ultra 3-kDa-molecular-mass-cutoff 4-ml centrifugal filters (catalog no. UFC800308). DNase I-digested EPS samples were analyzed in a 1% agarose gel with 0.5 μg/ml ethidium bromide. Gels were imaged on a Bio-Rad MP imaging system.
RESULTS
EPS characterization.
Crude carbohydrate, protein, and DNA were quantified for all extractions (Table 1). The average total mass of EPS per biofilm was greater for S. aureus extractions than for A. baumannii extractions. Proportional yields for crude carbohydrate (0.14 and 0.21), protein (0.84 and 0.78), and DNA (0.01 and 0.02) did not differ greatly between EPS extractions from A. baumannii and S. aureus, respectively.
TABLE 1.
EPS component mass per milligram of biomass from EPS extractions from A. baumannii biofilms (n = 26) and S. aureus biofilms (n = 3)
| Species | Avg mass (μg/mg) ± SD |
|||
|---|---|---|---|---|
| Carbohydrate | Protein | DNA | Total EPSa | |
| A. baumannii | 1.9 ± 1.3 | 11 ± 5.1 | 0.16 ± 0.12 | 13 ± 5.5 |
| S. aureus | 7.78 ± 1.0 | 29 ± 4.7 | 0.61 ± 0.18 | 38 ± 4.0 |
The laEPS and bEPS from the extraction processes were combined for the total EPS.
Comparing EDTA and heat extraction methods.
Typically, laEPS is collected by centrifugation, and this was the technique used here. For our experiments, it was important that the bEPS experiments be devoid of added chemicals, both to limit chemical modification of the bEPS and to limit the carryover of compounds that might impact the subsequent bioassays. Consequently, we used two techniques to ensure that the final bEPSs were not significantly different. Bacteria from a single biofilm were first subjected to laEPS extraction and then split into two samples of equal mass. The two samples were treated with different bEPS extraction methods (heat or EDTA). Heat treatment yielded a higher biomass than did EDTA (1,875 μg versus 290.1 μg, respectively). There was little difference in total DNA yields (14.5 μg for heat treatment versus 16.6 μg for EDTA), suggesting that there was no significant difference in cell lysis during treatment. Furthermore, the EPS sample was split into two, and one fraction was heated before being added to a culture while the other was not. These were added to two sets of planktonic A. baumannii cultures. Heated EPS culture growth rates were not significantly different from unheated EPS growth rates (0.079 ± 0.008 OD/h and 0.071 ± 0.001 OD/h, respectively).
Effect of EPS on bacterial growth in the presence of antibiotic.
The MIC of tobramycin for A. baumannii strain BAA-1605 and S. aureus strain BAA-1747 was 1.0 μg/ml. We combined A. baumannii (from overnight culture) with tobramycin alone (1.0 μg/ml), with EPS from a separate A. baumannii extraction (50:50, vol/vol), or with both tobramycin and EPS. As expected, A. baumannii culture failed to grow with tobramycin alone, but the pattern of culture growth for EPS combined with tobramycin was identical to that of the no-treatment control (Fig. 2a). Importantly, the addition of S. aureus EPS resulted in a different response (Fig. 2b). Tobramycin (1.0 μg/ml) alone prevented the growth of A. baumannii, and the addition of EPS extracted from S. aureus had no impact on the inhibitory effects of tobramycin (Fig. 2b). This was true for both A. baumannii and S. aureus planktonic cultures. The average growth rates for A. baumannii culture with A. baumannii EPS (n = 26) or S. aureus EPS (n = 3) can be seen in Fig. 2c. To determine whether this difference between EPS treatments was due to a generalized Gram-negative versus Gram-positive bacterial interaction, we repeated the experiment using E. coli (strain K-12; MIC, 2.0 μg/ml). The addition of EPS extracted from A. baumannii protected E. coli from tobramycin, but EPS from S. aureus provided no protection (see Fig. S2 in the supplemental material).
FIG 2.
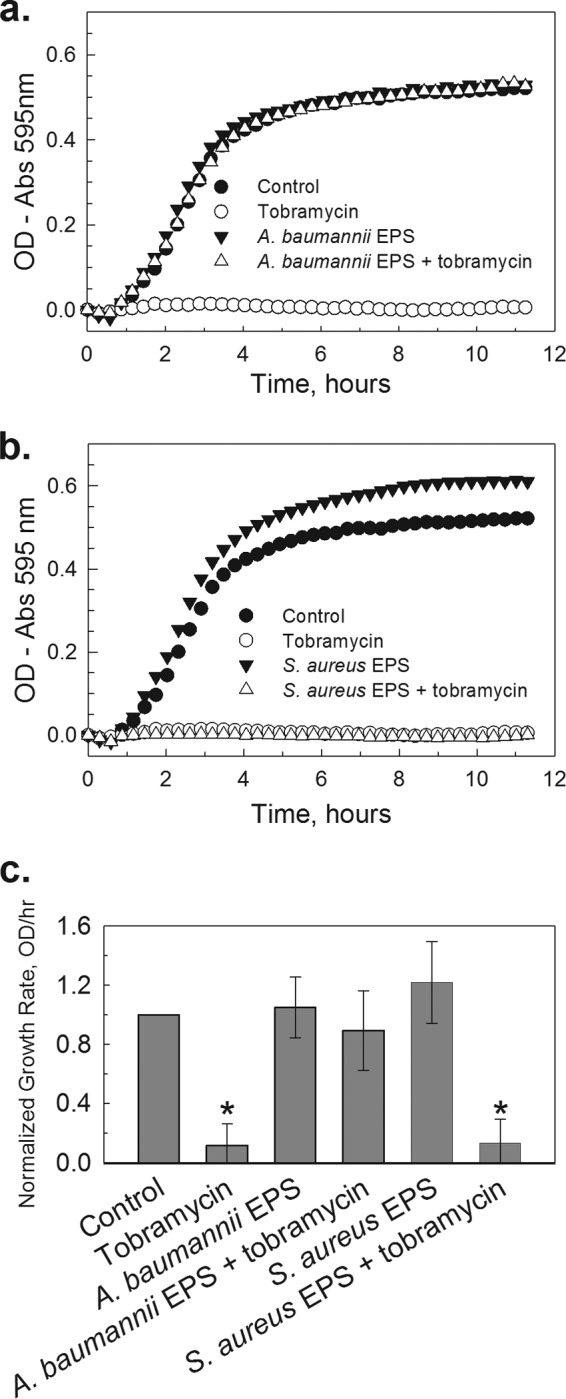
Addition of EPS from Acinetobacter baumannii, but not of EPS from Staphylococcus aureus, protects bacteria from tobramycin treatment. (A) Representative growth curves for A. baumannii that was cultured in 0.33× TSB alone (●), with 1 μg/ml tobramycin (○), with 50% (vol/vol) A. baumannii EPS (▼), or with both tobramycin and EPS (△). (B) Representative growth curves for S. aureus that was cultured in 0.33× TSB alone (●), with 1 μg/ml tobramycin (○), with 50% (vol/vol) A. baumannii EPS (▼), or with both tobramycin and EPS (△). (C) Normalized growth rates of cultures challenged with tobramycin, with EPS, or with both tobramycin and EPS. EPSs from both A. baumannii and S. aureus were used, but only EPS from A. baumannii protected against a tobramycin challenge (*, P < 0.001, n = 22). Error bars represent 1 standard deviation.
Dose-dependent protection from A. baumannii EPS.
If EPS from A. baumannii interferes with the bacteriostatic effects of tobramycin, then there should be a dose-dependent response. Consequently, while holding extracted EPS volume constant (50%), we added increasing concentrations of tobramycin (0.0 to 3.5 μg/ml) and monitored the impact on the rate of growth (Fig. 3). Across this range of concentrations, there was a clear dose response consistent with an adsorption mechanism (r2 = 0.91, n = 10). There was no significant correlation between growth rate and crude concentrations of carbohydrates, proteins, or DNA (P > 0.05; n = 21).
FIG 3.
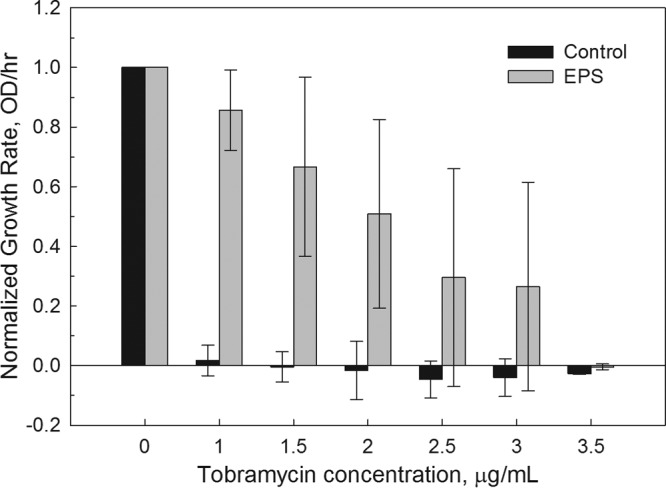
The growth rate of A. baumannii culture with 50% (vol/vol) extracted A. baumannii EPS responded in a dose-dependent manner to tobramycin (gray bars). Normalized growth rates are reported in OD/h. The controls (black bars) did not include EPS. The results show the average values for eight independent replicates. Error bars represent 1 standard deviation.
Protein and DNA do not contribute to EPS protection against tobramycin.
To investigate different mechanisms of interaction between the EPS and the antibiotic, we first subjected extracted EPS to proteinase K digest that was confirmed using SDS-PAGE (see Fig. S3 in the supplemental material). Both protease-treated EPS and untreated EPS blocked tobramycin activity (see Fig. S4). We attempted to eliminate DNA from the EPS by digestion with DNase I, but subsequent gel electrophoresis demonstrated that we were unable to digest DNA fully using a variety of approaches. This outcome is consistent with the probability of the EPS chelating the cations needed for DNase I activity. Consequently, we supplemented A. baumannii culture with tobramycin (1.0 μg/ml) and DNA. A final concentration of 0.39 μg/ml DNA, equivalent to that of the EPS in the culture, had no protective effect against tobramycin (0.1659 ± 0.004 OD/h versus −0.002 ± 0.0004 OD/h for DNA with and without 1 μg/ml tobramycin, respectively).
Addition of Mg2+ or Ca2+ cations decreases protection from A. baumannii EPS.
To determine whether the protective mechanism of A. baumannii EPS was due to the adsorption of tobramycin to charged binding sites of the EPS, we added Mg2+ and Ca2+ as “blocking agents” (28). When these cations were added to the cell culture, the protective ability of EPS was compromised, allowing the dose of tobramycin to become more effective (Fig. 4). The growth rates for the corresponding tobramycin concentrations decreased when Mg2+ or Ca2+ was present. The addition of Ca2+ in the same concentrations as Mg2+ showed a stronger effect, suppressing the growth of the cultures with the antibiotic (Fig. 4).
FIG 4.
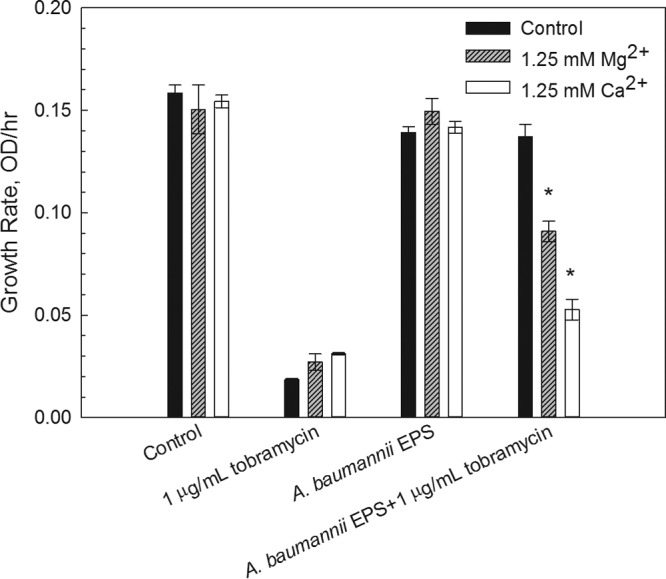
Average growth rates of A. baumannii cultures with extracted A. baumannii EPS and 1 μg/ml tobramycin with added Mg2+ or Ca2+. The decrease in the protective ability of EPS can be seen by comparing the growth rates for the EPS with those of tobramycin cultures. When 1.25 mM Mg2+ or Ca2+ was added, the growth was inhibited. Two-way analysis of variance confirms that Mg2+ and Ca2+ are statistically significant in the presence of A. baumannii EPS and 1 μg/ml tobramycin (*, P < 0.001). Error bars represent 1 standard deviation.
DISCUSSION
It is clear from these studies that, unlike EPS extracted from S. aureus biofilms, EPS extracted from A. baumannii biofilms protects biofilm communities in part by adsorption. When A. baumannii EPS was added to cell cultures along with inhibiting levels of tobramycin, the cell growth was similar to that of cultures without antibiotic treatment. When the tobramycin concentrations were increased, the antibiotic inhibition overcame the protective effects of the EPS, decreasing culture growth rate at higher tobramycin concentrations. This decrease in growth was linearly correlated with the increase in tobramycin concentration (r2 = 0.91 ± 0.05). The protective effects of the A. baumannii EPS can be transferred to cultures of other bacteria. When cultures of S. aureus and E. coli K-12 were tested, the same protection from tobramycin was observed. In contrast, S. aureus EPS was extracted using the same methods and did not exhibit any protection against tobramycin in cultures of S. aureus, A. baumannii, or E. coli.
Past studies of biofilm EPS focused on an alginate-producing bacterium, P. aeruginosa, as the organism of interest. These studies explored the role of alginate in antibiotic interactions. Alginate, a highly anionic polysaccharide, is now credited with only a minor role in the ability of EPS to protect biofilm communities from antibiotic exposure (21, 29). We have demonstrated that A. baumannii, a non-alginate-producing bacterium, synthesizes EPS that provides protection for cells against tobramycin after being extracted from the biofilm and added to planktonic cultures.
It is possible that bEPS may play a role in protection against antibiotics as well. The bEPS of P. aeruginosa is polysaccharide rich and consequently was thought to provide protection by decreasing diffusivity or otherwise excluding antibiotic access to the cell (30). P. aeruginosa bEPS is also effective in counteracting chlorine and monochloramine disinfectants, reducing their availability and prolonging cell life (31). A multidrug-resistant strain of A. baumannii produces bEPS in abundance compared to a drug-sensitive strain (32). While the bEPS fraction appears significant for protection inside the biofilm, it was not a significant factor in our ex situ experiments against tobramycin. In our tests, the addition of bEPS was not necessary for the protective effects seen from EPS in cultures. No difference in protection was seen when laEPS was used without the addition of the bEPS from the extraction (see Fig. S5 in the supplemental material).
Studies have encountered the complexity of EPS-antibiotic interactions. Biofilms from mucoid (alginate-producing) and nonmucoid strains of P. aeruginosa show similar tolerances to tobramycin (33, 34). However, studies of slime (polysaccharide)-producing and non-slime-producing staphylococcus biofilms found that the slime-producing strain exhibited a much higher resistance to aminoglycosides while maintaining a similar resistance to the other antibiotics (35).
Until recently, many of these studies of EPS were carried out inside a biofilm, i.e., based on a fully formed biofilm encasing viable bacteria. In situ studies of this nature are confounded by multiple variables, including diffusion, phenotypic changes of the biofilm cells, interactions in the EPS, and enzyme activity against the tested antibiotics. By using exogenously added EPS, we can largely rule out contributions from these other factors. By doing so, we have shown that the EPS per se is responsible for the protective effect against tobramycin.
The exact mechanism of the EPS-antibiotic interaction is still debated, with some studies supporting non-charge-based interactions (31) while others suggest an ionic interaction (28, 36). A recent study showed that the addition of divalent metal cations supports a greater penetration of tobramycin into a biofilm of P. aeruginosa microcolonies (28). Our study verifies this but also brings a novel approach by studying EPS-antibiotic interactions in a homogeneous system without mass transfer limitations that are normally caused by the biofilm. When the concentration of Ca2+ or Mg2+ was increased in our experiments, the protective effect of EPS decreased. By using exogenously added EPS, we were able to rule out the effects of diffusion on ion concentration and the ionic strength of solution. Consequently, we hypothesize that the protective effect of EPS is caused, in part, by physical binding of tobramycin to the EPS matrix and that the addition of cations reduces the protective effect of EPS by blocking these binding sites.
We attempted to remove two EPS components, proteins and DNA, to study the effects of the resulting EPS. Proteases, including proteinase K, are capable of degrading the majority of proteins in the EPS (37, 38), and SDS-PAGE confirmed that the proteins in the EPS were degraded in our study (see Fig. S3 in the supplemental material). When the digested EPS sample was added to cell culture, it showed no change from undigested EPS. We can therefore conclude that proteins play a minimal role in the protective ability of EPS.
After the attempted removal of DNA from the EPS, our results showed an incomplete DNA digestion by DNase I. Positive controls of λ DNA and digested λ DNA showed no effect on the antibiotic activity from the resultant DNA fragments in solution. More importantly, the addition of λ DNA into the bulk of A. baumannii culture with or without EPS added no protective benefits. These results are in contrast to those from a previous study in which P. aeruginosa biofilms grew in the presence of supplemented salmon sperm DNA and this appeared to make a more antibiotic-resistant biofilm in situ (36). Most eDNA resembles chromosomal DNA, although shorter polymers can be secreted by vesicles into the EPS or the DNA can be naturally degraded or modified after secretion (39, 40). This leads us to believe that λ DNA can accurately represent the whole DNA present in the EPS. Calf thymus DNA has previously been shown to sequester aminoglycoside antibiotics in solution (41). However, in another study, sputum samples, calf thymus DNA, and a mock sputum solution made using calf thymus DNA were subjected to digestion by recombinant human DNase (rhDNase) (42). These additives were applied to planktonic P. aeruginosa cell cultures. In concurrence with our results, the rhDNase digestion of these three additives showed no significant difference in the resulting bioavailability of tobramycin to the cells. Furthermore, if divalent metal cations were binding to the available sites in the EPS provided by eDNA, then we expected DNA added to the culture to also bind to these cations, thereby reducing their binding in the matrix. This was not seen for either Ca2+ or Mg2+, both of which are modeled as binding DNA (Ca2+ > Mg2+) (43).
To apply these findings, it is important to recognize that the amount of adsorption of antibiotic to the EPS is likely to control the penetration of antibiotics into the biofilm. This could be an important finding because blocking these binding sites (e.g., with divalent cations) would limit adsorption at the surface of the biofilm and permit further antibiotic penetration into the biofilm (28). In contrast, when EPS binds an antibiotic such as tobramycin at the surface, this likely creates a diffusion barrier, thereby preventing further movement of the antibiotic into the biofilm; in this scenario, bacteria at the bottom of the biofilm will be protected from the antibiotic. This idea could be tested by measuring the penetration of antibiotic into a biofilm before and after a blocking treatment with divalent cations. Following this work, we suggest that genetic knockouts be used to investigate which component of EPS plays a critical role in the interaction with antibiotics.
Our study was motivated by the lack of information on the interactions that biofilm EPS has with antibiotic treatments in a well-controlled system. Studying EPS outside the biofilm, in a homogeneous, controlled system, is beneficial to probing the interactions of EPS; it allows us to isolate and control the variables in the system. Biofilms of two prevalent nosocomial pathogens, A. baumannii and S. aureus, were grown, and EPS was harvested. We showed that EPS synthesized by A. baumannii acted as a “universal” protector, while S. aureus EPS did not protect any culture tested. These protective effects of EPS had an antibiotic capacity, observed by adding increasing amounts of tobramycin and observing a linear decrease in culture growth. The protective ability of EPS was decreased by the addition of Mg2+ or Ca2+ ions, while adding DNA to the cultures did not counteract the effects of adding tobramycin and/or Mg2+ or Ca2+. By digesting the proteins in the EPS, we showed that they were not significant players in the interactions with tobramycin. These results give us valuable information about the complexity of the interactions that biofilm EPS has with antibiotics, and they pave the way for further simplified studies of EPS.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NSF career award 0954186, and Emily K. Davenport was supported by NIH training grant 5T32GM008336-24.
Footnotes
Published ahead of print 9 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03071-14.
REFERENCES
- 1.Donlan RM. 2002. Biofilms: microbial life on surfaces. Emerg. Infect. Dis. 8:881–890. 10.3201/eid0809.020063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinberger R, Holden P. 2005. Extracellular DNA in single-and multiple-species unsaturated biofilms. Appl. Environ. Microbiol. 71:5404–5410. 10.1128/AEM.71.9.5404-5410.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flemming H-C, Wingender J. 2010. The biofilm matrix. Nat. Rev. Microbiol. 8:623–633. 10.1038/nrmicro2415 [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. 2013. Antibiotic resistance threats in the United States, 2013. Centers for Disease Control and Prevention, Atlanta, GA [Google Scholar]
- 5.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis. 39:309–317. 10.1086/421946 [DOI] [PubMed] [Google Scholar]
- 6.Lindsay D, von Holy A. 2006. Bacterial biofilms within the clinical setting: what healthcare professionals should know. J. Hosp. Infect. 64:313–325. 10.1016/j.jhin.2006.06.028 [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez-Baño J, Martí S, Soto S, Fernández-Cuenca F, Cisneros JM, Pachón J, Pascual A, Martínez-Martínez L, McQueary C, Actis LA, Vila J, Spanish Group for the Study of Nosocomial Infections (GEIH) 2008. Biofilm formation in Acinetobacter baumannii: associated features and clinical implications. Clin. Microbiol. Infect. 14:276–278. 10.1111/j.1469-0691.2007.01916.x [DOI] [PubMed] [Google Scholar]
- 8.Karageorgopoulos DE, Falagas ME. 2008. Current control and treatment of multidrug-resistant Acinetobacter baumannii infections. Lancet Infect. Dis. 8:751–762. 10.1016/S1473-3099(08)70279-2 [DOI] [PubMed] [Google Scholar]
- 9.Peleg AY, Seifert H, Paterson DL. 2008. Acinetobacter baumannii: emergence of a successful pathogen. Clin. Microbiol. Rev. 21:538–582. 10.1128/CMR.00058-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stewart PS. 1996. Theoretical aspects of antibiotic diffusion into microbial biofilms. Antimicrob. Agents Chemother. 40:2517–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denkhaus E, Meisen S, Telgheder U, Wingender J. 2007. Chemical and physical methods for characterisation of biofilms. Microchim. Acta 158:1–27. 10.1007/s00604-006-0688-5 [DOI] [Google Scholar]
- 12.McKenney D. 1999. Broadly protective vaccine for Staphylococcus aureus based on an in vivo-expressed antigen. Science 284:1523–1527. 10.1126/science.284.5419.1523 [DOI] [PubMed] [Google Scholar]
- 13.Izano EA, Amarante MA, Kher WB, Kaplan JB. 2008. Differential roles of poly-N-acetylglucosamine surface polysaccharide and extracellular DNA in Staphylococcus aureus and Staphylococcus epidermidis biofilms. Appl. Environ. Microbiol. 74:470–476. 10.1128/AEM.02073-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cramton S, Ulrich M, Götz F, Döring G. 2001. Anaerobic conditions induce expression of polysaccharide intercellular adhesin in Staphylococcus aureus and Staphylococcus epidermidis. Infect. Immun. 69:4079–4085. 10.1128/IAI.69.6.4079-4085.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russo TA, Luke NR, Beanan JM, Olson R, Sauberan SL, MacDonald U, Schultz LW, Umland TC, Campagnari AA. 2010. The K1 capsular polysaccharide of Acinetobacter baumannii strain 307-0294 is a major virulence factor. Infect. Immun 78:3993–4000. 10.1128/IAI.00366-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Riordan K, Lee J. 2004. Staphylococcus aureus capsular polysaccharides. Clin. Microbiol. Rev. 17:218–234. 10.1128/CMR.17.1.218-234.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fregolino E, Gargiulo V, Lanzetta R, Parrilli M, Holst O, Castro CD. 2011. Identification and structural determination of the capsular polysaccharides from two Acinetobacter baumannii clinical isolates, MG1 and SMAL. Carbohydr. Res. 346:973–977. 10.1016/j.carres.2011.03.024 [DOI] [PubMed] [Google Scholar]
- 18.Vu B, Chen M, Crawford RJ, Ivanova EP. 2009. Bacterial extracellular polysaccharides involved in biofilm formation. Molecules 14:2535–2554. 10.3390/molecules14072535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Bishop PL. 2003. Biodegradability of biofilm extracellular polymeric substances. Chemosphere 50:63–69. 10.1016/S0045-6535(02)00319-3 [DOI] [PubMed] [Google Scholar]
- 20.Bordi C, de Bentzmann S. 2011. Hacking into bacterial biofilms: a new therapeutic challenge. Ann. Intensive Care 1:19. 10.1186/2110-5820-1-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nichols WW, Dorrington SM, Slack MP, Walmsley HL. 1988. Inhibition of tobramycin diffusion by binding to alginate. Antimicrob. Agents Chemother. 32:518–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodges N, Gordon C. 1991. Protection of Pseudomonas aeruginosa against ciprofloxacin and beta-lactams by homologous alginate. Antimicrob. Agents Chemother. 35:2450–2452. 10.1128/AAC.35.11.2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donlan RM. 2000. Role of biofilms in antimicrobial resistance. ASAIO J. 46:S47–S52. 10.1097/00002480-200011000-00037 [DOI] [PubMed] [Google Scholar]
- 24.Mulcahy H, Charron-Mazenod L, Lewenza S. 2008. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 4:e1000213. 10.1371/journal.ppat.1000213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao B, Shi L, Brown RN, Xiong Y, Fredrickson JK, Romine MF, Marshall MJ, Lipton MS, Beyenal H. 2011. Extracellular polymeric substances from Shewanella sp. HRCR-1 biofilms: characterization by infrared spectroscopy and proteomics. Environ. Microbiol. 13:1018–1031. 10.1111/j.1462-2920.2010.02407.x [DOI] [PubMed] [Google Scholar]
- 26.Cao B, Ahmed B, Kennedy DW, Wang Z, Shi L, Marshall MJ, Fredrickson JK, Isern NG, Majors PD, Beyenal H. 2011. Contribution of extracellular polymeric substances from Shewanella sp. HRCR-1 biofilms to U(VI) immobilization. Environ. Sci. Technol. 45:5483–5490. 10.1021/es200095j [DOI] [PubMed] [Google Scholar]
- 27.Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. 1956. Colorimetric method for determination of sugars and related substances. Anal. Chem. 28:350–356. 10.1021/ac60111a017 [DOI] [Google Scholar]
- 28.Tseng BS, Zhang W, Harrison JJ, Quach TP, Song JL, Penterman J, Singh PK, Chopp DL, Packman AI, Parsek MR. 2013. The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycin. Environ. Microbiol. 15:2865–2878. 10.1111/1462-2920.12155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drenkard E. 2003. Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect. 5:1213–1219. 10.1016/j.micinf.2003.08.009 [DOI] [PubMed] [Google Scholar]
- 30.Slack M, Nichols W. 1982. Antibiotic penetration through bacterial capsules and exopolysaccharides. J. Antimicrob. Chemother. 10:368–372 [DOI] [PubMed] [Google Scholar]
- 31.Xue Z, Hessler CM, Panmanee W, Hassett DJ, Seo Y. 2013. Pseudomonas aeruginosa inactivation mechanism is affected by capsular extracellular polymeric substances reactivity with chlorine and monochloramine. FEMS Microbiol. Ecol. 83:101–111. 10.1111/j.1574-6941.2012.01453.x [DOI] [PubMed] [Google Scholar]
- 32.Chopra S, Ramkissoon K, Anderson DC. 2013. A systematic quantitative proteomic examination of multidrug resistance in Acinetobacter baumannii. J. Proteomics 84:17–39. 10.1016/j.jprot.2013.03.008 [DOI] [PubMed] [Google Scholar]
- 33.Nichols WW, Evans MJ, Slack MP, Walmsley HL. 1989. The penetration of antibiotics into aggregates of mucoid and non-mucoid Pseudomonas aeruginosa. J. Gen. Microbiol. 135:1291–1303 [DOI] [PubMed] [Google Scholar]
- 34.Wozniak DJ, Wyckoff TJO, Starkey M, Keyser R, Azadi P, O'Toole GA, Parsek MR. 2003. Alginate is not a significant component of the extracellular polysaccharide matrix of PA14 and PAO1 Pseudomonas aeruginosa biofilms. Proc. Natl. Acad. Sci. U. S. A. 100:7907–7912. 10.1073/pnas.1231792100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gristina AG, Jennings RA, Naylor PT, Myrvik QN, Webb LX. 1989. Comparative in vitro antibiotic resistance of surface-colonizing coagulase-negative staphylococci. Antimicrob. Agents Chemother. 33:813–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiang W-C, Nilsson M, Jensen PØ, Høiby N, Nielsen TE, Givskov M, Tolker-Nielsen T. 2013. Extracellular DNA shields against aminoglycosides in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 57:2352–2361. 10.1128/AAC.00001-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molobela I, Cloete T, Beukes M. 2010. Protease and amylase enzymes for biofilm removal and degradation of extracellular polymeric substances (EPS) produced by Pseudomonas fluorescens bacteria. Afr. J. Microbiol. Res. 4:1515–1524 [Google Scholar]
- 38.Adav SS, Lee D-J, Tay J-H. 2008. Extracellular polymeric substances and structural stability of aerobic granule. Water Res. 42:1644–1650. 10.1016/j.watres.2007.10.013 [DOI] [PubMed] [Google Scholar]
- 39.Allesen-Holm M, Barken KB, Yang L, Klausen M, Webb JS, Kjelleberg S, Molin S, Givskov M, Tolker-Nielsen T. 2006. A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol. Microbiol. 59:1114–1128. 10.1111/j.1365-2958.2005.05008.x [DOI] [PubMed] [Google Scholar]
- 40.Wu J, Xi C. 2009. Evaluation of different methods for extracting extracellular DNA from the biofilm matrix. Appl. Environ. Microbiol. 75:5390–5395. 10.1128/AEM.00400-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tannenbaum CS, Hastie AT, Higgins TML, Kueppers F, Weinbaum G. 1984. Inability of purified Pseudomonas aeruginosa exopolysaccharide to bind selected antibiotics. Antimicrob. Agents Chemother. 25:673–675. 10.1128/AAC.25.6.673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hunt B, Weber A, Berger A. 1995. Macromolecular mechanisms of sputum inhibition of tobramycin activity. Antimicrob. Agents Chemother. 39:34–39. 10.1128/AAC.39.1.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Korolev N, Lyubartsev AP, Rupprecht A, Nordenskiöld L. 1999. Competitive binding of Mg2+, Ca2+, Na+, and K+ ions to DNA in oriented DNA fibers: experimental and Monte Carlo simulation results. Biophys. J. 77:2736–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.