

Abstract
Radicicol, an antifungal antibiotic, was previously identified as a compound having antimalarial activity. However, its mechanism of action in Plasmodium falciparum was not elucidated. While characterizing its antimalarial function, we observed that radicicol manifested two distinct developmental defects in cultured P. falciparum in a concentration-dependent manner. At a low concentration of radicicol, a significant percentage of drug-treated parasites were arrested at the schizont stage, while at a higher concentration, the parasites were unable to multiply from schizont to ring. Also, the newly formed rings and trophozoites were extremely delayed in development, eventually leading to cell death. We intended to characterize the potential molecular target of radicicol at its sublethal doses. Our results demonstrated that radicicol specifically impaired mitochondrial replication. This decrement was associated with a severalfold increment of the topoisomerase VIB transcript as well as protein in treated cells over that of untreated parasites. Topoisomerase VIB was found to be localized in the organelle fraction. Our docking study revealed that radicicol fits into the Bergerat fold of Pf topoisomerase VIB present in its ATPase domain. Altogether, these data allow us to conclude that P. falciparum topoisomerase VIB might be one of the targets of radicicol causing inhibition of mitochondrial replication. Hence, radicicol can be suitably employed to explore the mitochondrial physiology of malaria parasites.
INTRODUCTION
The protozoan parasite Plasmodium falciparum causes the disease malaria, which is responsible for 200 million illnesses per year and kills nearly 1.2 million people annually. A recent report in Lancet claims that the death rate due to malaria is hugely underestimated and may be twice as high as previously estimated (see http://www.bbc.co.uk/news/health-16854026). Resistance to the antimalarial drug chloroquine makes P. falciparum a potential life-threatening parasite. According to a World Health Organization update in April 2012 (see http://www.who.int/malaria/areas/treatment/withdrawal_of_oral_artemisinin_based_monotherapies/en/), there is a threat of P. falciparum resistance to artemisinin. The discovery of efficacious drug targets is urgently required to battle against drug-resistant malaria.
During its life cycle, Plasmodium increases its numbers by geometric progression, which occurs at the schizont stage. Parasites strategically use this stage to multiply their number by 16 to 32 times, which is crucial for its infectivity. This event is known as endoreduplication or schizogony, where it duplicates its chromosome without cell division. A similar sort of cell cycle is also present in plant cells, where they skip the entire M phase and continue on to the S phase (endoreduplication) (1). Several genes that direct endoreduplication in Arabidopsis have been identified, and it has been revealed that the topoisomerase VI complex (a heterotetramer composed of topoisomerase VIA2 [TopoVIA2] and TopoVIB2) is an essential component for the decatenation of the replicated chromosome during endoreduplication (2, 3). Mutation in these genes causes a dwarf phenotype in Arabidopsis along with reduced ploidy (4, 5). Plasmodium carries genes encoding both of the subunits of archaeal DNA topoisomerase VI (6) and it might have a role in endoreduplication. However, no work has been reported till now regarding its biological function in Plasmodium or any related Apicomplexa.
Topoisomerase VI is a type II topoisomerase, which is present in Archaea and plants and absent from most of the animal kingdom except for the Apicomplexa. Type II topoisomerase relaxes both positively and negatively supercoiled DNA in the presence of ATP and divalent cations and catalyzes the passage of one DNA duplex through another (7, 8, 9, 10). This class of enzymes is classified into two evolutionary distinct protein families, type IIA and type IIB (11), depending on their structural differences.
Radicicol has been shown to inhibit the growth of Archaea, which possess topoisomerase VIB (12). Biochemical experiments confirm that radicicol inhibits decatenation and relaxation activities of Sulfolobus shibatae topoisomerase VIB. X-ray crystallographic analysis shows that radicicol binds to the ATP-binding pocket of this protein (13). Radicicol has also been reported to inhibit a wide variety of tumor cell lines by targeting heat shock protein 90 (Hsp90) (14). Radicicol binding to the ATPase domain of Hsp90 prevents maturation of Hsp90 clients, leading to proteasomal degradation (15). X-ray crystallographic analysis of yeast Hsp90 N-terminal domain-bound radicicol (16) identifies the key aspect of its nucleotide mimetic interactions. Another study in a breast cancer cell line shows that radicicol increases steady-state levels of Hsp90 protein similarly to a stress response (17) and destabilizes Hsp90-dependent proteins.
Earlier, radicicol extracted from a soil strain, FO-4910, collected from Oklahoma, showed antimalarial activity on the NIHJ strain (18). However, its cellular target and the mechanism of action remained elusive. To characterize the antimalarial mechanisms of radicicol, we evaluated its activity on an in vitro culture of P. falciparum 3D7. We report a detailed study on the effects of radicicol on Plasmodium developmental stages, ploidy, and replication. We evaluated the effects of radicicol on the expression of two putative target genes, Hsp90 and topoisomerase VIB. Our results demonstrated that radicicol had no effect on nuclear and apicoplast DNA but targeted DNA in the mitochondria and caused upregulation of topoisomerase VIB both at the transcript level and the protein level. Further, we performed an in silico analysis of the complexes between radicicol and Plasmodium TopoVIB and Hsp90. Our evidence suggested that topoisomerase VIB might be one of the targets of radicicol, due to the presence of the enzyme in organelle fractions.
MATERIALS AND METHODS
Materials.
Radicicol was purchased from Sigma-Aldrich. SYBR green I nucleic acid-staining dye (10,000× stock concentration) was purchased from Molecular Probes, Inc.
Parasite.
P. falciparum 3D7 cultures were maintained by the bell jar candle method at 37°C in RPMI 1640 medium supplemented with 1% Albumax (Invitrogen) and 0.005% hypoxanthine containing 5% red blood cells (RBC). Gametocyte cultures were maintained according to standard procedure as described previously (19). Cultures rich in stage III and stage IV gametocytes were harvested for RNA preparation.
RNA isolation.
The parasite culture having 10% parasitemia was synchronized using 5% sorbitol, and ring, trophozoite, and schizont stages were harvested. For harvesting the gametocyte-specific stage, we followed the protocol as mentioned in Methods in Malaria Research (20). Total RNA was isolated according to the protocol previously reported (21).
Semiquantitative and real-time RT-PCR.
Equal amount of RNA measured by spectroscopic analysis (JASCO spectrophotometer EMC-709) was subjected to DNase I (Fermentas) digestion for 15 min at room temperature. DNase I was finally inactivated by incubating with 25 mM EDTA at 65°C for 10 min. Complete removal of contaminating genomic DNA was verified by the absence of any amplification product in the absence of the reverse transcriptase step. About 10 μg of total RNA from each sample was then reverse transcribed with oligodeoxythymidylic acid [oligo(dT)] primer (Sigma-Aldrich) using reverse transcriptase (Omniscript kit; Qiagen). The resulting cDNA was subjected to semiquantitative reverse transcription (RT)-PCR to detect the transcripts of P. falciparum TopoVIA (PfTopoVIA) and -VIB. For real-time PCR, cDNA was diluted (1:50) and used for PCR using an RT-PCR kit (Roche). The real-time analysis was done using the Applied Biosystems 7500 Fast Real-Time PCR system. The threshold cycle (CT) value of the ARP transcript of each sample was used to normalize the corresponding CT values of the PfTopoVIA, PfTopoVIB, or PfHsp90 transcripts. The normalized CT values of PfTopoVIB (or of PfTopoVIA or PfHsp90) from different samples were compared to obtain ΔCT values. The relative levels of mRNA were deduced from the formula (change in mRNA level, 2ΔCT). The mean values (± standard deviation [SD]) from three independent experiments were plotted using GraphPad Prism 6 software.
The primers OSB66 (5′-AAG GAT CAA ACC TCT GTT G-3′) and OMKB52 (5′-CAT GAT ATC ATT TAT TTC-3′) were designed to amplify a 279-bp gene-specific region of PfTopoVIB, OSB67 (5′-TGA TAT GTC CAT CGA GAA TC-3′) and OMKB50 (5′-TAA AAG CTC CTT AAT GCG-3′) were used to amplify a 284-bp gene-specific region of PfTopoVIA, OSB96 (5′-ATG GAA ATC AAT GCT CGT CAC-3′) and OMKB120 (5′-GAC CCC GGG TTA GTC AAC TTC TTC CAT TTT AG-3′) were used to amplify a 282-bp fragment of PfHSP90, and OSB94 (5′-CTG TAA CAC ATA ATA GAT CCG AC-3′) and OSB95 (5′-TTA ACC ATC GTT ATC ATC ATT ATT TC-3′) were used to amplify a 300-bp gene-specific regions of P. falciparum aspartate-rich protein (PfARP).
Radicicol inhibition assay.
A synchronized P. falciparum culture arrested primarily at the schizont stage and having 1.45% parasitemia was used for the radicicol inhibition assay. For each experiment, we segregated the culture into four parts and centrifuged at 3,000 rpm for 10 min at 25°C. The supernatant was aspirated and the cultures were resuspended in increasing amounts of radicicol-containing medium, followed by incubation at 37°C. Our experimental conditions comprised four different radicicol concentrations, i.e., 0.5 μM, 1.0 μM, 1.5 μM, and 0 μM (positive control). At every 10-h interval, 5-μl samples were taken out to make a smear, which was fixed with methanol and stained with Giemsa and observed under the microscope.
For calculation of the 50% inhibitory concentration (IC50), we exposed the synchronized schizont-stage parasites at nine increasing concentration of radicicol (0.25 μM, 0.75 μM, 2 μM, 4 μM, 6 μM, 8 μM, 10 μM, 14 μM, and 17 μM) for 48 h. The parasitemia was measured at each concentration after 48 h. For the standard Giemsa counting assay we counted (100 to 200) parasite-infected RBC per slide at each radicicol concentration as well as repeating each assay two times for reproducibility. The IC50 was calculated using Graph Pad version 6.0. For light microscopy, thin smears were prepared. They were fixed with methanol followed by staining with Giemsa.
The SYBR green I-based assay was done using 96-well plates with nine concentrations of radicicol ranging from 0.25 μM to 18 μM. After 48-h incubation with the drug, 100 μl of SYBR green-containing lysis buffer (20 mM Tris-HCl [pH 7.5], 5 mM EDTA, 0.008% [wt/vol] saponin, and 0.08% [vol/vol] Triton X-100) was added to each well (22) and incubated at room temperature in the dark for 1 h. Fluorescence was measured using a Bio-Tek Synergy Mx microplate reader using an excitation wavelength at 485 nm. The emission was measured at 530 nm. The background reading for parasite-free medium was subtracted to obtain the correct fluorescence value. Triplicate wells for each dilution of radicicol were measured, and from these values the percentage of parasite inhibition was calculated and plotted against the log of the radicicol concentration and the IC50 was determined after curve fitting using nonlinear regression.
Southern hybridization.
One-percent synchronized schizont-stage parasites were incubated with or without radicicol for 40 h. A constant drug pressure of 1.5 μM was maintained at every 10-h interval in the radicicol-treated sample. After 40 h, the genomic DNA was isolated from radicicol-treated and untreated parasites. Likewise, 1% synchronized schizont-stage parasites were incubated with and without 17-allylamino-17-demethoxygeldanamycin (17AAG) for 48 h. A constant drug pressure of 170 nM was maintained at every 12 h interval in 17AAG-treated sample. After 48 h, genomic DNA was isolated from drug-treated and untreated samples. Equal amounts of genomic DNA from treated and untreated samples were digested with HindIII. Subsequently, the genomic DNA was electrophoresed on a 0.8% agarose gel and transferred to a nitrocellulose membrane. For probe preparation, we amplified 1,391-bp PfOrc1 (nuclear genome) using the primers 5′-TGA CGA AAG CGA TGA ACA ACG-3′ and 5′-GGA TAT CTC CTG ATA CAT TAG C-3′. Similarly, 1,626-bp PfCox3 (mitochondrial genome) was amplified using the primers 5′-CAT CCC ATA GCA AGT ATC ATA G-3′ and 5′-GCT TCT GAT ATT ATG ATA GAT AAC-3′, and 1,651-bp PfSufB (apicoplast genome) was amplified using the primers 5′-ATG AGA TAG TTT GAC TGG GGC-3′ and 5′-CGA ACC TAC TTC AAC TTG TAT C-3′. The probes were gel purified and labeled with [α-32P]dATP using a DecaLabel DNA labeling kit (Fermentas). The purified probe was denatured and hybridized with the blot overnight at 65°C and analyzed by phosphorimaging (Amersham).
Western blot analysis.
The ring-, trophozoite-, and schizont-specific fractions as well as the radicicol-treated and -untreated fractions were first analyzed by SDS-polyacrylamide gel electrophoresis (10%), and proteins were transferred to a polyvinylidene difluoride (PVDF) membrane for Western blot analysis. A unique peptide sequence (CLLNLFTRVKEEYPDEFESI) of topoisomerase VIB with high antigenicity was chosen, and primary antibody against that peptide was generated in rabbits. Primary antibody was used at 1:1,000 dilutions, and horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (Santa Cruz Biotechnology, Inc., CA) was used as a secondary antibody at a 1:20,000 dilution. The Western blots were developed using a chemiluminescent detection system (Pierce). For detection of the cytoplasmic fraction, we used antibody to PfHsp70 at a 1:1,000 dilution and HRP-conjugated anti-mouse secondary antibody.
Subcellular fractionation of parasite.
For subcellular fractionation we used the standard procedure (23) with minor modifications. Twenty-five milliliters of 8% trophozoite-specific parasitized erythrocyte was harvested and treated with 0.15% saponin to free parasites from RBC. The pellet containing parasites was washed three times with phosphate-buffered saline (PBS) buffer followed by 5 ml buffer I (containing 0.34 M sucrose,15 mM NaCl, 0.2 mM EDTA, 0.2 mM EGTA, 15 mM Tris-HCl [pH 7.4], and 0.2 mM phenylmethylsulfonyl fluoride [PMSF]). The pellet was resuspended in 5 ml buffer II (buffer I containing 1% Triton X-100) and incubated at 4°C for 30 min. Afterward, it was homogenized using a Dounce homogenizer with 25 complete strokes. The nuclei were pelleted at 600 × g for 5 min. The nuclear pellet was first washed with buffer I and then resuspended in 50 μl low-salt buffer (containing 1.5 mM MgCl2, 0.2 mM EDTA, 20 mM HEPES [pH 7.9], 25% glycerol, and 1× protease inhibitor cocktail). The nuclei were extracted using 30 μl of high-salt buffer (containing 1.2 M KCl) by slow addition for 30 min with gentle mixing at 4°C. The extracted nuclei were centrifuged at 12,000 × g for 30 min at 4°C, and supernatant was used as the nuclear extract.
The supernatant obtained after Dounce homogenization was centrifuged at 12,000 × g for 30 min and the pellet was used as the organellar fraction. The supernatant containing the cytoplasmic fraction was concentrated 20 times using Amicon Ultra-4 centrifugal filter units (Merck Millipore) before Western blot analysis.
Docking analysis.
The P. falciparum topoisomerase VIB sequence was downloaded from the NCBI website with the GenBank accession number CAD52775.1. No crystal or solution structure of this protein had been documented previously. PfTopoVIB was modeled based on a Plasmodium Hsp90 template (Protein Data Bank [PDB] accession no. 3IED) using SWISS-MODEL in an automated mode with a high E value score of 3.6 × 10−11. Ramachandran plot generation using Procheck identified 77.4% of residues in the most favored region. Energy minimizations using 100 steps of the steepest descent algorithm and a distance-dependent dielectric constant of 4r in AMBER forcefield implemented in Schrodinger's MacroModel were done to lower the molecule's energy and strain without causing any significant distortion to the binding pocket. Molecular docking studies with this energy-minimized model and the drug radicicol and 17AAG were done using AutoDock 4.2 implemented within the PyRx virtual screening tool with default settings. A Lamarckian genetic algorithm was chosen for the conformer search. 17AAG was derived from a 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17DMAG) ligand bound to human Hsp90 in the crystal structure (PDB accession no. 1OSF). The protein receptor was treated as rigid and the grid centering was done at the radicicol-binding pocket with a box having the dimensions 21.4 Å × 35.7 Å × 14.4 Å, with a spacing of 0.3750 Å. As required by AutoDock, the internal degrees of freedom in the ring structures were fixed. The two torsional degrees of freedom in the ligand molecule were the dihedral angles for the hydroxy groups on the aromatic ring. Graphical visualizations were done using Accelrys' Discovery Studio.
In an independent study, Plasmodium Hsp90 was also analyzed with respect to its ability to bind radicicol and 17AAG. For this analysis, the PfHsp90 structure with PDB accession no. 3IED had several missing residues in its N-terminal region, and therefore, another structure with the PDB accession no. 3K60 was downloaded from the PDB website. All the steps in the P. falciparum topoisomerase VIB-radicicol docking procedure were followed, except for autogrid values being kept at the default numbers of 73.3 Å × −25.9 Å × 11.8 Å with spacing of 0.3750 Å.
RESULTS
Effects of radicicol on Plasmodium-infected erythrocyte culture.
In order to determine the IC50 of radicicol inhibition, we exposed a synchronized late-schizont culture of the P. falciparum 3D7 strain to nine different concentrations of radicicol (ranging from 0.25 μM to 18 μM) for 48 h. Radicicol showed a dose-dependent inhibitory effect on parasite growth (Fig. 1A). For determination of parasitemia, we employed two methods, (i) the standard assay of staining the parasites with Giemsa and counting parasites under the microscope and (ii) an SYBR green I-based fluorescence assay. In the case of the fluorescence assay, the variability in the results between triplicate fluorescence wells was low. The dose-response curve as determined by Giemsa-stained parasitemia yielded an IC50 concentration of 8.563 μM (Fig. 1B), which was very much comparable to that obtained by the fluorescence assay (9.029 μM) (Fig. 1C). Figure 1D shows a linear relationship (r2 = 0.9659) between parasitemia and relative fluorescence units determined by our assay. For a control, we tested chloroquine in 3D7 by the fluorescence method and the IC50 obtained was 16 ± 3 nM.
FIG 1.

Radicicol-induced death of Plasmodium falciparum. (A) Structure of radicicol showing its functional groups. (B) Inhibition of P. falciparum 3D7 by radicicol. Synchronized schizont stage-specific parasites were incubated with increasing doses of radicicol for 48 h. Parasitemia levels were determined by microscopy using Giemsa stain, and percent inhibition was calculated. Mean and standard errors from two experiments were plotted. (C) Parasitemia was determined using SYBR green I-based fluorescence measurement (repeated two times) as described in Materials and Methods. (D) Linear relationship between parasitemia and fluorescence values as used in this study.
Higher concentration of radicicol causes inefficient schizont-to-ring transition.
We aimed to monitor the stage-specific development of parasites at an 8 μM radicicol concentration, based on IC50 determination. We started with 1% parasitemia and parasites synchronized at the schizont stage and treated with 8 μM radicicol-containing medium and allowed them to grow for 50 h, replenishing the drug at 10-h intervals. The growth of the parasites was measured by Giemsa staining. Untreated parasites developed into rings at the end of the first 10 h (from 0.8% schizont to 4% ring). Interestingly, radicicol-treated parasites remained arrested at the schizont stage and eventually (at 20 h) very few schizonts developed into the ring stage (0.8% ring up to 30 h) (Fig. 2). This experiment was repeated three times and we observed similar phenotypes. Thus, these results showed that radicicol delayed the schizont-to-ring transition.
FIG 2.
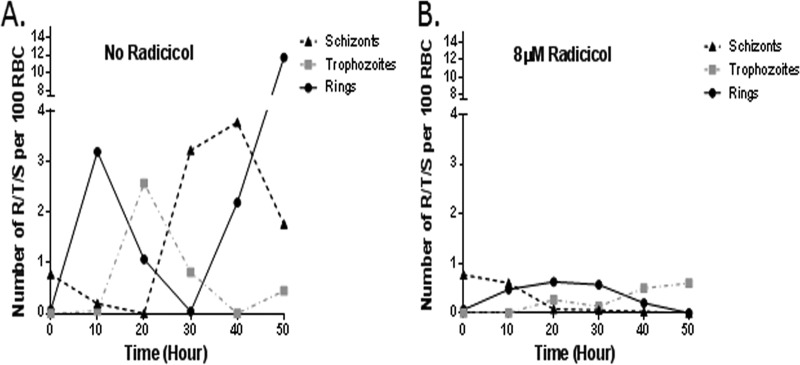
Higher concentration of radicicol induces inefficient schizont-to-ring transition. A synchronized schizont stage 3D7 culture (0.8%) was grown for 50 h in the presence or absence of radicicol (8 μM). Parasitemia at every 10th hour was determined by counting a total of 2,000 erythrocytes, using Giemsa staining. The results from one of the two experiments with similar trends are shown. R, rings; T, trophozoites; S, schizonts.
Lower concentration of radicicol delays schizont-to-ring transition in a reversible manner.
To better understand the effect of radicicol on parasite development, we performed a similar experiment at sublethal doses of radicicol. In a typical assay, the late-schizont-stage-synchronized culture was exposed to constant drug pressure at every 10th hour, and we followed parasite growth for a span of 60 h. We carried out the experiment with three different concentrations of radicicol (0.5 μM, 1.0 μM, and 1.5 μM) and closely monitored the distribution of ring, trophozoite, and schizont stages at every 10-h interval. At these concentrations of radicicol (from 0.5 μM to 1.5 μM), the decline in parasitemia was not drastic. We did not observe any change in the immediate transition of schizont to ring stage as opposed to the drastic effect observed with a higher dose of radicicol (8 μM concentration). However, during the 2nd round of endoreduplication, i.e., from the 30th hour onward, we observed that a significant population of parasites was arrested at the schizont stage. There was a striking difference in the distribution of rings/trophozoites/schizonts at the 50th hour. While untreated parasites were found to be predominantly at ring stage, a large population of the treated parasites was found to be arrested at the schizont stage. Even after 50 or 60 h, the majority of the radicicol-treated parasites were detained at the schizont stage and only a small fraction of treated schizonts made it to the ring stage. However, total parasitemia did not show any significant reduction. Our study showed that even 0.5 μM radicicol delayed the schizont-to-ring transition by 20 h compared to that of the untreated parasites (Fig. 3A). Thus, it appeared that radicicol treatment captured parasites at the mid-schizont stage. To understand whether this phenotype was strictly drug induced or not, we washed out the drug after 20 and 40 h of treatment and examined the distribution of rings/trophozoites/schizonts at every 10th hour thereafter. We observed that while the 1.5 μM radicicol-treated schizonts were still arrested as before, after washing (at the 20th or 40th hour) a marked rise in the ring population was observed at the 50th hour, similar to that of the untreated sample (Fig. 3B). Thus, it seemed that radicicol treatment trapped the parasite at a specific developmental stage in a reversible manner.
FIG 3.

Lower concentration of radicicol causes arrest of schizonts in erythrocytic stage of P. falciparum in a reversible manner. (A) Schizont stage-synchronized 3D7 culture was exposed to constant drug pressure of three different concentrations of radicicol (0.5 μM, 1 μM, and 1.5 μM). The Giemsa-stained parasites are morphologically characterized as rings, trophozoites, and schizonts. (B) Radicicol was washed out at 20- and 40-h intervals, and distributions of rings, trophozoites, and schizonts were compared with those in the unwashed sample. These experiments were repeated three times with similar trends, and the results for one typical experiment are shown.
Radicicol targets mitochondrial DNA replication.
Since parasitic DNA replication took place during the schizont stage, we sought to monitor the effects of sublethal doses of radicicol (1.5 μM) on nuclear, mitochondrial, and apicoplast DNA replication. The arrested schizont stage parasites showed no detectable changes in morphology compared to the wild type (Fig. 4A). We counted the number of nuclei present in each schizont in both treated and untreated parasites and did not observe any significant differences in the ploidy levels in radicicol-treated versus untreated parasitic schizonts. The majority of the treated and untreated parasites exhibited 16-nuclei content. However, both the treated and untreated parasite populations also contained 8 and 32 nuclei (Fig. 4B). Thus, the distribution of ploidy levels was unaltered with radicicol treatment, suggesting no apparent effect on nuclear replication.
FIG 4.

Characterization of radicicol-treated parasites. (A) Morphology of parasites treated with 1.5 μM radicicol showed no change compared to the untreated parasites. (B) The ploidy of the treated parasites is comparable to that of the untreated parasites. A minimum of 100 parasitic schizont nuclei in each case (untreated and radicicol treated) were counted. (C) Synchronized schizont-stage parasites were treated with 1.5 μM radicicol for 40 h and genomic DNA samples from treated and untreated cultures were subjected to Southern hybridization using PfOrc1 (nuclear probe), PfCox3 (mitochondrial probe), or PfSufB (apicoplast probe). The experiment was repeated two times. One of the Southern blots is shown. (D) Synchronized schizont-stage parasites were treated with 170 nM 17AAG for 48 h and genomic DNA from treated and untreated cultures was subjected to Southern hybridization in a similar fashion using the above-mentioned nuclear, mitochondrial, and apicoplast probes.
It is known that mitochondrial and apicoplast genome replication begins at the mid-schizont stage of parasite development. We designed a time course experiment similar to the previous one. We started with late-schizont culture and maintained constant drug pressure (growth medium supplemented with 1.5 μM radicicol at every 10-h interval). Parasites were harvested at 40-h time points. We compared the replication of nuclear, apicoplast, and mitochondrial genomes under radicicol-treated and untreated conditions by Southern hybridization. The genomic DNA isolated from treated and untreated samples at different time points was probed with a nuclear probe (ORC1), a mitochondrial probe (COX3), and an apicoplast probe (SUFB) (Fig. 4C). The Southern hybridization data clearly indicated that the relative intensities of nuclear and apicoplast genomes appeared to be the same in radicicol-treated and -untreated samples. However, there was a noticeable difference in the band intensities corresponding to the mitochondrial genome between radicicol-untreated versus -treated samples. These results indicated that radicicol-treated parasites possessed reduced amounts of mitochondrial genome but that radicicol treatment had no effect on the nuclear or apicoplast genome. In order to determine whether decrement of mitochondrial replication is specific to radicicol treatment, we performed a similar study by exposing the parasites with 17AAG, a potent Hsp90 inhibitor. It was previously reported that 17AAG inhibits the growth of 3D7 with an IC50 of 160 nM when treated for 48 h (24). With our experimental conditions, we determined the IC50 of 17AAG as 170 nM, which is very close to the previously reported value. Hence, we exposed parasites to 170 nM 17AAG for 48 h (with a constant drug pressure maintained every 12 h) and performed Southern hybridization in a fashion similar to that described above. We observed that there were no alterations in the relative intensities of the CoxB-probed mitochondrial genomes in the 17AAG-treated and -untreated samples (Fig. 4D). Thus, it can be concluded that impairment of mitochondrial DNA is specific to radicicol action and not to other Hsp90 inhibitors such as 17AAG.
Stage-specific expression of PfTopoVIB and PfTopoVIA.
Based on the presence of the Bergerat fold, there are two putative targets of radicicol, namely, PfHsp90 and PfTopoVIB. In order to investigate whether these two genes were expressed at the schizont stage, we performed semiquantitative RT-PCR as well as real-time RT-PCR analyses on RNA isolated from four different stages (ring, trophozoite, schizont, and gametocyte). We also tested the expression of PfTopoVIA, since PfTopoVIA and PfTopoVIB were expected to form the active enzyme together. Aspartate-rich protein (ARP), which is constitutively expressed at all developmental stages, was used as a loading control (25) to normalize the relative abundance of PfTopoVIA, PfTopoVIB, and PfHsp90 transcripts. Semiquantitative RT-PCR showed high levels of PfTopoVIB and PfTopoVIA transcripts at the schizont stage (Fig. 5A). The PfHsp90 transcript was found at all the stages, albeit lower at the schizont stage. Real-time RT-PCR data analysis revealed 65× more TopoVIB and 2× more TopoVIA in the schizonts than in the rings (Fig. 5B). On the other hand, PfHsp90 expression was dominant in the trophozoite and gametocyte stages compared to the ring and schizont stages. Antibody against TopoVIB was raised against a C-terminal peptide. As expected, the immune sera showed a PfTopoVIB-specific band near 61 kDa. The immunoblot analysis confirmed the presence of PfTopoVIB mainly in the schizont stage (Fig. 5C).
FIG 5.

Stage-specific expression of PfTopoVIA and PfTopoVIB. (A) RT-PCR analysis on RNA isolated from parasites synchronized at ring (R), trophozoite (T), schizont (S), and gametocyte (G) stages. PCR amplification was done using PfTopoVIA-, PfTopoVIB-, and PfHsp90-specific primer pairs to yield 300-bp amplicon in each case. Aspartate-rich protein (ARP) transcript was used as a loading control. PCR without reverse transcription (−RT) shows no amplification of PfTopoVIB. Genomic DNA (gDNA) sample acted as a positive control. (B) Real-time RT-PCR reveals quantitative abundance of the TopoVIB, TopoVIA, and Hsp90 transcripts at various stages of parasite. (C) Western blot analysis was performed with antibody against PfTopoVIB protein to compare its stage-specific expression. The positions of molecular weight markers (in thousands) are indicated to the left of the blot. PfHsp70 acts as a loading control.
Radicicol causes upregulation of topoisomerase VIB.
In the breast cancer cell line SKBR3, it was observed that treatment with geldanamycin and radicicol resulted in an increase in Hsp90 synthesis (17). In order to explore whether a similar effect occurred in Plasmodium, we treated 3D7 cells with 1.5 μM radicicol for a period of 40 h (maintaining a constant dose by supplementing fresh radicicol-containing media at regular intervals of 10 h) and then extracted RNA from treated as well as untreated cells. Semiquantitative RT-PCR showed that the Hsp90 transcript was not significantly affected; however, the TopoVIB transcript was found to be markedly upregulated (Fig. 6A). Quantitative analysis by real-time RT-PCR revealed about a 2-fold increase in TopoVIB transcripts in radicicol-treated parasites compared to untreated parasites, whereas there was no significant alteration of the Hsp90 transcripts in these samples (Fig. 6B). We also looked for protein expression changes, and our studies revealed that TopoVIB was upregulated significantly in treated parasites (Fig. 6C).
FIG 6.

Radicicol-induced upregulation of P. falciparum topoisomerase VIB. Synchronized schizont-specific cultures were divided into two groups and grown for 40 h in the absence or presence of radicicol (1.5 μM). (A) Semiquantitative RT-PCR analysis of PfTopoVIB, PfHsp90, and PfARP genes in the absence (−) and presence (+) of radicicol. ARP acted as a loading control. PCR without reverse transcription (−RT) shows no amplification of PfTopoVIB. Genomic DNA (gDNA) sample acted as a positive control. (B) Real-time RT-PCR reveals quantitative abundance of the PfTopoVIB and PfHsp90 transcripts in radicicol-treated and -untreated cells. (C) Western blot analysis on parasite lysates isolated from radicicol-untreated (−) and -treated (+) cultures using anti-PfTopoVIB antibody. PfHsp70 acts as a loading control.
Subcellular localization of Plasmodium topoisomerase VIB.
As radicicol treatment seemed to have an effect at the level of mitochondria, we speculated that the drug target might be localized in the mitochondria. Unfortunately, the antipeptide antibody for PfTopoVIB did not work in the indirect immunofluorescence experiment (data not shown). Subcellular fractionation of parasite-infected erythrocytes yielded a nuclear fraction, cytoplasmic fraction, and organelle fraction (containing both mitochondria and apicoplasts). Western blot analysis of the parasite organelle, nuclear, and cytoplasmic fractions revealed that cytoplasm was devoid of detectable TopoVIB, whereas the protein was abundantly present in both nuclear as well as organelle fractions (Fig. 7). PfHsp70 was used as a control, and its presence in the cytoplasmic and organelle fractions was shown.
FIG 7.
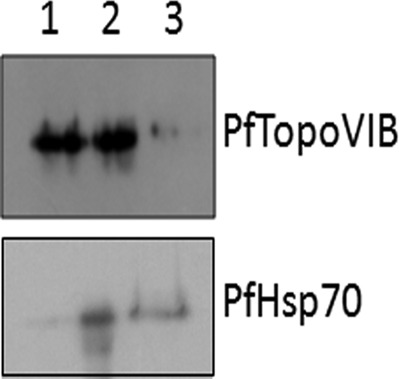
Subcellular localization of topoisomerase VIB. Western blot analysis of subcellular fractions of infected RBC using antibodies against PfTopoVIB and PfHsp70. Lane 1 corresponds to the nuclear fraction, lane 2 corresponds to the organelle fraction, and lane 3 corresponds to the cytoplasmic fraction.
In silico analysis shows docking of radicicol into the Bergerat fold of topoisomerase VIB.
Since no crystal or solution structure of this protein has been determined so far, we have done homology modeling of this protein and then looked for radicicol binding. Amino acid analysis of TopoVIB reveals the presence of a GHKL ATPase domain that forms a Bergerat fold, which is the ATP-binding pocket. A crystal structure of radicicol binding to Sulfolobus shibatae topoisomerase VIB (PDB accession no. 2HKJ) exists in the PDB and was used to decipher the correct orientation and binding modes of radicicol to PfTopoVIB and PfHsp90.
The criteria for selection of the docking mode was the proximity of the ligand's epoxide oxygen to a conserved residue (Asp43 in PfTopoVIB is conserved with the Asp45 residue of Sulfolobus TopoVIB) and bent toward the longer alpha helix in the active site, as observed in the crystal structure of Sulfolobus TopoVIB bound to radicicol. The aromatic ring was focused in the binding cavity formed by Leu37, Asn40, Ser41, Ala44, Asp43, Asn47, Asn48, Asp92, Gly94, and Lys95. The ligand was seen hydrogen bonded to Asn40, Asn47, and Asp92 (Fig. 8A). Radicicol was also docked in the same orientation to PfHsp90 as in the above structure (Fig. 8B). Its epoxide oxygen was placed near the Asp40 residue of Hsp90, which aligns with the Asp43 residue of PfTopoVIB (Fig. 8C). The ligand was seen hydrogen bonded to Leu34, Asn37, Lys44, Asp79, and Thr171 residues. It was observed that the top-scoring docking mode had the same orientation, and the epoxide oxygen of radicicol was placed near Asp43 of PfTopoVIB (Fig. 8D).
FIG 8.

Binding mode of radicicol with PfTopoVIB and PfHsp90. (A) Hydrogen-bonding interactions between radicicol and PfTopoVIB. (B) Hydrogen-bonding interactions between radicicol and PfHsp90. (C) Docking mode with the protein (shown as surface) and radicicol (as stick). Radicicol docked into PfHsp90. (D) Radicicol docked into PfTopoVIB, near the Asp43 residue, which is labeled. The epoxide oxygen of radicicol near the Asp40 residue of PfHsp90 aligns with the Asp43 residue of PfTopoVIB. (E) 17AAG docked into the binding site of PfHsp90, and its orientation is the same as in the crystal structure shown in panel F. (F) Crystal structure of human Hsp90 bound to 17DMAG (PDB accession no. 1OSF). (G) 17AAG failed to dock into the binding site of PfTopoVIB in its proper orientation.
In silico analyses predicted that 17AAG was docked into the binding site of PfHsp90 in the same orientation (Fig. 8E) as 17-N, N-dimethyl ethylene diamine-geldanamycin (17DMAG) was bound to human Hsp90 in the crystal structure (Fig. 8F). However, the orientation of 17AAG docked into the binding cavity of PfTopoVIB was different from that of the above two (Fig. 8G). Thus, while 17AAG may inhibit PfHsp90, it may not be inhibiting PfTopoVIB, as it may need proper orientation for binding.
DISCUSSION
Here we report that radicicol inhibits intraerythrocytic growth of P. falciparum with an IC50 near the range of 8 μM. We employed two independent assays to determine IC50s. First, we performed traditional parasite counting under the microscope, and then, we validated the counts by a very sensitive SYBR green I-based fluorescence assay. IC50s determined by the two assays were comparable. We also tested the inhibitory effects of another drug, chloroquine (CQ), on our 3D7 strain and observed that it exhibited IC50 values of 16 ± 3 nM, which are comparable to earlier reported values in the literature. The radicicol IC50 determined in our work apparently differs from previously reported antimalarial effects of radicicol, which demonstrated a much lower IC80 value of 32.3 nM (26). In the previous work, the IC80 value was determined after 4 days of radicicol treatment as opposed to 48 h of treatment in our assay. Interestingly, in the previously reported study no significant effect of radicicol was observed after 2 days of radicicol treatment (up to a 2.75 μM concentration). Thus, our results are in general agreement with previous findings that the inhibitory concentration of radicicol when tested on one asexual developmental cycle (48 h) is in the micromolar range.
We observed that two distinct phenotypes of radicicol-treated parasites were present in a concentration-dependent manner. At a higher concentration (8 μM), significant reduction of the parasite count was observed, whereas at a lower concentration (1.5 μM) radicicol did not manifest any parasiticidal activity. At a lower concentration of the drug, the immediate first cycle of schizont-to-ring conversion remained unaffected; at a higher concentration, even the first cycle of schizont-to-ring transition was severely affected. One of the possible reasons for such discrepancy might be that at lower concentrations the available intracellular concentration of the drug might not reach effective levels. Once the parasites were released into the medium in the form of merozoites, they were exposed to the drug (as we kept constant drug pressure at every 10 h), and thus, in the 2nd cycle of schizont-to-ring transition, the effect could be more pronounced. However, at high doses the intracellular concentration of the drug might be optimum, leading to the severe outcome manifested even at the first cycle of the schizont-to-ring transition. Another equally likely possibility is that radicicol has more than one cellular target, one with higher affinity than the other. The inactivation of the high-affinity target was not lethal for the parasite but affected some aspect of schizogonic development (for example, mitochondrial replication). On the other hand, the low-affinity target could be an essential gene product and its inhibition at higher radicicol concentrations leads to parasitic cell death. Identification and biochemical characterization of such cellular target(s) of radicicol would be necessary to test these possibilities in future. It has been reported that camptothecin (an inhibitor of topoisomerase I) (27) irreversibly binds to the target protein, leading to cell death. However, here we find that radicicol binding to the target protein is reversible in nature, as the impasse of the schizont-to-ring transition is released once the radicicol is washed off.
Our study revealed that radicicol treatment led to parasite growth arrest at the schizont stage and that it affected mitochondrial DNA. Earlier work had established that synthesis of mitochondrial DNA occurs in a stage-specific manner, being at a maximum during the schizont stage (28). Thus, radicicol-mediated schizont stage-specific arrest of the parasite is in good agreement with inhibition of mitochondrial replication. Previously, the inhibitory effect of radicicol on parasite culture was correlated with free radical generation, which was found to be reversed by the presence of radical scavengers (26). Since mitochondrial dysfunction can also lead to increased free radical formation, these findings were consistent. Our data did not support primary action of radicicol on nuclear or apicoplast genome, as the replications of those genomes were not altered even with up to 40 h of drug treatment.
We intended to predict the putative target of radicicol in this parasite. We observed that the schizogony development (equivalent to endoreduplication) of the parasite was affected by radicicol treatment. Our docking study predicted that radicicol fitted comfortably in the ATP-binding pocket/Bergerat fold present in both PfTopoVIB and PfHsp90. Our results established that radicicol treatment caused upregulation of TopoVIB at both the transcript and protein levels, without any significant alteration of the Hsp90 transcript. Since radicicol was predicted to bind to the Bergerat fold of TopoVIB, the increase in the level of the TopoVIB transcript could be related to a compensatory mechanism to maintain the level of active TopoVIB in the cell. In an earlier study it has been demonstrated that novobiocin treatment leads to upregulation of its target gene gyrB transcription in Synechocystis (29). The presence of abundant TopoVIB transcripts as well as proteins mainly at the schizont stage is consistent with the notion that TopoVIB is likely to be a target of radicicol. Western blot analysis established the localization of topoisomerase VIB in the nuclear and organelle fractions; however, topoisomerase VIB localization was negative for the cytoplasmic fraction. The nuclear function of topoisomerase VIB might be redundant, as we did not observe any change in nuclear replication on radicicol treatment. However, its presence in the organelle fraction (mitochondria) supported our view that TopoVIB might be one of the targets of radicicol in parasites.
In vitro biochemical characterization of PfTopoVIB activity and its inhibition by radicicol would provide direct evidence on whether or not PfTopoVIB is a target of radicicol. Unfortunately, all our attempts to express recombinant PfTopoVIB in Escherichia coli (as an His-tagged, maltose-binding protein [MBP]-tagged, and glutathione S-transferase [GST]-tagged protein, as well as a truncated protein containing the ATPase domain alone) went in vain. We had tried a number of various bacterial host systems (BL21, pLysS, BL21 star, Rosetta, and codon plus) but failed in spite of several attempts.
Our docking study reveals that radicicol binds to the Bergerat fold of PfTopoVIB as well as PfHsp90. Therefore, PfHsp90 or PfTopoVIB is the potential target of radicicol in the parasite. To identify the target responsible for inhibition of mitochondrial replication, we used a potent Hsp90 inhibitor, 17AAG. The docking study revealed that binding of 17AAG to PfHsp90 is very similar to its binding with human Hsp90; however, it fails to dock with PfTopoVIB in proper orientation, indicating that 17AAG might not inhibit the function of PfTopoVIB. Also, no bioinformatics or experimental evidence so far has indicated that 17AAG binds to topoisomerase VIB in any organisms. Southern hybridization data have demonstrated that 17AAG treatment does not cause any alteration of mitochondrial replication. This observation gave us the unique opportunity to demarcate the possible target of radicicol through which the effect on mitochondrial replication is exerted. Since the effect on mitochondrial replication is observed only by radicicol and not by 17AAG, it is highly unlikely that PfHsp90 is the affected molecule. Moreover, PfHsp90 has not been reported in Plasmodium mitochondria, whereas PfTopoVIB is found in the mitochondrial fraction. Previous work with geldanamycin, a potent drug that blocks the ATPase domain of Hsp90, has revealed that parasite growth is affected mostly at the ring-to-trophozoite transition, where Hsp90 is profusely expressed (30). Another study using yeast as a surrogate model system documented that geldanamycin is much more potent in blocking PfHsp90 function than is radicicol (31). It is not unlikely that radicicol has different binding affinities for PfHsp90 and PfTopoVIB. For example, at lower concentrations it could be a potent inhibitor of PfTopoVIB, whereas at higher concentrations it might target PfHsp90, causing drastic cell lethality. This notion is consistent with the fact that in other organisms TopoVIB knockouts are viable while Hsp90 knockouts are not.
It has been demonstrated that novobiocin, an inhibitor of gyrase B (topoisomerase II) causes parasite death by blocking apicoplast replication, and it does not have any effect on mitochondrial replication (32). On the other hand, our study shows that radicicol does not encompass a significant inhibitory effect on apicoplast replication, suggesting that it has a different target which is accountable for mitochondrial replication. Thus, it appears that these two type II topoisomerases have their specific roles during organelle DNA replication in Plasmodium. While TopoVIB is involved in mitochondrial DNA replication, gyrase B is involved in apicoplast DNA replication. Our work suggests a link between mitochondrial DNA replication and topoisomerase VIB inhibition, which needs to be explored in detail in the future.
ACKNOWLEDGMENTS
This work was supported by grants from the Council of Scientific and Industrial Research, government of India [37(1549)/12/EMR-II] (to S.B.), and from UPE-II (University of Hyderabad) (to M.K.B.). S.C. is supported by a Senior Research Fellowship from the University Grants Commission, government of India. Computational work incorporated in the article was carried out at the DBT-funded Bioinformatics Infrastructure Facility, University of Hyderabad.
We thank Nirbhay Kumar, Tulane University, for providing PfHsp70 antibody.
Footnotes
Published ahead of print 19 May 2014
REFERENCES
- 1.Joubès J, Chevalier C. 2000. Endoreduplication in higher plants. Plant Mol. Biol. 43:735–745. 10.1023/A:1006446417196 [DOI] [PubMed] [Google Scholar]
- 2.Sugimoto-Shirasu K, Stacey NJ, Corsar J, Roberts K, McCann MC. 2002. DNA topoisomerase VI is essential for endoreduplication in Arabidopsis. Curr. Biol. 12:1782–1786. 10.1016/S0960-9822(02)01198-3 [DOI] [PubMed] [Google Scholar]
- 3.Kirik V, Schrader A, Uhrig JF, Hulskamp M. 2007. MIDGET unravels functions of the Arabidopsis topoisomerase VI complex in DNA endoreduplication, chromatin condensation, and transcriptional silencing. Plant Cell 19:3100–3110. 10.1105/tpc.107.054361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breuer C, Stacey NJ, West CE, Zhao Y, Chory J, Tsukaya H, Azumi Y, Maxwell A, Roberts K, Sugimoto-Shirasu K. 2007. BIN4, a novel component of the plant DNA topoisomerase VI complex, is required for endoreplication in Arabidopsis. Plant Cell 19:3655–3668. 10.1105/tpc.107.054833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sugimoto-Shirasu K, Roberts GR, Stacey NJ, McCann MC, Maxwell A, Roberts K. 2005. RHl1 is an essential component of the plant DNA topoisomerase VI complex and is required for ploidy-dependent cell growth. Proc. Natl. Acad. Sci. U. S. A. 51:18736–18741. 10.1073/pnas.0505883102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aravind L, Iyer LM, Wellems TE, Miller LH. 2003. Plasmodium biology: genomic gleanings. Cell 115:771–785. 10.1016/S0092-8674(03)01023-7 [DOI] [PubMed] [Google Scholar]
- 7.Wang JC. 1996. DNA topoisomerases. Annu. Rev. Biochem. 65:635–692. 10.1146/annurev.bi.65.070196.003223 [DOI] [PubMed] [Google Scholar]
- 8.Nitiss JL. 1998. Investigating the biological functions of DNA topoisomerases in eukaryotic cells. Biochim. Biophys. Acta 1400:63–81. 10.1016/S0167-4781(98)00128-6 [DOI] [PubMed] [Google Scholar]
- 9.Champoux JJ. 2001. DNA topoisomerases: structure, function and mechanism. Annu. Rev. Biochem. 70:369–413. 10.1146/annurev.biochem.70.1.369 [DOI] [PubMed] [Google Scholar]
- 10.Wang JC. 1998. Moving one DNA double helix through another by a type II DNA topoisomerase: the story of a simple molecular machine. Q. Rev. Biophys. 31:107–144. 10.1017/S0033583598003424 [DOI] [PubMed] [Google Scholar]
- 11.Bergerat A, deMassy B, Gadelle D, Varoutas PC, Nicolas A, Forterre P. 1997. An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature 386:414–417. 10.1038/386414a0 [DOI] [PubMed] [Google Scholar]
- 12.Gadelle D, Bocs C, Graille M, Forterre P. 2005. Inhibition of archaeal growth and DNA topoisomerase VI activities by the Hsp90 inhibitor radicicol. Nucleic Acids Res. 33:2310–2317. 10.1093/nar/gki526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corbett KD, Berger JM. 2006. Structural basis for topoisomerase VI inhibition by the anti-Hsp90 drug radicicol. Nucleic Acids Res. 34:4269–4277. 10.1093/nar/gkl567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soga S, Neckers LM, Schulte TW, Shiotsu Y, Akasaka K, Narumi H, Agatsuma T, Ikuina Y, Murakata C, Tamaoki T, Akinaga S. 1999. KF25706, a novel oxime derivative of radicicol, exhibits in vivo antitumor activity via selective depletion of Hsp90 binding signaling molecules. Cancer Res. 59:2931–2938 [PubMed] [Google Scholar]
- 15.Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. 1997. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 89:239–250. 10.1016/S0092-8674(00)80203-2 [DOI] [PubMed] [Google Scholar]
- 16.Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, Pearl LH. 1999. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 42:260–266. 10.1021/jm980403y [DOI] [PubMed] [Google Scholar]
- 17.Schulte TW, Akinaga S, Soga S, Sullivan W, Stensgard B, Toft D, Neckers LM. 1998. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones 3:100–108. 10.1379/1466-1268(1998)003<0100:ARBTTN>2.3.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanaka Y, Shiomi K, Kamei K, Sugoh-Hagino M, Enomoto Y, Fang F, Yamaguchi Y, Masuma R, Zhang CG, Zhang XW, Omura S. 1998. Antimalarial activity of radicicol, heptelidic acid and other fungal metabolites. J. Antibiot. (Tokyo) 51:153–160. 10.7164/antibiotics.51.153 [DOI] [PubMed] [Google Scholar]
- 19.Bhattacharyya MK, Kumar N. 2001. Effects of xanthurenic acid on infectivity of Plasmodium falciparum to Anophiles stephensi. Int. J. Parasitol. 31:1129–1133. 10.1016/S0020-7519(01)00222-3 [DOI] [PubMed] [Google Scholar]
- 20.Moll K, Ljungstrom I, Perlmann H, Scherf A, Wahlgren M. 2008. Methods in malaria research, 5th ed. Malaria Research and Reference Reagent Resource Center/ATCC, Manassas, VA [Google Scholar]
- 21.Bhattacharyya MK, Kumar N. 2003. Identification and molecular characterization of DNA damaging agent induced expression of Plasmodium falciparum recombination protein PfRad51. Int. J. Parasitol. 33:1385–1392. 10.1016/S0020-7519(03)00212-1 [DOI] [PubMed] [Google Scholar]
- 22.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. 2004. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 48:1803–1806. 10.1128/AAC.48.5.1803-1806.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vardharajan S, Sagar BKC, Rangarajan PN, Padmanaban G. 2004. Localization of ferrochelatase in Plasmodium falciparum. Biochem. J. 384:429–436. 10.1042/BJ20040952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pallavi R, Roy N, Nageshan RK, Talukdar P, Pavithra SR, Reddy R, Venketesh S, Kumar R, Gupta AK, Singh RK, Yadav SC, Tatu U. 2010. Heat shock protein 90 as a drug target against protozoan infections: biochemical characterization of Hsp90 from Plasmodium falciparum and Trypanosoma evansi and evaluation of its inhibitor as a candidate drug. J. Biol. Chem. 285:36964–36975. 10.1074/jbc.M110.155317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dobson S, Kumar R, Bracchi-Ricard V, Freeman S, Al-Murrani SW, Johnson C, Damuni Z, Chakrabarti D, Barik S. 2003. Characterization of a unique aspartate-rich protein of the SET/TAF-family in the human malaria parasite, Plasmodium falciparum, which inhibits protein phosphatase 2A. Mol. Biochem. Parasitol. 126:239–250. 10.1016/S0166-6851(02)00293-1 [DOI] [PubMed] [Google Scholar]
- 26.Tanaka Y, Kamei K, Otoguro K, Omura S. 1999. Heme-dependent radical generation: possible involvement in antimalarial action of nonperoxide microbial metabolites, nanaomycin A and radicicol. J. Antibiot. 52:880–888. 10.7164/antibiotics.52.880 [DOI] [PubMed] [Google Scholar]
- 27.Hertzberg RP, Busby RW, Caranfa MJ, Holden KG, Johnson RK, Hecht SM, Kingsbury WD. 1990. Irreversible trapping of the DNA-topoisomerase I covalent complex. Affinity labeling of the camptothecin binding site. J. Biol. Chem. 265:19287–19295 [PubMed] [Google Scholar]
- 28.Preiser PR, Wilson RJM, Moore PW, McCready S, Hajibagheri MAN, Blight KJ, Strath M, Williamson DH. 1996. Recombination associated with replication of malarial mitochondrial DNA. EMBO J. 15:684–693 [PMC free article] [PubMed] [Google Scholar]
- 29.Prakash JS, Sinetova M, Zorina A, Kupriyanova E, Suzuki I, Murata N, Los DA. 2009. DNA supercoiling regulates the stress-inducible expression of genes in the cyanobacterium Synechocystis. Mol. Biosyst. 5:1904–1912. 10.1039/b903022k [DOI] [PubMed] [Google Scholar]
- 30.Kumar R, Musiyenko A, Barik S. 2003. The heat shock protein 90 of Plasmodium falciparum and antimalarial activity of its inhibitor, geldanamycin. Malar. J. 2:30. 10.1186/1475-2875-2-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wider D, Péli-Gulli MP, Briand PA, Tatu U, Picard D. 2009. The complementation of yeast with human or Plasmodium falciparum Hsp90 confers differential inhibitor sensitivities. Mol. Biochem. Parasitol. 164:147–152. 10.1016/j.molbiopara.2008.12.011 [DOI] [PubMed] [Google Scholar]
- 32.RaghuRam EVS, Kumar A, Biswas S, Kumar A, Chaubey S, Siddiqi MI, Habib S. 2007. Nuclear gyrB encodes a functional subunit of the Plasmodium falciparum gyrase that is involved in apicoplast DNA replication. Mol. Biochem. Parasitol. 154:30–39. 10.1016/j.molbiopara.2007.04.001 [DOI] [PubMed] [Google Scholar]