

Abstract
Pseudomonas aeruginosa is a major cause of hospital-acquired infections, particularly in mechanically ventilated patients, and it is the leading cause of death in cystic fibrosis patients. A key virulence factor associated with disease severity is the P. aeruginosa type III secretion system (T3SS), which injects bacterial toxins directly into the cytoplasm of host cells. The PcrV protein, located at the tip of the T3SS injectisome complex, is required for T3SS function and is a well-validated target in animal models of immunoprophylactic strategies targeting P. aeruginosa. In an effort to identify a highly potent and protective monoclonal antibody (MAb) that inhibits the T3SS, we generated and characterized a panel of novel anti-PcrV MAbs. Interestingly, some MAbs exhibiting potent inhibition of T3SS in vitro failed to provide protection in a mouse model of P. aeruginosa infection, suggesting that effective in vivo inhibition of T3SS with anti-PcrV MAbs is epitope dependent. V2L2MD, while not the most potent MAb as assessed by in vitro cytotoxicity inhibition assays, provided strong prophylactic protection in several murine infection models and a postinfection therapeutic model. V2L2MD mediated significantly (P < 0.0001) better in vivo protection than that provided by a comparator antibody, MAb166, a well-characterized anti-PcrV MAb and the progenitor of a clinical candidate, KB001-A. The results described here support further development of a V2L2MD-containing immunotherapeutic and may suggest even greater potential than was previously recognized for the prevention and treatment of P. aeruginosa infections in high-risk populations.
INTRODUCTION
Pseudomonas aeruginosa infections impose a significant burden on the health care system (1) and have a high mortality rate, particularly when comorbidities are present (2, 3). The spread of multidrug-resistant P. aeruginosa further compounds the problem, leaving few effective treatment options available for this pathogen (4). In an era of rising drug resistance among bacterial pathogens, due in large part to the empirical use of broad-spectrum antibiotics, mechanistically distinct and pathogen-specific approaches are badly needed. Explorations of antibody-based approaches for the prevention or treatment of serious bacterial infections, including those caused by P. aeruginosa, are gaining momentum (5–8). Given the potential for specific and targeted therapies with distinct mechanisms of action, monoclonal antibodies (MAbs) may represent a paradigm-shifting opportunity for the development of novel antimicrobial therapies that will not encourage further antibiotic resistance (9, 10).
A bacterial virulence mechanism common to some virulent Gram-negative pathogens is the type III secretion system (T3SS). This macromolecular delivery system for the intracellular injection of bacterial toxins and effector molecules has been under intense scrutiny as a target for vaccines and antibodies (11–13). Investigations of P. aeruginosa T3SS gene expression in human disease isolates reveal a correlation between exotoxin expression/transport and increased disease severity and poor clinical outcomes (14–17). The P. aeruginosa T3SS is a well-validated target for intervention in infections caused by this opportunistic pathogen. Both active vaccination with T3SS component proteins and passive immunotherapy targeting PcrV strongly attenuate P. aeruginosa disease in animal models (18–22). In fact, a pegylated Fab fragment of an anti-PcrV MAb is currently in development for preventing P. aeruginosa respiratory infections in mechanically ventilated patients (11, 23). This drug candidate is based on the PcrV-specific mouse monoclonal antibody MAb166. While effective in blocking P. aeruginosa T3SS in vitro, MAb166 requires relatively high antibody doses for protection in animal models (18, 19). Whether this is due to its modest PcrV-binding affinity (11) or results from the mechanism by which its specific binding to PcrV inhibits the T3SS is unclear.
With the goal of identifying an anti-PcrV MAb that exhibits enhanced inhibition of the T3SS, we generated a panel of novel antibodies. Here, we describe the selection and characterization of a candidate with clinical potential. Our screening strategy selected MAbs that bound to PcrV and potently neutralized PcrV-dependent cytotoxicity in vitro. Interestingly, we observed that only a subset of these MAbs significantly improved survival when administered prior to infection in a murine pneumonia model. In fact, the MAb exhibiting the most potent T3SS inhibition in vitro provided poor protection in vivo. In contrast, a MAb exhibiting less potent in vitro activity and that bound a distinct epitope had highly protective activity in multiple P. aeruginosa infection models. The therapeutic potential of this MAb, V2L2MD, was also assessed by comparing its activity to that of the well-studied anti-PcrV monoclonal antibody MAb166, the progenitor of the promising clinical candidate KB001-A. V2L2MD exhibited superior potency in cell-based assays of T3SS intoxication and in multiple mouse models of P. aeruginosa infection. Our results indicate that targeting PcrV may offer greater potential than was previously demonstrated and that V2L2MD may be a promising component of an antibody-based approach for combating P. aeruginosa infections in high-risk patients.
MATERIALS AND METHODS
Bacterial strains and culture.
P. aeruginosa strains 6077, 6206, and 6294 were provided by J. B. Goldberg (University of Virginia, Charlottesville, VA). The strains were propagated in 2× YT medium (16 g/liter tryptone, 10 g/liter yeast extract, 5.0 g/liter NaCl) (Difco) or on tryptic soy agar plates (BBL).
Expression of recombinant PcrV.
The pcrV open reading frame was PCR amplified from the genomic DNA of P. aeruginosa strain PAO1. The product was cloned into expression vector pET-26b(+) (Novagen) and verified by sequencing. The construct was transformed into Escherichia coli BL21(DE3) and expression induced by overnight culture in Magic medium (Invitrogen). The harvested cells were disrupted using a fixed-geometry fluid processor (Microfluidics) and soluble recombinant PcrV purified by anion-exchange chromatography.
Vaccination of VelocImmune mice and hybridoma generation.
Recombinant PcrV protein was used to immunize VelocImmune mice using a modified Repetitive Immunizations Multiple Sites (RIMMS) protocol (24). The mice were sacrificed, and B cells from the spleen and lymph nodes were first selected for antigen binding before fusion with P3X myeloma for hybridoma generation.
RBC lysis inhibition assay.
Red blood cells (RBCs) were prepared from fresh whole rabbit blood (Pel-Freez) by centrifugation and multiple phosphate-buffered saline (PBS) washes. Washed RBCs (2.5% [vol/vol] final) in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal bovine serum (FBS) (Invitrogen) and anti-PcrV hybridoma supernatant or purified IgG diluted in PBS were combined into wells of a round-bottom 96-well plate. P. aeruginosa strain 6077 was grown to mid-log phase in 2× YT medium (Difco), harvested by centrifugation, and resuspended in DMEM-fetal bovine serum (FBS) at an optical density at 600 nm (OD600) of 0.15. Ten microliters of bacterial suspension was added to the RBC-antibody mixture, mixed by agitation, and incubated 2 h at 37°C. The plates were briefly centrifuged to pellet the intact RBCs, the supernatants transferred to a flat-bottom 96-well plate, and the OD405 measured.
A549 cell lysis inhibition assay.
Antibodies were added to the human bronchoepithelial cell line A549 seeded in white 96-well plates (Nunc Nunclon Delta) in DMEM plus 10% fetal bovine serum. Log-phase strain 6077 was added at a multiplicity of infection (MOI) of 10 and incubated for 2 h at 37°C and 5% CO2. Lactate dehydrogenase (LDH) activity released from lysed cells was quantified using the CytoTox-ONE kit (Promega).
Competition ELISA.
A matrix capture enzyme-linked immunosorbent assay (ELISA) was used for the MAb competition studies. ELISA plates (Nunc MaxiSorp) were coated with purified murine anti-PcrV monoclonal antibodies at 100 ng/well in PBS and incubated overnight at 4°C. The plates were washed with PBS supplemented with 0.1% Tween 20 (PBS-T) and blocked with PBS-T plus 2% bovine serum albumin (BSA) for 1 h at room temperature, followed by washing with PBS-T. Biotinylated recombinant PcrV (rPcrV) was premixed with a 500-fold excess of the competing anti-PcrV monoclonal antibodies, and the mixture was added to the wells and incubated at room temperature (RT) for 1 h. Bound biotinylated rPcrV was detected with streptavidin-horseradish peroxidase (HRP) (GE Healthcare Life Sciences) diluted to 1:30,000. The plates were developed with SureBlue 3,3′,5,5′-tetramethylbenzidine (TMB) (KPL) and read at 450 nm.
Affinity measurements.
The recombinant PcrV-binding kinetics of MAbs was measured by surface plasmon resonance (SPR) using a Bio-Rad ProteOn XPR36 instrument. The MAbs were captured on a GLC biosensor chip using anti-mouse IgG and anti-human IgG reagents. Purified rPcrV protein was injected at multiple concentrations, followed by the dissociation phase for 600 s. The data were captured and equilibrium dissociation constant (KD) values were determined using the ProteOn Manager software.
Affinity optimization of V2L2.
The V2L2 variable regions were fully germlined by constructing and screening a combinatorial library containing all possible combinations of germ line and wild-type residues of V2L2. The parsimonious mutagenesis approach was used for affinity maturation and optimization of V2L2. Each amino acid of a complementarity determining region (CDR) was randomized one at a time using QuikChange mutagenesis (Agilent Technologies) to construct a sublibrary for each CDR. All six CDR sublibraries were expressed and screened using bacterial and mammalian cell IgG expression and a competition-based screening platform to identify affinity-improved single mutations. A combinatorial library encoding all possible combinations of affinity-improved single mutations was generated and screened in the same manner as the CDR sublibraries. Clones with improved affinity to PcrV were selected by competition ELISA as above, and binding kinetics were confirmed using SPR.
V2L2MD-N297Q construction.
PCR mutagenesis was used to modify the human IgG1 heavy-chain-coding sequence of V2L2MD to substitute a glutamine for asparagine at residue 297, thereby eliminating the N297 glycosylation site.
Murine infection models.
All procedures were performed in accordance with federal, state, and institutional guidelines and were approved by the MedImmune Institutional Animal Care and Use Committee in an Association for Assessment and Accreditation of Laboratory Animal Care International-accredited facility. Antibodies or PBS was administered intraperitoneally (i.p.) at 24 h before infection (prophylaxis) or intravenously (i.v.) at 1 h postinfection (treatment). A P. aeruginosa acute pneumonia model was performed as described previously (25), with modifications. In the acute pneumonia model, BALB/c mice (The Jackson Laboratory) were infected with P. aeruginosa strains suspended in a 50-μl inoculum. For the organ burden experiments, acute pneumonia was induced in mice, followed by harvesting of the lungs, spleens, and kidneys 24 h after infection for determination of CFU. For the intraperitoneal (i.p.) infection model, P. aeruginosa grown to lawn on tryptic soy agar (TSA) plates was suspended in PBS and washed prior to preparing the inoculum. The cells were adjusted with PBS to the desired density, and the indicated inoculum was injected i.p. in a 0.5-ml volume.
RESULTS
To generate fully human antibodies targeting PcrV, VelocImmune mice, which express human antibody variable regions, were immunized with purified recombinant PcrV protein. B cells expressing PcrV-binding antibodies were enriched by antigen affinity selection, and the hybridomas were subsequently generated. Hybridoma supernatants were initially screened for rPcrV binding by ELISA, and supernatants from binding-positive wells were subsequently assayed for the inhibition of T3SS-mediated rabbit red blood cell (RBC) lysis by exoU+ P. aeruginosa strain 6077. Strongly inhibitory candidates were cloned by limiting dilution, and all were determined to be of the same isotype, IgG1(κ). The candidate IgGs were purified, and their T3SS inhibitory activities were compared (Fig. 1A).
FIG 1.
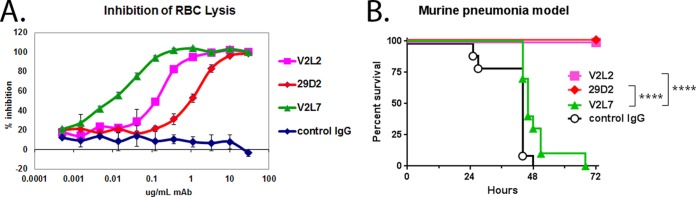
Evaluation of purified IgG from anti-PcrV hybridoma clones vitro and in vivo. (A) To quantitatively compare the ability of anti-PcrV hybridoma clones to inhibit P. aeruginosa T3SS in vitro, washed rabbit red blood cells (RBC) were incubated with log-phase P. aeruginosa strain 6077 in the presence of the indicated concentrations of purified IgG. The percent inhibition was determined by comparing the OD values from the test wells to the OD value in the control wells, which received no IgG. Error bars represent the standard deviations for four replicate samples. (B) The protective activities of anti-PcrV hybridoma clones were evaluated in a murine acute pneumonia model. BALB/c mice (n = 10) were treated with control IgG or the indicated clone at 25 mg/kg at 24 h before intranasal infection with strain 6077 (1e6 CFU). The animals were monitored for survival up to 72 h after infection. The results are represented as Kaplan-Meier survival curves. Differences in survival were calculated by the log rank test for V2L2 versus V2L7 (P < 0.0001) and for 29D2 versus V2L7 (P < 0.0001). Significance calculations for figures were performed with GraphPad Prism software, which assigns a number of asterisks to symbolically represent minimal significance (*) to high significance (****).
A subset of MAbs exhibiting in vitro T3SS inhibition activity were evaluated in a murine model of P. aeruginosa pneumonia. Surprisingly, some anti-PcrV MAbs that had exhibited potent inhibition of T3SS-dependent cytotoxicity in vitro did not provide protection in this infection model. In particular, MAb V2L7, the most potent MAb as assessed in vitro, did not provide significant protection in vivo. In contrast, MAbs V2L2 and 29D2, while less potent in vitro, provided complete protection at a dose of 25 mg/kg of body weight (Fig. 1B). To better understand this discrepancy between the in vitro and in vivo activities, we used a competitive binding assay to determine that V2L7, V2L2, and 29D2 did not compete with each other for binding to rPcrV, indicating they each bound to distinct regions of the PcrV protein (data not shown). Taken together, these findings indicate that in vivo protection by anti-PcrV MAbs may not directly correlate with in vitro activity and suggest that this relative incongruity is due to the MAbs binding to distinct epitopes on PcrV.
The protective activities of the candidates V2L2 and 29D2, which were active in vivo, were then compared by dose titration in the pneumonia prophylaxis model. V2L2 provided complete protection down to 1 mg/kg, whereas 29D2 did not (Fig. 2). MAb V2L2 was therefore selected to advance as the progenitor to a potential human clinical candidate molecule and was converted to a human IgG by genetically fusing its variable heavy (VH)- and variable light (VL)-coding regions to human IgG1 Fc- and C-kappa-coding regions. The V2L2 variable region-coding sequences were also modified at several residues to approximate the closest human germ line sequence, so as to minimize its immunogenic potential in humans. Finally, affinity optimization was accomplished by systematic mutation of the V2L2 complementarity determining regions (CDRs), followed by competition-based selections resulting in the fully human V2L2 derivative, V2L2MD, which exhibited 2-fold-higher affinity for rPcrV than the parent molecule while retaining similar T3SS inhibitory activity in vitro and protective activity in vivo.
FIG 2.
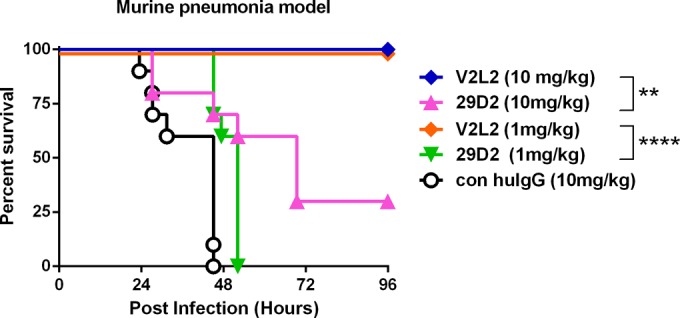
Protective activity of candidate anti-PcrV MAbs in a murine pneumonia model. The MAbs 29D2 and V2L2 were evaluated in a murine acute pneumonia model. BALB/c mice (n = 10) were treated with control human IgG (con huIgG) or the indicated MAb at 10 or 1 mg/kg at 24 h prior to intranasal infection with strain 6077 (1e6 CFU). The animals were monitored for survival up to 96 h postinfection. The results are represented as Kaplan-Meier survival curves. Differences in survival were calculated by the log rank test for V2L2 versus 29D2 at 10 mg/kg (P = 0.0014) and at 1 mg/kg (P < 0.0001).
Given that an anti-PcrV Fab fragment is currently in clinical development (23), we compared the T3SS inhibition activity of a Fab fragment prepared from V2L2MD to the IgG form of V2L2MD using the RBC lysis assay. When normalized for PcrV-binding site concentration, the V2L2MD IgG and monovalent Fab fractions exhibited identical inhibitory potency, indicating that neither bivalency of PcrV binding nor the antibody Fc domain is required for in vitro inhibition of PcrV function (Fig. 3). To further investigate any potential role of Fc function in the protection provided by V2L2MD in vivo, we constructed an Fc-modified variant, V2L2MD-N297Q. The N297Q mutation, which prevents glycosylation at this position, has been demonstrated to ablate the Fc effector function of IgG by eliminating binding to both Fcγ-RI and C1q, while retaining normal serum half-life in mice (26). In a comparison using the murine pneumonia model, V2L2MD-N297Q provided a protective benefit indistinguishable from that of V2L2MD, indicating no apparent role of Fc effector function in V2L2MD-mediated protection in this infection model (Fig. 4).
FIG 3.
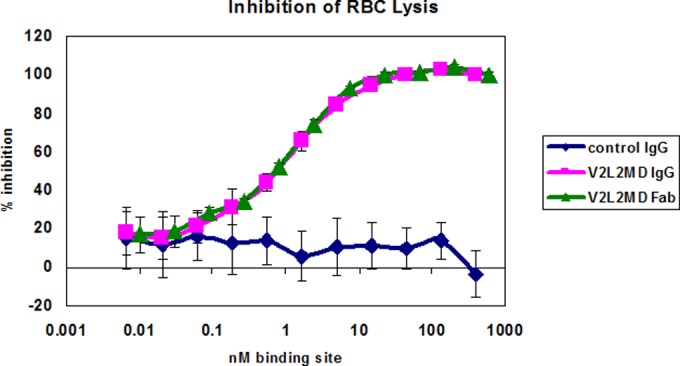
Comparison of in vitro inhibition of T3SS by V2L2MD IgG and Fab fractions. Washed rabbit red blood cells (RBC) were incubated with log-phase P. aeruginosa strain 6077 in the presence of the indicated concentrations of purified V2L2MD IgG or Fab. Released hemoglobin was measured spectrophotometrically. The percent inhibition was determined by comparing the OD values from the test wells to the OD value in the control wells, which received no antibody. Error bars represent the standard deviations for four replicate samples.
FIG 4.
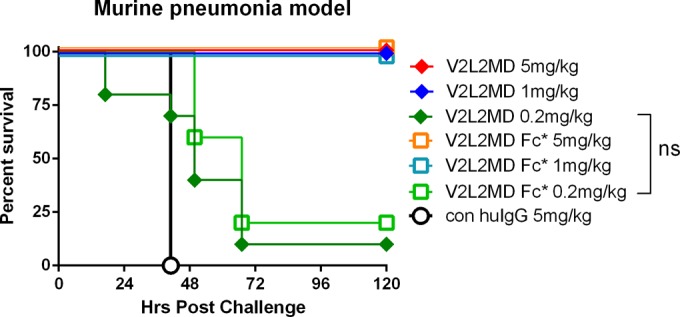
Relative protective activity of MAb V2L2MD with Fc N297Q mutant in murine pneumonia model. Fc*, N297Q mutant version of V2L2MD. BALB/c mice (n = 10) were treated with control IgG or the indicated MAb at 5, 1, or 0.2 mg/kg at 24 h before intranasal infection with strain 6077 (1e6 CFU). The animals were monitored for survival up to 120 h postinfection. The results are represented as Kaplan-Meier survival curves. Differences in survival were calculated by the log rank test at 0.2 mg/kg (P = 0.22). ns, not significant.
The therapeutic potential of V2L2MD was further evaluated both in vitro and in additional infection models. As a comparator MAb for these studies, we expressed a version of MAb166, the well-characterized anti-PcrV MAb and progenitor of the clinical candidate KB001-A, which has been shown to inhibit the P. aeruginosa T3SS in vitro and in vivo (18, 19). The MAb166 VH and VL coding sequences (27) were fused in frame to mouse IgG2a Fc- and C-kappa-coding regions to generate MAb166.2a. The mouse IgG2a format was chosen, as it is the homologous isotype to human IgG1 (28, 29), the isotype of V2L2MD. The subsequent binding and functional analysis with MAb166.2a described below revealed activities very close to the published results for MAb166.
Using surface plasmon resonance-based measurements, the relative affinities of MAb166.2a and V2L2MD for rPcrV were compared. The calculated KD value of 2.08 nM for MAb166.2a is very similar to the published value of 1.1 nM for MAb166 (11). The KD of V2L2MD binding to rPcrV was determined to be 0.04 nM, reflecting a ∼50-fold-greater affinity for rPcrV, though preliminary binding studies suggest that V2L2MD binds a region of PcrV distinct from that bound by MAb166.2a (data not shown).
The relative T3SS inhibitory activities of V2L2MD and MAb166.2a MAbs were evaluated in the RBC lysis inhibition assay (Fig. 5A). The 50% inhibitory concentrations (IC50s) for V2L2MD and MAb166.2a in this assay were determined to be 0.37 μg/ml and 3.7 μg/ml, respectively. An additional in vitro functional comparison of the MAbs was performed by assessing their ability to block T3SS-dependent P. aeruginosa cytotoxicity toward the human bronchoepithelial cell line A549. The lysis of A549 cells cocultured with strain 6077 (exoU+) was monitored as LDH activity released into the culture supernatant. V2L2MD inhibited A549 lysis in this assay, with an IC50 of 0.1 μg/ml, compared to an IC50 of 2.8 μg/ml for MAb166.2a (Fig. 5B).
FIG 5.
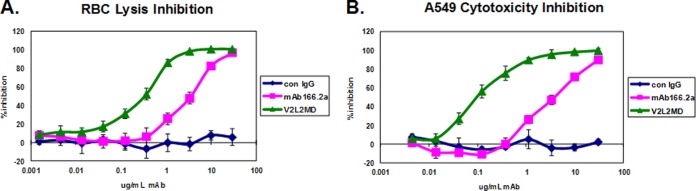
Comparison of in vitro inhibition of T3SS by V2L2MD and MAb166.2a IgG. (A) Rabbit red blood cells (RBC) were incubated with log-phase P. aeruginosa strain 6077 in the presence of the indicated concentrations of purified IgG. The released hemoglobin in the reaction mixture supernatants was measured spectrophotometrically. The percent inhibition was determined by comparing the OD values from the test wells to the OD value in the control wells, which received no IgG. (B) A549 cells were incubated with log-phase P. aeruginosa strain 6077 in the presence of the indicated concentrations of purified IgG. LDH activity released from lysed cells was quantified. The percent inhibition was determined by comparing the values from the test wells to the value in the control wells, which received no IgG. Each data point represents the average of 4 replicates. The IC50s were determined by nonlinear curve fitting. Differences in the IC50s for V2L2MD and MAb166.2a in both assays were determined to be significant (P < 0.05) by unpaired t test. Error bars represent the standard deviations for four replicate samples.
The abilities of V2L2MD and MAb166.2a to protect against P. aeruginosa infection following prophylactic administration were compared in multiple mouse infection models. Multiple P. aeruginosa challenge strains were utilized to assess the strain coverage of V2L2MD-mediated protection. In the acute lethal pneumonia model, different doses of V2L2MD, MAb166.2a, or a control IgG were administered 24 h prior to intranasal P. aeruginosa challenge, and survival was monitored. First, mice were challenged with cytotoxic (exoU+) strain 6077 at approximately 2× the 100% lethal dose (LD100) (1e6 CFU/mouse). While both V2L2MD and MAb166.2a provided protection at the highest doses tested (Fig. 6A), V2L2MD provided significantly greater in vivo protection at lower doses (P = 0.025 at 5 mg/kg and P = 0.037 at 1 mg/kg). Next, the mice were intranasally challenged with twice the LD100 (1.5e7 CFU/mouse) of the noncytotoxic (ExoS+) strain 6294 to evaluate MAb-mediated protection against a strain requiring a much larger inoculum to achieve lethality (Fig. 6B). In this model, V2L2MD again afforded a survival benefit that was significantly greater than that provided by the comparator MAb166.2a at an equivalent dose (P < 0.0001 at 1 mg/kg).
FIG 6.
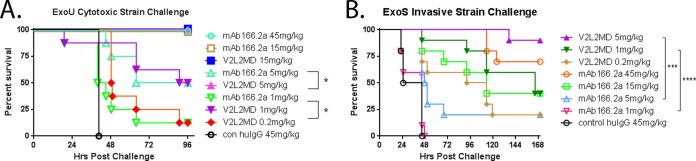
Protective activity of anti-PcrV MAbs V2L2MD and MAb166.2a in the acute murine pneumonia model. BALB/c mice (n = 10) were injected i.p. with V2L2MD, MAb166.2a, or control (con) IgG at the indicated concentrations 24 h before intranasal infection with ∼2× the LD100 dose of P. aeruginosa. The animals were monitored for survival up to 96 h. The results are represented as Kaplan-Meier survival curves; differences in survival were calculated by the log rank test for V2L2MD versus MAb166.2a. (A) Infection with cytotoxic exoU strain 6077 (1.0e6 CFU/mouse); 5 mg/kg, P = 0.025; 1 mg/kg, P = 0.037. (B) Infection with invasive exoS strain 6294 (1.5e7 CFU/mouse); 5 mg/kg, P = 0.0007; 1 mg/kg, P < 0.0001.
An additional infection model, looking at survival following acute bacteremia, was also utilized to evaluate V2L2MD-mediated protection and to investigate protection against the highly virulent cytotoxic P. aeruginosa strain 6206 (Fig. 7). Again, V2L2MD provided significantly greater protection than did MAb166.2a at equivalent doses (P = 0.0004 at 5 mg/kg and P = 0.0002 at 1 mg/kg). In fact, MAb166.2a provided no significant protection even at a dose of 15 mg/kg, suggesting that the relative potency of the anticytotoxic mechanism of V2L2MD versus that of MAb166.2a is even greater than that observed in the acute pneumonia model. The effect of V2L2MD on P. aeruginosa proliferation and dissemination from the lungs to other organs in the acute pneumonia model was also assessed. Mice were pretreated with V2L2MD doses ranging from 0.2 to 5 mg/kg at 24 h prior to intranasal infection, with an LD100 dose of strain 6206. An analysis of bacterial burdens in the lungs, spleen, and kidneys at 24 h postinfection revealed ≥2 log reductions in CFU in all three organs in comparison to the levels in control IgG-treated animals (Fig. 8). These reductions were also significant (Dunn's multiple comparison test) relative to the reductions achieved by prophylaxis with the comparator MAb166.2a, even when administered at the much higher dose of 15 mg/kg.
FIG 7.
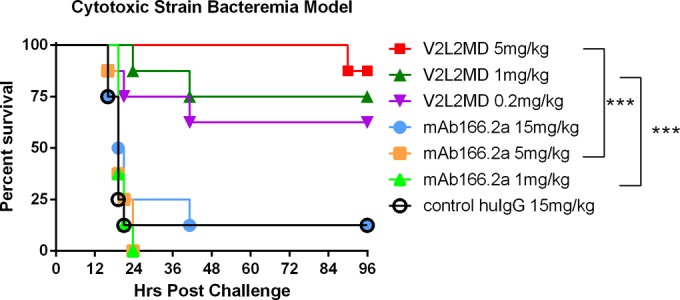
Protective activity of anti-PcrV MAbs V2L2MD and MAb166.2a in the murine bacteremia model. BALB/c mice (n = 10) were injected i.p. with V2L2MD, MAb166.2a, or control IgG at the indicated concentrations 24 h before i.p. infection with ∼2× the LD100 dose (2e7 CFU/mouse) of P. aeruginosa cytotoxic exoU strain 6206. The animals were monitored for survival up to 96 h. The results are represented as Kaplan-Meier survival curves; differences in survival were calculated by the log rank test for V2L2MD versus MAb166.2a: 5 mg/kg, P = 0.0004; 1 mg/kg, P = 0.0002.
FIG 8.
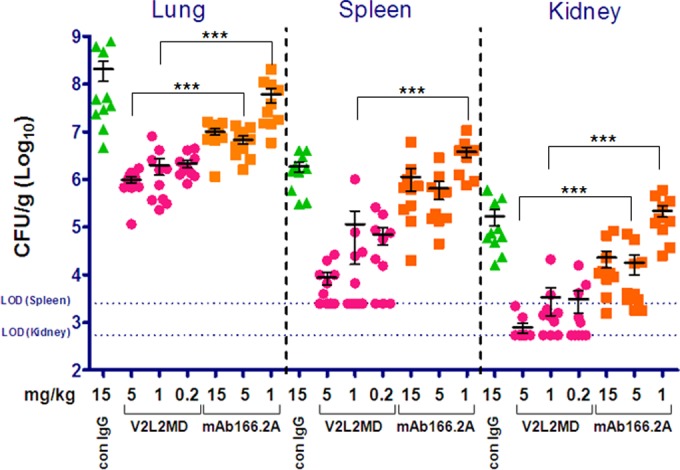
MAb V2L2MD mediates superior organ burden reduction following the induction of acute pneumonia. BALB/c mice (n = 10) were injected i.p. with V2L2MD, MAb166, or control (con) IgG at the indicated concentrations 24 h before intranasal infection with ∼1× the LD100 doses of P. aeruginosa strain 6206. At 24 h after infection, the animals were euthanized. The organs were harvested and homogenized, and dilutions were plated on LB agar to enumerate the viable CFU/g tissue. The standard error of the mean for each group is displayed. The limit of detection (LOD) values are indicated for spleen and kidney. One-way analysis of variance (ANOVA) of the equivalent MAb dose groups achieved significance where indicated (***).
Finally, to extend our investigations beyond prophylactic protection against P. aeruginosa infection, we evaluated the protective benefit of V2L2MD as a treatment delivered 1 h after bacterial challenge. The mice were infected intranasally (i.n.) with twice the LD100 of strain 6077 and then injected intravenously (i.v.) with V2L2MD, MAb166.2a, or a control IgG at 1 h postinfection, and survival was monitored. V2L2MD provided a dose-dependent survival benefit in this therapeutic modality (Fig. 9). The level of protection provided by V2L2MD was significantly greater than the protection provided by MAb166.2a at all MAb doses tested (P = 0.0067 at 15 mg/kg, P = 0.0003 at 5 mg/kg, and P < 0.0001 at 1 mg/kg).
FIG 9.
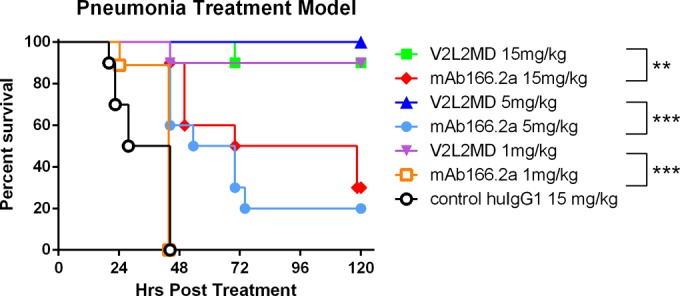
Postinfection protective activity of anti-PcrV MAbs V2L2MD and MAb166.2a in the acute murine pneumonia model. BALB/c mice (n = 10) were administered i.v. with V2L2MD, MAb166.2a, or control IgG at the indicated concentrations 1 h after i.n. infection with ∼2× the LD100 dose (1e6 CFU/mouse) of P. aeruginosa cytotoxic exoU strain 6077. The animals were monitored for survival up to 96 h. The results are represented as Kaplan-Meier survival curves; differences in survival were calculated by the log rank test for V2L2MD versus MAb166.2: P = 0.0067, 15 mg/kg; P = 0.0003, 5 mg/kg; and P < 0.0001, 1 mg/kg.
DISCUSSION
The extensive empirical use of antibiotics has selected for widespread resistance to virtually all small-molecule antibacterial drug classes. As a result, effective antibiotic treatment options are rapidly diminishing, particularly for serious Gram-negative pathogens, including P. aeruginosa. In this challenging environment, a monoclonal antibody-based strategy for the prevention of infection, or adjunctive use with antibiotics to treat serious bacterial infections, is an approach with considerable potential (10, 23, 25, 30). Monoclonal antibodies have been demonstrated to enhance bacterial clearance, prevent colonization and invasion, and prevent damage caused by cytotoxic or hyperinflammatory factors (8, 31, 32). These mechanisms are distinct from those of small-molecule antibiotics and, as a result, antibody-based antibacterial agents do not encourage cross-resistance to small-molecule antibiotics. In fact, they can actually complement antibiotic effectiveness and thus reduce the potential for antibiotic overuse (30, 33, 34). This high specificity of MAbs minimizes off-target interactions and allows for the selective treatment of only the disease-causing bacteria without impacting the beneficial microbiota of the host. In addition, the half-lives of antibodies far exceed those of small-molecule antibiotics, and the potential for drug-drug interactions between antibodies and small molecules is negligible. These advantageous features make antibodies highly compatible with concurrent small-molecule antibiotic therapy, particularly in intensive care settings where patients frequently receive antibiotics in addition to other drug treatments.
The type III secretion system (T3SS) plays a critical role in P. aeruginosa pathogenesis and is a well-studied and validated virulence factor in multiple bacterial species and animal models of infection. This macromolecular syringe-like complex is located on the bacterial surface and delivers exotoxins into the cytoplasm of host cells. Injected P. aeruginosa exotoxins (ExoS, T, U, and Y) can disrupt signal transduction, subvert bacterial phagocytosis, or directly degrade cell membranes (16). Located at the injectisome needle tip, PcrV is thought to act as a chaperone or scaffold for the insertion of the proteins PopB and PopD into the host cell membranes, where they form a pore, or translocon, through which exotoxins are injected into the target cells (31). By inhibiting PcrV function with specific antibodies, translocon formation and exotoxin injection are blocked (35).
In the present study, anti-PcrV MAbs were derived from rPcrV-immunized mice expressing human antibody variable regions. A subset of anti-PcrV MAbs inhibited T3SS function in a red blood cell lysis assay and were prioritized for evaluation of protective activity in a P. aeruginosa mouse acute pneumonia model. Two novel MAbs, 29D2 and V2L2MD, provided potent protection against P. aeruginosa in animal models of infection. Interestingly, several anti-PcrV MAbs exhibited in vitro inhibition of T3SS comparable to or greater than that of V2L2MD, but they provided no survival benefit when administered prophylactically in the pneumonia infection model. This incongruity was surprising given that the function-based MAb screening assay involved live bacteria injecting toxins into eukaryotic cells and was expected to be quite correlative of T3SS function despite its in vitro setting. The MAb V2L2MD, while identified as less potent using this same assay and binding an epitope distinct from that bound by MAbs that are inactive in vivo, was found to exhibit superior potency in an in vivo infection model. Given our observation that these MAbs bind different regions of PcrV, these results suggest that only a subset of inhibitory epitopes on PcrV is present or accessible in vivo. Epitope modifications in the infected host or blocking by host factors might also account for these observations. Alternate explanations are certainly possible, and further structural and mechanistic studies on MAb-mediated inhibition of the T3SS are warranted as the focus of a separate investigation. Given that multiple independent MAbs with unique sequences and epitope specificities exhibited this disparity between in vitro and in vivo activities, the importance of testing the in vivo activities of potential therapeutic MAbs as early as practical in the selection process cannot be overstated.
With an anti-PcrV pegylated Fab currently in clinical trials (23), we sought to better understand the role of bivalency and Fc function in the activities of our novel MAb. The V2L2MD Fab displayed in vitro T3SS inhibition activity identical to that of the IgG form. Similarly, the effector function-negative N297Q version of V2L2MD exhibited in vivo protective activity that was indistinguishable from that of the parent molecule. While the mechanisms of some antibacterial MAbs require Fc-mediated functions, such as opsonization or toxin clearance (5, 36), our results strongly suggest that the protection provided by V2L2MD-mediated T3SS inhibition does not require Fc functions.
To understand the properties and performance of our novel anti-PcrV MAbs in the context of the well-studied and promising benchmark antibody MAb166, we expressed an IgG with the MAb166 VH and VL regions, termed MAb166.2a, for use as a comparator molecule in our studies. Both the measured affinity of MAb166.2a for rPcrV and its inhibition of P. aeruginosa-mediated red blood cell lysis closely matched the published values for MAb166 for these activities (11, 35), supporting its utility in our experiments.
V2L2MD compared quite favorably to MAb166.2a when its T3SS inhibition activity was assayed in vitro in two cell-based assays, with V2L2MD exhibiting IC50s ≥10-fold lower than those observed for MAb166.2a. In addition, V2L2MD exhibits a 50-fold-higher affinity for PcrV than MAb166.2a, though based on our preliminary analysis, it appears that the two MAbs bind to distinct and likely conformational epitopes on PcrV (data not shown). More importantly, V2L2MD provided remarkable protective potency in multiple mouse models of P. aeruginosa infection. The survival benefit afforded by V2L2MD was observed in both the acute pneumonia and acute bacteremia models. This protection was observed for both the cytotoxic (ExoU-expressing) strains 6077 and 6206 and for the noncytotoxic (ExoS-expressing) strain 6294, underscoring the potential for broad-strain coverage of V2L2MD-mediated protection. Further, V2L2MD (at doses as low as 0.2 mg/kg) afforded strong protection in the murine bacteremia model and dramatically inhibited bacterial proliferation and dissemination in the murine pneumonia model compared to those in the untreated controls. In contrast, MAb166.2a had a more modest effect on lung bacterial burden and provided no observable protection in the bacteremia model, even at doses up to 15 mg/kg. These results further support the hypothesis that the P. aeruginosa T3SS function impairs phagocytic clearance of bacteria in the lungs in this model (37) and that V2L2MD blocks this effect. Further, the T3SS has also been shown to play a role in the disruption of epithelial barriers, leading to the dissemination of infection (38). The marked reduction in spread from the lungs to the spleen and kidneys observed with V2L2MD-treated animals is consistent with this mechanism. Thus, V2L2MD likely provides enhanced protection in the survival models by both preserving innate host bacterial clearance mechanisms and preventing bacterial dissemination by neutralizing P. aeruginosa T3SS-mediated epithelial disruption.
A previous study described a modest survival benefit when multiple doses of MAb166 were administered beginning at the time of P. aeruginosa infection in a murine thermal injury model (39). To investigate the potential of V2L2MD for treating existing infections, we adapted a therapeutic dosing modality similar to that in our murine pneumonia model. i.v. administration of V2L2MD at 1 h postinfection provided a significant (P < 0.0001) survival benefit. These results, taken together with those of a recent study demonstrating the benefit of combining anti-PcrV MAb administration with antibiotics (34), suggest that the addition of V2L2MD to an existing antipseudomonal antibiotic regimen might hold substantial promise for the treatment of P. aeruginosa infections.
In summary, we have derived and characterized a novel anti-PcrV MAb that exhibits potent inhibition of the P. aeruginosa T3SS in vitro and provides significantly greater protection against P. aeruginosa infection in multiple animal models than does MAb166.2a, the progenitor of the clinical candidate KB001-A. While this study is not a direct comparison of V2L2MD to KB001-A, published data indicate that this humanized pegylated Fab version of MAb166 shows only a very modest performance advantage over MAb166 IgG in murine infection models (11). Notwithstanding the indirect nature of the comparison to KB001-A, the data for V2L2MD provided in the present study suggest that targeting PcrV has even greater potential for preventing and treating P. aeruginosa disease than was previously recognized and support V2L2MD as a promising component in a new strategy for controlling P. aeruginosa infections.
ACKNOWLEDGMENTS
This work was funded by MedImmune, LLC, a wholly owned subsidiary of AstraZeneca Pharmaceuticals.
All authors are employees of MedImmune.
Footnotes
Published ahead of print 19 May 2014
REFERENCES
- 1.Jones RN. 2010. Microbial etiologies of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia. Clin. Infect. Dis. 51(Suppl 1):S81–S87. 10.1086/653053 [DOI] [PubMed] [Google Scholar]
- 2.Nseir S, Di Pompeo C, Soubrier S, Cavestri B, Jozefowicz E, Saulnier F, Durocher A. 2005. Impact of ventilator-associated pneumonia on outcome in patients with COPD. Chest 128:1650–1656. 10.1378/chest.128.3.1650 [DOI] [PubMed] [Google Scholar]
- 3.Trouillet JL, Vuagnat A, Combes A, Kassis N, Chastre J, Gibert C. 2002. Pseudomonas aeruginosa ventilator-associated pneumonia: comparison of episodes due to piperacillin-resistant versus piperacillin-susceptible organisms. Clin. Infect. Dis. 34:1047–1054. 10.1086/339488 [DOI] [PubMed] [Google Scholar]
- 4.Michalopoulos A, Fotakis D, Virtzili S, Vletsas C, Raftopoulou S, Mastora Z, Falagas ME. 2008. Aerosolized colistin as adjunctive treatment of ventilator-associated pneumonia due to multidrug-resistant Gram-negative bacteria: a prospective study. Respir. Med. 102:407–412. 10.1016/j.rmed.2007.10.011 [DOI] [PubMed] [Google Scholar]
- 5.DiGiandomenico A, Warrener P, Hamilton M, Guillard S, Ravn P, Minter R, Camara MM, Venkatraman V, Macgill RS, Lin J, Wang Q, Keller AE, Bonnell JC, Tomich M, Jermutus L, McCarthy MP, Melnick DA, Suzich JA, Stover CK. 2012. Identification of broadly protective human antibodies to Pseudomonas aeruginosa exopolysaccharide Psl by phenotypic screening. J. Exp. Med. 209:1273–1287. 10.1084/jem.20120033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marozsan AJ, Ma D, Nagashima KA, Kennedy BJ, Kang YK, Arrigale RR, Donovan GP, Magargal WW, Maddon PJ, Olson WC. 2012. Protection against Clostridium difficile infection with broadly neutralizing antitoxin monoclonal antibodies. J. Infect. Dis. 206:706–713. 10.1093/infdis/jis416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Secher T, Fauconnier L, Szade A, Rutschi O, Fas SC, Ryffel B, Rudolf MP. 2011. Anti-Pseudomonas aeruginosa serotype O11 LPS immunoglobulin M monoclonal antibody panobacumab (KBPA101) confers protection in a murine model of acute lung infection. J. Antimicrob. Chemother. 66:1100–1109. 10.1093/jac/dkr038 [DOI] [PubMed] [Google Scholar]
- 8.Tkaczyk C, Hua L, Varkey R, Shi Y, Dettinger L, Woods R, Barnes A, MacGill RS, Wilson S, Chowdhury P, Stover CK, Sellman BR. 2012. Identification of anti-alpha toxin monoclonal antibodies that reduce the severity of Staphylococcus aureus dermonecrosis and exhibit a correlation between affinity and potency. Clin. Vaccine Immunol. 19:377–385. 10.1128/CVI.05589-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oleksiewicz MB, Nagy G, Nagy E. 2012. Anti-bacterial monoclonal antibodies: back to the future? Arch. Biochem. Biophys. 526:124–131. 10.1016/j.abb.2012.06.001 [DOI] [PubMed] [Google Scholar]
- 10.Saylor C, Dadachova E, Casadevall A. 2009. Monoclonal antibody-based therapies for microbial diseases. Vaccine 27(Suppl 6):G38–G46. 10.1016/j.vaccine.2009.09.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baer M, Sawa T, Flynn P, Luehrsen K, Martinez D, Wiener-Kronish JP, Yarranton G, Bebbington C. 2009. An engineered human antibody Fab fragment specific for Pseudomonas aeruginosa PcrV antigen has potent antibacterial activity. Infect. Immun. 77:1083–1090. 10.1128/IAI.00815-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez-Becerra FJ, Kissmann JM, Diaz-McNair J, Choudhari SP, Quick AM, Mellado-Sanchez G, Clements JD, Pasetti MF, Picking WL. 2012. Broadly protective Shigella vaccine based on type III secretion apparatus proteins. Infect. Immun. 80:1222–1231. 10.1128/IAI.06174-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quenee LE, Berube BJ, Segal J, Elli D, Ciletti NA, Anderson D, Schneewind O. 2010. Amino acid residues 196–225 of LcrV represent a plague protective epitope. Vaccine 28:1870–1876. 10.1016/j.vaccine.2009.11.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El Solh AA, Akinnusi ME, Wiener-Kronish JP, Lynch SV, Pineda LA, Szarpa K. 2008. Persistent infection with Pseudomonas aeruginosa in ventilator-associated pneumonia. Am. J. Respir. Crit. Care Med. 178:513–519. 10.1164/rccm.200802-239OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Solh AA, Hattemer A, Hauser AR, Alhajhusain A, Vora H. 2012. Clinical outcomes of type III Pseudomonas aeruginosa bacteremia. Crit. Care Med. 40:1157–1163. 10.1097/CCM.0b013e3182377906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hauser AR. 2009. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 7:654–665. 10.1038/nrmicro2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roy-Burman A, Savel RH, Racine S, Swanson BL, Revadigar NS, Fujimoto J, Sawa T, Frank DW, Wiener-Kronish JP. 2001. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. J. Infect. Dis. 183:1767–1774. 10.1086/320737 [DOI] [PubMed] [Google Scholar]
- 18.Faure K, Fujimoto J, Shimabukuro DW, Ajayi T, Shime N, Moriyama K, Spack EG, Wiener-Kronish JP, Sawa T. 2003. Effects of monoclonal anti-PcrV antibody on Pseudomonas aeruginosa-induced acute lung injury in a rat model. J. Immune Based Ther. Vaccines 1:2. 10.1186/1476-8518-1-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frank DW, Vallis A, Wiener-Kronish JP, Roy-Burman A, Spack EG, Mullaney BP, Megdoud M, Marks JD, Fritz R, Sawa T. 2002. Generation and characterization of a protective monoclonal antibody to Pseudomonas aeruginosa PcrV. J. Infect. Dis. 186:64–73. 10.1086/341069 [DOI] [PubMed] [Google Scholar]
- 20.Holder IA, Neely AN, Frank DW. 2001. PcrV immunization enhances survival of burned Pseudomonas aeruginosa-infected mice. Infect. Immun. 69:5908–5910. 10.1128/IAI.69.9.5908-5910.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawa T, Yahr TL, Ohara M, Kurahashi K, Gropper MA, Wiener-Kronish JP, Frank DW. 1999. Active and passive immunization with the Pseudomonas V antigen protects against type III intoxication and lung injury. Nat. Med. 5:392–398. 10.1038/7391 [DOI] [PubMed] [Google Scholar]
- 22.Shime N, Sawa T, Fujimoto J, Faure K, Allmond LR, Karaca T, Swanson BL, Spack EG, Wiener-Kronish JP. 2001. Therapeutic administration of anti-PcrV F(ab′)2 in sepsis associated with Pseudomonas aeruginosa. J. Immunol. 167:5880–5886. 10.4049/jimmunol.167.10.5880 [DOI] [PubMed] [Google Scholar]
- 23.François B, Luyt CE, Dugard A, Wolff M, Diehl JL, Jaber S, Forel JM, Garot D, Kipnis E, Mebazaa A, Misset B, Andremont A, Ploy MC, Jacobs A, Yarranton G, Pearce T, Fagon JY, Chastre J. 2012. Safety and pharmacokinetics of an anti-PcrV PEGylated monoclonal antibody fragment in mechanically ventilated patients colonized with Pseudomonas aeruginosa: a randomized, double-blind, placebo-controlled trial. Crit. Care Med. 40:2320–2326. 10.1097/CCM.0b013e31825334f6 [DOI] [PubMed] [Google Scholar]
- 24.Kilpatrick KE, Wring SA, Walker DH, Macklin MD, Payne JA, Su JL, Champion BR, Caterson B, McIntyre GD. 1997. Rapid development of affinity matured monoclonal antibodies using RIMMS. Hybridoma 16:381–389. 10.1089/hyb.1997.16.381 [DOI] [PubMed] [Google Scholar]
- 25.DiGiandomenico A, Rao J, Harcher K, Zaidi TS, Gardner J, Neely AN, Pier GB, Goldberg JB. 2007. Intranasal immunization with heterologously expressed polysaccharide protects against multiple Pseudomonas aeruginosa infections. Proc. Natl. Acad. Sci. U. S. A. 104:4624–4629. 10.1073/pnas.0608657104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tao MH, Morrison SL. 1989. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. 143:2595–2601 [PubMed] [Google Scholar]
- 27.Frank DW. March 2005. Method and compositions for immunization with the Pseudomonas V antigen. US Patent 7,494,653
- 28.Bruhns P. 2009. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 119:5640–5649. 10.1182/blood-2012-01-380121 [DOI] [PubMed] [Google Scholar]
- 29.Overdijk MB, Verploegen S, Ortiz Buijsse A, Vink T, Leusen JH, Bleeker WK, Parren PW. 2012. Crosstalk between human IgG isotypes and murine effector cells. J. Immunol. 189:3430–3438. 10.4049/jimmunol.1200356 [DOI] [PubMed] [Google Scholar]
- 30.Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, Nichol G, Thomas WD, Jr, Leney M, Sloan S, Hay CA, Ambrosino DM. 2010. Treatment with monoclonal antibodies against Clostridium difficile toxins. N. Engl. J. Med. 362:197–205. 10.1056/NEJMoa0907635 [DOI] [PubMed] [Google Scholar]
- 31.Goure J, Pastor A, Faudry E, Chabert J, Dessen A, Attree I. 2004. The V antigen of Pseudomonas aeruginosa is required for assembly of the functional PopB/PopD translocation pore in host cell membranes. Infect. Immun. 72:4741–4750. 10.1128/IAI.72.8.4741-4750.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horn MP, Zuercher AW, Imboden MA, Rudolf MP, Lazar H, Wu H, Hoiby N, Fas SC, Lang AB. 2010. Preclinical in vitro and in vivo characterization of the fully human monoclonal IgM antibody KBPA101 specific for Pseudomonas aeruginosa serotype IATS-O11. Antimicrob. Agents Chemother. 54:2338–2344. 10.1128/AAC.01142-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karginov VA, Robinson TM, Riemenschneider J, Golding B, Kennedy M, Shiloach J, Alibek K. 2004. Treatment of anthrax infection with combination of ciprofloxacin and antibodies to protective antigen of Bacillus anthracis. FEMS Immunol. Med. Microbiol. 40:71–74. 10.1016/S0928-8244(03)00302-X [DOI] [PubMed] [Google Scholar]
- 34.Song Y, Baer M, Srinivasan R, Lima J, Yarranton G, Bebbington C, Lynch SV. 2011. PcrV antibody-antibiotic combination improves survival in Pseudomonas aeruginosa-infected mice. Eur. J. Clin. Microbiol. Infect. Dis. 31:1837–1845. 10.1007/s10096-011-1509-2 [DOI] [PubMed] [Google Scholar]
- 35.Goure J, Broz P, Attree O, Cornelis GR, Attree I. 2005. Protective anti-V antibodies inhibit Pseudomonas and Yersinia translocon assembly within host membranes. J. Infect. Dis. 192:218–225. 10.1086/430932 [DOI] [PubMed] [Google Scholar]
- 36.Cheng LW, Stanker LH, Henderson TD, Jr, Lou J, Marks JD. 2009. Antibody protection against botulinum neurotoxin intoxication in mice. Infect. Immun. 77:4305–4313. 10.1128/IAI.00405-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howell HA, Logan LK, Hauser AR. 2013. Type III secretion of ExoU is critical during early Pseudomonas aeruginosa pneumonia. mBio 4(2):e00032-13. 10.1128/mBio.00032-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vance RE, Rietsch A, Mekalanos JJ. 2005. Role of the type III secreted exoenzymes S, T, and Y in systemic spread of Pseudomonas aeruginosa PAO1 in vivo. Infect. Immun. 73:1706–1713. 10.1128/IAI.73.3.1706-1713.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neely AN, Holder IA, Wiener-Kronish JP, Sawa T. 2005. Passive anti-PcrV treatment protects burned mice against Pseudomonas aeruginosa challenge. Burns 31:153–158. 10.1016/j.burns.2004.09.002 [DOI] [PubMed] [Google Scholar]