

Abstract
Methicillin-resistant Staphylococcus aureus (MRSA) has acquired the mecA gene encoding a peptidoglycan transpeptidase, penicillin binding protein 2a (PBP2a), which has decreased affinity for β-lactams. Quickly spreading and highly virulent community-acquired (CA) MRSA strains recently emerged as a frequent cause of infection in individuals without exposure to the health care system. In this study, we found that the inactivation of the components of the ClpXP protease substantially increased the β-lactam resistance level of a CA-MRSA USA300 strain, suggesting that the proteolytic activity of ClpXP controls one or more pathways modulating β-lactam resistance. These pathways do not involve the control of mecA expression, as the cellular levels of PBP2a were unaltered in the clp mutants. An analysis of the cell envelope properties of the clpX and clpP mutants revealed a number of distinct phenotypes that may contribute to the enhanced β-lactam tolerance. Both mutants displayed significantly thicker cell walls, increased peptidoglycan cross-linking, and altered composition of monomeric muropeptide species compared to those of the wild types. Moreover, changes in Sle1-mediated peptidoglycan hydrolysis and altered processing of the major autolysin Atl were observed in the clp mutants. In conclusion, the results presented here point to an important role for the ClpXP protease in controlling cell wall metabolism and add novel insights into the molecular factors that determine strain-dependent β-lactam resistance.
INTRODUCTION
The rapid spread of methicillin-resistant Staphylococcus aureus (MRSA) has made the treatment of staphylococcal infections increasingly difficult (1, 2). Community-acquired (CA) MRSA strains of the USA300 type cause particular concern because of their frequent isolation and the severity of infection they cause (3). Methicillin and other β-lactam antibiotics inhibit the growth of S. aureus by covalently binding to the transpeptidase domain of penicillin binding proteins (PBPs), which cross-link the polypeptide chains of the cell wall component peptidoglycan. The resistance of MRSA strains is caused by the acquisition of the mecA gene encoding the alternative transpeptidase penicillin binding protein 2a (PBP2a), with very low affinity for almost all β-lactam antibiotics (4–7). Recently, anti-MRSA β-lactams, such as ceftaroline and ceftobiprole, with stronger binding to PBP2a, have been discovered.
Clinical MRSA isolates exhibit highly variable resistance levels toward methicillin, with MICs ranging from <3 μg/ml (comparable to those of susceptible strains) to 1,600 μg/ml in highly resistant strains (8). The mechanisms underlying this variation remain poorly understood, but the lack of correlation between the resistance level and the level of mecA expression suggests that factors other than PBP2a modulate the strain-specific level of β-lactam resistance (8–11). Indeed, genetic screens have identified a number of auxiliary factors in addition to PBP2a that are critical for resistance to β-lactam antibiotics (12, 13). Examples include cell division proteins, endogenous PBPs, and enzymes involved in the synthesis of teichoic acids and peptidoglycan precursors (5, 14–21). Intriguingly, the realization that β-lactam resistance depends on auxiliary factors opens up new possibilities for the treatment of MRSA infections, as drugs that inhibit the functions of auxiliary factors are predicted and have been shown to work synergistically with β-lactams to kill MRSA (22, 23).
Intracellular proteolysis carried out by energy-dependent proteases is one of the most conserved biological processes. During the infection process, bacterial pathogens depend on energy-dependent proteases for both the general turnover of damaged and nonfunctional proteins and the degradation of short-lived regulatory proteins (24). Accordingly, the highly conserved ClpXP protease is essential for the virulence of S. aureus in both systemic and abscess models of infection (25, 26), and it has also been implicated in the virulence of other pathogens, such as Listeria monocytogenes, Salmonella, and Bacillus anthracis (24, 27). The ClpXP protease is composed of proteolytic and ATPase subunits. Fourteen ClpP subunits constitute a proteolytic chamber that is accessible only through a narrow pore, which prevents the entrance of native folded proteins (reviewed in reference 24). ClpX serves to specifically recognize, unfold, and translocate substrates into the ClpP proteolytic chamber. ClpX belongs to the family of closely related Clp ATPases and can also function independently of ClpP as a molecular chaperone (28). The treatment of MRSA infections with daptomycin in vivo or vancomycin in vitro has been shown to select for mutants that carry loss-of-function mutations in clpX or clpP, suggesting that ClpX and ClpP are involved in the response of S. aureus to antibiotics that are active against the cell wall (29–31). This finding prompted us to investigate if inactivating the components of the ClpXP protease modulates the susceptibility of S. aureus to antibiotics targeting the cell wall. We found that inactivating clpX or clpP increased the level of resistance to β-lactam antibiotics in a CA-MRSA USA300 strain and at the same time affected a number of cell envelope properties associated with resistance.
MATERIALS AND METHODS
Strains and culture conditions.
S. aureus strains (Table 1) were cultured in tryptic soy broth (TSB) (Oxoid) at 37°C with aeration. S. aureus strain JE2-derived strains were obtained from the Network of Antimicrobial Resistance in Staphylococcus aureus (NARSA) program (supported under NIAID/NIH contract HHSN272200700055C).
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Genotype/description | Source or reference |
|---|---|---|
| S. aureus strains | ||
| 8325-4 | MSSA strain cured of prophages | S. Foster, University of Sheffield, 73 |
| HI2209 | 8325-4ΔclpX | 25 |
| HI2300 | 8325-4ΔclpP | 25 |
| HI2304 | 8325-4ΔclpC | 42 |
| SA564 | Low-passage clinical isolate | 74 |
| HI2781 | SA564ΔclpX | 36 |
| HI2726 | SA564ΔclpP | 64 |
| JE2 | CA-MRSA strain USA300 LAC cured of plasmids | NARSA (http://www.narsa.net) (75) |
| HI3393 | JE2 ΔclpX | This study |
| NE912 | JE2 clpP::ΦNΣ | Nebraska Transposon Mutant Library (http://www.narsa.net) |
| KB1399 | JE2 clpP::ΦNΣ, transduced from NE912 | This study |
| NE699 | JE2 clpC::ΦNΣ | Nebraska Transposon Mutant Library (http://www.narsa.net) |
| COL | Hospital-acquired MRSA strain | 76 |
| HI3469 | COLΔclpX | This study |
| HI2570 | COLΔclpP | 64 |
| RN4220 | Restriction-deficient derivative of strain 8325-4 | R. Novick, New York University |
| KB1257 | 8325-4ΔclpX ermB | This study |
| ANG406 | SEJ1Δatl | 39 |
| SEJ1 | RN4220Δspa | 77 |
| ATCC 29213 | Control strain for MIC testing | |
| Plasmids | ||
| pBT2 | Allelic exchange vector | 33 |
| pAM367 | Plasmid containing ermB marker on BamHI fragment | W. L. Kelley and A. Monod (University of Geneva), unpublished data |
| pKB1221 | Plasmid to insert ermB near clpX | This study |
Construction of S. aureus mutants. (i) USA300 JE2 and COL ΔclpX mutants.
The clpX gene was deleted in S. aureus strains JE2 and COL by transduction with bacteriophage Φ11, using an ermB-tagged S. aureus 8325-4ΔclpX mutant (KB1257) as the donor strain for the transduction. KB1257 was constructed by inserting an ermB marker 8 kb from clpX between the convergently transcribed genes with locus tags SAOUHSC_1768 and SAOUHSC_1769. The ermB marker was inserted into the chromosome as follows: first, two PCR fragments were amplified from strain 8325-4 using the primer pairs KB65F (5′-CATTTCCATTTGCGGTACCTCC-3′) and KB65R (5′-GCATTAACTTGGATCCTTAACTTATG-3′), and KB66F (5′-CATAAGTTAAGGATCCAAGTTAATGC-3′) and KB67R (5′-TTAGGTCTGCAGATCGGATCAAC-3′) for the upstream and downstream fragments, respectively. The primers KB65R and KB66F are overlapping and contain substitutions that introduce a BamHI restriction site. The PCR fragments were joined by the splicing-by-overlap-extension PCR method (32) using KB65F, introducing a KpnI site, and KB67R, introducing a PstI site, and the resulting fragment was cloned into the KpnI and PstI sites of the allelic exchange vector pBT2 (33), resulting in plasmid pKB1217. A BamHI fragment containing ermB was excised from the plasmid pAM367 (W. L. Kelley, personal communication) and cloned into the BamHI site of pKB1217. The resulting plasmid, pKB1221, was then transformed into S. aureus strain RN4220 and subsequently introduced into 8325-4. The resulting strain was grown in the presence of erythromycin at the nonpermissive temperature for plasmid replication to select for double crossover and insertion of the ermB marker into the chromosome, as well as loss of the plasmid, as described previously (34). A plasmid-cured resistant mutant was selected (KB1239), and the presence of the ermB marker was verified by PCR with the primer pair KB76F (5′-GTTTCATTTCCATTTGCTATACCTCC-3′) and KB76R (5′-TATTCATCGCACGTATTACTTCCA-3′). The ermB marker was introduced into strain 8325-4ΔclpX by transduction using bacteriophage Φ11, yielding strain KB1244 containing ermB and the clpX deletion. From this strain, ΔclpX and the ermB marker were cotransduced into wild-type 8325-4, giving rise to KB1257, the donor strain for the transduction of the ΔclpX-ermB region into strains JE2 and COL.
(ii) JE2 clpP::ΦNΣ.
The clpP gene disrupted by the erythromycin resistance-coding transposon, ΦNΣ, was moved by transduction with bacteriophage Φ11 from NE912 into a clean JE2 wild-type background, yielding strain S. aureus KB1399.
Susceptibility testing.
Etest strips (bioMérieux) were used to determine the MICs of ertapenem, daptomycin, and vancomycin for all strains, as well as those of other β-lactams (except ceftaroline and ceftobiprole) for the 8325-4, SA564, and JE2 strains. Broth microdilution assays were used according to the guidelines of the Clinical and Laboratory Standards Institute (35) to determine the MICs of oxacillin, imipenem, cefoxitin, and cefuroxime (all from Sigma) for the COL strains. The Trek Sensititre Vizion broth microdilution system (Thermo Fisher Scientific) was used to determine the MICs of linezolid, ceftaroline, and ceftobiprole for all strains. S. aureus strain ATCC 29213 was included as a reference in all MIC determinations.
Western blotting.
S. aureus cultures were grown in TSB at 37°C. At an optical density at 600 nm (OD600) of 0.5, the cultures were split, and 16 μg/ml oxacillin was added to one of the culture aliquots. All cultures were grown for an additional 30 min. The cells were harvested by centrifugation at 5,000 × g, the cellular extracts were prepared by lysostaphin treatment, and the volumes normalized to the optical density of the initial culture were loaded onto 4 to 12% SDS-PAGE gels (NuPAGE). Western blotting was performed as previously described (36) using an antibody against PBP2a (37). The representative blots from three independent experiments are shown (see Fig. 2). In the first experiment, a range of different oxacillin concentrations, from 4 to 256 μg/ml, was tested without any difference in the observed PBP2a levels (data not shown), and 16 μg/ml was chosen as the concentration for all further experiments.
FIG 2.
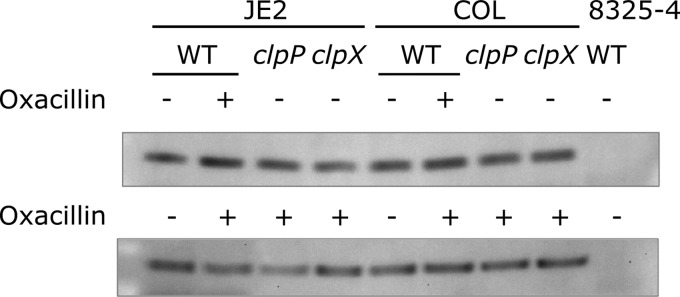
Cellular concentration of PBP2a in the presence (+) or absence (−) of inducer (16 mg/liter oxacillin) determined by Western blotting. PBP2a levels were similar in the wild type and clp mutants and in JE2 and COL. Strain 8325-4 is included as a strain not expressing PBP2a. Representative blots from three independent experiments are shown.
Population analysis profiles.
Antibiotic resistance profiles were determined by plating appropriate dilutions of an overnight culture on tryptic soy agar plates containing increasing concentrations of oxacillin, meropenem, or cefoxitin (all from Sigma), or on brain heart infusion agar (Oxoid) plates containing 50 μg/ml CaCl2 and increasing daptomycin (Sigma) concentrations, as described previously (38). The plates were incubated at 37°C, and the number of CFU per ml was determined after 48 h.
Autolytic activity.
An analysis of Triton X-100-induced autolysis was performed as described previously (39). In short, overnight cultures were diluted into fresh TSB medium and grown to an OD600 of 1. The cells were harvested at 8,000 × g and washed twice in 10 mM sodium phosphate buffer (pH 7.0) and twice in ice-cold double-distilled water (ddH2O), before being suspended in 10 mM sodium phosphate buffer (pH 7.0) containing 0.05% (vol/vol) Triton X-100. The suspensions were incubated at 37°C and autolysis was monitored by measuring the OD600 values at the indicated time points.
Zymographic analyses were performed essentially as described previously (39). Cultures were grown and harvested as described above, and the pellet was suspended in SDS sample buffer and boiled for 20 min. The boiled samples were centrifuged at 17,000 × g, and volumes of supernatant normalized to the optical density of the initial culture were loaded onto 7.5% SDS-PAGE gels containing 3% (wt/vol) heat-killed S. aureus SA564 cells. The gels were washed twice in ddH2O before overnight incubation in 0.2 M phosphate buffer (pH 7.0) at 37°C. The gels were subsequently stained with 0.1% methylene blue.
Electron microscopy and image analysis.
Overnight cultures of S. aureus strains SA564, SA564ΔclpX, SA564ΔclpP, JE2, JE2ΔclpX, and JE2clpP::ΦNΣ were diluted 1:200 into 40 ml of fresh TSB and grown at 37°C to an OD600 of 1.0. Bacteria from a 10-ml culture aliquot were collected by centrifugation at 8,000 × g, and the cell pellets were suspended in fixation solution (2.5% glutaraldehyde in 0.1 M cacodylate buffer [pH 7.4]) and incubated overnight at 4°C. The fixed cells were further treated with 2% osmium tetroxide, followed by 0.25% uranyl acetate for contrast enhancement. The pellets were dehydrated in increasing concentrations of ethanol, followed by pure propylene oxide, and then embedded in Epon resin. Thin sections for electron microscopy were stained with lead citrate and observed in a Philips CM100 BioTWIN transmission electron microscope fitted with an Olympus Veleta camera with a resolution of 2,048 by 2,048 pixels. For quantitative analysis, the images were acquired in an unbiased fashion by using the multiple image alignment function in the ITEM software (Olympus). Sample processing and microscopy were performed at the Center for Integrated Microscopy, Faculty of Health and Medical Sciences, University of Copenhagen.
An analysis of cell size was performed in Fiji (ImageJ) by running a script that automatically measures the area of the particles that fit predefined criteria on circularity and minimum size. The lower cutoff for size was chosen to be 40% of the average of the 10 largest particles in each frame. At least 300 cells were measured in each strain. Transmission electron microscopy (TEM) shows only a thin two-dimensional slice of the cells, and the measured areas will therefore show considerable variability in size, and the mean area size will be an underestimate of the true cell size. Cell wall thickness was measured in the ITEM software (Olympus). At least 25 cells distributed on ≥3 image frames were measured, and 3 to 5 measurements of cell wall thickness were performed on each cell. Student's two-sample t test was used to calculate statistical significance between the mutants and their cognate wild types.
Muropeptide analysis by HPLC.
One liter TSB medium was inoculated with overnight cultures of S. aureus strains SA564, SA564ΔclpX, SA564ΔclpP, JE2, JE2ΔclpX, and JE2clpP::ΦNΣ to an OD600 of 0.06 and grown at 37°C to late-log phase (OD600, approximately 1.5). The cultures were then cooled on ice and the bacteria were subsequently collected by centrifugation. Peptidoglycan was purified, digested with mutanolysin, and analyzed by high-performance liquid chromatography (HPLC), as described previously (39). Muropeptide profiles were determined for each strain in duplicate, and HPLC chromatograms from one representative experiment are shown (see Fig. 4). Data analysis was carried out in the software environment R, using the function package zoo for calculations of the area under the elution curve (40, 41).
FIG 4.

Altered peptidoglycan structure in clpX and clpP mutants. HPLC chromatograms of mutanolysin-digested peptidoglycan purified from the indicated strains. The peak numbering is according to de Jonge et al. (44). The table shows the muropeptide composition expressed as a percentage of the total area under the curve for each strain. The dark grey lines indicate baselines used for calculations of area under the curves. Representative results of two individual experiments are shown.
RESULTS
Loss of ClpX or ClpP increases β-lactam resistance in CA-MRSA.
To investigate if the ClpXP protease has an impact on the resistance to cell wall-active antibiotics, we inactivated clpX or clpP in four different S. aureus strains: 8325-4 (methicillin-sensitive S. aureus [MSSA], standard laboratory strain), SA564 (MSSA, low-passage clinical isolate), COL (MRSA, early clinical isolate), and USA300 JE2 (a plasmid-cured derivative of the CA-MRSA strain USA300 LAC). Susceptibilities to antibiotics acting on the cell wall (vancomycin, daptomycin, and β-lactam antibiotics) were tested by Etest or broth microdilution assays, and for comparison, the protein synthesis inhibitor linezolid was included. We found that while inactivation of clpX or clpP did not alter the susceptibility to linezolid or vancomycin, inactivation of clpX caused slightly increased daptomycin MICs in all four strain backgrounds (Table 2). The putative role of ClpX in controlling daptomycin susceptibility was supported by the finding that the clpX (but not the clpP) deletion caused a susceptibility shift in population analyses (Fig. 1A). However, the most striking result was that the inactivation of clpP or clpX had a significant strain-dependent effect on the susceptibility to β-lactams: in the MSSA strains 8325-4 and SA564, disruption of clpX and clpP slightly decreased susceptibility to oxacillin but not to other β-lactams tested (Table 2). In the MRSA strain USA300 JE2, however, the disruption of clpX or clpP resulted in a drastic increase in the MICs of all tested β-lactams, except ceftaroline and ceftobiprole, which have high affinity for PBP2a (Table 2). In particular, the deletion of clpP had a substantial effect on β-lactam resistance, with a 32-fold increase in the MICs of ertapenem and imipenem and a >10-fold increase in the MICs of cefoxitin and cefuroxime. The effect of deleting clpX was less pronounced, with around 5-fold increases in the MICs of oxacillin, ertapenem, imipenem, and cefoxitin, and a >10-fold increase in the MIC of cefuroxime. In S. aureus, ClpP can associate with an alternative substrate recognition factor, ClpC (42). The observation that the lack of ClpP had a greater effect on the resistance level than did the lack of ClpX might indicate that ClpP controls resistance partly via ClpC. However, this does not seem to be the case, as the inactivation of clpC did not change the MICs of any of the tested antibiotics (Table 2). USA300 strains exhibit a relatively low level of resistance compared to that of other MRSA strains. COL is an example of a highly resistant MRSA strain, and in this background, we did not see an effect of the clp mutations on the resistance level to any of the tested antibiotics (Table 2). We conclude that ClpX and ClpP control one or more pathways that determine the β-lactam resistance level of the clinically important CA-MRSA clone USA300 but not in the highly resistant COL strain.
TABLE 2.
Antibiotic susceptibility of clpX, clpP, and clpC mutants in different S. aureus strain backgrounds
| Antibiotic | MIC (mg/liter) for indicated S. aureus strain or variant: |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8325-4 |
SA564 |
JE2 |
COL |
|||||||||||
| WTa | ΔclpX | ΔclpP | ΔclpC | WT | ΔclpX | ΔclpP | WT | ΔclpX | clpP::ΦNΣ | clpC::ΦNΣ | WT | ΔclpX | ΔclpP | |
| Oxacillin | 0.19–0.25 | 0.38–0.5 | 1 | 0.25 | 0.38–0.5 | 1.5–4 | 0.75 | 32–48 | 192–256 | >256 | 48 | 256 | 256 | 256 |
| Ertapenem | 0.19 | 0.19 | 0.19 | 0.125 | 0.19 | 0.25 | 0.19 | 0.5–0.75 | 2–3 | 16–24 | 0.5–0.75 | >32 | >32 | >32 |
| Imipenem | 0.047 | 0.047 | 0.047 | 0.064 | 0.064 | 0.064 | 0.094 | 0.125 | 0.5–0.75 | 3–4 | 0.13–0.19 | 32 | 16 | 16 |
| Cefoxitin | 3 | 3 | 3 | 3 | 4 | 4 | 4 | 24 | 32–256 | >256 | 24–32 | 256 | 256 | 256 |
| Cefuroxime | 1.5 | 1.5 | 1.5 | 1.5 | 3 | 3 | 1.5 | 24 | >256 | >256 | 24–32 | 2,048 | 2,048 | 2,048 |
| Ceftaroline | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.5 | 0.5 | 1 | 1 | 0.5 | 2 | 2 | 2 |
| Ceftobiprole | 0.25 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 1 | 2 | 2 | 1 | 2 | 2 | 2 |
| Linezolid | 2 | 2 | <1 | 2 | 4 | 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Vancomycin | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1 | 1.5 | 1 | 1.5 | 1.5 | 1.5 | 1.5 | 3 | 1.5 |
| Daptomycin | 0.38–0.75 | 0.75–1 | 0.38–0.5 | 0.38 | 0.125–0.25 | 0.38 | 0.25–0.38 | 0.19–0.38 | 0.38 | 0.5–0.75 | 0.25 | 1 | 1.5 | 1 |
WT, wild type.
FIG 1.

Population analysis profiles. JE2 wild type (WT), JE2ΔclpX, and JE2 clpP::ΦNΣ were plated on increasing concentrations of daptomycin (A), oxacillin (B), meropenem (C), and cefoxitin (D), as indicated. Representative data from three individual experiments are shown.
Inactivation of clpX or clpP converts USA300 from a heterogeneously resistant to a homogeneously resistant MRSA strain without affecting PBP2a expression.
Cultures of CA-MRSA strains often display heterogeneity with respect to β-lactam susceptibility, meaning that the majority of cells exhibit a low level of antibiotic resistance, while a minority of cells are highly resistant (43). To assess if an MRSA strain is hetero- or homogeneously resistant, a population analysis profile (PAP) can be obtained by plating dilutions of a stationary culture on plates containing a wide range of antibiotic concentrations to determine the colony count at each concentration (38). In this analysis, JE2 exhibited a typical heterogeneous profile, with the majority of cells being killed by low concentrations of oxacillin, meropenem, and cefoxitin, while a small subpopulation survived much higher concentrations (Fig. 1). Strikingly, the deletion of clpX or clpP transformed JE2 into a homogeneously highly resistant strain. This effect of ClpXP inactivation was seen for all three β-lactam antibiotics but was most pronounced for oxacillin (Fig. 1). Although the β-lactam MIC values determined for the clpP mutant were higher than those for the clpX mutant (Table 2), the clpP mutant population showed some sensitivity to sub-MICs of meropenem and cefoxitin, compared to the clpX mutant, which showed a more homogeneous resistance pattern toward these antibiotics (Fig. 1). A simple explanation for the high homogenous β-lactam resistance in JE2 upon inactivation of ClpX or ClpP is that the cellular amount of PBP2a is increased. However, no differences in PBP2a levels were observed between the wild-type JE2 strain and the clp mutants (Fig. 2), and we concluded that ClpXP influences β-lactam resistance independently of the level of PBP2a.
Loss of ClpX or ClpP causes substantial changes in the cell wall structure.
The decreased susceptibilities of the clp mutants prompted us to investigate if the lack of ClpX or ClpP causes changes in the cell envelope structure. To this end, wild-type and mutant SA564 and JE2 strains were analyzed by transmission electron microscopy (TEM) (Fig. 3). Measurements of cell wall thickness revealed that the cell walls of clpX and in particular clpP mutant cells were thicker than the cell walls of the respective wild-type cells. We also noted that the outer edge of the cell wall appeared less distinct and fuzzier in the clpX and clpP mutants than in the wild-type cells (Fig. 3). Furthermore, quantitative analysis of the electron microscopy images revealed that the SA564 clpX and clpP cells were significantly smaller than the wild-type cells (Fig. 3). The smaller diameter of the SA564 clpX and clpP cells corresponds to a calculated average cell volume that is approximately half the volume of the wild-type cells. In the JE2 background, however, wild-type and clpP mutant cells were of approximately the same size, whereas only clpX mutants were slightly smaller (Fig. 3). In conclusion, the TEM analysis showed that the disruption of clpX or clpP led to increased cell wall thickness and changes in the appearance of the cell wall, regardless of the strain background.
FIG 3.

Representative TEM images of SA564 (A), SA564ΔclpX (B), SA564ΔclpP (C), JE2 (D), JE2ΔclpX (E), and JE2 clpP::ΦNΣ (F). The scale bar corresponds to 500 nm. The table shows the results of the measurements of cell diameter and cell wall thickness for various S. aureus strains and variants. P values show the results of Student's t tests between mutants and their cognate wild types.
To gain a deeper insight into the cell wall properties of the clp mutants, we purified the peptidoglycan from SA564, JE2, and their cognate clpX and clpP mutants, and determined the muropeptide composition by HPLC following mutanolysin digestion. A comparison of the HPLC profiles showed that the deletion of clpP caused a drastic change in the peptidoglycan compositions of both the MSSA and the MRSA strains (Fig. 4). A substantial increase in the abundance of muropeptides with long retention times was observed, indicating a hyper-cross-linked peptidoglycan layer for the clpP mutants in both the SA564 and JE2 strain backgrounds. As shown in Fig. 4, oligomeric muropeptides with a retention time of >104 min, corresponding to ≥9-mers, constituted approximately 57% of the peptidoglycan in the clpP mutants compared to approximately 48% in the wild-type strains. Even more highly cross-linked muropeptides with retention times of >120 min constituted up to three times more of the peptidoglycan in the cells that lacked ClpP. In contrast, there was a decrease in the abundance of trimeric to octameric muropeptides (retention times, 78 to 104 min) in the clpP mutants in both strain backgrounds. No effect was observed on the abundance of monomeric (retention times, 19 to 54 min) or dimeric (retention times, 54 to 78 min) muropeptides. The deletion of clpX resulted in only slight alterations in the peptidoglycan composition; while no change in the level of cross-linking was observed in the SA564 strain background, a small increase in the concentrations of highly cross-linked muropeptides was seen in the clpX mutant in the JE2 background (Fig. 4).
In addition to the observed changes in the abundances of late eluting muropeptides, we noted differences in the relative abundances of the monomeric peaks. Thus, in both strain backgrounds, the peak eluting at 27.1 min was notably higher in the clpX and clpP mutants than in the wild-type strains, and the integration of the monomeric peaks showed that this peak was approximately three times more abundant relative to the total abundance of monomeric muropeptides in the clpX and clpP mutants than in the parental strains. Based on the relative retention times of the monomeric peaks, we suggest that this peak most likely corresponds to peak 2, using the numbering of de Jonge et al. (44), and to our knowledge, no chemical structure has been assigned to this peak.
Altered autolysin activity in the clpX and clpP mutants.
Increased autolytic activity has been shown to correlate with increased β-lactam resistance (39, 45, 46); hence, we measured the rate of Triton X-100-induced autolysis in the clpX and clpP mutants. The deletion of clpX or clpP in the MSSA strain backgrounds had no effect on the autolytic rate (Fig. 5A), whereas a slight increase was observed for the clpX or clpP mutants in the JE2 strain background. We also examined if the clp mutations altered the amounts or activities of the autolytic enzymes by analyzing cell wall hydrolytic enzymes from wild-type and mutant cells on zymograms. As can be seen in Fig. 5B, the disruption of clpX or clpP in both SA564 and JE2 altered the intensities of several bands. The low-molecular-weight band was reported to be caused by the activity of the 32-kDa peptidoglycan hydrolase Sle1 (47). Interestingly, increased Sle1 activity was observed in extracts derived from the clp mutants, which is consistent with our recent finding that Sle1 is a substrate of ClpXP (48). The high-molecular-weight bands are due to the activity of the major autolysin Atl, as these bands are absent in extracts derived from an atl mutant strain (data not shown). Atl is a bifunctional murein hydrolase that is produced as a 138-kDa precursor protein, sequentially cleaved to generate 115- and 85-kDa intermediate products, and further processed to generate a 62-kDa N-acetylmuramyl-l-alanine amidase and a 51-kDa N-acetylglucosaminidase (49, 50). In both the wild type and the clp mutants, we identified Atl-specific bands of all the expected sizes, except the 51-kDa band. Notably, the 85-kDa Atl intermediate accumulated in extracts from the clp mutants, indicating that the processing of the 85-kDa product is slowed down in the mutants. ClpXP is an intracellular protease and therefore is likely to impact Atl processing indirectly via its effect on the expression of extracellular proteases (25). Alternatively, the association of the 85-kDa product with the cell wall is stronger in the clp mutants than in the wild type.
FIG 5.

Effect of lack of ClpX or ClpP on autolytic activity. (A) Triton X-100-induced autolysis. Mean and standard deviation (SD) values of three independent experiments are shown for the various strains. (B) Zymogram showing cell wall-associated proteins run on an SDS gel containing heat-killed S. aureus SA564 wild-type cells. Representative results of three independent experiments are shown. The positions of the molecular mass standards are indicated on the left (in kilodaltons).
DISCUSSION
The ClpXP protease is essential for virulence of S. aureus (25, 26). The inhibition of ClpXP proteolytic activity has therefore been suggested as a potential antimicrobial therapeutic strategy, and compounds have recently been identified that target the activity of ClpXP (51, 52). Several findings, however, indicate that the role of ClpXP during infection may be complex in S. aureus. In one study, MRSA was isolated from a patient before and at different time points after the start of treatment with daptomycin. One of the mutations acquired in these clinical isolates was a loss-of-function mutation in clpX (30). Similarly, S. aureus strains selected for daptomycin or vancomycin resistance in the laboratory were shown to harbor mutations in clpP together with mutations in other genes, such as walK and agrA (29, 31). Thus, it seems that S. aureus may under some circumstances benefit from shutting down Clp proteolytic activity during treatment with antibiotics that target the cell wall.
The results of the present study emphasize the existence of a molecular link between the activity of the ClpXP protease and the susceptibility to antibiotics acting on the cell wall. First, we show that S. aureus strains belonging to the prevalent MRSA clone USA300 can become highly β-lactam resistant if the ClpXP protease is inactivated. Second, the clpX mutants examined in our study were less sensitive to daptomycin in all tested strain backgrounds, suggesting that the ClpX chaperone controls pathways involved in daptomycin susceptibility independently of ClpP. The development of daptomycin nonsusceptibility is often accompanied by a concomitant fall in β-lactam resistance, a phenomenon designated the daptomycin/β-lactam “seesaw effect” (53, 54). Our results indicate, however, that clpX decreases daptomycin susceptibility by a mechanism not leading to the seesaw effect.
The increased resistance to β-lactam antibiotics in USA300 caused by the loss of ClpX or ClpP underscores the complexity of the expression of β-lactam resistance in MRSA. Only a few other genes have been reported to increase β-lactam resistance when inactivated (39, 55–57). Most notably, the disruption of gdpP increased resistance to penicillin as much as 32-fold in the USA300 strain LAC*, and it was also reported that the gdpP mutant strain shows an increase in cross-linked peptidoglycan and a decrease in cell size (39, 46, 58). gdpP encodes a phosphodiesterase that specifically cleaves cyclic diadenylate monophosphate (c-di-AMP), a newly discovered essential secondary messenger in S. aureus (39). In several studies, c-di-AMP has now been linked to cell wall properties and β-lactam resistance (59). Another nucleotide messenger, ppGpp, the effector molecule of the stringent response, was also shown to greatly influence the level of β-lactam resistance exhibited by MRSA strains (46, 60, 61). It may be speculated that ClpXP exerts its effect on β-lactam resistance, at least partly, via c-di-AMP or ppGpp; notably, we show that clpX and clpP mutants share some phenotypes with a gdpP mutant, namely, a decrease in cell size and an increase in peptidoglycan cross-linking, although the cross-linking is much more pronounced in the clpP mutants than in a gdpP mutant (39). Increased levels of c-di-AMP and ppGpp have been implicated in the conversion from low-level heterogeneous resistance to high-level homogeneous resistance in MRSA (46, 61). For ppGpp, the shift to homogeneous resistance was shown to be mediated via the increased expression of PBP2a (61). However, as we show here, this was not the case in the clp mutants. Furthermore, similar levels of PBP2a were observed in the JE2 and COL strains, consistent with previous observations that the strain-specific level of resistance is not associated with the cellular amount of PBP2a (8).
Both the structure and thickness of the cell wall were altered substantially by the loss of ClpX or ClpP, showing that the activities of ClpX and ClpP take part in controlling cell wall synthesis in S. aureus. In support of this conclusion, we recently identified several enzymes in the peptidoglycan-synthesis pathway, including GlmS, MurI, FemA, FemB, MurE, MurC, and PBP2, as potential substrates of the ClpXP or ClpCP protease (48). Interestingly, all of these factors have been identified as auxiliary factors that are required for β-lactam resistance (13), and it is conceivable that ClpP-mediated proteolysis may influence a number of enzymatic steps in this pathway. As β-lactams inhibit PBP activity by competitively binding to the active site, an increase in the substrate concentrations in clpX or clpP mutants would in theory increase the concentration of β-lactams required for growth inhibition and thereby increase the β-lactam resistance level.
The largest increase in β-lactam MICs was observed in the JE2ΔclpP mutant for imipenem and ertapenem preferentially binding to PBP1 (62). Interestingly, the inhibition of PBP1 can turn on the expression of major virulence factors, like the Panton-Valentine leukocidin (63). The underlying mechanism is unknown, but it has been hypothesized that alterations in the composition of peptidoglycan caused by PBP1 dysfunction may be compensated for by repressing the expression of autolytic genes, and this signal transduction pathway involves global transcriptional regulators, like SarA and Rot, which also control the expression of virulence regulation (63). We now speculate that there may be a causal link between the altered cell wall metabolism and the changed expression of a number of global regulators (including Agr, Rot, and MgrA) observed in cells devoid of ClpXP (36, 64).
Although the loss of ClpX or ClpP gave rise to higher β-lactam resistance, increased cell wall thickness, and an altered peptidoglycan structure, the phenotypes were much more pronounced in the clpP mutants than in the clpX mutants. Consequently, ClpX or ClpP must have additional roles independent of the other in modulating β-lactam resistance and cell wall properties in S. aureus. We ruled out the involvement of the ClpCP protease, as a clpC mutant did not show an increase in β-lactam resistance. However, it is possible that ClpCP and ClpXP have partially redundant functions such that the loss of both, which is the case in the clpP mutant, gives rise to more severe phenotypes than those caused by a loss of the ClpXP protease alone. Alternatively, the chaperone activity of ClpX may affect β-lactam resistance and cell wall structure properties in an opposite manner from the activity of ClpXP, resulting in an intermediate phenotype in the absence of ClpX. Consistent with this hypothesis is the ClpP-independent role of ClpX in stress response and gene regulation of S. aureus (25, 36).
The degree of cell wall changes in the clpX and clpP mutants, respectively, correlated with the level of β-lactam resistance in the USA300 background, indicating that the underlying pathways are linked in this strain. The increase in higher-order cross-linking combined with the concomitant decrease in lower-order cross-linking and the unchanged abundance of monomeric muropeptides in the clp mutants are in accordance with the random addition model for S. aureus peptidoglycan synthesis, in which oligomers are formed by random linkage between less-cross-linked muropeptides rather than by sequential addition of monomers (65). Higher-order peptidoglycan cross-linking in S. aureus is catalyzed by the monofunctional nonessential transpeptidase PBP4 (14, 66, 67), which in MRSA functions in concert with PBP2 and PBP2a to build highly cross-linked peptidoglycan (14). PBP4 has been shown to affect the β-lactam resistance levels in MRSA strains (19); however, the requirement for PBP4 in the expression of β-lactam resistance is again strain dependent, as PBP4 is required in the CA-MRSA strains USA300 and MW2 (19) but not in the hospital-acquired (HA)-MRSA strain COL (68). It may be hypothesized that ClpXP affects β-lactam resistance levels through the modulation of PBP4 activity, which correlates with the absence of an effect of deleting clpXP in COL on β-lactam resistance. Alternatively, the increased peptidoglycan cross-linking or altered muropeptide composition of the clpXP mutants may affect the activity or localization of PBP2a in MRSA such that it becomes less sensitive to β-lactams. This possibility is especially intriguing, given that PBP2a has an allosteric domain (69, 70) with which peptidoglycan can interact to change the affinity of β-lactams to the active site (69–71). It is therefore plausible that the ability of β-lactams to inhibit PBP2a is highly influenced by the altered peptidoglycan structure observed in the absence of ClpXP, whereas the β-lactam sensitivities of the MSSA PBPs are less affected due to their lack of allosteric regulation (71, 72). The virtually unchanged resistance to ceftaroline and ceftobiprole that we found for the clp mutants may reflect the ability of these antibiotics to bind to the allosteric domain of PBP2a and thereby predispose it to β-lactam inhibition (71, 72).
In summary, we have shown that a lack of the ClpXP protease converts the quickly spreading heterogeneously resistant CA-MRSA clone USA300 into a homogeneously highly resistant strain, and this change is unrelated to the amount of PBP2a. We investigated the effect of inactivating clpX and clpP on cell wall appearance, cell size, peptidoglycan composition, and autolytic activity in both USA300 and an MSSA strain. Two major conclusions can be drawn: first, while β-lactam susceptibility is only very minimally decreased in clp mutants in MSSA strains, cell wall thickness, peptidoglycan composition, and autolysin activity are affected in both MRSA and MSSA strains. Second, since deleting clpP has a larger effect on β-lactam resistance, cell wall thickness, and peptidoglycan cross-linking than does deleting clpX, ClpX and ClpP have additional cellular functions, which contribute to the modulation of the cell wall properties in S. aureus.
ACKNOWLEDGMENTS
This work was supported by the Danish Council for Independent Research (postdoctoral grant 12-132527 to K.T.B.), the European Research Council (starter grant 260371 to A.G.), and the Nordic Joint Committee for Agricultural Research (to D.F.).
We thank Rebecca Corrigan (Imperial College London) for helpful assistance with the autolysis assays, Ewa Kuninska (University of Copenhagen) for technical assistance, Janne Kudsk Klitgaard (University of Southern Denmark) for providing the PBP2a antibody, and Bill Kelley (University of Geneva) for providing plasmid pAM367.
Footnotes
Published ahead of print 27 May 2014
REFERENCES
- 1.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. 2010. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375:1557–1568. 10.1016/S0140-6736(09)61999-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Graveland H, Duim B, van Duijkeren E, Heederik D, Wagenaar J. 2011. Livestock-associated methicillin-resistant Staphylococcus aureus in animals and humans. Int. J. Med. Microbiol. 301:630–634. 10.1016/j.ijmm.2011.09.004 [DOI] [PubMed] [Google Scholar]
- 3.Otto M. 2010. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu. Rev. Microbiol. 64:143–162. 10.1146/annurev.micro.112408.134309 [DOI] [PubMed] [Google Scholar]
- 4.Hartman B, Tomasz A. 1984. Low-affinity penicillin-binding protein associated with beta-lactam resistance in Staphylococcus aureus. J. Bacteriol. 158:513–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinho M, de Lencastre H, Tomasz A. 2001. An acquired and a native penicillin-binding protein cooperate in building the cell wall of drug-resistant staphylococci. Proc. Natl. Acad. Sci. U. S. A. 98:10886–10891. 10.1073/pnas.191260798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chambers HF, Sachdeva MJ, Hackbarth CJ. 1994. Kinetics of penicillin binding to penicillin-binding proteins of Staphylococcus aureus. Biochem. J. 301(Pt 1):139–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu WP, Sun Y, Bauer MD, Paule S, Koenigs PM, Kraft WG. 1999. Penicillin-binding protein 2a from methicillin-resistant Staphylococcus aureus: kinetic characterization of its interactions with beta-lactams using electrospray mass spectrometry. Biochemistry (Wash.) 38:6537–6546. 10.1021/bi990025e [DOI] [PubMed] [Google Scholar]
- 8.Parvez M, Shibata H, Nakano T, Niimi S, Fujii N, Arakaki N, Higuti T. 2008. No relationship exists between PBP 2a amounts expressed in different MRSA strains obtained clinically and their beta-lactam MIC values. J. Med. Invest. 55:246–253. 10.2152/jmi.55.246 [DOI] [PubMed] [Google Scholar]
- 9.Hososaka Y, Hanaki H, Endo H, Suzuki Y, Nagasawa Z, Otsuka Y, Nakae T, Sunakawa K. 2007. Characterization of oxacillin-susceptible mecA-positive Staphylococcus aureus: a new type of MRSA. J. Infect. Chemother. 13:79–86. 10.1007/s10156-006-0502-7 [DOI] [PubMed] [Google Scholar]
- 10.Murakami K, Tomasz A. 1989. Involvement of multiple genetic determinants in high-level methicillin resistance in Staphylococcus aureus. J. Bacteriol. 171:874–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartman B, Tomasz A. 1986. Expression of methicillin resistance in heterogeneous strains of Staphylococcus aureus. Antimicrob. Agents Chemother. 29:85–92. 10.1128/AAC.29.1.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roemer T, Schneider T, Pinho M. 2013. Auxiliary factors: a chink in the armor of MRSA resistance to β-lactam antibiotics. Curr. Opin. Microbiol. 16:1–11. 10.1016/j.mib.2013.03.001 [DOI] [PubMed] [Google Scholar]
- 13.Berger-Bächi B, Rohrer S. 2002. Factors influencing methicillin resistance in staphylococci. Arch. Microbiol. 178:165–171. 10.1007/s00203-002-0436-0 [DOI] [PubMed] [Google Scholar]
- 14.Leski T, Tomasz A. 2005. Role of penicillin-binding protein 2 (PBP2) in the antibiotic susceptibility and cell wall cross-linking of Staphylococcus aureus: evidence for the cooperative functioning of PBP2, PBP4, and PBP2A. J. Bacteriol. 187:1815–1824. 10.1128/JB.187.5.1815-1824.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pinho M, Ludovice A, Wu S, De Lencastre H. 1997. Massive reduction in methicillin resistance by transposon inactivation of the normal PBP2 in a methicillin-resistant strain of Staphylococcus aureus. Microb. Drug Resist. 3:409–413. 10.1089/mdr.1997.3.409 [DOI] [PubMed] [Google Scholar]
- 16.Campbell J, Singh A, Santa Maria JP, Jr, Kim Y, Brown S, Swoboda J, Mylonakis E, Wilkinson B, Walker S. 2011. Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem. Biol. 6:106–116. 10.1021/cb100269f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maki H, Yamaguchi T, Murakami K. 1994. Cloning and characterization of a gene affecting the methicillin resistance level and the autolysis rate in Staphylococcus aureus. J. Bacteriol. 176:4993–5000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee S, Jarantow L, Wang H, Sillaots S, Cheng H, Meredith T, Thompson J, Roemer T. 2011. Antagonism of chemical genetic interaction networks resensitize MRSA to β-lactam antibiotics. Chem. Biol. 18:1379–1389. 10.1016/j.chembiol.2011.08.015 [DOI] [PubMed] [Google Scholar]
- 19.Memmi G, Filipe S, Pinho M, Fu Z, Cheung A. 2008. Staphylococcus aureus PBP4 is essential for beta-lactam resistance in community-acquired methicillin-resistant strains. Antimicrob. Agents Chemother. 52:3955–3966. 10.1128/AAC.00049-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atilano M, Pereira P, Yates J, Reed P, Veiga H, Pinho M, Filipe S. 2010. Teichoic acids are temporal and spatial regulators of peptidoglycan cross-linking in Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 107:18991–18996. 10.1073/pnas.1004304107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farha M, Leung A, Sewell E, D'Elia M, Allison S, Ejim L, Pereira P, Pinho M, Wright G, Brown E. 2013. Inhibition of WTA synthesis blocks the cooperative action of PBPs and sensitizes MRSA to β-lactams. ACS Chem. Biol. 8:226–233. 10.1021/cb300413m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan CM, Therien AG, Lu J, Lee SH, Caron A, Gill CJ, Lebeau-Jacob C, Benton-Perdomo L, Monteiro JM, Pereira PM, Elsen NL, Wu J, Deschamps K, Petcu M, Wong S, Daigneault E, Kramer S, Liang L, Maxwell E, Claveau D, Vaillancourt J, Skorey K, Tam J, Wang H, Meredith TC, Sillaots S, Wang-Jarantow L, Ramtohul Y, Langlois E, Landry F, Reid JC, Parthasarathy G, Sharma S, Baryshnikova A, Lumb KJ, Pinho MG, Soisson SM, Roemer T. 2012. Restoring methicillin-resistant Staphylococcus aureus susceptibility to β-lactam antibiotics. Sci. Transl. Med. 4:126ra135. 10.1126/scitranslmed.3003592 [DOI] [PubMed] [Google Scholar]
- 23.Wang H, Gill CJ, Lee SH, Mann P, Zuck P, Meredith TC, Murgolo N, She X, Kales S, Liang L, Liu J, Wu J, Santa Maria J, Su J, Pan J, Hailey J, McGuinness D, Tan CM, Flattery A, Walker S, Black T, Roemer T. 2013. Discovery of wall teichoic acid inhibitors as potential anti-MRSA β-lactam combination agents. Chem. Biol. 20:272–284. 10.1016/j.chembiol.2012.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frees D, Brøndsted L, Ingmer H. 2013. Bacterial proteases and virulence. Subcell. Biochem. 66:161–192. 10.1007/978-94-007-5940-4_7 [DOI] [PubMed] [Google Scholar]
- 25.Frees D, Qazi SN, Hill P, Ingmer H. 2003. Alternative roles of ClpX and ClpP in Staphylococcus aureus stress tolerance and virulence. Mol. Microbiol. 48:1565–1578. 10.1046/j.1365-2958.2003.03524.x [DOI] [PubMed] [Google Scholar]
- 26.Farrand AJ, Reniere ML, Ingmer H, Frees D, Skaar EP. 2013. Regulation of host hemoglobin binding by the Staphylococcus aureus Clp proteolytic system. J. Bacteriol. 195:5041–5050. 10.1128/JB.00505-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frees D, Savijoki K, Varmanen P, Ingmer H. 2007. Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria. Mol. Microbiol. 63:1285–1295. 10.1111/j.1365-2958.2007.05598.x [DOI] [PubMed] [Google Scholar]
- 28.Wawrzynow A, Wojtkowiak D, Marszalek J, Banecki B, Jonsen M, Graves B, Georgopoulos C, Zylicz M. 1995. The ClpX heat-shock protein of Escherichia coli, the ATP-dependent substrate specificity component of the ClpP-ClpX protease, is a novel molecular chaperone. EMBO J. 14:1867–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shoji M, Cui L, Iizuka R, Komoto A, Neoh H, Watanabe Y, Hishinuma T, Hiramatsu K. 2011. walK and clpP mutations confer reduced vancomycin susceptibility in Staphylococcus aureus. Antimicrob. Agents Chemother. 55:3870–3881. 10.1128/AAC.01563-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peleg A, Miyakis S, Ward D, Earl A, Rubio A, Cameron D, Pillai S, Moellering RC, Jr, Eliopoulos G. 2012. Whole genome characterization of the mechanisms of daptomycin resistance in clinical and laboratory derived isolates of Staphylococcus aureus. PLoS One 7:e28316. 10.1371/journal.pone.0028316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song Y, Rubio A, Jayaswal R, Silverman J, Wilkinson B. 2013. Additional routes to Staphylococcus aureus daptomycin resistance as revealed by comparative genome sequencing, transcriptional profiling, and phenotypic studies. PLoS One 8:e58469. 10.1371/journal.pone.0058469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horton RM, Ho SN, Pullen JK, Hunt HD, Cai Z, Pease LR. 1993. Gene splicing by overlap extension. Methods Enzymol. 217:270–279. 10.1016/0076-6879(93)17067-F [DOI] [PubMed] [Google Scholar]
- 33.Brückner R. 1997. Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. FEMS Microbiol. Lett. 151:1–8. 10.1016/S0378-1097(97)00116-X [DOI] [PubMed] [Google Scholar]
- 34.Renzoni A, Kelley WL, Barras C, Monod A, Huggler E, François P, Schrenzel J, Studer R, Vaudaux P, Lew DP. 2009. Identification by genomic and genetic analysis of two new genes playing a key role in intermediate glycopeptide resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 53:903–911. 10.1128/AAC.01287-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clinical and Laboratory Standards Institute. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; ap-proved standard–9th ed. CLSI document M07-A9 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 36.Jelsbak L, Ingmer H, Valihrach L, Cohn MT, Christiansen MH, Kallipolitis BH, Frees D. 2010. The chaperone ClpX stimulates expression of Staphylococcus aureus protein A by Rot dependent and independent pathways. PLoS One 5:e12752. 10.1371/journal.pone.0012752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klitgaard JK, Skov MN, Kallipolitis BH, Kolmos HJ. 2008. Reversal of methicillin resistance in Staphylococcus aureus by thioridazine. J. Antimicrob. Chemother. 62:1215–1221. 10.1093/jac/dkn417 [DOI] [PubMed] [Google Scholar]
- 38.Sieradzki K, Villari P, Tomasz A. 1998. Decreased susceptibilities to teicoplanin and vancomycin among coagulase-negative methicillin-resistant clinical isolates of staphylococci. Antimicrob. Agents Chemother. 42:100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corrigan RM, Abbott JC, Burhenne H, Kaever V, Gründling A. 2011. c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog. 7:e1002217. 10.1371/journal.ppat.1002217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeileis A, Grothendieck G. 2005. zoo: S3 infrastructure for regular and irregular time series. J. Stat. Softw. 14:1–27 http://www.jstatsoft.org/v14/i06/paper [Google Scholar]
- 41.The R Core Team. 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://web.mit.edu/r_v3.0.1/fullrefman.pdf [Google Scholar]
- 42.Frees D, Chastanet A, Qazi S, Sørensen K, Hill P, Msadek T, Ingmer H. 2004. Clp ATPases are required for stress tolerance, intracellular replication and biofilm formation in Staphylococcus aureus. Mol. Microbiol. 54:1445–1462. 10.1111/j.1365-2958.2004.04368.x [DOI] [PubMed] [Google Scholar]
- 43.Tomasz A, Nachman S, Leaf H. 1991. Stable classes of phenotypic expression in methicillin-resistant clinical isolates of staphylococci. Antimicrob. Agents Chemother. 35:124–129. 10.1128/AAC.35.1.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Jonge BL, Chang YS, Gage D, Tomasz A. 1992. Peptidoglycan composition of a highly methicillin-resistant Staphylococcus aureus strain. The role of penicillin binding protein 2A. J. Biol. Chem. 267:11248–11254 [PubMed] [Google Scholar]
- 45.Gustafson JE, Berger-Bächi B, Strässle A, Wilkinson BJ. 1992. Autolysis of methicillin-resistant and -susceptible Staphylococcus aureus. Antimicrob. Agents Chemother. 36:566–572. 10.1128/AAC.36.3.566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dengler V, McCallum N, Kiefer P, Christen P, Patrignani A, Vorholt J, Berger-Bächi B, Senn M. 2013. Mutation in the c-di-AMP cyclase dacA affects fitness and resistance of methicillin resistant Staphylococcus aureus. PLoS One 8:e73512. 10.1371/journal.pone.0073512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kajimura J, Fujiwara T, Yamada S, Suzawa Y, Nishida T, Oyamada Y, Hayashi I, Yamagishi J, Komatsuzawa H, Sugai M. 2005. Identification and molecular characterization of an N-acetylmuramyl-l-alanine amidase Sle1 involved in cell separation of Staphylococcus aureus. Mol. Microbiol. 58:1087–1101. 10.1111/j.1365-2958.2005.04881.x [DOI] [PubMed] [Google Scholar]
- 48.Feng J, Michalik S, Varming AN, Andersen JH, Albrecht D, Jelsbak L, Krieger S, Ohlsen K, Hecker M, Gerth U, Ingmer H, Frees D. 2013. Trapping and proteomic identification of cellular substrates of the ClpP protease in Staphylococcus aureus. J. Proteome Res. 12:547–558. 10.1021/pr300394r [DOI] [PubMed] [Google Scholar]
- 49.Komatsuzawa H, Sugai M, Nakashima S, Yamada S, Matsumoto A, Oshida T, Suginaka H. 1997. Subcellular localization of the major autolysin, ATL and its processed proteins in Staphylococcus aureus. Microbiol. Immunol. 41:469–479. 10.1111/j.1348-0421.1997.tb01880.x [DOI] [PubMed] [Google Scholar]
- 50.Oshida T, Sugai M, Komatsuzawa H, Hong Y, Suginaka H, Tomasz A. 1995. A Staphylococcus aureus autolysin that has an N-acetylmuramoyl-l-alanine amidase domain and an endo-beta-N-acetylglucosaminidase domain: cloning, sequence analysis, and characterization. Proc. Natl. Acad. Sci. U. S. A. 92:285–289. 10.1073/pnas.92.1.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kirstein J, Hoffmann A, Lilie H, Schmidt R, Rübsamen-Waigmann H, Brötz-Oesterhelt H, Mogk A, Turgay K. 2009. The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol. Med. 1:37–49. 10.1002/emmm.200900002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McGillivray SM, Tran DN, Ramadoss NS, Alumasa JN, Okumura CY, Sakoulas G, Vaughn MM, Zhang DX, Keiler KC, Nizet V. 2012. Pharmacological inhibition of the ClpXP protease increases bacterial susceptibility to host cathelicidin antimicrobial peptides and cell envelope-active antibiotics. Antimicrob. Agents Chemother. 56:1854–1861. 10.1128/AAC.05131-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang SJ, Xiong YQ, Boyle-Vavra S, Daum R, Jones T, Bayer AS. 2010. Daptomycin-oxacillin combinations in treatment of experimental endocarditis caused by daptomycin-nonsusceptible strains of methicillin-resistant Staphylococcus aureus with evolving oxacillin susceptibility (the “seesaw effect”). Antimicrob. Agents Chemother. 54:3161–3169. 10.1128/AAC.00487-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berti AD, Sakoulas G, Nizet V, Tewhey R, Rose WE. 2013. β-Lactam antibiotics targeting PBP1 selectively enhance daptomycin activity against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 57:5005–5012. 10.1128/AAC.00594-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakao A, Imai S, Takano T. 2000. Transposon-mediated insertional mutagenesis of the d-alanyl-lipoteichoic acid (dlt) operon raises methicillin resistance in Staphylococcus aureus. Res. Microbiol. 151:823–829. 10.1016/S0923-2508(00)01148-7 [DOI] [PubMed] [Google Scholar]
- 56.Brown S, Xia G, Luhachack L, Campbell J, Meredith T, Chen C, Winstel V, Gekeler C, Irazoqui J, Peschel A, Walker S. 2012. Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proc. Natl. Acad. Sci. U. S. A. 109:18909–18914. 10.1073/pnas.1209126109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fujimura T, Murakami K. 1997. Increase of methicillin resistance in Staphylococcus aureus caused by deletion of a gene whose product is homologous to lytic enzymes. J. Bacteriol. 179:6294–6301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Griffiths J, O'Neill A. 2012. Loss of function of the gdpP protein leads to joint β-lactam/glycopeptide tolerance in Staphylococcus aureus. Antimicrob. Agents Chemother. 56:579–581. 10.1128/AAC.05148-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Corrigan RM, Gründling A. 2013. Cyclic di-AMP: another second messenger enters the fray. Nat. Rev. Microbiol. 11:513–524. 10.1038/nrmicro3069 [DOI] [PubMed] [Google Scholar]
- 60.Pozzi C, Waters EM, Rudkin JK, Schaeffer CR, Lohan AJ, Tong P, Loftus BJ, Pier GB, Fey PD, Massey RC, O'Gara JP. 2012. Methicillin resistance alters the biofilm phenotype and attenuates virulence in Staphylococcus aureus device-associated infections. PLoS Pathog. 8:e1002626. 10.1371/journal.ppat.1002626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim C, Mwangi M, Chung M, Milheirço C, de Lencastre H, Tomasz A. 2013. The mechanism of heterogeneous beta-lactam resistance in MRSA: key role of the stringent stress response. PLoS One 8:e82814. 10.1371/journal.pone.0082814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang Y, Bhachech N, Bush K. 1995. Biochemical comparison of imipenem, meropenem and biapenem: permeability, binding to penicillin-binding proteins, and stability to hydrolysis by beta-lactamases. J. Antimicrob. Chemother. 35:75–84. 10.1093/jac/35.1.75 [DOI] [PubMed] [Google Scholar]
- 63.Dumitrescu O, Choudhury P, Boisset S, Badiou C, Bes M, Benito Y, Wolz C, Vandenesch F, Etienne J, Cheung AL, Bowden MG, Lina G. 2011. Beta-lactams interfering with PBP1 induce Panton-Valentine leukocidin expression by triggering sarA and rot global regulators of Staphylococcus aureus. Antimicrob. Agents Chemother. 55:3261–3271. 10.1128/AAC.01401-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frees D, Andersen JH, Hemmingsen L, Koskenniemi K, Bæk KT, Muhammed MK, Gudeta DD, Nyman TA, Sukura A, Varmanen P, Savijoki K. 2012. New insights into Staphylococcus aureus stress tolerance and virulence regulation from an analysis of the role of the ClpP protease in the strains Newman, COL, and SA564. J. Proteome Res. 11:95–108. 10.1021/pr200956s [DOI] [PubMed] [Google Scholar]
- 65.Snowden MA, Perkins HR. 1990. Peptidoglycan cross-linking in Staphylococcus aureus. An apparent random polymerisation process. Eur. J. Biochem. 191:373–377 [DOI] [PubMed] [Google Scholar]
- 66.Wyke AW, Ward JB, Hayes MV, Curtis NA. 1981. A role in vivo for penicillin-binding protein-4 of Staphylococcus aureus. Eur. J. Biochem. 119:389–393. 10.1111/j.1432-1033.1981.tb05620.x [DOI] [PubMed] [Google Scholar]
- 67.Sieradzki K, Pinho MG, Tomasz A. 1999. Inactivated pbp4 in highly glycopeptide-resistant laboratory mutants of Staphylococcus aureus. J. Biol. Chem. 274:18942–18946. 10.1074/jbc.274.27.18942 [DOI] [PubMed] [Google Scholar]
- 68.Katayama Y, Zhang HZ, Chambers HF. 2003. Effect of disruption of Staphylococcus aureus PBP4 gene on resistance to beta-lactam antibiotics. Microb. Drug Resist. 9:329–336. 10.1089/107662903322762752 [DOI] [PubMed] [Google Scholar]
- 69.Fuda C, Hesek D, Lee M, Morio K, Nowak T, Mobashery S. 2005. Activation for catalysis of penicillin-binding protein 2a from methicillin-resistant Staphylococcus aureus by bacterial cell wall. J. Am. Chem. Soc. 127:2056–2057. 10.1021/ja0434376 [DOI] [PubMed] [Google Scholar]
- 70.Villegas-Estrada A, Lee M, Hesek D, Vakulenko SB, Mobashery S. 2008. Co-opting the cell wall in fighting methicillin-resistant Staphylococcus aureus: potent inhibition of PBP 2a by two anti-MRSA beta-lactam antibiotics. J. Am. Chem. Soc. 130:9212–9213. 10.1021/ja8029448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Otero LH, Rojas-Altuve A, Llarrull LI, Carrasco-López C, Kumarasiri M, Lastochkin E, Fishovitz J, Dawley M, Hesek D, Lee M, Johnson JW, Fisher JF, Chang M, Mobashery S, Hermoso JA. 2013. How allosteric control of Staphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proc. Natl. Acad. Sci. U. S. A. 110:16808–16813. 10.1073/pnas.1300118110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Banerjee R, Gretes M, Basuino L, Strynadka N, Chambers HF. 2008. In vitro selection and characterization of ceftobiprole-resistant methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 52:2089–2096. 10.1128/AAC.01403-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bæk KT, Frees D, Renzoni A, Barras C, Rodriguez N, Manzano C, Kelley WL. 2013. Genetic variation in the Staphylococcus aureus 8325 strain lineage revealed by whole-genome sequencing. PLoS One 8:e77122. 10.1371/journal.pone.0077122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Somerville GA, Beres SB, Fitzgerald JR, DeLeo FR, Cole RL, Hoff JS, Musser JM. 2002. In vitro serial passage of Staphylococcus aureus: changes in physiology, virulence factor production, and agr nucleotide sequence. J. Bacteriol. 184:1430–1437. 10.1128/JB.184.5.1430-1437.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4(1):e00537–12. 10.1128/mBio.00537-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dyke KG, Jevons MP, Parker MT. 1966. Penicillinase production and intrinsic resistance to penicillins in Staphylococcus aureus. Lancet i:835–838 [DOI] [PubMed] [Google Scholar]
- 77.Gründling A, Schneewind O. 2007. Genes required for glycolipid synthesis and lipoteichoic acid anchoring in Staphylococcus aureus. J. Bacteriol. 189:2521–2530. 10.1128/JB.01683-06 [DOI] [PMC free article] [PubMed] [Google Scholar]