

Abstract
Colistin is a key drug for the treatment of infections caused by extensively drug-resistant strains of Enterobacteriaceae producing carbapenemases. However, the emergence of colistin resistance is being increasingly reported, especially among Klebsiella pneumoniae strains producing KPC-type carbapenemases (KPC-KP). In this work, we investigated colistin-susceptible (KPB-1) and colistin-resistant (KPB-2) sequential isolates obtained from a patient with a KPC-KP infection before and after low-dosage colistin treatment, respectively. By using a next-generation sequencing approach and comparative genomic analysis of the two isolates, we detected in KPB-2 a nonsynonymous nucleotide substitution in the gene encoding the PmrB sensor kinase, resulting in a leucine-to-arginine substitution at amino acid position 82. Compared with KPB-1, KPB-2 exhibited upregulated transcription of pmrA and of pmrK, which is part of the pmrHFIJKLM operon responsible for modification of the colistin lipopolysaccharide target. Complementation with wild-type pmrB in KPB-2 restored colistin susceptibility and reduced the transcription of pmrA and pmrK to basal levels, while expression of PmrBL82R in KPB-1 did not alter colistin susceptibility or upregulate pmrA and pmrK expression, confirming the dominance of wild-type PmrB versus the PmrBL82R mutant. The present results indicated that PmrB mutations mediating colistin resistance may be selected during low-dosage colistin treatment. The colistin-resistant phenotype of KPB-2 was stable for up to 50 generations in the absence of selective pressure and was not associated with a significant fitness cost in a competition experiment.
INTRODUCTION
Klebsiella pneumoniae strains producing KPC-type carbapenemases (KPC-KP) are among the most challenging multidrug-resistant pathogens that have emerged in recent years, due to their extensively drug-resistant phenotypes. KPC-KP strains belonging in some high-risk clonal lineages, such as sequence type 258 (ST258) and ST512, have proved capable of rapid epidemic diffusion in health care settings, causing large outbreaks in many areas (1–4). Also in Italy, KPC-KP strains have recently undergone epidemic diffusion, resulting in a condition of high-level endemicity (5).
Polymyxins (polymyxin B and colistin) are among the few antimicrobial agents that retain activity, being key components of anti-KPC-KP antimicrobial regimens (6, 7). However, the emergence of colistin-resistant KPC-KP is increasingly being reported, with sizeable resistance rates in some areas (5, 8, 9).
Recently, we demonstrated that inactivation of the mgrB gene, which encodes a negative-feedback regulator of the PhoQ/PhoP signaling system (10), is a genetic mechanism responsible for acquired colistin resistance in KPC-KP strains of ST258 following exposure to colistin treatment (11), and a similar resistance mechanism was subsequently reported for a colistin-resistant clinical isolate of K. pneumoniae of ST37 that produces an AmpC-type β-lactamase (12). This mechanism leads to upregulation of the PhoQ/PhoP system, which eventually activates the PmrHFIJKLM system responsible for modification of the lipid A colistin target by decoration with 4-amino-4-deoxy-l-arabinose (l-Ara4N), which neutralizes the negative charge and decreases colistin binding (11, 13, 14).
In this work, we show that the in vivo emergence of colistin-resistant KPC-KP strains following colistin exposure can be associated with alteration of the PmrB sensor kinase of the PmrA/PmrB signaling system, which also controls lipopolysaccharide (LPS) modification (13, 14). PmrB is normally activated by various environmental signals, including some trivalent cations (Fe3+ and Al3+) and mild acidic pH, which trigger autophosphorylation of the cytoplasmic domain. The activated PmrB in turn activates (by transphosphorylation) the PmrA response regulator, which then promotes transcription of the PmrA regulon, including (among others) the pmrCAB and pmrHFIJKLM genes involved in LPS modification (14).
MATERIALS AND METHODS
Bacterial strains.
The KPC-KP strains studied in this work were a pair of sequential isolates, namely, KPB-1 (from pleural fluid) and KPB-2 (from blood drawn via a central venous catheter), obtained from an inpatient at the St. Orsola-Malpighi University Hospital (Bologna, Italy) in early 2011. KPB-1 was colistin susceptible, while KPB-2 was colistin resistant; KPB-2 had been isolated 12 days later, after 9 days of colistin treatment. During the period of colistin administration, the patient had demonstrated normal renal function, with creatinine clearance values within the normal range and had also received additional antibiotics (meropenem, teicoplanin, and trimethoprim-sulfamethoxazole) to which KPB-1 was resistant. Multilocus sequence typing (MLST) and genotyping by pulsed-field gel electrophoresis (PFGE) of the KPC-KP strains were performed as described previously (11). Escherichia coli DH5α (15) was used as the host for recombinant plasmids.
In vitro susceptibility testing.
Colistin MICs were determined by reference broth microdilution following the CLSI methodology (16) and were interpreted accordingly to the EUCAST guidelines (see http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/Breakpoint_table_v_4.0.pdf). Escherichia coli ATCC 25922 (colistin-susceptible) and K. pneumoniae KKBO-4 (colistin-resistant) (11) were used as quality control strains.
Whole-genome sequencing and sequence analysis.
Whole-genome sequencing (WGS) of KPB-1 and KPB-2 was performed at an external facility (IGA Technology Services, Udine, Italy), using the HiSeq2000 Illumina platform (Illumina Inc., San Diego, CA, USA) and a paired-end protocol with an average insert size of 350 bp. Reads were assembled into contigs using the ABySS de novo assembler (17), yielding 260 contigs for KPB-1 and 295 contigs for KPB-2, with N50 (length-weighted median) values of 130,840 bp (KPB-1) and 133,427 bp (KPB-2), mean contig sizes of 34,400 bp (KPB-1) and 35,958 bp (KPB-2), maximum contig lengths of 501,165 bp (KPB-1) and 545,254 bp (KPB-2), and contig sums of 5,779,306 bp (KPB-1) and 5,825,236 bp (KPB-2). Sequence data were analyzed following the bioinformatics approach described previously (11).
Recombinant DNA methodology.
Genomic DNA was purified using a DNeasy blood and tissue kit (Qiagen GmbH, Hilden, Germany), and plasmid DNA was purified as reported previously (11). Restriction endonucleases were purchased from Promega (Madison, WI). Electroporation of plasmids into E. coli and K. pneumoniae was carried out as described previously (11). The plasmids pACYC-pmrB and pACYC-pmrBT245G, which were used for complementation experiments, were derivatives of pACYC184 (18) containing the wild-type pmrB gene and the pmrBT245G gene, respectively. These plasmids were constructed by amplification of the pmrB gene and flanking regions from strains KPB-1 and KPB-2, using the primers AvaI_pmrAB_F and EcoRI_pmrAB _R (Table 1 and Fig. 1). The resulting 1,834-bp PCR products, covering the complete pmrB gene with its putative terminator and part of the upstream pmrA gene, were digested with AvaI and EcoRI and cloned into pACYC184 digested with the same enzymes. The authenticity of the cloned fragments was confirmed by sequencing of both strands at an external sequencing facility (Macrogen Inc., Seoul, South Korea).
TABLE 1.
Primers and PCR conditions used in this work
| Assay type and primer | Sequence (5′ to 3′)a | Cycling conditionsb |
|---|---|---|
| Conventional PCR | ||
| AvaI_pmrAB_F | TCCCTCGGGTCATTACAGCCTGATCGTGCTGGATCTCG | 95°C for 30 s, 62°C for 30 s, 72°C for 180 s |
| EcoRI_pmrAB _R | CCGGAATTCTCGTCCTGCTTGCCAGATAACAAACATTT | |
| qRT-PCR | ||
| pmrA_F | GCAGGGGTTAATTCTGGCGATGC | 95°C for 10 s, 52°C for 5 s, 72°C for 5 s |
| pmrA_R | CGATAGCGCGGCTTCGTGC | |
| pmrB_F | GGCCGTCGTCTCTGGCGATG | 95°C for 10 s, 52°C for 5 s, 72°C for 5 s |
| pmrB_R | GGGCTGTAGCGGTGAGCATTC | |
| pmrK_F2 | CGCTGAATATGCTCGACCCAGAAG | 95°C for 10 s, 52°C for 5 s, 72°C for 5 s |
| pmrK_R2 | GCTGGCGGTAATCGTCTGTACG | |
| rpsL13_F | GCCGTACTTGGAGCGAGCCTG | 95°C for 10 s, 52°C for 5 s, 72°C for 5 s |
| rpsL14_F | CCGTGGCGGTCGTGTTAAAGA |
Restriction sites added for cloning purposes in some primers are underlined.
All conventional PCRs included an initial denaturation step of 180 s at 95°C, 30 cycles of denaturation, annealing, and extension at the reported temperatures for the reported times, and a final extension step of 300 s at 72°C; all qRT-PCRs included an initial denaturation step of 300 s at 95°C and 40 cycles of amplification.
FIG 1.
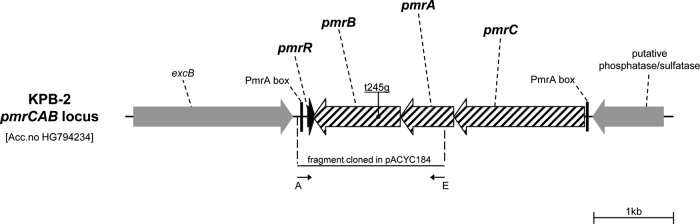
Organization of the pmrCAB operon and flanking regions in K. pneumoniae KPB-2 and location of the T245G mutation in the pmrB gene, leading to the L82R amino acid substitution in the transmembrane domain of the PmrB protein. The locations of primers used for PCR cloning of the pmrB gene for complementation experiments are indicated by thin black arrows. A, AvaI_pmrAB_F; E, EcoRI_pmrAB_R. In Salmonella enterica, the pmrC gene product catalyzes the addition of phosphoethanolamine to lipid A, which also could contribute to decreasing the negative charge of lipid A (14). The role of pmrC in K. pneumoniae has not been specifically reported, but given the conservation of pmrC and of the whole Pmr system, it likely retains the same function.
Quantitative real-time PCR for transcriptional analysis.
RNA extractions were performed using the SV total RNA isolation kit (Promega), and the corresponding cDNAs were obtained using the Improm-II reverse transcriptase kit (Promega). The relative levels of expression of the pmrA and pmrB genes of the pmrCAB operon and of pmrK (a member of the pmrHFIJKLM operon) were assessed by quantitative real-time PCR (qRT-PCR) using a LightCycler instrument (Roche Applied Science, Mannheim, Germany) and the primers and conditions reported in Table 1. Normalization was performed with the rpsL gene as an internal standard (11), using the ΔΔCT method (19), and the values obtained were then normalized against the value obtained with the susceptible KPB-1 strain.
Stability of the colistin-resistant phenotype in strain KPB-2.
To assess the stability of the colistin-resistant phenotype, the KPB-2 strain was grown in the absence of antibiotic selection, in cation-adjusted Mueller-Hinton (MH) broth (bioMérieux, Florence, Italy), at 37°C aerobically (10 ml of culture medium in 50-ml flasks, in an orbital shaker operating at 180 rpm) for approximately 50 generations, by subculturing the strain (1:100 dilution) every day for 5 days. After this period, the culture was plated onto MH agar without antibiotic, to obtain individual colonies, and the proportion of colistin-resistant colonies was enumerated by replica plating onto selective medium (MH agar supplemented with 4 μg/ml of colistin; upon replica plating, the KPB-2 strain was found to be able to grow on MH agar containing this colistin concentration).
Experiments for analysis of fitness cost.
Growth curves for KPB-1 and KPB-2 were evaluated in triplicate, either with strains growing separately or with strains growing together in a competition experiment, as described previously (20). Growth was carried out in cation-adjusted MH broth (bioMérieux) at 37°C aerobically (50 ml of culture medium in 250-ml flasks, in an orbital shaker operating at 180 rpm) for 24 h. The viable cell counts were determined at various time points by plating, in triplicate, serial dilutions of the cultures onto either MH agar or MH agar supplemented with colistin (3 μg/ml). The competition index (CI), defined as the colistin-resistant strain CFU/colistin-susceptible strain CFU ratio, normalized against the total amount of CFU inoculated for each strain, was calculated as described previously (21).
Nucleotide sequence accession numbers.
The nucleotide sequence of the pmrCAB operon and flanking regions from strain KPB-2 has been deposited at DDBJ/EMBL/GenBank under accession number HG794234. Genome projects for strains KPB-1 and KPB-2 have been registered as NCBI BioProjects PRJNA225494 and PRJNA225495, respectively. The draft genome projects for strains KPB-1 and KPB-2 have been deposited at DDBJ/EMBL/GenBank under accession numbers AYOV00000000 and AYOW00000000, respectively; the versions described in this paper are the first versions of these entries.
RESULTS AND DISCUSSION
Identification of a PmrB mutation as a putative mechanism for colistin resistance in a KPC-KP strain isolated after in vivo colistin exposure.
Two sequential KPC-KP isolates, KPB-1 (colistin susceptible) and KPB-2 (colistin resistant, isolated 12 days later, after colistin treatment), from a patient with an invasive KPC-KP infection were investigated in this work. The colistin MIC of KPB-2 was 4 μg/ml, being 64-fold higher than that of KPB-1 (Table 2). The colistin dosage given to the patient (2 million units twice a day [b.i.d.], intravenously) was significantly lower than the currently recommended dosage of 4.5 million units b.i.d. (7). It should be noted that, when this case was treated (in early 2011), the experience with colistin use for the treatment of carbapenem-resistant K. pneumoniae infections was still limited, while the label indications foresaw a maximum of 3 million units per day. Both strains carried a blaKPC-3 gene and belonged in ST512, and they exhibited identical XbaI PFGE profiles (data not shown).
TABLE 2.
Colistin MICs and expression levels of pmrA and pmrK genes of KPB-1 and KPB-2 and of the corresponding transformants carrying the pACYC184, pACYC-pmrB, and pACYC-pmrBT245G plasmidsa
| Strain | Chromosomal pmrB status | pACYC184 pmrB status | Colistin MIC (μg/ml) | Relative expression level |
||
|---|---|---|---|---|---|---|
| pmrA | pmrB | pmrK | ||||
| KPB-1 | WTb | 0.06 | 1 | 1 | 1 | |
| KPB-1(pACYC-pmrBT245G) | WT | T245G (L82R) | 0.125 | 1.06 ± 0.07 | NDc | 0.89 ± 0.02 |
| KPB-2 | T245G (L82R) | 4 | 10.23 ± 1.02 | 4.53 ± 0.92 | 4.33 ± 0.08 | |
| KPB-2(pACYC184) | T245G (L82R) | 4 | 11.51 ± 0.32 | ND | 3.85 ± 0.11 | |
| KPB-2(pACYC-pmrB) | T245G (L82R) | WT | 0.125 | 0.98 ± 0.21 | ND | 0.68 ± 0.05 |
| KPB-2(pACYC184-pmrBT245G) | T245G (L82R) | T245G (L82R) | 64 | 11.23 ± 0.61 | ND | 4.78 ± 0.13 |
Expression levels for the pmrA and pmrK genes were normalized against the values obtained with KPB-1. For each strain, the chromosomal status of pmrB and of the pmrB allele harbored by pACYC184 are also indicated.
WT, wild-type.
ND, not determined.
Comparative analysis of the two draft genomes confirmed the highly conserved genomic structure of the two strains and revealed two nonsynonymous single-nucleotide variations (SNVs) in KPB-2, i.e., (i) a SNV at nucleotide 245 (T245G) in the pmrB gene, leading to an L-to-R substitution at amino acid position 82 of the PmrB protein, and (ii) a SNV at nucleotide 1702 (C1702T) in the wzc gene, leading to an H-to-Y substitution at amino acid position 568 of the Wzc protein. Wzc is a bacterial tyrosine-type protein tyrosine kinase that, in E. coli, was shown to be involved in the biosynthesis of extracellular polysaccharide but apparently is not involved in the expression of polymyxin resistance (22, 23). On the other hand, PmrB is the sensor kinase of the conserved PmrA/PmrB signaling system, which, in Enterobacteriaceae and other Gram-negative organisms, is involved in responses to low pH and some cations (e.g., Fe3+ and Al3+) and controls the modification of lipid A via upregulation of various genes that are part of the PmrA regulon (14). In fact, PmrB mutations have already been associated with colistin resistance in other species, including Acinetobacter baumannii (24–26) and Pseudomonas aeruginosa (27). Recently, PmrB mutations were also reported in colistin-resistant K. pneumoniae mutants selected in vitro (28). Based on these findings, we hypothesized that expression of the PmrBL82R mutant protein could be the cause of the colistin-resistant phenotype in the KPB-2 isolate. Indeed, analysis of the expression of pmrA (a member of the pmrCAB operon) and of pmrK (a member of the pmrHFIJKLM operon) by qRT-PCR revealed increased levels in KPB-2 compared with KPB-1 (Table 2), confirming that both the PmrA/PmrB signaling system and the PmrHFIJKLM lipid A modification system were upregulated in KPB-2 and supporting the previous hypothesis.
Restoration of colistin susceptibility in the PmrBL82R mutant with wild-type pmrB complementation.
Expression of wild-type PmrB in the KPB-2 background was able to restore colistin susceptibility, with an MIC value comparable to that of KPB-1 (Table 2). In KPB-2 (pACYC-pmrB), the expression levels of pmrA and pmrK were also reduced to basal levels (Table 2). Expression of the PmrBL82R mutant in the KPB-1 background, on the other hand, did not result in an increased colistin MIC or in upregulation of expression of the pmrA and pmrK genes (Table 2). Finally, expression of the PmrBL82R mutant in the KPB-2 background resulted in further increases in the colistin MIC and expression of the pmrA and pmrK genes (Table 2). Together, these findings confirmed the role of the PmrBL82R mutation in colistin resistance through upregulation of the PmrA/PmrB signaling system and of the PmrHFIJKLM LPS modification system, and they also indicated that wild-type PmrB is dominant versus PmrBL82R in regulation of the LPS modification system and in autoregulation of the PmrA/PmrB signaling system.
The mechanisms by which the L82R amino acid substitution in PmrB activates the PmrA/PmrB signaling system in the absence of the physiological activating signals (e.g., Fe3+, Al3+, and mildly acidic pH) and by which this activation is suppressed in the presence of the wild-type PmrB protein remain to be clarified and will require additional investigation. The most likely hypothesis is that the L82R amino acid substitution, which is located within the transmembrane domain of the protein (29), might result in some activation (autophosphorylation) of PmrBL82R in the absence of the physiological activating signals, which might be suppressed by the phosphatase activity of wild-type PmrB when the latter protein is coexpressed with PmrBL82R in the absence of activating signals.
Stability and fitness cost of the PmrBL82R mutation.
In order to evaluate the stability of the colistin-resistant phenotype mediated by the PmrB alteration, KPB-2 was cultured for approximately 50 generations in the absence of antibiotic, and the resulting bacterial population was tested for colistin resistance. The results of this experiment showed that 100% of the tested cells were still resistant to colistin, suggesting that the colistin-resistant phenotype conferred by this mutation was very stable, even in the absence of selective pressure.
In order to evaluate whether the colistin-resistant phenotype mediated by the PmrB alteration was associated with a fitness burden, growth curves for KPB-1 and KPB-2 were compared, either with the strains growing separately or with the strains growing together in a competition experiment. No significant differences were observed between the growth curves for KPB-1 and KPB-2, grown either separately (data not shown) or together (Fig. 2). In the competition experiment, the CI values remained stable overall (Fig. 2). Together, these results suggested that the colistin-resistant phenotype mediated by the PmrBL82R alteration was not associated with a significant fitness cost. Interestingly, these results are different from those observed previously with colistin-resistant pmrB or pmrA mutants of A. baumannii, which consistently exhibited reductions in fitness in comparison with the colistin-susceptible progenitors (20, 21, 25, 30).
FIG 2.
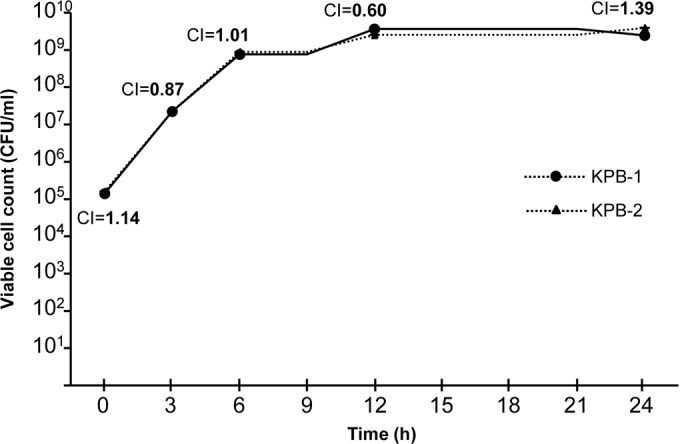
Growth curves for the KPB-1 (colistin-susceptible) and KPB-2 (colistin-resistant) K. pneumoniae isolates obtained in the competition experiment. The competition index (CI) values were determined at various time points, as previously reported (21).
It should be noted that, in the KPB-2 colistin-resistant mutant investigated in this work, a missense mutation was also detected in the wzc gene, which was assumed to be not involved in colistin resistance and was not investigated further in this work. Whether the wzc mutation might play a role in compensating for any potential fitness defect associated with expression of the PmrBL82R mutant in this context remains to be clarified and will be the subject of future investigation.
Conclusions.
The emergence of colistin resistance in KPC-KP strains is a matter of major concern, due to the very few therapeutic options for treatment of infections caused by similar strains (2, 7). An additional concern with colistin-resistant strains is that the LPS modification responsible for the resistance phenotype can be associated with increased resistance to the antimicrobial peptides of human polymorphonuclear leukocytes (14, 31).
Mutations in the PmrA/PmrB signaling system, which normally is involved in the responses to various environmental signals and controls the system responsible for modification of the colistin LPS target (14), were reported previously for colistin-resistant mutants of K. pneumoniae selected in vitro, although mutations were found at different positions (28). Our findings showed that colistin resistance due to PmrB alterations could be selected in vivo following colistin treatment, and they revealed an additional type of PmrB mutation associated with a colistin-resistant phenotype. In this case, a relatively low dosage of colistin was used for treatment (2 million units b.i.d., compared with the currently recommended dosage of 4.5 million units b.i.d.), in the absence of any additional active antibiotic. This might have played a role in selection of the resistant mutant and is consistent with the high mutant prevention concentration reported previously for colistin and K. pneumoniae clinical isolates (28). Studying the possible role of multistep mutations in the evolution of colistin resistance could be of interest and currently is a subject of investigation.
ACKNOWLEDGMENTS
The valuable scientific advice of Cesira Galeotti is gratefully acknowledged.
This work was supported by a grant from FP7 projects from EvoTAR (grant HEALTH-F3-2011-2011-282004) to G.M.R.
Footnotes
Published ahead of print 19 May 2014
REFERENCES
- 1.Nordmann P, Naas T, Poirel L. 2011. Global spread of carbapenemase-producing Enterobacteriaceae. Emerg. Infect. Dis. 17:1791–1798. 10.3201/eid1710.110655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tzouvelekis LS, Markogiannakis A, Psichogiou M, Tassios PT, Daikos GL. 2012. Carbapenemases in Klebsiella pneumoniae and other Enterobacteriaceae: an evolving crisis of global dimensions. Clin. Microbiol. Rev. 25:682–707. 10.1128/CMR.05035-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Canton R, Akova M, Carmeli Y, Giske CG, Glupczynski Y, Gniadkowski M, Livermore DM, Miriagou V, Naas T, Rossolini GM, Samuelsen O, Seifert H, Woodford N, Nordmann P, European Network on Carbapenemases 2012. Rapid evolution and spread of carbapenemases among Enterobacteriaceae in Europe. Clin. Microbiol. Infect. 18:413–431. 10.1111/j.1469-0691.2012.03821.x [DOI] [PubMed] [Google Scholar]
- 4.Munoz-Price LS, Poirel L, Bonomo RA, Schwaber MJ, Daikos GL, Cormican M, Cornaglia G, Garau J, Gniadkowski M, Hayden MK, Kumarasamy K, Livermore DM, Maya JJ, Nordmann P, Patel JB, Paterson DL, Pitout J, Villegas MV, Wang H, Woodford N, Quinn JP. 2013. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect. Dis. 13:785–796. 10.1016/S1473-3099(13)70190-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giani T, Pini B, Arena F, Conte V, Bracco S, Migliavacca R, AMCLI-CRE Survey Participants. Pantosti A, Pagani L, Luzzaro F, Rossolini GM. 2013. Epidemic diffusion of KPC carbapenemase-producing Klebsiella pneumoniae in Italy: results of the first countrywide survey, 15 May to 30 June 2011. Euro Surveill. 18:pii=20489 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20489 [PubMed] [Google Scholar]
- 6.Livermore DM, Warner M, Mushtaq S, Doumith M, Zhang J, Woodford N. 2011. What remains against carbapenem-resistant Enterobacteriaceae? Evaluation of chloramphenicol, ciprofloxacin, colistin, fosfomycin, minocycline, nitrofurantoin, temocillin and tigecycline. Int. J. Antimicrob. Agents. 37:415–419. 10.1016/j.ijantimicag.2011.01.012 [DOI] [PubMed] [Google Scholar]
- 7.Petrosillo N, Giannella M, Lewis R, Viale P. 2013. Treatment of carbapenem-resistant Klebsiella pneumoniae: the state of the art. Expert Rev. Anti Infect. Ther. 11:159–177. 10.1586/eri.12.162 [DOI] [PubMed] [Google Scholar]
- 8.Ah YM, Kim AJ, Lee JY. 8 April 2014. Colistin resistance in Klebsiella pneumoniae. Int. J. Antimicrob. Agents. 10.1016/j.ijantimicag.2014.02.016 [DOI] [PubMed] [Google Scholar]
- 9.Zagorianou A, Sianou E, Iosifidis E, Dimou V, Protonotariou E, Miyakis S, Roilides E, Sofianou D. 2012. Microbiological and molecular characteristics of carbapenemase-producing Klebsiella pneumoniae endemic in a tertiary Greek hospital during 2004–2010. Euro Surveill. 17:pii=20088 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20088 [PubMed] [Google Scholar]
- 10.Lippa AM, Goulian M. 2009. Feedback inhibition in the PhoQ/PhoP signaling system by a membrane peptide. PLoS Genet. 5:e1000788. 10.1371/journal.pgen.1000788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cannatelli A, D'Andrea MM, Giani T, Di Pilato V, Arena F, Ambretti S, Gaibani P, Rossolini GM. 2013. In vivo emergence of colistin resistance in Klebsiella pneumoniae producing KPC-type carbapenemases mediated by insertional inactivation of the PhoQ/PhoP mgrB regulator. Antimicrob. Agents Chemother. 57:5521–5526. 10.1128/AAC.01480-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.López-Camacho E, Gómez-Gil R, Tobes R, Manrique M, Lorenzo M, Galván B, Salvarelli E, Moatassim Y, Salanueva IJ, Pareja E, Codoñer FM, Alvarez-Tejado M, Garcillán-Barcia MP, De la Cruz F, Mingorance J. 2014. Genomic analysis of the emergence and evolution of multidrug resistance during a Klebsiella pneumoniae outbreak including carbapenem and colistin resistance. J. Antimicrob. Chemother. 69:632–636. 10.1093/jac/dkt419 [DOI] [PubMed] [Google Scholar]
- 13.Cheng HY, Chen YF, Peng HL. 2010. Molecular characterization of the PhoPQ-PmrD-PmrAB mediated pathway regulating polymyxin B resistance in Klebsiella pneumoniae CG43. J. Biomed. Sci. 17:60–76. 10.1186/1423-0127-17-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen HD, Groisman EA. 2013. The biology of the PmrA/PmrB two-component system: the major regulator of lipopolysaccharide modifications. Annu. Rev. Microbiol. 67:83–112. 10.1146/annurev-micro-092412-155751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 16.Clinical and Laboratory Standards Institute. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard—9th ed. CLSI document M07-A9. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 17.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res. 19:1117–1123. 10.1101/gr.089532.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang AC, Cohen SN. 1978. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 134:1141–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 20.Beceiro A, Moreno A, Fernández N, Vallejo JA, Aranda J, Adler B, Harper M, Boyce JD, Bou G. 2014. Biological cost of different mechanisms of colistin resistance and their impact on virulence in Acinetobacter baumannii. Antimicrob. Agents Chemother. 58:518–526. 10.1128/AAC.01597-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.López-Rojas R, McConnell MJ, Jiménez-Mejías ME, Domínguez-Herrera J, Fernández-Cuenca F, Pachón J. 2013. Colistin resistance in a clinical Acinetobacter baumannii strain appearing after colistin treatment: effect on virulence and bacterial fitness. Antimicrob. Agents Chemother. 57:4587–4589. 10.1128/AAC.00543-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lacour S, Doublet P, Obadia B, Cozzone AJ, Grangeasse C. 2006. A novel role for protein-tyrosine kinase Etk from Escherichia coli K-12 related to polymyxin resistance. Res. Microbiol. 157:637–641. 10.1016/j.resmic.2006.01.003 [DOI] [PubMed] [Google Scholar]
- 23.Lacour S, Bechet E, Cozzone AJ, Mijakovic I, Grangeasse C. 2008. Tyrosine phosphorylation of the UDP-glucose dehydrogenase of Escherichia coli is at the crossroads of colanic acid synthesis and polymyxin resistance. PLoS One 3:e3053. 10.1371/journal.pone.0003053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams MD, Nickel GC, Bajaksouzian S, Lavender H, Murthy AR, Jacobs MR, Bonomo RA. 2009. Resistance to colistin in Acinetobacter baumannii associated with mutations in the PmrAB two-component system. Antimicrob. Agents Chemother. 53:3628–3634. 10.1128/AAC.00284-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lesho E, Yoon EJ, McGann P, Snesrud E, Kwak Y, Milillo M, Onmus-Leone F, Preston L, St Clair K, Nikolich M, Viscount H, Wortmann G, Zapor M, Grillot-Courvalin C, Courvalin P, Clifford R, Waterman PE. 2013. Emergence of colistin-resistance in extremely drug-resistant Acinetobacter baumannii containing a novel pmrCAB operon during colistin therapy of wound infections. J. Infect. Dis. 208:1142–1151. 10.1093/infdis/jit293 [DOI] [PubMed] [Google Scholar]
- 26.Beceiro A, Llobet E, Aranda J, Bengoechea JA, Doumith M, Hornsey M, Dhanji H, Chart H, Bou G, Livermore DM, Woodford N. 2011. Phosphoethanolamine modification of lipid A in colistin-resistant variants of Acinetobacter baumannii mediated by the pmrAB two-component regulatory system. Antimicrob. Agents Chemother. 55:3370–3379. 10.1128/AAC.00079-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moskowitz SM, Brannon MK, Dasgupta N, Pier M, Sgambati N, Miller AK, Selgrade SE, Miller SI, Denton M, Conway SP, Johansen HK, Høiby N. 2012. PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients. Antimicrob. Agents Chemother. 56:1019–1030. 10.1128/AAC.05829-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi MJ, Ko KS. 2014. Mutant prevention concentrations of colistin for Acinetobacter baumannii, Pseudomonas aeruginosa and Klebsiella pneumoniae clinical isolates. J. Antimicrob. Chemother. 69:275–277. 10.1093/jac/dkt315 [DOI] [PubMed] [Google Scholar]
- 29.Wösten MM, Kox LF, Chamnongpol S, Soncini FC, Groisman EA. 2000. A signal transduction system that responds to extracellular iron. Cell 103:113–125. 10.1016/S0092-8674(00)00092-1 [DOI] [PubMed] [Google Scholar]
- 30.López-Rojas R, Domínguez-Herrera J, McConnell MJ, Docobo-Peréz F, Smani Y, Fernández-Reyes M, Rivas L, Pachón J. 2011. Impaired virulence and in vivo fitness of colistin-resistant Acinetobacter baumannii J. Infect. Dis. 203:545–548. 10.1093/infdis/jiq086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Llobet E, Campos MA, Giménez P, Moranta D, Bengoechea JA. 2011. Analysis of the networks controlling the antimicrobial-peptide-dependent induction of Klebsiella pneumoniae virulence factors. Infect. Immun. 79:3718–3732. 10.1128/IAI.05226-11 [DOI] [PMC free article] [PubMed] [Google Scholar]