

Abstract
Genome editing using zinc finger nucleases (ZFNs) has been successfully applied to disrupt CCR5 or CXCR4 host factors and inhibit viral entry and infection. Gene therapy using ZFNs to modify the PSIP1 gene, which encodes the lens epithelium-derived growth factor (LEDGF) protein, might restrain an early step of the viral replication cycle at the integration level. ZFNs targeting the PSIP1 gene (ZFNLEDGF) were designed to specifically recognize the sequence after the integrase binding domain (IBD) of the LEDGF/p75 protein. ZFNLEDGF successfully recognized the target region of the PSIP1 gene in TZM-bl cells by heteroduplex formation and DNA sequence analysis. Gene editing induced a frameshift of the coding region and resulted in the abolishment of LEDGF expression at the mRNA and protein levels. Functional assays revealed that infection with the HIV-1 R5 BaL or X4 NL4-3 viral strains was impaired in LEDGF/p75 knockout cells regardless of entry tropism due to a blockade in HIV-1 proviral integration into the host genome. However, residual infection was detected in the LEDGF knockout cells. Indeed, LEDGF knockout restriction was overcome at a high multiplicity of infection, suggesting alternative mechanisms for HIV-1 genome integration rather than through LEDGF/p75. However, the observed residual integration was sensitive to the integrase inhibitor raltegravir. These results demonstrate that the described ZFNLEDGF effectively targets the PSIP1 gene, which is involved in the early steps of the viral replication cycle; thus, ZFNLEDGF may become a potential antiviral agent for restricting HIV-1 integration. Moreover, LEDGF knockout cells represent a potent tool for elucidating the role of HIV integration cofactors in virus replication.
INTRODUCTION
Human immunodeficiency virus (HIV) requires the host cellular machinery in order to successfully replicate (1). The development of genome-editing tools, such as zinc finger endonucleases (ZFNs) (2), transcription activator-like effector nucleases (TALEN) (3), and clustered regularly interspaced short palindromic repeats (CRISPR) (4–6), has introduced a promising alternative for the modification of essential host factors along the replication cycle of HIV (7, 8). ZFNs have demonstrated their applicability to reproduce the CCR5Δ32 phenotype in vitro by successfully cleaving the CCR5 gene, generating human CD4+ T cells refractory to HIV-1 infection (9–12). Similarly, a ZFN approach successfully cleaved the alternative HIV-1 coreceptor CXCR4 in CD4+ T cells in a humanized mouse model, resulting in impaired HIV-1 infection (13). Genome editing as anti-HIV therapy is currently under study in at least 2 or 3 clinical trials using ZFNs targeting CCR5. However, similar strategies targeting host cellular factors affecting the later steps of the virus replication cycle have not been evaluated.
A crucial step of the viral replication cycle is exerted by the lens epithelium-derived growth factor (LEDGF), a member of the hepatoma-derived growth factor (HDGF)-related protein (HRP) family. HRPs are characterized by a conserved N-terminal PWWP domain, a 90- to 135-amino-acid module found in a variety of nuclear proteins (14). Six human HRP family members have been described: HDGF, HRP1, HRP2, HRP3, LEDGF/p75, and LEDGF/p52. Two of them, LEDGF/p75 and HRP2, possess affinity for HIV-1 integrase (IN), given by a second evolutionarily conserved domain within their C termini that mediates the interaction with HIV-1 IN, hence the term IN-binding domain (IBD) (15). Initially identified as an IN-associated protein (16), LEDGF/p75 was revealed to be a lentivirus-specific cellular cofactor required for HIV integration into the host genome (see references 17 to 19 for review). LEDGF/p75 directly interacts with viral HIV-IN, tethering a viral preintegration complex into the active transcription units of the cellular chromatin. The role of LEDGF/p75 in HIV-1 replication was studied using RNA interference (RNAi) targeting LEDGF/p75 and LEDGF knockout (KO) mouse embryonic fibroblasts (MEFs). Although both strategies potently downregulated or completely abolished LEDGF/p75 expression, residual replication was observed. Thus, all studies point to a key but not essential role for LEDGF/p75 in lentiviral replication and suggest that the existence of alternative cellular cofactors, such as HRP2, are responsible for the residual replication observed in the absence of LEDGF/p75 (20, 21).
Nevertheless, the LEDGF/p75 interaction with HIV IN has been suggested as a valid target for antiviral therapy (22–24). In that sense, recently developed allosteric LEDGF/p75-IN interaction inhibitors (LEDGINs and ALLINIS) have been proven to target the LEDGF/p75 binding pocket of HIV IN and to inhibit the catalytic activity of the IN. Moreover, LEDGINs and ALLINIS also exert antiviral activity by promoting IN multimerization. Aberrant IN complexes lead to the formation of defective regular cores during the maturation process, resulting in an impaired infectivity in the new viral particles (25–27).
On the other hand, a series of PSIP1 single nucleotide polymorphisms (SNP) were associated with HIV-1 disease progression in cohorts of African and Caucasian HIV-1-positive individuals (28, 29). In addition, two missense mutations were identified in two samples belonging to a long-term nonprogression (LTNP) cohort (30). All identified missense mutations are located in the helix-turn-helix (HTH) motifs at the C-terminal region of the protein, which is after the IBD. Although none of the mutations restricted HIV replication in vitro (29, 31, 32), these findings suggest that genetic variation in PSIP1 may influence susceptibility to HIV-1 infection and disease progression.
Here, we describe a novel genome-editing ZFN that specifically disrupts the PSIP1 gene encoding LEDGF/p75 in its C terminus, which is found after all relevant functional domains and near the missense mutations that have been described in patients (ZFNLEDGF). ZFNLEDGF was able to generate LEDGF/p75 cells expressing a truncated protein that becomes refractory to HIV-1 integration. The LEDGF/p75 knockout cells generated represent a potent tool for further investigation of the function of the LEDGF/p75 protein, and they may help to elucidate the role of HIV integration cofactors in virus replication. Moreover, ZFNLEDGF may become an antiviral strategy for restricting HIV-1 integration and virus replication in vivo.
MATERIALS AND METHODS
Vectors.
CompoZr knockout zinc finger nucleases targeting the PSIP1 gene (ZFNLEDGF) were obtained from Sigma-Aldrich Biotechnology (St. Louis, MO, USA). Briefly, ZFNLEDGF were designed to target the sequence AACATGTTCTTGGTTGGTGAAGGAGATTCCGTG (Fig. 1a), according to the guidelines of Mussolino and Cathomen (33), to ensure minimal homology of the ZFN DNA-binding domain to any other site in the genome. The FokI and zinc finger domains of each ZFN pair were assembled as previously described (34). Next, the ZFNLEDGF pairs were cloned into the pLVX-IRES-ZsGreen1 vector (Clontech), by replacing the EcoRI-XbaI fragment, to obtain ZFNLEDGF tagged with the green fluorescent reporter gene (pLVX-ZFNLEDGF-ZsGreen).
FIG 1.

Experimental design and specificity of ZFNLEDGF targeting the PSIP1 gene. (a) Schematic representation of LEDGF/p75 protein highlighting the position of the ZFNLEDGF targeted region. The cutting site of ZFNLEDGF is located near the sequence coding for the integrase binding domain (IBD) of the LEDGF/p75. NLS, nuclear localization signal; AT, AT-hook-like domains. (b) Experimental design/process used to generate and evaluate LEDGF/p75 knockout cells. (c) Flow cytometry plots of wild-type cells, mock-treated cells, and cells transfected with ZFNLEDGF plasmids and challenged with VSV-pseudotyped NL4-3 GFP-expressing virus. WT, wild type; MOCKtr, mock-transfected cells; FSC, forward scatter; FITC, fluorescein isothiocyanate. (d) Gene editing by ZFNLEDGF induces heteroduplex formation determined by the surveyor mutation assay (CEL-I). After genomic DNA extraction, heteroduplex formation due to the generation of insertions or deletions was assessed by the surveyor mutation assay. DNA fragments were resolved in a 10% Tris-borate-EDTA (TBE)-PAGE gel. The lower migrating products (arrows) are a direct measure of ZFN-mediated gene disruption. +/−, clone 1 (heterozygous); −/−, clone 2 (homozygous).
Cells.
Human K562 cells were obtained from the ATCC (CCL-243) and grown in Iscove's modified Dulbecco's medium supplemented with 10% fetal bovine serum (FBS) and 2 mM l-glutamine. The TZM-bl cell line (NIH AIDS Research and Reference Program) was grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco, Life Technologies, Madrid, Spain), supplemented with 10% heat-inactivated fetal calf serum (FCS) (Gibco, Thermo Fisher, Madrid, Spain) and antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin [Life Technologies]), and maintained at 37°C in a 5% CO2 incubator.
ZFN transfection.
The TZM-bl cells were transfected with ZFNLEDGF-expressing plasmids, as described previously (35, 36). Briefly, 1.5 × 105 cells were seeded in 24-well plates. After overnight culture, 0.5 μg of each ZFNLEDGF plasmid was mixed with Lipofectamine 2000 reagent (Invitrogen) in the serum-free medium Opti-MEM (Invitrogen) and then added to previously washed cells. The medium was replaced by fresh DMEM 4 h after transfection and left in the incubator for 3 days, when ZsGreen-positive cells were sorted using a FACSAria II (BD Biosciences) flow cytometer. Single-cell clones were obtained by limiting dilution seeding of the sorted cells in 96-well plates.
Analysis of PSIP1 disruption by CEL-I assay.
The effect of ZFN on the PSIP1 alleles was assessed by performing PCR on the region surrounding the ZFN target site, followed by digestion with the Surveyor (CEL-1) nuclease assay (Transgenomic, Omaha, NE, USA), which cleaves DNA heteroduplex formations at the mismatch sites. The generated fragments were resolved by 10% polyacrylamide electrophoresis, as previously described (10, 12).
Sequence analysis of targeted PSIP1 gene in TZM-bl cells.
Genomic DNA from the sorted cell clones was extracted using the QIAamp DNA blood minikit (Qiagen, Barcelona, Spain). Extracted DNA was used to amplify the LEDGF gene using the Expand high-fidelity PCR system (Roche, Barcelona, Spain) and the forward primer 5′-TTCAAGTCATGTGGATTCTTTGA-3′ and reverse primer 5′-TCTAGCTTTTTGTTTGGCCC-3′. The PCR products were cloned into the pGEM-T Easy vector system (Promega, Madrid, Spain), according to the manufacturer's instructions. Plasmid sequencing was carried out by the Macrogen Genomic Division, Seoul, South Korea, using ABI Prism BigDye Terminator cycle sequencing technology (Applied Biosystems), with the following internal primers of the PSIP1 genomic sequence: forward, 5′-TTGGAAACGATCTTTAGAAACAGA-3′, and reverse, 5′-CAGTGAAACTATGTATGAAAGCCATT-3′. When bigger deletions were observed, the sequences were obtained directly from the mRNA sequence (see above). The sequences were analyzed with the Sequencher 4.5 software (Gene Codes Corporation, Ann Arbor, MI, USA). In addition, a bioinformatics algorithm (Sigma) was used to evaluate the putative off-target effects for ZFNLEDGF throughout the human genome.
In silico prediction of the mRNA and protein sequences of the edited cell clones was performed using the ExPASy SIB bioinformatics resource portal (37).
Quantitative RT-PCR and mRNA expression assessment.
Relative mRNA quantification of PSIP1 expression was assessed by reverse transcription-quantitative PCR (RT-qPCR), as previously described (38, 39). Briefly, RNA was extracted using the Qiagen RNeasy extraction minikit (Qiagen), according to the manufacturer's instructions, including the DNase I treatment step. Reverse transcription was performed using the High-Capacity cDNA reverse transcription kit (Life Technologies, Madrid, Spain). To assess the presence of the predicted PSIP1 mRNA truncated forms, the mRNA sequence was divided into two fragments of similar size, which were PCR amplified with primers for the 5′-containing fragment: forward primer 5′-GGCAAACCAAATAAAAGAAAAGG-3′ and reverse primer 5′-CTTGCTTGCGTTTTCGATCT-3′; the primers for the 3′-containing fragment were forward primer 5′-AAAAGGTGGGAGGAACTTTCA-3′ and reverse primer 5′-GCAGTCTATTTCAAATGAAAACCAT-3′ Relative gene expression of the PSIP1 wild-type form was also measured by two-step quantitative RT-PCR and normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene expression using the delta-delta threshold cycle (CT) method. The primers and DNA probes were purchased from Life Technologies.
Western blot assay.
The treated cells were rinsed in ice-cold phosphate-buffered saline (PBS), nuclear proteins were enriched using the CelLytic NuCLEAR extraction kit (Sigma), and extracts were prepared in lysis buffer (50 mM Tris-HCl [pH 7.5], 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 10 mM Na β-glycerophosphate, 50 mM NaF, 5 mM Na pyrophosphate, 270 mM sucrose, and 1% Triton X-100) supplemented with protease inhibitor cocktail (Roche) and 1 mM phenylmethylsulfonyl fluoride. The samples were run using a NuPAGE 4 to 12% Bis-Tris gel (Novex, Life Technologies) and blotted onto nitrocellulose membranes. The blocked membranes were incubated overnight with monoclonal antibodies (MAbs) against the human C-terminal LEDGF protein (C57G11; Cell Signaling Technologies), human N-terminal LEDGF protein (clone 26, 611714; BD Transduction Laboratories), and β-actin (Sigma-Aldrich) at 4°C. After washing, the membranes were incubated with a secondary horseradish peroxidase-conjugated antibody for 1 h at room temperature and then revealed with SuperSignal West Pico chemiluminescent substrate (Pierce Chemical, Rockford, IL).
Virus production and infections.
Vesicular stomatitis virus (VSV)-pseudotyped NL4-3 green fluorescent protein (GFP)-expressing virus (40) was produced as described previously (39). The HIV-1 viral strains BaL (R5 tropic) and NL4-3 (X4 tropic) were obtained from the MRC Centre for AIDS Reagents (London, United Kingdom). The BaL and NL4-3 strains were grown in peripheral blood mononuclear cells (PBMCs) or a lymphoid MT-4 cell line, respectively. Both viral stocks were titrated for use in TZM-bl cells. For infections, 1.5 × 104 TZM-bl cells were seeded in 96-well plates and infected with the BaL and NL4-3 viral strains at a multiplicity of infection of 0.01. The CXCR4 antagonist AMD3100 (Sigma-Aldrich), the reverse transcriptase inhibitor 3-azido-3-deoxythymidine (zidovudine [AZT]; Sigma-Aldrich), and the IN strand transfer inhibitor raltegravir (RAL) (Merck) were used as controls. For the β-galactosidase assays, the cells were lysed 72 h after infection and kept frozen until β-galactosidase determination.
To assess the effect of ZFNLEDGF on viral integration, the cells were infected with NL4-3, and after 8 h of infection, the cells were lysed and DNA was harvested to measure the amount of viral DNA. To determine viral integration, the cells were infected, and 8 h after infection, fresh growth medium supplemented with 10 μg/ml of the neutralizing anti-gp120 MAb IgG b12 (Polymun Scientific) was added and left in the incubator for 24 h, when DNA was harvested and stored at −20°C until the integration of viral DNA was determined.
β-Galactosidase detection assay.
β-Galactosidase activity in 30-μl cell extracts was quantified by a colorimetric assay, as described elsewhere (39, 41). The absorbances (405 to 620 nm) of the noninfected samples were subtracted from those of the rest of the samples, and the values were expressed as a percentage of β-galactosidase activity relative to the non-drug-treated control.
Viral and integrated DNA determination.
Viral DNA was extracted using a QIAamp DNA extraction kit (QIAamp DNA blood minikit; Qiagen). Total viral DNA was quantified by amplifying a Gag fragment, as described elsewhere (forward primer, 5′-CAAGCAGCCATGCAAATGTT-3′; reverse primer, 5′-TGCACTGGATGCAATCTATCC-3′; probe, 5′-FAM-AAAGAGACCATCAATGAGGAAGCTGCAGA-TAMRA-3′ [FAM, 6-carboxyfluorescein; TAMRA, 6-carboxytetramethylrhodamine]) (39, 41). Integration was detected by Alu-long terminal repeat (LTR) preamplification (Alu forward primers 5′-GCCTCCCAAAGTGCTGGGATTACAG-3′ and LTR reverse primer 5′-AGGGTTCCTTTGGTCCTTGT-3′, followed by Gag quantitative PCR (qPCR).
Statistical analyses.
The experimental data are presented as means ± standard deviation (SD). A paired Student's t test was used for comparing two groups, using the GraphPad Prism software (GraphPad Software, San Diego, CA, USA). A P value of 0.05 was considered statistically significant.
RESULTS
Design and efficiency of ZFNLEDGF.
ZFNs targeting PSIP1 were designed to specifically disrupt the C-terminal region where mutations in HIV patients have been identified, which is found after the sequence coding for the IBD of LEDGF/p75 (Fig. 1a) (30, 32). The endonuclease activity of ZFNLEDGF was determined in vitro in K562 cells and by assessing heteroduplex formation in the surrounding region of the targeted DNA sequence within the PSIP1 gene. Endonuclease activity in human K562 cells was estimated to be 6.6% when transfecting plasmids encoding the ZFN, increasing to 12% when using ZFN mRNA. The applicability of ZFNs is hampered by the potential risk of off-site cutting events leading to undesired side effects (42). Since off-target events have been reported for the previously described ZFNs targeting the CCR5 gene (43, 44), the possibility of off-site cutting events for ZFNLEDGF was assessed in silico. The in silico analysis predicted up to 19 potential off-target sites for ZFNLEDGF, all with ≥4 mismatches compared to the target sequence and corresponding to 9 coding regions distributed in 7 different chromosomes of the human genome (see Table S1 in the supplemental material).
To improve ZFN efficiency and speed up the selection of single-cell clones edited by ZFNLEDGF, the sequence encoding ZFNLEDGF was cloned in plasmids carrying a ZsGreen fluorescence reporter gene, whose expression was transient. After transfection, 15% of the cells were ZsGreen positive, a population that was enriched to 95% ZsGreen-positive cells by cell sorting. Monoclonal cell populations of ZFNLEDGF-treated cells were obtained by limiting dilution, resulting in 38 single clonal cell lines, of which 6 were discarded due to slow growth kinetics. Thus, further functional characterization was performed in 32 clonal cell lines.
The ZFNLEDGF-treated clonal cell lines were screened for their capacity to restrict single-round HIV-1 infection by measuring the fluorescence induced by a VSV-pseudotyped NL4-3 GFP-expressing virus. Although different degrees of HIV infection were identified, 25 of 32 cell lines blocked HIV replication >50% compared to the wild-type control cells, suggesting that the PSIP1 gene has been effectively targeted and genomically edited (Table 1). In only 7 cell lines were residual or no effects on HIV replication observed, representing an efficiency of 70% for the overall experimental procedure. Next, three of the cell lines were chosen for further validation, taking into account their different degrees of HIV infection impairment (Fig. 1c). The deletions/insertions induced by ZFNLEDGF were additionally confirmed using the heteroduplex formation assay in selected cell lines (Fig. 1d).
TABLE 1.
Inhibition of HIV replication of all monoclonal cell lines obtaineda
| Cell line | Mean GFP (%) | Rel % replication | Rel % inhibition |
|---|---|---|---|
| 1 | 0.6 | 14 | 86 |
| 2 | 0.1 | 2 | 98 |
| 3 | 0.2 | 5 | 95 |
| 4 | 3.9 | 93 | 7 |
| 5 | 0.5 | 12 | 88 |
| 6 | 2 | 48 | 52 |
| 7 | 1.7 | 40 | 60 |
| 8 | 0.6 | 14 | 86 |
| 9 | 1.1 | 26 | 74 |
| 10 | 1.8 | 43 | 57 |
| 11 | 5.2 | 124 | −24 |
| 12 | 5.1 | 121 | −21 |
| 13 | 0.7 | 17 | 83 |
| 14 | ND | ND | ND |
| 15 | 2.7 | 64 | 36 |
| 16 | 1.7 | 40 | 60 |
| 17 | 0.8 | 19 | 81 |
| 18 | 5.5 | 131 | −31 |
| 19 | 0.6 | 14 | 86 |
| 20 | 0.8 | 19 | 81 |
| 21 | 3.8 | 90 | 10 |
| 22 | ND | ND | ND |
| 23 | 0.8 | 19 | 81 |
| 24 | ND | ND | ND |
| 25 | 4.1 | 98 | 2 |
| 26 | ND | ND | ND |
| 27 | 0.9 | 21 | 79 |
| 28 | 1.1 | 26 | 74 |
| 29 | ND | ND | ND |
| 30 | 1 | 24 | 76 |
| 31 | 1.1 | 26 | 74 |
| 32 | 1.1 | 26 | 74 |
| 33 | 2 | 48 | 52 |
| 34 | ND | ND | ND |
| 35 | 0.6 | 14 | 86 |
| 36 | 1.4 | 33 | 67 |
| 37 | 0.7 | 17 | 83 |
| 38 | 0.6 | 14 | 86 |
| Control | 4.2 | 100 | 0 |
High efficacy (roughly 70% effective impairment of HIV replication) is observed as a consequence of ZNFLEDGF treatment. Values represent the mean of two independent determinations performed in duplicate. Rel % replication, % replication relative to control cell line; Rel % inhibition, % inhibition relative to control cell line; ND, not determined.
Molecular characterization of ZFNLEDGF-edited cell lines.
To characterize the genomic defects introduced by ZFNLEDGF, the PSIP1 genomic sequence surrounding the target site of the selected clonal cell lines was amplified, cloned, and sequenced. At least 16 different PSIP1 sequences were obtained from each selected cell line, and the sequences were aligned, thus confirming that ZFNLEDGF successfully recognized and cleaved the target sequence. Two cell lines harbored large modifications in both alleles (LEDGF−/− KO, cell lines 2 and 3), whereas another cell line, together with a large modification, presented a 3-bp deletion that led to a single amino acid deletion at the protein level, thus resembling a heterozygotic phenotype (LEDGF+/−, cell line 1) (Fig. 2a). Sequencing analysis and alignment confirmed that ZFNLEDGF introduced genomic defects located in exon 13, in most cases occurring after the IBD. The genomic defects detected were three deletions of 3 bp, 41 bp, and 17 bp, one insertion of 155 bp, and two very large deletions that could not be well characterized at the genomic level but were characterized at the mRNA level (Fig. 2a and data not shown). In silico prediction of the putative mRNA and protein sequences of the LEDGF−/− KO cell lines suggested the presence of a truncated protein with a conserved functional PWWP domain (both alleles of cell lines 1, 2, and 3) and IBD (cell line 1, allele A and cell line 2, allele B) but lacking the C-terminal part (Fig. 2b and c).
FIG 2.

Generation and phenotypic characterization of LEDGF/p75 knockout cells. (a) Sequence analysis of the insertions and deletions identified in the three cell lines selected after ZFNLEDGF treatment. At least 16 different sequences from each selected cell line were sequenced and aligned. The consensus sequences of the modifications identified in each of the two alleles of the selected cell lines are depicted as allele A and allele B of cell lines 1 to 3. WT, wild type. (b and c) Protein alignments of in silico-predicted sequences based on the sequencing data obtained from the two alleles of the ZFNLEDGF cell lines that introduce a premature stop codon (b) or of cell line 1 allele B that harbors an in-frame deletion of 3 exons (c). The IBD is highlighted by a red box. (d) Gene expression of PSIP1 mRNA corresponding to the 5′ and 3′ regions. Shown is an agarose gel with which the presence or absence of 5′ and 3′ fragments of PSIP1 mRNA was identified. In the case of cell line 1, the full-length and a truncated form of the 3′ mRNA fragment were identified. Ctrl, control. (e) Quantification of gene expression (mRNA) corresponding to the 3′ region of PSIP1 in the selected cell lines. Expression of LEDGF mRNA was completely inhibited in LEDGF−/− KO cell lines tested compared to untreated or mock-transfected (MOCKtr) cells. LEDGF+/− (cell line 1) showed a 50% decrease of the LEDGF mRNA compared to control cells. The mean ± SD values of three independent determinations are shown. (f) Assessment of protein levels of LEDGF/p75 determined by Western blot assay in the selected LEDGF/p75+/− and LEDGF/p75−/− KO cell lines compared to the control cells (upper panel, antibody recognizing the C terminus [C-ter] of LEDGF/p75 protein; lower panel, antibody recognizing the N terminus [N-ter] of the LEDGF/p75 and LEDGF/p52 proteins. Molecular weight (in thousands) markers are depicted. The red arrows indicate putative truncated proteins.
The effect of ZFNLEDGF on gene expression was assessed at both the mRNA and LEDGF/p75 protein levels using RT-qPCR and Western blot assay, respectively. In silico prediction of a truncated mRNA was confirmed by testing mRNA expression of the 5′ and 3′ fragments of the PSIP1 gene. Expression of the PSIP1 5′ mRNA fragment (from PSIP1 mRNA positions +204 to +951) was detected in cDNA samples of all selected cell lines (Fig. 2d). Conversely, expression of the PSIP1 3′ mRNA (from PSIP1 mRNA position +864 to position *66 after the stop codon), which included the ZFNLEDGF cutting site, was not detected in the LEDGF−/− KO cell lines with larger deletions (cell lines 2 and 3), but it was present in cell line 1, resembling a heterozygote compared to the wild-type or mock-transfected controls (Fig. 2d). Cell line 1 also presented a smaller mRNA fragment, indicative of a large deletion, the boundaries of which cannot be characterized at the genomic level. However, we identified an mRNA form lacking exons 12, 13, and 14 by cloning and sequencing the cDNA products, indicating that the induced deletion expanded over at least the 700-bp genomic region that includes all 3 exons (Fig. 2c).
To confirm mRNA truncation, a quantitative analysis of gene expression corresponding to the C-terminal end of the PSIP1 gene was performed, confirming a significant decrease in C-terminal mRNA expression in ZFNLEDGF-treated cells compared to the wild-type and mock-transfected cells (60% reduction in heterozygotic cells, residual expression in homozygotes; P < 0.01; Fig. 2d). Similarly, when using an antibody specifically recognizing the LEDGF/p75 C terminus, no protein expression was observed in cell lines 2 and 3, but it was partly detected in cell line 1, further confirming that ZFNLEDGF induces the formation of a truncated sequence that leads to a potent abrogation of complete LEDGF/p75 expression at the mRNA and protein levels (Fig. 2d, e, and upper panel of f).
Deletions in the IBD C terminus of LEDGF have been reported to affect protein stability and reduce the solubility of the protein (15). Thus, to determine whether the predicted truncated proteins were effectively expressed, a monoclonal antibody recognizing the N-terminal region of LEDGF was used. Expression of the full-length p75 and p52 isoforms was detected in the control cell lines (wild type and mock transfected; Fig. 2f, lower panel, first two lanes), as well as in edited cell line 1 (Fig. 2f, lane 4), whereas only the p52 isoform was detected in cell lines 2 and 3 (Fig. 2f, lanes 3 and 5), confirming the results obtained with the C-terminal antibody. However, a band of intermediate molecular weight (MW) appeared in cell lines 1 and 3 (Fig. 2f, red arrows), which was in accordance with our in silico predictions (predicted molecular mass: LEDGF/p75, 60.1 kDa; LEDGF/p52, 37 kDa; and truncated proteins, 47 to 48 kDa; based on ExPASy prediction tools [37]) and therefore suggested the expression of LEDGF-truncated proteins. No clear protein expression was detected in cell line 2, suggesting that the genomic modifications induced in this cell line compromised protein stability, solubility, and/or folding, as previously reported for other truncated LEDGF constructs (15).
HIV-1 infection.
Functional assays to establish the effect of ZFNLEDGF were performed with the LEDGF+/− (cell line 1) cells and one homozygote LEDGF−/− KO cell (cell line 2). No differences in the growth kinetics of the cell lines compared to that of the control parental cells were observed (data not shown). The LEDGF+/− and LEDGF−/− KO cells were challenged with either HIV-1 X4-tropic NL4-3 or R5-tropic BaL strains, and infection was monitored by β-galactosidase (β-Gal) production (Fig. 3). The LEDGF+/− cell line slightly inhibited HIV-1 infection compared to the wild-type and mock-transfected controls (24% and 45% inhibition for the HIV-1 X4 NL4-3 and R5 BaL strains, respectively) (Fig. 3a and b). Conversely, HIV-1 infection in the LEDGF−/− KO cells was strongly inhibited compared to that in the wild type and mock-transfected control, regardless of the viral strain used (up to 70%, P < 0.01) (Fig. 3a and b). Nevertheless, and in line with previous reports, infection was not completely abolished in the KO cell line, and residual infection persisted, accounting for roughly 30% of the infections for both the R5 BaL and X4 NL4-3 viral strains (Fig. 3a and b). To evaluate the effect of antiviral compounds in the ZFNLEDGF cell lines, the antiviral activities of the X4 entry inhibitor AMB3100, the reverse transcriptase inhibitor AZT, and the integrase inhibitor raltegravir were determined (Fig. 3c and d). No significant differences were observed in the efficacies of the different compounds for any of the drugs tested. Interestingly, raltegravir was able to inhibit the residual replication observed in the LEDGF −/− KO cells, consistent with previous reports (20, 21) and confirming the different mechanism of action for the two strategies.
FIG 3.

Infectivity of HIV in ZFNLEDGF-treated cells and susceptibility to antiretroviral compounds. (a and b) Relative (Rel.) infection of ZFNLEDGF-treated cells compared to wild-type (WT) and mock-transfected (MOCKtr) controls for the NL4-3 (a) and BaL (b) viral strains. The mean ± SD values of three independent experiments are shown. ***, P < 0.001; **, P < 0.01; *, P < 0.05. (c) Percentage of HIV X4 replication using strain NL4-3 in LEDGF+/− and LEDGF−/− KO mutants relative to the results for the wild-type and mock-treated controls treated or not with the reverse transcriptase inhibitor AZT, the CXCR4 antagonist AMD3100, and the IN strand transfer inhibitor RAL. The mean ± SD values of three independent experiments are shown. (d) Percentage of HIV R5 replication using strain BaL in LEDGF+/− and LEDGF−/− KO mutants relative to the results for the wild-type and mock-treated controls treated or not with the reverse transcriptase inhibitor AZT, the CXCR4 antagonist AMD3100, and the IN strand transfer inhibitor RAL. The mean ± SD values of three independent experiments are shown. WT, wild-type.
To further characterize the functionality of ZFNLEDGF, viral and integrated viral DNA were determined in the LEDGF−/− KO cell line. No differences were observed in the HIV-1 DNA between the mock-transfected cells and LEDGF−/− KO cells when challenged with HIV-1 X4 NL4-3 (Fig. 4a). Conversely, integrated viral DNA was inhibited up to 87% in the LEDGF−/−KO cells compared to the viral integration in the mock-transfected control (P < 0.001, Fig. 4b). Residual viral integration was also observed in the LEDGF−/−KO cells (approximately 12% of integration, P < 0.001) (Fig. 4b), which was fully inhibited by the IN inhibitor raltegravir (Fig. 4b).
FIG 4.
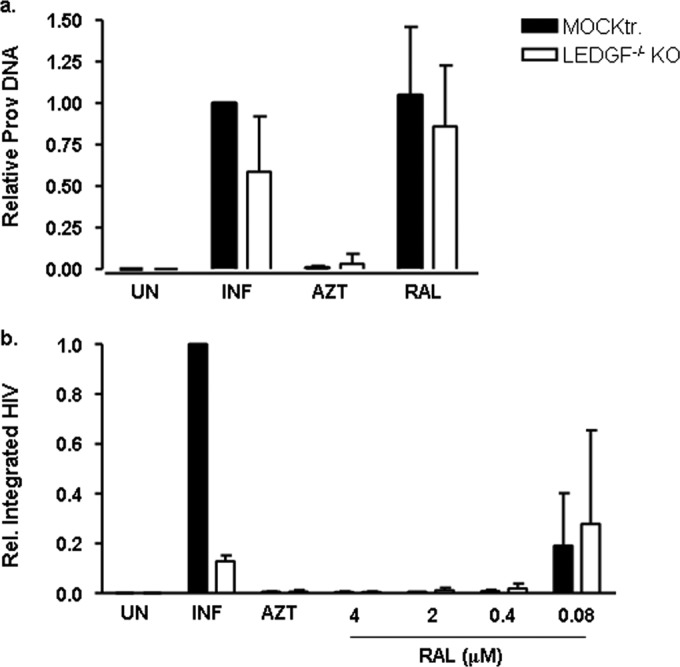
Integration of HIV NL4-3 is impaired in LEDGF−/− KO cells. (a) Viral DNA in LEDGF−/− KO cells (white bars) and mock-treated cells (black bars) with or without the reverse transcriptase inhibitor AZT (4 μM) and the IN inhibitor raltegravir (RAL) (2 μM). The values are expressed relative to those of the mock-transfected cells. The mean ± SD values of three independent experiments are shown. (b) Integrated viral DNA in LEDGF−/− KO cells (white bars) and mock-treated cells (black bars) with or without the RT inhibitor AZT (4 μM) and increasing concentrations of the IN inhibitor raltegravir. The values are expressed relative to those of the mock-transfected cells. The mean ± SD values of three independent experiments are shown. Prov, total proviral DNA; UN, uninfected; INF, infected.
Altogether, these results suggest that a complete LEDGF/p75 protein, including the C-terminal domain, is necessary to successfully tether the HIV preintegration complex to the active transcriptional units. Consistent with previous reports (19–21), LEDGF−/− KO cells were able to support inefficient but detectable viral integration and produce new viral particles, confirming the presence of alternative pathways for HIV-1 replication in the absence of LEDGF/p75.
DISCUSSION
Genome editing is an emerging strategy for studying virus-host interactions and to combat and cure HIV-1 infection (7, 8). An ideal therapy for HIV or other chronic viral infections that course with latent reservoirs is believed to involve the generation of a source of long-lived, self-renewing, and multilineage hematopoietic stem cells that repopulate the host with genetically modified cells that are refractory to infection (45, 46). Since the unique and exceptional case of an HIV-1 sterilizing cure of a patient due to bone marrow transplantation with a matched donor homozygote for the CCR5Δ32 mutation (47, 48), alternative strategies have aimed to reproduce the CCR5Δ32 phenotype using genome-editing tools. Indeed, ZFNs targeting the HIV coreceptor gene have been successfully developed to generate human CD4+ T cells and human embryonic cell precursors and induce pluripotent stem cells that were refractory to HIV-1 infection in different mouse models (9–13).
Here, we evaluated the feasibility and efficacy of generating a cell line with knockout of LEDGF/p75, a key factor for the integration of viral DNA into the host genome, using ZFNs targeting the C-terminal region of the LEDGF/p75 protein, outside the best-described functional PWWP and IBD. LEDGF/p75 has already been validated as a candidate for gene therapy in a model in which engraftment of lentivirus-transduced CD4+ T cells overexpressing a truncated form of LEDGF (LEDGF325–530) induced a 3-log reduction in plasma viral load of HIV-1-infected mice (23). Overexpression of the deficient mutant LEDGF325–530 in primary CD4+ T cells impeded but did not completely block viral replication, due to minimal wild-type LEDGF/p75 expression. Thus, the use of ZFNLEDGF might be advantageous in gene therapy settings, as it confers a permanent disruption of the target gene, avoiding the presence of residual levels of the wild-type LEDGF/p75 form that might be hijacked by the HIV IN to successfully replicate.
Here, the use of ZFNLEDGF generated TZM-bl cell lines carrying a truncated form of PSIP1 at the genomic region encoding the protein C-terminal region that preserves the N-terminal functional domains. Although the C-terminal region of LEDGF/p75 is poorly functionally characterized, several genetic variants in HIV-infected patients have been identified outside the known functional domains, variants that might be related to different susceptibilities to HIV infection and disease outcomes (28–30). Although the identified LEDGF/p75 variants support efficient HIV-1 infection ex vivo, the C-terminal amino acid positions involved are well conserved throughout the evolution, suggesting an important role for protein functionality (31, 32). The induction of large deletions in the sequence of the PSIP1 gene coding for the C-terminal region of the LEDGF/p75 protein resulted in an LEDGF−/− KO phenotype and provided a genetic barrier to HIV-1 infection in vitro.
A previous work reported a decrease in protein stability, expression levels, and solubility of recombinant LEDGF/p75 mutants lacking an IBD and/or C terminus (15). In accordance, a stable expression of truncated proteins was detected in only 2 of the 5 edited alleles harboring large modifications, pointing to a need for the LEDGF/p75 C-terminal domain for warranting protein stability and/or correct folding. Our data reinforce the relevance of the C-terminal-end region for LEDGF function and HIV infection outcome, even if the N-terminal functional domains and the IBD of the LEDGF/p75 remain intact. The inhibition of the HIV-1 infection in our ZFNLEDGF clones might seem less apparent than those of previously described LEDGF/p75 knockout models using mouse embryonic fibroblasts (MEFs) (19) and/or LEDGF KO in human somatic NALM-6 cells (pre-B acute lymphoblastic leukemia cell line) (20). However, LEDGF/p75 KO cells were generated by genetic modifications at exons 2 and 3 of the PSIP1 gene in the MEF model (19) and in human somatic NALM-6 cells by homologous recombination of exons 11 to 14 containing the IBD (20). Contrary to previous models, our ZFNLEDGF was not designed against the functional domains of the LEDGF protein, strengthening the relevance of the C-terminal end of the protein.
The modulation of LEDGF/p75 expression by different approaches involving RNA interference (49), short-hairpin RNA knockdown (50), and knockout models (19, 20) has provided strong evidence of how LEDGF/p75 interacts with the HIV IN and tethers HIV proviral DNA to the host chromatin; however, in accordance with our results, it also indicates that residual HIV infection occurred even in the absence of LEDGF/p75. Indeed, HRP2, which has similar structural features as LEDGF/p75, has been postulated as an alternative factor triggering HIV integration (21, 51). Nevertheless, double PSIP1/HRP2 KO mouse cells are still able to support HIV-1 integration (21); thus, alternative pathways that might allow virus to overcome LEDGF/p75 deficiency cannot be excluded. In that sense, and given that the TZM-bl cell line is a widely accepted model of HIV infection research, our recently developed TZM-bl LEDGF/p75−/− KO model is a helpful tool for elucidating the host factors involved in HIV integration.
In summary, we describe the generation of LEDGF/p75 knockout cells using a ZFN that successfully recognizes and disrupts the sequence of the PSIP1 gene coding for the C-terminal end of the LEDGF/p75 protein. The truncation of the C-terminal end of the LEDGF/p75 protein results in reduced stability, which leads to the generation of KO cells with an impaired HIV-1 replication independent of genetic modification concerning the N-terminal functional domains or the IBD of the LEDGF protein. Further studies must be carried out to elucidate the functional roles of genetic variants in the coding regions of the PSIP1 gene in vivo. Our results confirm previous data indicating that pathways other than LEDGF/p75 might allow for HIV integration. Finally, ZFNLEDGF provides a new cellular model for studying the host factors involved in the HIV-1 integration process.
Supplementary Material
ACKNOWLEDGMENTS
Cells, drugs, and viruses were received from the EU Programme EVA Centralised Facility for AIDS Reagents, NIBSC, United Kingdom, and the National Institutes of Health (AIDS Research and Reference Reagent Program).
This work was supported by the Spanish MINECO projects BFU2012-06958 and FIS PI/01083 and the Gala Contra la SIDA.
Footnotes
Published ahead of print 12 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02690-14.
REFERENCES
- 1.Ballana E, Esté JA. 2013. Insights from host genomics into HIV infection and disease: identification of host targets for drug development. Antiviral Res. 100:473–486. 10.1016/j.antiviral.2013.09.017 [DOI] [PubMed] [Google Scholar]
- 2.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. 2010. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 11:636–646. 10.1038/nrg2842 [DOI] [PubMed] [Google Scholar]
- 3.Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ. 2010. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 29:143–148. 10.1038/nbt.1755 [DOI] [PubMed] [Google Scholar]
- 4.Gaj T, Gersbach CA, Barbas CF., III 2013. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31:397–405. 10.1016/j.tibtech.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiedenheft B, Sternberg SH, Doudna JA. 2012. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482:331–338. 10.1038/nature10886 [DOI] [PubMed] [Google Scholar]
- 6.Gijsbers R, Ronen K, Vets S, Malani N, De Rijck J, McNeely M, Bushman FD, Debyser Z. 2010. LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Mol. Ther. 18:552–560. 10.1038/mt.2010.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchholz F, Hauber J. 2012. Engineered DNA modifying enzymes: components of a future strategy to cure HIV/AIDS. Antiviral Res. 97:211–217. 10.1016/j.antiviral.2012.12.017 [DOI] [PubMed] [Google Scholar]
- 8.Manjunath N, Yi G, Shankar P. 2013. Newer gene editing technologies toward HIV gene therapy. Viruses 5:2748–2766. 10.3390/v5112748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, Guschin DY, Rupniewski I, Waite AJ, Carpenito C, Carroll RG, Orange JS, Urnov FD, Rebar EJ, Ando D, Gregory PD, Riley JL, Holmes MC, June CH. 2008. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 26:808–816. 10.1038/nbt1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, Crooks GM, Kohn DB, Gregory PD, Holmes MC, Cannon PM. 2010. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 28:839–847. 10.1038/nbt.1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao Y, Nashun B, Zhou T, Qin L, Qin L, Zhao S, Xu J, Esteban MA, Chen X. 2011. Generation of CD34+ cells from CCR5-disrupted human embryonic and induced pluripotent stem cells. Hum. Gene Ther. 23:238–242. 10.1089/hum.2011.126 [DOI] [PubMed] [Google Scholar]
- 12.Badia R, Riveira-Muñoz E, Clotet B, Esté J, Ballana E. 20 March 2014. Gene editing using a zinc finger nuclease mimicking CCR5 Delta32 mutation shows HIV-1 resistance. J. Antimicrob. Chemother. 10.1093/jac/dku072 [DOI] [PubMed] [Google Scholar]
- 13.Wilen CB, Wang J, Tilton JC, Miller JC, Kim KA, Rebar EJ, Sherrill-Mix SA, Patro SC, Secreto AJ, Jordan AP, Lee G, Kahn J, Aye PP, Bunnell BA, Lackner AA, Hoxie JA, Danet-Desnoyers GA, Bushman FD, Riley JL, Gregory PD, June CH, Holmes MC, Doms RW. 2011. Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog. 7:e1002020. 10.1371/journal.ppat.1002020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engelman A, Cherepanov P. 2008. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 4:e1000046. 10.1371/journal.ppat.1000046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cherepanov P, Devroe E, Silver PA, Engelman A. 2004. Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J. Biol. Chem. 279:48883–48892. 10.1074/jbc.M406307200 [DOI] [PubMed] [Google Scholar]
- 16.Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, De Clercq E, Debyser Z. 2003. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 278:372–381. 10.1074/jbc.M209278200 [DOI] [PubMed] [Google Scholar]
- 17.Marshall HM, Ronen K, Berry C, Llano M, Sutherland H, Saenz D, Bickmore W, Poeschla E, Bushman FD. 2007. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One 2:e1340. 10.1371/journal.pone.0001340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Astiazaran P, Bueno MT, Morales E, Kugelman JR, Garcia-Rivera JA, Llano M. 2011. HIV-1 integrase modulates the interaction of the HIV-1 cellular cofactor LEDGF/p75 with chromatin. Retrovirology 8:27. 10.1186/1742-4690-8-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shun MC, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, Engelman A. 2007. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 21:1767–1778. 10.1101/gad.1565107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schrijvers R, De Rijck J, Demeulemeester J, Adachi N, Vets S, Ronen K, Christ F, Bushman FD, Debyser Z, Gijsbers R. 2012. LEDGF/p75-independent HIV-1 replication demonstrates a role for HRP-2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog. 8:e1002558. 10.1371/journal.ppat.1002558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H, Jurado KA, Wu X, Shun MC, Li X, Ferris AL, Smith SJ, Patel PA, Fuchs JR, Cherepanov P, Kvaratskhelia M, Hughes SH, Engelman A. 2012. HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic Acids Res. 40:11518–11530. 10.1093/nar/gks913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christ F, Debyser Z. 2013. The LEDGF/p75 integrase interaction, a novel target for anti-HIV therapy. Virology 435:102–109. 10.1016/j.virol.2012.09.033 [DOI] [PubMed] [Google Scholar]
- 23.Vets S, Kimpel J, Volk A, De Rijck J, Schrijvers R, Verbinnen B, Maes W, Von Laer D, Debyser Z, Gijsbers R. 2012. Lens epithelium-derived growth factor/p75 qualifies as a target for HIV gene therapy in the NSG mouse model. Mol. Ther. 20:908–917. 10.1038/mt.2012.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greene WC, Debyser Z, Ikeda Y, Freed EO, Stephens E, Yonemoto W, Buckheit RW, Esté JA, Cihlar T. 2008. Novel targets for HIV therapy. Antiviral Res. 80:251–265. 10.1016/j.antiviral.2008.08.003 [DOI] [PubMed] [Google Scholar]
- 25.Desimmie BA, Schrijvers R, Demeulemeester J, Borrenberghs D, Weydert C, Thys W, Vets S, Van Remoortel B, Hofkens J, De Rijck J, Hendrix J, Bannert N, Gijsbers R, Christ F, Debyser Z. 2013. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 10:57. 10.1186/1742-4690-10-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel B, Strelkov SV, De Maeyer M, Chaltin P, Debyser Z. 2010. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 6:442–448. 10.1038/nchembio.370 [DOI] [PubMed] [Google Scholar]
- 27.Jurado KA, Engelman A. 2013. Multimodal mechanism of action of allosteric HIV-1 integrase inhibitors. Expert Rev. Mol. Med. 15:e14. 10.1017/erm.2013.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madlala P, Gijsbers R, Christ F, Hombrouck A, Werner L, Mlisana K, An P, Abdool Karim SS, Winkler CA, Debyser Z, Ndung'u T. 2011. Association of polymorphisms in the LEDGF/p75 gene (PSIP1) with susceptibility to HIV-1 infection and disease progression. AIDS 25:1711–1719. 10.1097/QAD.0b013e328349c693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Messiaen P, De Spiegelaere W, Alcami J, Vervisch K, Van Acker P, Verhasselt B, Meuwissen P, Calonge E, Gonzalez N, Gutierrez-Rodero F, Rodriguez-Martín C, Sermijn E, Poppe B, Vogelaers D, Verhofstede C, Vanderkerckhove L. 2012. Characterization of LEDGF/p75 genetic variants and association with HIV-1 disease progression. PLoS One 7:e50204. 10.1371/journal.pone.0050204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ballana E, Gonzalo E, Grau E, Iribarren JA, Clotet B, Esté JA. 2012. Rare LEDGF/p75 genetic variants in white long-term nonprogressor HIV+ individuals. AIDS 26:527–528. 10.1097/QAD.0b013e32834fa194 [DOI] [PubMed] [Google Scholar]
- 31.Schrijvers R, Demeulemeester J, De Rijck J, Christ F, Gérard M, Debyser Z, Gijsbers R. 2013. Characterization of rare lens epithelium-derived growth factor/p75 genetic variants identified in HIV-1 long-term nonprogressors. AIDS 27:539–543. 10.1097/QAD.0b013e32835d0d86 [DOI] [PubMed] [Google Scholar]
- 32.Koh Y, Ballana E, Esté J, Engelman A. 2013. Polymorphic LEDGF/p75 variants support efficient HIV-1 infection ex vivo. AIDS 27:665–667. 10.1097/QAD.0b013e32835af34c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mussolino C, Cathomen T. 2011. On target? Tracing zinc-finger-nuclease specificity. Nat. Methods 8:725–726. 10.1038/nmeth.1680 [DOI] [PubMed] [Google Scholar]
- 34.Hansen K, Coussens MJ, Sago J, Subramanian S, Gjoka M, Briner D. 2012. Genome editing with CompoZr custom zinc finger nucleases (ZFNs). J. Vis. Exp. 2012(64):e3304. 10.3791/3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ballana E, Pauls E, Clotet B, Perron-Sierra F, Tucker GC, Esté JA. 2010. β5 integrin is the major contributor to the αVintegrin-mediated blockade of HIV-1 replication. J. Immunol. 186:464–470. 10.4049/jimmunol.1002693 [DOI] [PubMed] [Google Scholar]
- 36.Ballana E, Senserrich J, Pauls E, Faner R, Mercader JM, Uyttebroeck F, Palou E, Mena MP, Grau E, Clotet B, Ruiz L, Telenti A, Ciuffi A, Esté JA. 2010. ZNRD1 (zinc ribbon domain-containing 1) is a host cellular factor that influences HIV-1 replication and disease progression. Clin. Infect. Dis. 50:1022–1032. 10.1086/651114 [DOI] [PubMed] [Google Scholar]
- 37.Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, Duvaud S, Flegel V, Fortier A, Gasteiger E, Grosdidier A, Hernandez C, Ioannidis V, Kuznetsov D, Liechti R, Moretti S, Mostaguir K, Redaschi N, Rossier G, Xenarios I, Stockinger H. 2012. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 40:W597–W603. 10.1093/nar/gks400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ballana E, Riveira-Munoz E, Pou C, Bach V, Parera M, Noguera M, Santos JR, Badia R, Casadellà M, Clotet B, Paredes R, Martinez MA, Brander C, Esté JA. 2012. HLA class I protective alleles in an HIV-1-infected subject homozygous for CCR5-Δ32/Δ32. Immunobiology 218:543–547. 10.1016/j.imbio.2012.06.012 [DOI] [PubMed] [Google Scholar]
- 39.Pauls E, Jimenez E, Ruiz A, Permanyer M, Ballana E, Costa H, Nascimiento R, Parkhouse RM, Peña R, Riveiro-Muñoz E, Martinez MA, Clotet B, Esté JA, Bofill M. 2013. Restriction of HIV-1 replication in primary macrophages by IL-12 and IL-18 through the upregulation of SAMHD1. J. Immunol. 190:4736–4741. 10.4049/jimmunol.1203226 [DOI] [PubMed] [Google Scholar]
- 40.Clouser CL, Patterson SE, Mansky LM. 2010. Exploiting drug repositioning for discovery of a novel HIV combination therapy. J. Virol. 84:9301–9309. 10.1128/JVI.01006-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ballana E, Pauls E, Senserrich J, Clotet B, Perron-Sierra F, Tucker GC, Esté JA. 2009. Cell adhesion through alphaV-containing integrins is required for efficient HIV-1 infection in macrophages. Blood 113:1278–1286. 10.1182/blood-2008-06-161869 [DOI] [PubMed] [Google Scholar]
- 42.Palpant NJ, Dudzinski D. 2013. Zinc finger nucleases: looking toward translation. Gene Ther. 20:121–127. 10.1038/gt.2012.2 [DOI] [PubMed] [Google Scholar]
- 43.Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, Nowrouzi A, Bartholomae CC, Wang J, Friedman G, Holmes MC, Gregory PD, Glimm H, Schmidt M, Naldini L, von Kalle C. 2011. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 29:816–823. 10.1038/nbt.1948 [DOI] [PubMed] [Google Scholar]
- 44.Pattanayak V, Ramirez CL, Joung JK, Liu DR. 2011. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat. Methods 8:765–770. 10.1038/nmeth.1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kiem HP, Jerome KR, Deeks SG, McCune JM. 2012. Hematopoietic-stem-cell-based gene therapy for HIV disease. Cell Stem Cell 10:137–147. 10.1016/j.stem.2011.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoxie JA, June CH. 2012. Novel cell and gene therapies for HIV. Cold Spring Harb. Perspect. Med. 2:a007179. 10.1101/cshperspect.a007179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K, Schneider T, Hofmann J, Kücherer C, Blau O, Blau IW, Hofmann WK, Thiel E. 2009. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 360:692–698. 10.1056/NEJMoa0802905 [DOI] [PubMed] [Google Scholar]
- 48.Allers K, Hütter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, Schneider T. 2011. Evidence for the cure of HIV infection by CCR5Δ32/Δ32 stem cell transplantation. Blood 117:2791–2799. 10.1182/blood-2010-09-309591 [DOI] [PubMed] [Google Scholar]
- 49.Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. 2006. An essential role for LEDGF/p75 in HIV integration. Science 314:461–464. 10.1126/science.1132319 [DOI] [PubMed] [Google Scholar]
- 50.Vandekerckhove L, Christ F, Van Maele B, De Rijck J, Gijsbers R, Van den Haute C, Witvrouw M, Debyser Z. 2006. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J. Virol. 80:1886–1896. 10.1128/JVI.80.4.1886-1896.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schrijvers R, Vets S, De Rijck J, Malani N, Bushman FD, Debyser Z, Gijsbers R. 2012. HRP-2 determines HIV-1 integration site selection in LEDGF/p75 depleted cells. Retrovirology 9:84. 10.1186/1742-4690-9-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.