

Abstract
Bacterial growth in biofilms is the major cause of recalcitrant biofouling in industrial processes and of persistent infections in clinical settings. The use of bacteriophage treatment to lyse bacteria in biofilms has attracted growing interest. In particular, many natural or engineered phages produce depolymerases to degrade polysaccharides in the biofilm matrix and allow access to host bacteria. However, the phage-produced depolymerases are highly specific for only the host-derived polysaccharides and may have limited effects on natural multispecies biofilms. In this study, an engineered T7 bacteriophage was constructed to encode a lactonase enzyme with broad-range activity for quenching of quorum sensing, a form of bacterial cell-cell communication via small chemical molecules (acyl homoserine lactones [AHLs]) that is necessary for biofilm formation. Our results demonstrated that the engineered T7 phage expressed the AiiA lactonase to effectively degrade AHLs from many bacteria. Addition of the engineered T7 phage to mixed-species biofilms containing Pseudomonas aeruginosa and Escherichia coli resulted in inhibition of biofilm formation. Such quorum-quenching phages that can lyse host bacteria and express quorum-quenching enzymes to affect diverse bacteria in biofilm communities may become novel antifouling and antibiofilm agents in industrial and clinical settings.
INTRODUCTION
Bacteria in natural and industrial environments mostly grow as biofilms attached to surfaces or associated with interfaces, where bacterial cells are encased in a self-produced extracellular matrix known as extracellular polymeric substances (EPS), composed of polysaccharides, DNA, proteins, and lipids (1). EPS in biofilms provide a local environment that protects the bacterial cells against harm from antibiotics and other antimicrobial treatments. This enables bacteria within biofilms to be the cause of persistent infections of live tissues and contaminations of medical device surfaces (2). This also enables biofilm growth to be the main cause of recalcitrant biofouling in industrial settings, which affects the normal functioning of pipes, food-processing equipment, membrane filters, and condenser tubes of electric power stations (3).
Interactions of bacteriophages and bacteria in biofilms have attracted growing interest in recent years, as part of the “renaissance” of phage research in the Western world during the last decade (4, 5). Phages are capable of lysing host bacteria in experimental single- and mixed-species bacterial biofilms (6, 7). Phages have been explored as antibiofilm agents in diverse industrial and clinical settings, including phage therapy (4), biofilm-infected medical devices (8–11), and filtration membranes (12). In many biofilm systems, however, EPS still pose a challenge, limiting the bactericidal effect of phages (13, 14). One strategy adopted by phages during the phage-bacterium evolutionary arms race (15) is the production of polysaccharide depolymerases to degrade EPS and allow phages to access encased bacterial cells. Such phages expressing free or phage-bound polysaccharide depolymerases show characteristic semitransparent halos around phage plaques on the lawns of host bacteria on solid culture media (5, 16). To mimic the natural process, T7 phage, which does not contain polysaccharide depolymerase genes, was genetically engineered to incorporate the polysaccharide depolymerase gene dspB, originating from Actinobacillus actinomycetemcomitans, under the control of the T7 Φ10 promoter. Such an engineered phage reduced the bacterial count in a single-species E. coli biofilm 100-fold more than the wild-type T7 phage did (17). However, the enzyme DspB and other polysaccharide depolymerases, in general, have narrow substrate specificities. Each enzyme at most degrades a few related polysaccharides, which constitute a small proportion of the pool of polysaccharides in natural multispecies biofilms. Thus, the antibiofilm efficacy of these phages is limited (1, 18).
Despite this limitation, natural and engineered phages producing polysaccharide depolymerases have provided a paradigm of biofilm control that inspired us to design an engineered phage producing an enzyme with broad-range antibiofilm effects. Specifically, a gene encoding an enzyme that interferes with quorum sensing among diverse bacteria was incorporated into the T7 genome. Quorum sensing is a general cell-cell communication mechanism in the bacterial kingdom, occurring via small chemical molecules, termed autoinducers, as chemical “languages” to coordinate bacterial population behaviors. One such population behavior is biofilm formation (19, 20). In the model Gram-negative bacterium Pseudomonas aeruginosa, biofilm formation is mediated by the autoinducers acyl homoserine lactones (AHLs or acyl HSLs). Interference with quorum sensing, i.e., quorum quenching, via enzymes that degrade AHLs has been shown to modulate biofilm formation. For example, AHL lactonases encoded by the aiiA genes of Bacillus spp. have broad-range specificity for cleaving the lactone rings of diverse AHLs (21), leading to inhibition of formation of P. aeruginosa biofilms (22). The enzyme acylase from porcine kidneys, which cleaves acyl moieties of AHLs, was reported to decrease EPS production and to mitigate biofilm formation and membrane biofouling in a membrane bioreactor inoculated with bacterial communities from activated sludge of wastewater treatment plants (23, 24). In this paper, we report that an engineered T7 phage incorporating the AHL lactonase aiiA gene from Bacillus anthracis degraded AHLs from diverse bacteria and caused inhibition of biofilm formation in a mixed-species biofilm composed of P. aeruginosa and Escherichia coli. This enables the engineered quorum-quenching T7 phage to be a promising antibiofilm agent.
MATERIALS AND METHODS
Bacterial strains.
Escherichia coli strains BL21 and TG1(lacI::kan) were kindly provided by Timothy K. Lu of the Massachusetts Institute of Technology. E. coli BL21(DE3) and P. aeruginosa PAO1 were provided by Tao Wei and Robert Renthal, respectively, of the University of Texas at San Antonio. E. coli MG1655 was provided by Thomas Wood of Pennsylvania State University. Agrobacterium tumefaciens strain A136(pCF218)(pCF372) and strain KYC6 were provided by R. J. McLean of Texas State University. All bacterial strains were cultured at 37°C in Miller Luria-Bertani (LB) broth (Thermo Fisher Scientific, Waltham, MA) or on agar medium with no antibiotics added, except for Agrobacterium tumefaciens A136(pCF218)(pCF372), which was grown with spectinomycin (50 μg/ml) and tetracycline (4.5 μg/ml).
Construction of engineered phage.
The engineered T7 phage was generated by inserting the aiiA gene into the T7select415-1 phage display vector (EMD Millipore, San Diego, CA), which contains a modified genome of the natural T7 phage, with deletion of a small portion of the nonessential early genes and incorporation of the multiple-cloning site (MCS) at the 3′ end of the 10B gene. The cloning process was composed of two stages. (i) The aiiA gene was first cloned into the NdeI and BamHI sites of the pET-9a vector (EMD Millipore), downstream of the in-vector T7 Φ10 promoter. (ii) The T7 Φ10 promoter-aiiA gene in the pET-9a vector was subcloned into the EcoRI and NotI sites in the MCS of the phage vector. During the subcloning process, stop codons in three frames were inserted upstream of the Φ10 promoter.
The aiiA gene from Bacillus anthracis strain Ames in plasmid pBA01 (25), kindly provided by Ricky L. Ulrich of the U.S. Army Medical Research Institute of Infectious Diseases, was amplified by PCR with the 1st primer set (forward primer, 5′-atataatcCATATGatgacagtaaagaagctttattt-3′ [the NdeI site is shown in uppercase italics, and the aiiA coding sequence is underlined]; and reverse primer, 5′-atatacGGATCCctatatatattccgggaacactt-3′ [the BamHI site is shown in uppercase italics, and the coding sequence is underlined]), which appended NdeI and BamHI recognition sites to the 5′ and 3′ ends (according to the coding strand), respectively, of the PCR product. The PCR product was digested with NdeI and BamHI and then ligated with the linearized product of the pET-9a plasmid precut with the NdeI and BamHI restriction enzymes. The ligated products were transformed into E. coli BL21(DE3) and screened using restriction enzyme digestion and DNA sequencing. The desired pET-9a-aiiA product contained the T7 Φ10 promoter-aiiA cassette, which, after being transformed into E. coli BL21(DE3) cells, can express the AiiA enzyme in the presence of isopropyl-β-d-thiogalactopyranoside (IPTG; 1 mM).
The pET-9a plasmid containing the T7 Φ10 promoter-aiiA cassette was used as the template for PCR amplification with the 2nd primer set (forward primer, 5′-gTAAcTAAcgaaattaatacgactcactatagg-3′ [stop codons are shown in uppercase, and the Φ10 promoter sequence is underlined], and reverse primer, 5′-atataaGCGGCCGCcaagcttctatatatattccgggaacactt-3′ [the NotI site is shown in uppercase italics, and the aiiA coding sequence is underlined]). The PCR product was then used as a template for amplification with the 3rd primer set (forward primer, 5′-tactcGAATTCtTAAgTAAcTAAcgaaattaatacgactc-3′ [the EcoRI site is shown in uppercase italics, stop codons are shown in uppercase, and the T7 Φ10 promoter sequence is underlined]; and reverse primer, 5′-atataaGCGGCCGCcaagcttctatatatattccgggaacactt-3′ [the NotI site is shown in uppercase italics, and the aiiA coding sequence is underlined]). The two successive PCR amplification reactions generated a final PCR product containing the following (5′ → 3′ end): EcoRI site-stop codons-Φ10 promoter-aiiA-NotI site. Though it is theoretically possible to obtain the final PCR product by performing one amplification reaction directly with the third set of primers, it is more advantageous to perform two successive amplification reactions, with the amplified product of the first reaction used as the template for the second reaction. The matches of the primers to their respective templates in each of the two reactions were higher than those for the one amplification reaction, thereby increasing the odds of success. The product was cut with EcoRI and NotI, and the largest fragment, termed fragment 1, was purified. The MCS of the T7select415-1 DNA was cut with EcoRI and NotI to produce 21.5-kb (termed fragment 2) and 15.8-kb (termed fragment 3) fragments, respectively. Fragments 2 and 3 were purified after agarose gel electrophoresis. A reaction for ligating fragments 1, 2, and 3 was set up, the product of which was packaged into T7 phage particles by using T7select packaging extract (EMD Millipore) according to the manufacturer's instructions. The desired T7 phage was screened by PCR as described previously (26). The phage genomic DNA was verified using restriction digestion with NdeI and by DNA sequencing of the aiiA gene and the flanking sequences.
Phage lysate preparation and phage count determination.
The methods for preparation of the phage lysates and determination of the phage counts in the lysates, expressed as numbers of PFU, were adapted from previously described methods (27). Briefly, upon inoculation of phage (100 μl phage lysate; about 1 × 1010 PFU/ml) into 100 ml E. coli BL21 host culture (1:1 mixture of stationary-phase culture and LB medium), the culture was rocked at 250 rpm for 3 to 4 h at 37°C to obtain cleared lysates. The lysates were centrifuged at 3,200 × g for 20 min, and the pellets, which contained unlysed bacterial cell debris, were discarded. No treatment with chloroform was performed to avoid potential denaturation of the AiiA enzyme in the T7aiiA lysates. The phage counts were determined by overlaying a mixture of 5 ml 6% melted soft LB agar (50°C), 250 μl T7 host E. coli BL21 overnight culture, and 100 μl phage lysate (serially diluted with LB liquid medium) on top of 100-mm LB agar plates. Typical phage counts of the freshly prepared phage lysates described above were on the order of 1010 PFU/ml for both T7wt and T7aiiA.
Agrobacterium tumefaciens-based AHL bioassay.
Both qualitative and quantitative AHL bioassays were performed to evaluate the AHL lactonase activity in the phage lysates. A. tumefaciens strain A136(pCF218)(pCF372) is a genetically modified reporter strain devoid of AHL synthesis genes and transformed with two plasmids: pCF218 and pCF372. This enables strain A136 to respond to a broad range of AHLs, with acyl side chains of different lengths and modifications (C5- to C10-HSLs and 3-oxo-C4- to 3-oxo-C18-HSLs) (28), by expressing the traI-lacZ fusion gene under the control of the tra promoter (29). A. tumefaciens strain KYC6, which produces 3-oxo-C8-HSL, and P. aeruginosa PAO1, which produces C4-HSL and 3-oxo-C12-HSL, were used as AHL producers. The presence of the AHL degradation activity in the T7aiiA lysates was evaluated in an AHL bioassay as described previously (30). Briefly, A. tumefaciens A136 and an AHL producer were streaked as two parallel lines about 1 cm apart on 100-mm LB agar plates with X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside). In such a setting, the AHL producer colony releases AHL, which diffuses across the plate to the A136 colony, induces lacZ expression in the A136 colony, and causes the A136 colony to turn blue (30). In parallel settings, T7wt or T7aiiA lysates were streaked between A136 and an AHL producer. The lysates were freshly prepared by phage-induced lysis of E. coli BL21 and titrated to determine the phage counts as described above. The phage counts in the lysates were adjusted to the same PFU by dilution with LB. One hundred microliters of each lysate was then carefully streaked between the two parallel lines of the A136 AHL reporter and the AHL producer and air dried. The plates were cultured at 37°C and observed after 24 h. To analyze the AiiA expression of plasmid pET-9a-aiiA transformed into E. coli BL21(DE3), with the resultant strain termed the E. coli-aiiA strain, the same procedure was performed, except that the E. coli-aiiA cells or the control E. coli cells were streaked as a line between the two parallel lines of A136 and KYC6.
Quantitative assay of the AHL activity in the T7aiiA lysates was performed using the Miller β-galactosidase assay (31). A culture of A. tumefaciens A136 and a culture of an AHL producer (either A. tumefaciens KYC6 or P. aeruginosa PAO1) were mixed at the exponential growth stage, at a 1:1 ratio. A 0.5-ml aliquot of the above mixture was added either to a mixture of 0.4 ml of E. coli BL21 culture (optical density at 600 nm [OD600] of 0.05 or 0.9) and 0.1 ml of 105-PFU/ml phage lysate (T7wt or T7aiiA) or to 0.5 ml LB medium (as the negative control). After 4 h of incubation at room temperature to induce LacZ expression in A136, the β-galactosidase activity assay was initiated by drawing 50-μl samples from the reaction mixtures. Each sample was mixed with 50 μl Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 2.8 μl/ml β-mercaptoethanol) in microcentrifuge tubes. Five microliters of 0.1% SDS and 10 μl chloroform were added to permeabilize the cells. After incubation at room temperature for 2 min, 20 μl of 4-mg/ml β-galactosidase substrate (o-nitrophenyl-β-d-galactoside) was added and incubated for 30 min. The reaction was stopped by adding 50 μl 1 M Na2CO3, and the amount of yellow coloration in the supernatant was determined by measuring the OD420. In addition, the OD550 and OD650 were measured to eliminate interference of bacterial cells with the OD420 value, and the activity was calculated as described previously (31).
Chromobacterium violaceum-based AHL bioassay.
An AHL bioassay based on violacein (a purple pigment) production by C. violaceum strain 12472 was performed as described previously (30). C. violaceum 12472 produces C4- and C6-HSLs to self-elicit violacein expression (30). P. aeruginosa PAO1 was used as the quorum-quenching positive control, as the AHLs released by P. aeruginosa can act as inhibitors to interfere with the effects of the structurally similar C4- and C6-HSLs released by C. violaceum (30). On day 1, P. aeruginosa was streaked at the center of LB agar plates. On day 2, when each colony of P. aeruginosa was grown (and AHLs were released), drops of 50°C melted LB soft agar (0.6% agar) were carefully added to cover the colonies and protect them from being flushed in the next step (soft agar overlay). Fifty microliters of a stationary-phase culture of C. violaceum 12472 was mixed with 5 ml of melted LB soft agar (0.3% agar), and the mixture was overlaid on the LB agar plates with P. aeruginosa colonies. Also on day 2, in parallel settings, 100-μl aliquots of freshly prepared T7wt or T7aiiA lysates were dripped on the center of unused LB agar plates and dried, and the areas containing the lysates were protected by adding drops of 50°C melted LB soft agar (0.6% agar) before the mixture of C. violaceum and soft agar was overlaid on the plates. The plates were cultured at 30°C and observed on day 3. Existence of a colorless area around the P. aeruginosa colony or the area containing phage lysates indicates the quorum-quenching activity that prevents violacein production in C. violaceum 12472.
Biofilm formation assay.
Overnight cultures of P. aeruginosa and E. coli TG1 were diluted 1:100 in fresh LB medium, unless otherwise indicated. The diluted cultures of P. aeruginosa and E. coli TG1 (25 μl each) were mixed, and then E. coli BL21 (50 μl at an OD600 of 0.05, unless otherwise noted), with or without phage (T7wt or T7aiiA; final concentration, 104 PFU/ml), was added. The mixture was plated in the wells of round-bottomed 96-well polyvinyl chloride (PVC) microtiter plates and incubated without shaking at 37°C to allow biofilm formation. In cases where single- or dual-species biofilms were studied, only the respective species were added, while the rest of the above-described system was replaced with LB medium to make up the 100-μl final volume in each well. Staining of biofilms with crystal violet was performed as described previously (32). Briefly, the medium inside the wells of a 96-well microtiter plate was dumped. The plate was washed three times with Milli-Q water and air dried for 20 min. Subsequently, 125 μl 0.1% crystal violet solution was added to the wells to stain the biofilms for 10 min. The stained wells were washed three times with Milli-Q water and dried for 20 min before 125 μl 30% acetic acid was added to dissolve the stained crystal violet. After incubation for 10 min, 100 μl of the solubilized crystal violet in each well was transferred to a flat-bottomed 96-well microtiter plate, and the OD600 was measured using an Epoch microplate spectrometer (BioTek Inc., Winooski, VT). All staining steps were performed at room temperature.
Determination of phage counts and bacterial cell counts in the biofilm system.
The phage counts in the liquid medium and in the biofilms of the wells of the microtiter plates were measured as described previously (33). Samples (50 μl) of the liquid medium in the wells at 8 h of biofilm growth were collected for determination of phage counts. The leftover medium was discarded. The wells were rinsed three times gently with 200 μl sterile phosphate-buffered saline (PBS), each followed by dumping out the PBS. After adding 150 μl PBS to each well, the plates were placed in a Bransonic ultrasonic cleaner sonicator (model 3510; Branson Ultrasonic Corporation, Danbury, CT) at room temperature for 10 min to dislodge the bacteria and the phage residing in the biofilms into the PBS. For the wells containing PBS with the suspended biofilm, samples (50 μl) were collected and the phage counts determined immediately. The phage counts in the samples of liquid medium and the samples from biofilms, expressed as numbers of PFU, were determined as described above for the phage counts in the phage lysates.
The bacterial cell counts in the biofilms were measured as described previously (33). The bacterial cell counts in the liquid medium of the wells were not determined, as the microtiter plates were incubated without shaking during biofilm growth and most bacteria were not expected to be detected in the liquid medium. From the wells containing the biofilm suspended in PBS as described above, samples (50 μl) were collected and immediately analyzed for bacterial cell counts. Briefly, after serial dilutions, the bacterial cells in the samples were plated on LB agar plates and incubated at 37°C. Bacterial cell counts, expressed as numbers of CFU, were determined after 24 h by counting the bacterial colonies.
Statistical test.
Statistical significance was determined by performing one-way analysis of variance (ANOVA) with the Tukey-Kramer multiple-comparison test, using the statistical software R, with P values of <0.05 indicating statistical significance.
RESULTS AND DISCUSSION
Construction of quorum-quenching bacteriophage.
To explore the antibiofilm potential of a phage producing a quorum-quenching enzyme, the AHL lactonase gene aiiA from Bacillus anthracis strain Ames (25) was inserted into the T7select415-1 phage vector. A DNA fragment containing the stop codons, the T7 Φ10 promoter, and the coding sequence of aiiA, listed in order from the 5′ to the 3′ end (the coding strand), was inserted into the MCS at the 3′ end of the T7 10B capsid gene (Fig. 1). The stop codons were used to terminate translation of the upstream gene (10B) near the 3′ end, and the T7 Φ10 promoter was used to drive high-level expression of the downstream gene (aiiA). Such a design enables the expression of the free (as opposed to phage-bound) AiiA enzyme in the extracellular milieu upon phage-induced bacterial cell lysis. The T7 DNA containing the aiiA gene was packaged to generate the T7aiiA phage. The T7select415-1 vector was used to produce the control, wild-type T7wt phage. The morphologies of the phage plaques formed on a lawn of E. coli BL21 were indistinguishable between the T7wt and T7aiiA phages. The growth rates of the T7wt and T7aiiA phages were indistinguishable, as the same number of phages T7wt and T7aiiiA inoculated into the E. coli BL21 culture resulted in similar PFU counts in the phage lysates.
FIG 1.
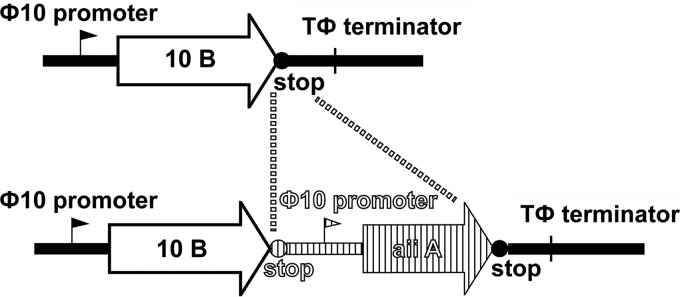
Design of the engineered phage. The engineered phage T7aiiA was generated by inserting the AHL lactonase gene aiiA, the upstream stop codons, and the promoter into the MCS at the 3′ end of the 10B gene of wild-type T7wt DNA.
AiiA activity in phage lysates.
To determine whether the aiiA gene was expressed upon phage lysis of the E. coli BL21 host, the enzyme activity of AiiA in the phage lysates was measured in an AHL bioassay, using Agrobacterium tumefaciens strain A136 as the AHL reporter and strain KYC6 as an AHL producer (30). A136 and KYC6 were streaked as two lines on LB agar plates containing X-Gal; after 24 h, A136 colonies turned blue as a result of induction of LacZ expression by the AHL from KYC6 (Fig. 2A). The presence of the fresh T7wt lysate streaked between the KYC6 and A136 lines did not change the blue color (Fig. 2B), while the presence of the T7aiiA lysate dramatically decreased the blue color intensity (Fig. 2C), suggesting that the T7aiiA lysate contained AiiA enzyme activity for degrading 3-oxo-C8-HSL from KYC6. This result is consistent with the report that AiiA from B. anthracis can effectively degrade AHLs with lengths of >7 carbons (25). Also, inhibition of the blue coloration of A136 colonies by the T7aiiA lysate persisted for at least 48 h after the first observation of inhibition. As AHLs were continuously being released from the KYC6 colony at high cell density (29), such observations implied that AiiA activity in the fresh lysates was stable. To quantify the AHL degradation activity in the phage lysates, a β-galactosidase assay was performed (31). The T7wt phage lysate did not significantly inhibit AHL-induced β-galactosidase activity, while the T7aiiA lysate obtained by lysing strain BL21 at an OD600 of 0.05 exhibited a significant (18%) inhibition of the β-galactosidase activity, and that obtained by lysing BL21 at an OD600 of 0.9 exhibited a stronger inhibition (65.7%) (Fig. 2D), consistent with the results from the agar plate-based bioassay (Fig. 2A to C). These results confirmed that the cloning procedure was successful and that the quorum-quenching enzyme AiiA was expressed upon phage-induced lysis of the E. coli BL21 host.
FIG 2.

T7aiiA phage degrades AHLs produced by A. tumefaciens. An AHL bioassay was performed on LB agar plates containing X-Gal as described in Materials and Methods. A. tumefaciens A136 (AHL reporter) and KYC6 (AHL producer) were streaked at the indicated positions, without phage (A) or with the T7wt (B) or T7aiiA (C) phage. (D) A quantitative AHL bioassay was performed in a liquid-based AHL bioassay system composed of A136 and KYC6, without phages or with the T7wt or T7aiiA phage at various dosages (by altering the host E. coli BL21 dosage). Each bar shows the mean ± standard deviation (SD) (n = 3). Asterisks indicate statistical significance (P < 0.05).
To demonstrate whether the T7aiiA lysate can degrade AHLs released by other bacteria, P. aeruginosa PAO1 was used as the AHL producer, in place of A. tumefaciens KYC6, in the same AHL bioassay. P. aeruginosa PAO1 produces a different set of AHLs, i.e., C4- and 3-oxo-C12-HSLs (19), among which C4-HSL cannot and 3-oxo-C12-HSL can be recognized by A136 (28). Consistent with such a report, it was observed that the A136 colony streaked next to the P. aeruginosa colony turned blue in the plate-based AHL bioassay (Fig. 3A). Similar to the results obtained using KYC6 as the AHL producer, the phage T7aiiA lysate, but not the T7wt lysate, inhibited P. aeruginosa AHL-elicited LacZ expression in A136 (Fig. 3B and C), and this was also confirmed by a quantitative β-galactosidase assay with two dosages of each phage (Fig. 3D). As mentioned above, only 3-oxo-C12-HSL, not C4-HSL, was responsible for inducing blue color formation in the A136 colony. Likewise, 3-oxo-C12-HSL, not C4-HSL, is degradable by the AiiA enzyme from B. anthracis expressed in the T7aiiA lysate (25). Thus, only 3-oxo-C12-HSL contributed to the results of this assay.
FIG 3.

T7aiiA phage degrades AHLs released by P. aeruginosa. An agar plate-based AHL activity bioassay was performed as described in Materials and Methods. A136 and P. aeruginosa were streaked at the indicated positions, without phages (A) or with the T7wt (B) or T7aiiA (C) phage. (D) A quantitative AHL bioassay was performed in a liquid-based AHL bioassay system composed of A136 and P. aeruginosa, without phages or with the T7wt or T7aiiA phage at various dosages (by altering the host E. coli BL21 dosage). Each bar shows the mean ± SD (n = 3). Asterisks indicate statistically significant differences (P < 0.05).
To further evaluate the scope of substrate specificity of AiiA, we determined whether the T7aiiA lysate could degrade AHLs released by another AHL producer, Chromobacterium violaceum 12472 (30). Chromobacterium violaceum 12472 produces C4- and C6-HSLs to self-elicit the expression of the purple pigment violacein. P. aeruginosa was used as a quorum-quenching control. LB soft agar medium containing C. violaceum 12472 was overlaid on the pregrown P. aeruginosa colony on an LB agar plate. As P. aeruginosa released AHLs which inhibited binding of C4- and C6-HSLs from C. violaceum to their receptors (30), a colorless zone in the vicinity of the P. aeruginosa colony within the purple background of the cocultured Chromobacterium violaceum 12472 was observed after 24 h (Fig. 4A). The T7aiiA or T7wt phage lysate smeared on the LB agar plate and then overlaid with C. violaceum 12472 cells did not cause the appearance of the colorless zone, suggesting that AiiA in the T7aiiA lysate could not affect AHL-induced violacein production in C. violaceum (Fig. 4B and C). This is consistent with the substrate specificity of AiiA from B. anthracis, which has no or weak activity toward C4- and C6-HSLs (25), which are produced by C. violaceum (30). Taken together, the results from the three AHL-producing bacterial species suggested that the engineered phage T7aiiA can quench quorum sensing of diverse bacteria (A. tumefaciens and P. aeruginosa), if not all bacteria (C. violaceum), consistent with the substrate specificity of AiiA used in this research.
FIG 4.
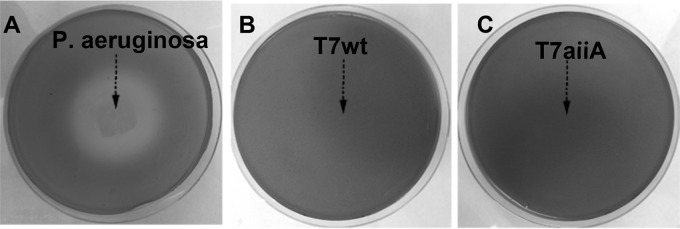
T7aiiA phage does not degrade AHLs released by C. violaceum. An AHL bioassay based on violacein production in C. violaceum 12472 was performed. (A) As a quorum-quenching positive control, P. aeruginosa PAO1, growing in the center of the LB agar plate (arrow), released AHLs to prevent the violacein production in C. violaceum. Phage T7wt (B) and T7aiiA (C) lysates were smeared at the center of LB agar plates, as indicated by arrows. C. violaceum cells in LB soft agar were overlaid on the preplated positive-control or phage lysates to examine their quorum-quenching effects.
Effects of quorum-quenching phage on formation of mixed- and single-species biofilms.
Upon confirming that the T7aiiA lysate contained AiiA enzyme activity, mixed-species biofilms composed of P. aeruginosa and E. coli were chosen for examination of the antibiofilm effects of the T7aiiA phage, for the following reasons. First, such a biofilm system mimics naturally occurring biofilms composed of E. coli and P. aeruginosa growing together in various settings, such as contaminated groundwater (34) and catheter surfaces (35). Second, biofilms composed of E. coli and P. aeruginosa have been examined to elucidate bacterium-phage ecology (18), which provides a defined context for evaluating the behavior of the engineered T7aiiA phage. Third, P. aeruginosa can synthesize and respond to AHLs (36), while E. coli cannot synthesize AHLs (37) but can respond to AHLs released by other bacteria (38). A biofilm system composed of P. aeruginosa and E. coli reflects the composition of many naturally occurring biofilms, containing both AHL-producing and nonproducing bacterial species. P. aeruginosa strain PAO1, which forms thick biofilms (39) and is not a host of T7 phages, was chosen for this research. E. coli strain TG1, which forms thick biofilms due to the presence of the conjugative F plasmid in TG1 (40) but is resistant to T7 phages due to the presence of the same F plasmid (41), was also chosen. To demonstrate whether the T7aiiA phage affected the mixed-species biofilm, the T7-susceptible E. coli strain BL21 was included as part of the biofilm. The biofilm quantification results, based on crystal violet staining of the EPS component of biofilms (32), indicated that P. aeruginosa PAO1, E. coli TG1, and E. coli BL21 caused formation of a much thicker biofilm at 4 and 8 h postplating than that formed by each individual bacterium (Fig. 5A). Among the individual bacteria, P. aeruginosa strain PAO1 and E. coli TG1 each formed thicker biofilms than did E. coli BL21. Such results suggested the existence of synergism among the biofilm-forming bacteria, which mechanistically may include metabolic cooperation, physical contact, and quorum sensing via autoinducers, such as AHLs, based on previous research (42).
FIG 5.

Effects of engineered phage on biofilms of mixed species of bacteria. (A) P. aeruginosa and E. coli TG1 cultures were plated with or without phage T7wt or T7aiiA in the presence of E. coli BL21 cells (OD600 = 0.05; 50 μl) in a 100-μl total volume in 96-well plates, and biofilm formation was quantified after 4 or 8 h of biofilm growth. (B) Altering the dosage of the T7wt and T7aiiA phages via altering the E. coli BL21 host cell quantity affected biofilm formation at 8 h postplating. (C) Effects of T7wt and T7aiiA phages on dual-species biofilms composed of P. aeruginosa and E. coli TG1, plated with or without phage T7wt or T7aiiA in 96-well microtiter plates. Each bar shows the mean ± SD (n = 5). Asterisks indicate statistical significance (P < 0.05).
To demonstrate the effects of phages in the mixed-species biofilm, T7wt or T7aiiA phage were added to the mixture of P. aeruginosa PAO1, E. coli TG1, and E. coli BL21. Both the T7wt and T7aiiA phages exhibited biofilm inhibition 4 and 8 h after plating. The T7aiiA phage caused significant reductions of the biofilm, by 74.9% and 65.9%, with regard to the no-phage control at 4 and 8 h postplating, respectively. In comparison, the T7wt phage caused reductions of only 23.8% and 31.7% at 4 and 8 h, respectively, compared to the no-phage control (Fig. 5A). An increase of the T7aiiA dosage by increasing the concentration of the host bacterium E. coli BL21 during plating, from an OD600 of 0.05 to 0.9, caused a statistically significant increase of biofilm inhibition (Fig. 5B). We attribute the antibiofilm effect of the T7wt phage to the phage-induced lysis of the E. coli BL21 component of the mixed-species biofilm. The enhanced antibiofilm effects of T7aiiA in comparison to T7wt (Fig. 5A) can be explained by the expression of AiiA upon T7aiiA-induced lysis of E. coli BL21 cells. AiiA may exert the antibiofilm action, likely through the following mechanism. AiiA in the T7aiiA lysates can degrade the AHLs produced by P. aeruginosa (Fig. 3), which leads to inhibition of P. aeruginosa biofilm, as AHLs produced by P. aeruginosa mediate biofilm formation (43). Among the two AHLs of P. aeruginosa, only 3-oxo-C12-HSL contributed to the results in Fig. 5, as it is the only AHL regulating biofilm formation (19) and also the only one degradable by AiiA (25). Note that phage were inoculated together with the biofilm-forming bacteria in this and subsequent experiments to study the effects of phage on biofilm formation. In the literature, such effects on biofilm formation have often been studied (9, 44), even though the effect on dispersion of established biofilms (6, 17) should also be explored for the T7aiiA phage in a separate study. The effects of the quorum-quenching phages on dual-species biofilms composed of P. aeruginosa PAO1 and E. coli TG1 (i.e., no E. coli BL21 was added) were measured; both T7wt and T7aiiA had no inhibitory effect on the dual-species biofilm (Fig. 5C). This is consistent with the fact that the T7 phages cannot infect P. aeruginosa PAO1 and E. coli TG1. For single-species P. aeruginosa biofilms, both T7wt and T7aiiA had no inhibitory effect (data not shown). In the single- and dual-species biofilm systems, no E. coli BL21 was present for phage T7aiiA to infect, and thus no new AiiA was synthesized. Though fresh T7aiiA lysates contained active AiiA capable of degrading AHLs from P. aeruginosa in the reporter bioassay (Fig. 3), the phage dosage in the biofilm systems was 1 × 104 PFU/ml, which is about 106-fold diluted relative to the fresh phage lysates, at about 1 × 1010 PFU/ml, used in the reporter bioassay. Thus, the AiiA in the T7aiiA lysate in the biofilm system was too diluted to be enzymatically active and exert antibiofilm effects. Also, both T7wt and T7aiiA had no effects on the single-species E. coli TG1 biofilm (data not shown). E. coli TG1 can bind T7 phages; however, the infection is arrested during phage DNA entry (41), leading to normal E. coli TG1 biofilm formation in the presence of various T7 phages. As E. coli BL21 formed negligible single-species biofilms, the effects of phages were not investigated.
The above-described bacteria in the single-species biofilms were either T7 insensitive or unable to form thick biofilms. To fully evaluate the antibiofilm properties of the T7aiiA phage, an E. coli host sensitive to T7 infection and capable of forming thick biofilms should be investigated. One potential target is E. coli MG1655, which is T7 sensitive and has no F plasmid (45). It has been reported that E. coli MG1655 forms weak (OD600 of <0.1, using crystal violet staining) (46) to medium (OD600 of >0.2) biofilms (47). Our initial results demonstrated that E. coli MG1655 biofilms in 96-well microtiter plates, obtained by plating a 1:100-diluted overnight culture of E. coli MG1655 similarly to the procedure for the biofilms described above, were weak (OD600 of <0.1). However, upon increasing the plating density of E. coli MG1655 to an OD600 of 0.05, maximal and significant amounts of biofilm (OD600 of >0.2) were obtained, though still less than the amount with E. coli TG1 (Fig. 5). Our results indicated that both the T7aiiA and T7wt phages caused significant inhibition of biofilm formation at 4 or 8 h postplating, and the inhibitory effects were indistinguishable (Fig. 6). This is consistent with the report that E. coli does not synthesize AHLs (37) for AiiA to degrade.
FIG 6.

Effects of phage T7aiiA on single-species E. coli MG1655 biofilms. Bacterial cultures (OD600 = 0.05) were plated with or without phage T7wt or T7aiiA in 96-well microtiter plates. Biofilm formation was quantified after 4 or 8 h. Each bar shows the mean ± SD (n = 5). Asterisks indicate statistical significance (P < 0.05).
To verify whether inhibition of biofilm formation by phages is accompanied by phage multiplication, the phage counts in the mixed-species biofilms composed of P. aeruginosa, E. coli TG1, and E. coli BL21 and in the liquid medium of the biofilm system were measured. Specifically, phage counts in the biofilms were determined after the biofilms in the wells were sonicated and suspended in PBS, while phage counts in the liquid medium were measured directly, as described in Materials and Methods. It was found that at 8 h postplating, the PFU counts for T7wt and T7aiiA in the liquid medium were, on average, 5.7 × 106 and 5.8 × 106 PFU, respectively, while PFU counts for T7wt and T7aiiA in the biofilm were 4.6 × 105 and 4.8 × 105 PFU, respectively. These counts were several orders of magnitude greater than the initially inoculated phage count of 1 × 103 PFU in each well (1 × 104 PFU/ml in a 100-μl total volume) (Fig. 7A), suggesting that phages indeed multiplied in the liquid medium and biofilms after plating. To verify whether inhibition of biofilm formation by phages is accompanied by bacterial cell death during biofilm formation, the bacterial cell counts were determined. The cell count of the E. coli BL21 culture with an OD600 of 0.05 inoculated in the beginning was 2.9 × 107 ± 1.7 × 107 CFU in each well. Since the inoculated phage count was 1 × 103 PFU, the multiplicity of infection (MOI) was ≈1:2.9 × 104. The initially inoculated total number of CFU for the mixture of P. aeruginosa PAO1, E. coli TG1, and E. coli BL21 was 5.0 × 107 ± 2.2 × 107. At 8 h postplating, the total cell counts in the biofilms were determined after sonicating the biofilms from the wells in PBS. At 8 h, the control biofilm with no phage treatment reached an average cell count per well of 8.5 × 108 CFU, while the T7wt- and T7aiiA-treated biofilms had average cell counts of 4.1 × 107 and 1.2 × 107 CFU, respectively (Fig. 7B). The bacterial counts of the biofilms without phage were significantly higher than those of the biofilms with phage, indicating that bacterial cells were lysed by the phages during the course of biofilm formation. The bacterial cell count in the biofilm treated with phage T7wt was significantly higher than that with phage T7aiiA (Fig. 7B), which is consistent with the result that a thicker biofilm (EPS stained by crystal violet) was formed with T7wt treatment (Fig. 5). With the low MOI of 1:2.9 × 104, excessive host E. coli BL21 cells not bound by phage were protected by the biofilm. The thicker biofilm occurring with T7wt treatment resulted in more protection of bacteria by EPS, more cell multiplication, and a larger total number of CFU.
FIG 7.

Effects of phage T7aiiA on cell counts and phage counts in mixed-species biofilms. Upon growth of mixed-species biofilms composed of P. aeruginosa, E. coli TG1, and E. coli BL21 for 8 h, as described in the legend to Fig. 5, the phage counts in the liquid medium and the biofilms (A) and the bacterial cell counts in the biofilms (B) were quantified. Each bar shows the mean ± SD (n = 5). Asterisks indicate statistical significance (P < 0.05).
Comparison of antibiofilm efficacies of T7aiiA phage and bacteria expressing AiiA.
It has been reported that free quorum-quenching acylase enzymes added to a membrane filtration system caused inhibition of biofilm formation and delay of membrane clogging (23). As direct addition of an enzyme is costly and unsustainable, an improvement was made to engineer bacteria to express quorum-quenching enzymes, such as AiiA, as antibiofilm agents (24). In light of the reports of antibiofilm effects of quorum-quenching bacteria, one question concerns the relative effectiveness of quorum-quenching phages versus quorum-quenching bacteria. To address this question, aiiA from B. anthracis was cloned into the pET-9a plasmid, which was transformed into E. coli BL21(DE3) containing a λDE3 prophage harboring the T7 RNA polymerase gene that drives high-level expression of aiiA in the presence of IPTG (termed the E. coli-aiiA strain). It is appropriate to evaluate the relative antibiofilm effects of the bacterium versus the phage, as the same quorum-quenching gene (aiiA) was used. Our results indicated that the E. coli-aiiA strain caused degradation of AHLs in the AHL reporter bioassay, while the control E. coli strain had no effect, suggesting that aiiA was expressed by E. coli-aiiA cells (Fig. 8A to C). Consistent with the role of AiiA in degrading AHLs, in the biofilm composed of P. aeruginosa and E. coli TG1, the E. coli-aiiA strain added to the mixture (50 μl of IPTG [1 mM]-containing medium with an OD600 of 0.05 in a total volume of 100 μl) caused a significant inhibition of biofilm formation (37.2% and 32.0% inhibition with respect to the control biofilm at 4 and 8 h postplating, respectively), whereas phage T7aiiA (50 μl of IPTG [1 mM]-containing medium with an OD600 of 0.05 [E. coli BL21] in a total volume of 100 μl) caused 75.0% and 65.9% inhibition at the same times. Increasing the concentration of the E. coli-aiiA strain to an OD600 of 0.9, however, slightly but significantly decreased the antibiofilm effect of the E. coli-aiiA strain, while increasing the dosage of phage T7aiiA significantly enhanced its antibiofilm effect (Fig. 8D). Similar trends were also identified for 4 h postplating (Fig. 8D). Overall, the results from using the E. coli-aiiA strain suggested that engineered phage T7aiiA outperformed the engineered E. coli-aiiA bacterium. The reason is likely that E. coli-aiiA cells directly participate in building up the multispecies biofilm, which antagonizes the antibiofilm effect of the expressed AiiA, while phage T7aiiA does not participate in building up the biofilm but lyses the bacterial cells (Fig. 7).
FIG 8.

Antibiofilm effects of the E. coli-aiiA strain and phage T7aiiA. An agar plate-based AHL reporter bioassay was performed without bacteria (A) or with E. coli (B) or the E. coli-aiiA strain (C), streaked at the indicated position between the AHL reporter A136 and the AHL producer KYC6. (D) Effects of increasing the dosage of the E. coli-aiiA strain versus increasing the dosage of T7aiiA phage on biofilm formation (by increasing the E. coli BL21 dosage from an OD600 of 0.05 to an OD600 of 0.9; phage level of 104 PFU/ml). Each bar shows the mean ± SD (n = 5). Asterisks indicate statistical significance (P < 0.05).
Antibiofilm potentials of quorum-quenching bacteriophages.
As part of the resurrection of phage research in Western countries to solve the problem of increases in antibiotic resistance (4, 48), bacteriophages which lyse host bacteria have aroused increasing interest for their potential application as antibiofilm agents (4, 8–12). Also, quorum quenching has been investigated as a biofilm control approach in many clinical (49, 50) and industrial (23, 24, 51) settings, due to the essential role of quorum sensing in regulating biofilm formation. In our research, phage treatment and quorum quenching were combined into a single entity, the quorum-quenching phage. Our results indicated that such a phage exerted a two-pronged effect of lysing the host bacteria in biofilms (Fig. 7) and expressing the AiiA enzyme to disrupt quorum sensing between bacteria (Fig. 2 and 3). As a result, compared to the wild-type phage, the engineered T7aiiA phage exhibited an increased antibiofilm effect in a mixed-species biofilm composed of P. aeruginosa PAO1, E. coli TG1, and E. coli BL21 (Fig. 5). The increased inhibition by T7aiiA was due to the expression of AiiA, as the antibiofilm effects of T7aiiA and T7wt were the same in a mixed-species biofilm composed of P. aeruginosa PAO1 and E. coli TG1 (Fig. 5). Compared to phages producing polysaccharide depolymerases, which also have two-pronged antibiofilm effects, the quorum-quenching phage provides advantages. The quorum-quenching phage circumvents not only the limitations of substrate specificity but also the host specificity of the phages with polysaccharide depolymerases. Specifically, the AiiA enzyme of B. anthracis expressed by T7aiiA can degrade different AHLs from multiple bacteria, including A. tumefaciens and P. aeruginosa (Fig. 2 and 3) but not C. violaceum (Fig. 4), in contrast to the polysaccharide depolymerases, which degrade one or, at most, a few related polysaccharides (1). The T7aiiA phage affected multiple bacteria in the mixed-species biofilm, including bacteria that are not hosts of T7 phages, such as P. aeruginosa and E. coli TG1 (Fig. 5), in contrast to the existing enzymatic phages affecting only the host bacteria.
Besides its immediate usefulness as an antibiofilm agent, the engineered T7 phage may serve as a proof of concept for constructing genomes of diverse phages to enhance their antibiofilm function. To custom build phage genomes, the modular design principle of synthetic biology (17) can be adopted. Three types of modules can be incorporated to fit diverse biofilm systems: (i) genes encoding quorum-quenching enzymes with custom substrate specificities, (ii) host-range-expanding modules to enable a phage to infect nonhost strains (17) and species (52), and (iii) genetic markers such as green fluorescent protein (53) for source tracking once the phage is released into the environment. For example, the gene encoding the AHL lactonase AiiA from Bacillus sp. strain 240, which is capable of degrading C4- to C12-HSLs (21), can be incorporated into the T7 genome to degrade AHLs released by and affect biofilms composed of A. tumefaciens, P. aeruginosa, and C. violaceum. We envision that such phages inoculated into a biofilm community can proliferate by lysing the host bacteria, even if their hosts account for only a minor proportion of the community. In the event that no or very few host bacteria preexist in the bacterial community, the host bacteria can be added, similar to the addition of E. coli BL21 together with the phage in this study (Fig. 5). The phages would release the enzymes into difficult-to-access sites inside biofilms to disrupt the bacterial cell-cell communication necessary for biofilm formation. Eventually, an equilibrium of engineered phage with the bacterial host in biofilms would be reached, with the overall biofilm reduced compared to the setting with the control wild-type phage.
Possible existence of natural quorum-quenching phages.
Since the engineered phage producing polysaccharide depolymerases (17) mimics natural phages (5, 16), does our engineered quorum-quenching phage mimic a natural phage? In other words, do natural quorum-quenching phages exist? The answer is likely yes. A φPLPE phage with a putative AHL acylase gene was isolated, whose host, Iodobacter sp. CDM7, expresses the purple pigment violacein controlled by AHL (54). On a lawn of Iodobacter, the φPLPE phage generates plaques with purple haloes (54), reminiscent of the haloed plaques of phages producing polysaccharide depolymerases (5, 16). Such haloes imply expression and diffusion of enzymes during phage lysis. If this is confirmed, natural quorum-quenching phages may be explored for antibiofilm function.
ACKNOWLEDGMENT
This study was supported by start-up funds from the University of Texas at San Antonio.
Footnotes
Published ahead of print 20 June 2014
REFERENCES
- 1.Flemming HC, Wingender J. 2010. The biofilm matrix. Nat. Rev. Microbiol. 8:623–633. 10.1038/nrmicro2415 [DOI] [PubMed] [Google Scholar]
- 2.Elias S, Banin E. 2012. Multi-species biofilms: living with friendly neighbors. FEMS Microbiol. Rev. 36:990–1004. 10.1111/j.1574-6976.2012.00325.x [DOI] [PubMed] [Google Scholar]
- 3.Rosenhahna A, Ederth T, Pettitt ME. 2008. Advanced nanostructures for the control of biofouling: the FP6 EU integrated project AMBIO. Biointerphases 3:IR1–IR5. 10.1116/1.2844718 [DOI] [PubMed] [Google Scholar]
- 4.Sulakvelidze A, Alavidze Z, Morris JG. 2001. Bacteriophage therapy. Antimicrob. Agents Chemother. 45:649–659. 10.1128/AAC.45.3.649-659.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sutherland IW, Hughes KA, Skillman LC, Tait K. 2004. The interaction of phage and biofilms. FEMS Microbiol. Lett. 232:1–6. 10.1016/S0378-1097(04)00041-2 [DOI] [PubMed] [Google Scholar]
- 6.Doolittle M, Cooney J, Caldwell D. 1995. Lytic infection of Escherichia coli biofilms by bacteriophage T4. Can. J. Microbiol. 41:12–18. 10.1139/m95-002 [DOI] [PubMed] [Google Scholar]
- 7.Sillankorva S, Neubauer P, Azeredo J. 2010. Phage control of dual species biofilms of Pseudomonas fluorescens and Staphylococcus lentus. Biofouling 26:567–575. 10.1080/08927014.2010.494251 [DOI] [PubMed] [Google Scholar]
- 8.Curtin JJ, Donlan RM. 2006. Using bacteriophages to reduce formation of catheter-associated biofilms by Staphylococcus epidermidis. Antimicrob. Agents Chemother. 50:1268–1275. 10.1128/AAC.50.4.1268-1275.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu WL, Forster T, Mayer O, Curtin JJ, Lehman SM, Donlan RM. 2010. Bacteriophage cocktail for the prevention of biofilm formation by Pseudomonas aeruginosa on catheters in an in vitro model system. Antimicrob. Agents Chemother. 54:397–404. 10.1128/AAC.00669-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merril CR, Scholl D, Adhya SL. 2003. The prospect for bacteriophage therapy in Western medicine. Nat. Rev. Drug Discov. 2:489–497. 10.1038/nrd1111 [DOI] [PubMed] [Google Scholar]
- 11.Liao K, Lehman S, Tweardy D, Donlan R, Trautner B. 2012. Bacteriophages are synergistic with bacterial interference for the prevention of Pseudomonas aeruginosa biofilm formation on urinary catheters. J. Appl. Microbiol. 113:1530–1539. 10.1111/j.1365-2672.2012.05432.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldman G, Starosvetsky J, Armon R. 2009. Inhibition of biofilm formation on UF membrane by use of specific bacteriophages. J. Membr. Sci. 342:145–152. 10.1016/j.memsci.2009.06.036 [DOI] [Google Scholar]
- 13.Doolittle MM, Cooney JJ, Caldwell DE. 1996. Tracing the interaction of bacteriophage with bacterial biofilms using fluorescent and chromogenic probes. J. Ind. Microbiol. 16:331–341. 10.1007/BF01570111 [DOI] [PubMed] [Google Scholar]
- 14.Tait K, Skillman L, Sutherland I. 2002. The efficacy of bacteriophage as a method of biofilm eradication. Biofouling 18:305–311. 10.1080/0892701021000034418 [DOI] [Google Scholar]
- 15.Shapiro OH, Kushmaro A. 2011. Bacteriophage ecology in environmental biotechnology processes. Curr. Opin. Biotechnol. 22:449–455. 10.1016/j.copbio.2011.01.012 [DOI] [PubMed] [Google Scholar]
- 16.Rydman PS, Bamford DH. 2002. The lytic enzyme of bacteriophage PRD1 is associated with the viral membrane. J. Bacteriol. 184:104–110. 10.1128/JB.184.1.104-110.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu TK, Collins JJ. 2007. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. U. S. A. 104:11197–11202. 10.1073/pnas.0704624104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kay MK, Erwin TC, McLean RJ, Aron GM. 2011. Bacteriophage ecology in Escherichia coli and Pseudomonas aeruginosa mixed-biofilm communities. Appl. Environ. Microbiol. 77:821–829. 10.1128/AEM.01797-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. 1998. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280:295–298. 10.1126/science.280.5361.295 [DOI] [PubMed] [Google Scholar]
- 20.Branda SS, Vik Å, Friedman L, Kolter R. 2005. Biofilms: the matrix revisited. Trends Microbiol. 13:20–26. 10.1016/j.tim.2004.11.006 [DOI] [PubMed] [Google Scholar]
- 21.Wang LH, Weng LX, Dong YH, Zhang LH. 2004. Specificity and enzyme kinetics of the quorum-quenching N-acyl homoserine lactone lactonase (AHL-lactonase). J. Biol. Chem. 279:13645–13651. 10.1074/jbc.M311194200 [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Dai Y, Zhang Y, Hu Y, Yang B, Chen S. 2007. Effects of quorum sensing autoinducer degradation gene on virulence and biofilm formation of Pseudomonas aeruginosa. Sci. China C Life Sci. 50:385–391. 10.1007/s11427-007-0044-y [DOI] [PubMed] [Google Scholar]
- 23.Yeon KM, Cheong WS, Oh HS, Lee WN, Hwang BK, Lee CH, Beyenal H, Lewandowski Z. 2009. Quorum sensing: a new biofouling control paradigm in a membrane bioreactor for advanced wastewater treatment. Environ. Sci. Technol. 43:380–385. 10.1021/es8019275 [DOI] [PubMed] [Google Scholar]
- 24.Oh HS, Yeon KM, Yang CS, Kim SR, Lee CH, Park SY, Han JY, Lee JK. 2012. Control of membrane biofouling in MBR for wastewater treatment by quorum quenching bacteria encapsulated in microporous membrane. Environ. Sci. Technol. 46:4877–4884. 10.1021/es204312u [DOI] [PubMed] [Google Scholar]
- 25.Ulrich RL. 2004. Quorum quenching: enzymatic disruption of N-acylhomoserine lactone-mediated bacterial communication in Burkholderia thailandensis. Appl. Environ. Microbiol. 70:6173–6180. 10.1128/AEM.70.10.6173-6180.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Israel DI. 1993. A PCR-based method for high stringency screening of DNA libraries. Nucleic Acids Res. 21:2627–2631. 10.1093/nar/21.11.2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 28.Zhu J, Beaber JW, Moré MI, Fuqua C, Eberhard A, Winans SC. 1998. Analogs of the autoinducer 3-oxooctanoyl-homoserine lactone strongly inhibit activity of the TraR protein of Agrobacterium tumefaciens. J. Bacteriol. 180:5398–5405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuqua C, Winans SC. 1996. Conserved cis-acting promoter elements are required for density-dependent transcription of Agrobacterium tumefaciens conjugal transfer genes. J. Bacteriol. 178:435–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu W, Vattem DA, Maitin V, Barnes MB, McLean RJ. 2011. Bioassays of quorum sensing compounds using Agrobacterium tumefaciens and Chromobacterium violaceum. Methods Mol. Biol. 692:3–19. 10.1007/978-1-60761-971-0_1 [DOI] [PubMed] [Google Scholar]
- 31.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 32.O'Toole GA, Pratt LA, Watnick PI, Newman DK, Weaver VB, Kolter R. 1999. Genetic approaches to study of biofilms. Methods Enzymol. 310:91–109. 10.1016/S0076-6879(99)10008-9 [DOI] [PubMed] [Google Scholar]
- 33.Corbin BD, McLean RJC, Aron GM. 2001. Bacteriophage T4 multiplication in a glucose-limited Escherichia coli biofilm. Can. J. Microbiol. 47:680–684. 10.1139/w01-059 [DOI] [PubMed] [Google Scholar]
- 34.Banning N, Toze S, Mee BJ. 2003. Persistence of biofilm-associated Escherichia coli and Pseudomonas aeruginosa in groundwater and treated effluent in a laboratory model system. Microbiology 149:47–55. 10.1099/mic.0.25938-0 [DOI] [PubMed] [Google Scholar]
- 35.Cerqueira L, Oliveira JA, Nicolau A, Azevedo NF, Vieira MJ. 2013. Biofilm formation with mixed cultures of Pseudomonas aeruginosa/Escherichia coli on silicone using artificial urine to mimic urinary catheters. Biofouling 29:829–840. 10.1080/08927014.2013.807913 [DOI] [PubMed] [Google Scholar]
- 36.Pearson JP, Passador L, Iglewski BH, Greenberg E. 1995. A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 92:1490–1494. 10.1073/pnas.92.5.1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahmer BM. 2004. Cell-to-cell signalling in Escherichia coli and Salmonella enterica. Mol. Microbiol. 52:933–945. 10.1111/j.1365-2958.2004.04054.x [DOI] [PubMed] [Google Scholar]
- 38.Dyszel JL, Soares JA, Swearingen MC, Lindsay A, Smith JN, Ahmer BMM. 2010. E. coli K-12 and EHEC genes regulated by SdiA. PLoS One 5:e8946. 10.1371/journal.pone.0008946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klausen M, Heydorn A, Ragas P, Lambertsen L, Aaes-Jørgensen A, Molin S, Tolker-Nielsen T. 2003. Biofilm formation by Pseudomonas aeruginosa wild type, flagella and type IV pili mutants. Mol. Microbiol. 48:1511–1524. 10.1046/j.1365-2958.2003.03525.x [DOI] [PubMed] [Google Scholar]
- 40.Ghigo JM. 2001. Natural conjugative plasmids induce bacterial biofilm development. Nature 412:442–445. 10.1038/35086581 [DOI] [PubMed] [Google Scholar]
- 41.Garcia LR, Molineux IJ. 1995. Incomplete entry of bacteriophage T7 DNA into F plasmid-containing Escherichia coli. J. Bacteriol. 177:4077–4083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burmølle M, Webb JS, Rao D, Hansen LH, Sørensen SJ, Kjelleberg S. 2006. Enhanced biofilm formation and increased resistance to antimicrobial agents and bacterial invasion are caused by synergistic interactions in multispecies biofilms. Appl. Environ. Microbiol. 72:3916–3923. 10.1128/AEM.03022-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hentzer M, Riedel K, Rasmussen TB, Heydorn A, Andersen JB, Parsek MR, Rice SA, Eberl L, Molin S, Høiby N, Kjelleberg S, Givskov M. 2002. Inhibition of quorum sensing in Pseudomonas aeruginosa biofilm bacteria by a halogenated furanone compound. Microbiology 148:87–102 [DOI] [PubMed] [Google Scholar]
- 44.Hosseinidoust Z, Tufenkji N, van de Ven TG. 2013. Formation of biofilms under phage predation: considerations concerning a biofilm increase. Biofouling 29:457–468. 10.1080/08927014.2013.779370 [DOI] [PubMed] [Google Scholar]
- 45.Moons P, Faster D, Aertsen A. 2013. Lysogenic conversion and phage resistance development in phage exposed Escherichia coli biofilms. Viruses 5:150–161. 10.3390/v5010150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jackson DW, Simecka JW, Romeo T. 2002. Catabolite repression of Escherichia coli biofilm formation. J. Bacteriol. 184:3406–3410. 10.1128/JB.184.12.3406-3410.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.del Mar Cendra M, Juárez A, Torrents E. 2012. Biofilm modifies expression of ribonucleotide reductase genes in Escherichia coli. PLoS One 7:e46350. 10.1371/journal.pone.0046350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu TK, Koeris MS. 2011. The next generation of bacteriophage therapy. Curr. Opin. Microbiol. 14:524–531. 10.1016/j.mib.2011.07.028 [DOI] [PubMed] [Google Scholar]
- 49.Hentzer M, Givskov M. 2003. Pharmacological inhibition of quorum sensing for the treatment of chronic bacterial infections. J. Clin. Invest. 112:1300–1307. 10.1172/JCI200320074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Loughlin CT, Miller LC, Siryaporn A, Drescher K, Semmelhack MF, Bassler BL. 2013. A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc. Natl. Acad. Sci. U. S. A. 110:17981–17986. 10.1073/pnas.1316981110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang W, Xia S, Liang J, Zhang Z, Hermanowicz SW. 2013. Effect of quorum quenching on the reactor performance, biofouling and biomass characteristics in membrane bioreactors. Water Res. 47:187–196. 10.1016/j.watres.2012.09.050 [DOI] [PubMed] [Google Scholar]
- 52.Tetart F, Repoila F, Monod C, Krisch H. 1996. Bacteriophage T4 host range is expanded by duplications of a small domain of the tail fiber adhesin. J. Mol. Biol. 258:726–731. 10.1006/jmbi.1996.0281 [DOI] [PubMed] [Google Scholar]
- 53.Jain P, Hartman TE, Eisenberg N, O'Donnell MR, Kriakov J, Govender K, Makume M, Thaler DS, Hatfull GF, Sturm AW. 2012. ϕ2GFP10, a high-intensity fluorophage, enables detection and rapid drug susceptibility testing of Mycobacterium tuberculosis directly from sputum samples. J. Clin. Microbiol. 50:1362–1369. 10.1128/JCM.06192-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leblanc C, Caumont-Sarcos A, Comeau AM, Krisch HM. 2009. Isolation and genomic characterization of the first phage infecting Iodobacteria: ϕPLPE, a myovirus having a novel set of features. Environ. Microbiol. Rep. 1:499–509. 10.1111/j.1758-2229.2009.00055.x [DOI] [PubMed] [Google Scholar]