

Abstract
Rhizobia induce nitrogen-fixing nodules on host legumes, which is important in agriculture and ecology. Lipopolysaccharide (LPS) produced by rhizobia is required for infection or bacteroid survival in host cells. Genes required for LPS biosynthesis have been identified in several Rhizobium species. However, the regulation of their expression is not well understood. Here, Sinorhizobium meliloti LsrB, a member of the LysR family of transcriptional regulators, was found to be involved in LPS biosynthesis by positively regulating the expression of the lrp3-lpsCDE operon. An lsrB in-frame deletion mutant displayed growth deficiency, sensitivity to the detergent sodium dodecyl sulfate, and acidic pH compared to the parent strain. This mutant produced slightly less LPS due to lower expression of the lrp3 operon. Analysis of the transcriptional start sites of the lrp3 and lpsCDE gene suggested that they constitute one operon. The expression of lsrB was positively autoregulated. The promoter region of lrp3 was specifically precipitated by anti-LsrB antibodies in vivo. The promoter DNA fragment containing TN11A motifs was bound by the purified LsrB protein in vitro. These new findings suggest that S. meliloti LsrB is associated with LPS biosynthesis, which is required for symbiotic nitrogen fixation on some ecotypes of alfalfa plants.
INTRODUCTION
Lipopolysaccharide (LPS) is required for Rhizobium infection or survival in host cells. It is composed of O-antigen, core oligosaccharide, and lipid A. LPS contributes to infection thread formation in symbioses between legumes, Rhizobium etli, and R. leguminosarum bv. trifolii and viciae (1–3). As demonstrated by Sinorhizobium fredii, S. meliloti, and Bradyrhizobium japonicum, bacteroid differentiation in root nodule cells requires appropriate LPS (4–7). LPS suppresses host defenses, enabling rhizobia to infect their hosts successfully and survive long term in host cells (8, 9).
In S. meliloti, several LPS biosynthesis genes have been identified, including lpsB, lpsCDE, lpsL, rkpK, and ddhB (10–14). The lpsB, lpsC, and lpsDE genes encode a type I glycosyltransferase, β-1,4-glycosyltransferase, and RfaG glycosyltransferases, respectively, which are involved in the biosynthesis of the core oligosaccharides. The lpsL gene encodes a UDP-glucuronate 5′-epimerase, rkpK encodes a UDP-glucose 6-dehydrogenase, and ddhB encodes a CDP-glucose 4,6-dehydratase to catalyze the synthesis of CDP-4-keto-6-deoxy–d-glucose from CDP–d-glucose, which is associated with O-antigen biosynthesis. Genetic evidence demonstrates that null mutants of lpsB, lpsC, rkpK, and lpsL in the S. meliloti 1021 background induce defective nitrogen fixation nodules on some ecotypes of Medicago sativa or Medicago truncatula A17 (11, 13). Root nodules hosting lpsB::TnphoA mutants from M. sativa cv. GT-13R+ or Iroquois show premature senescence, in which bacteroid differentiation is blocked at different stages and bacteroids appear to activate host innate immunity (4).
A few regulators involved in LPS biosynthesis have been identified in rhizobial strains. A small periplasmic regulator, SyrA, is coupled with the ExoS/ChvI two-component system to regulate expression of the LPS sulfotransferase gene, lpsS, in S. meliloti 1021 (12, 15). In S. fredii NGR234, the type III secretion system regulator TtsI controls the biosynthesis of rhamnose and a rhamnose-rich component (rhamnan) of LPS (16). In S. fredii HH103, the transcription elongation factor GreA could be involved in LPS production, because its null mutant synthesized defective LPS (7). In fact, some S. meliloti mutants also affect LPS biosynthesis (11). However, regulation of LPS core biosynthesis genes is not well understood.
LysR regulators constitute a large family of transcriptional factors in prokaryotes and play important regulatory roles in amino acid metabolism (LysR, ArgP, GltC, IlvR/Y, MetR, MtaR, GcvA, and CysB), carbon dioxide fixation (CbbR and CfxR), catechol catabolism (CatR/M), oxidative stress adaptation (OxyR), and bacterium-plant interactions (NodD, SyrM, OccR, NocR, GbpR, and PhcA) (17). As many of the rhizobial genomes have been sequenced, an increasing number of genes encoding LysR family regulators have been annotated. The sequence of the S. meliloti 1021 genome was published in 2001, and more than 90 genes encoding LysR regulatory proteins have been predicted (18). In our previous work, 83 genes of strain 1021 were mutated by generating plasmid insertions (19). Two mutants, lsrA1 and lsrB1, induced ineffective nodules on alfalfa (19). Here, an in-frame deletion of lsrB (lsrB1-2) was constructed in S. meliloti 1021 to investigate the role of LsrB in symbiosis. We found that LsrB positively regulates the expression of the lrp3-lpsCDE operon involved in LPS biosynthesis.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Escherichia coli DH5α, MT616 (20), and BL21(DE3) were grown in Luria-Bertani (LB) medium at 37°C (Table 1). S. meliloti 1021 (21) and the lsrB1-2 mutant (Table 1) were grown in LB medium supplemented with 2.5 mM MgSO4 and 2.5 mM CaCl2 (LB/MC) at 28°C (25). The following antibiotics were used: chloramphenicol, 10 μg ml−1; neomycin, 200 μg ml−1; kanamycin, 25 μg ml−1; spectinomycin, 100 μg ml−1; tetracycline, 10 μg ml−1; and streptomycin, 500 μg ml−1.
TABLE 1.
Strains and plasmids used in this study
| Strain/plasmid | Relevant property(ies) | Reference/source |
|---|---|---|
| Strains | ||
| E. coli DH5α | F− φ80lacZΔM15Δ(lacZYA-argF)U169 deoR recA1 endA1hsdR17 (rK−, mK+) phoA supE44 λ− thi-1 gyrA96 relA1 | Laboratory stock |
| E. coli MT616 | MM294 pRK600, Cmr | 20 |
| E. coli BL21(DE3) | F− dcm ompT hsdS(rB− mB−) gal λ(DE3), Kmr | Laboratory stock |
| S. meliloti 1021 or Rm1021 | SU47 but also str-2, Smr | 21 |
| S. meliloti lsrB1-2 | Rm1021 ΔlsrB, Smr | This study |
| Plasmid | ||
| pK18mobsacB | Suicide vector, Kmr | 22 |
| pLMG1 | pK18mobsacB carrying a 1,600-bp joint DNA fragment from S. meliloti trxB and SMc01226, Kmr | This study |
| pSRK-Tc | Expression vector under the control of lac promoter, Tcr | 23 |
| pLMG2 | pSRK-Tc carrying an S. meliloti lsrB gene, Tcr | This study |
| pRG960 | pRG930 containing the promoterless gusA with the start codon, Smr/Spr | 24 |
| pLMG3 | pRG960 carrying the 300-bp DNA fragment upstream of lsrB, Spr | This study |
| pLMG4 | pRG960 carrying the 300-bp DNA fragment upstream of trxB, Spr | This study |
| pLMG5 | pRG960 carrying the 300-bp DNA fragment upstream of lrp3, Spr | This study |
| pLMG6 | pRG960 carrying the 300-bp DNA fragment upstream of lpsC, Spr | This study |
| pMD-18T | Cloning vector, Apr | TaKaRa |
| pLMG7 | pMD-18T carrying an S. meliloti lsrB gene, Apr | This study |
| pET28b | Expression vector carrying His tags, Kmr | Novagen |
| pLMG8 | pET28b carrying an S. meliloti lsrB gene, Kmr | This study |
Construction of an lsrB in-frame deletion mutant.
Two 800-bp DNA fragments from the proximate regions of the lsrB open reading frame (ORF) on the genome of S. meliloti 1021 were amplified using KOD plus DNA polymerase (Toyobo, Osaka, Japan). Both PCR products were purified using the gel extraction system B kit (Biodev-Tech, Beijing, China). The purified PCR products (1:1) were employed to amplify a 1.6-kb joint fragment using TransStart Taq DNA polymerase (Transgen, Beijing, China). All four primers (P1 to P4) are listed in Table 2. The purified PCR product was digested by EcoRI and XbaI (TaKaRa, Dalian, China) at 37°C overnight. The digested DNA then was purified using the EasyPure PCR purification kit (Transgen, Beijing, China) and cloned into pK18mobsacB (22) with T4 DNA ligase (TaKaRa, Dalian, China) overnight at 16°C. The plasmid then was transformed into E. coli DH5α competent cells. The plasmid, named pLMG1 (Table 1), was purified using the EasyPure plasmid miniprep kit (Transgen, Beijing, China) and identified by EcoRI and XbaI digestion. It then was transferred into S. meliloti 1021 by triparental mating with the helper strain E. coli MT616 (20). Streptomycin- and neomycin-resistant colonies were spread onto LB/MC agar plates containing 5% sucrose and streptomycin. The resulting colonies were screened for sensitivity to neomycin and identified by PCR. The PCR product was sequenced (Invitrogen, Shanghai, China). The confirmed S. meliloti mutant was named the lsrB1-2 mutant. The lsrB ORF also was amplified using the P5 and P6 primers (Table 2), and the DNA was digested with NdeI and XbaI and cloned into pSRK-Tc (23). The recombinant plasmid pLMG2 (Table 1) was used to complement the lsrB1-2 mutant.
TABLE 2.
Primersa
| Name | Sequence | Purpose |
|---|---|---|
| P1 | 5′-GGAATTCCGACCTCGTAACCGATGTCGA-3′ | Construction of the lsrB deletion mutant |
| P2 | 5′-AAAGGCTCAGCCGGAGAAGC-3′ | Construction of the lsrB deletion mutant |
| P3 | 5′-CTCCGGCTGAGCCTTTCGATCCGTTCAATTGCCCG-3′ | Construction of the lsrB deletion mutant |
| P4 | 5′-GCTCTAGAGCACGGCGAGATGAGATGAAA-3′ | Construction of the lsrB deletion mutant |
| P5 | 5′-GGAATTCCATATGGGGGATTCTATGTCGCTGGACT-3′ | Cloning of lsrB gene |
| P6 | 5′-GCTCTAGATCAGAAGTTCCAGTTTCTCGCTTTA-3′ | Cloning of lsrB gene |
| P7 | 5′-TGCACTGCAGCCGGCGACGTGACCGAC-3′ | Cloning of lsrB promoter |
| P8 | 5′-CGGGATCCGGGCGTGCCGATGAAAGA-3′ | Cloning of lsrB promoter |
| P9 | 5′-TGCACTGCAGTTGCCCAAATCCCCAATCCT-3′ | Cloning of trxB promoter |
| P10 | 5′-CGGGATCCGCTTTTGTCCTTCGTCCGCC-3′ | Cloning of trxB promoter |
| P11 | 5′-CGGGATCCTTGCCCAAATCCCCAATCCT-3′ | Cloning of lrp3 promoter |
| P12 | 5′-TGCACTGCAGGCTTTTGTCCTTCGTCCGCC-3′ | Cloning of lrp3 promoter |
| P13 | 5′-TGCACTGCAGATTTTGTTATTGAAGAACTGACCG-3′ | Cloning of lpsC promoter |
| P14 | 5′-CGGGATCCGAGACTCTCGATGCACTTGCC-3′ | Cloning of lpsC promoter |
| P19 | 5′-TCCGGAAATCCAGGTACAGC-3′ | qRT-PCR of lsrB |
| P20 | 5′-ATGCGATATGCGCGGAT-3′ | qRT-PCR of lsrB |
| P21 | 5′-TCAGAAAGGGACCGACCTCTT-3′ | qRT-PCR of lpsB |
| P22 | 5′-CCAGTCGGGAATATTCGTGTG-3′ | qRT-PCR of lpsB |
| P23 | 5′-GCCTATCTCGGCAAGTGCAT-3′ | qRT-PCR of lpsC |
| P24 | 5′-GATGTTGAAGCACCAGGGCT-3′ | qRT-PCR of lpsC |
| P25 | 5′-ATCTTTGGCTGCTTGGCGA-3′ | qRT-PCR of lpsE |
| P26 | 5′-ATGAACCATTGCGGCCCTT-3′ | qRT-PCR of lpsE |
| P27 | 5′-ATCGTGCGCTTCTGAATGC-3′ | qRT-PCR of lrp3 |
| P28 | 5′-TCCTGAAAGCTCGCAAGGTT-3′ | qRT-PCR of lrp3 |
| P29 | 5′-AAGGTGATCTGGGACCACGA-3′ | qRT-PCR of trxB |
| P30 | 5′-GCGAAGTTTGCCCTTGAAGA-3′ | qRT-PCR of trxB |
| P31 | 5′-GATCGTCATGTAGCGCAGGA-3′ | qRT-PCR of rpsF |
| P32 | 5′-CCTCGCTCGGCAGGACAT-3′ | qRT-PCR of rpsF |
| P33 | 5′-CATGGCTACATGCTGACAGCCTA-3′ | 5′-RACE of outRAP |
| P34 | 5′-CGCGGATCCACAGCCTACTGATGATCAGTCGATGGAAA-3′ | 5′-RACE of inRAP |
| P35 | 5′-TTGTCGAGGATGAGCTGTAC-3′ | 5′-RACE of lsrB |
| P36 | 5′-TTGGCTTGTCCGTCGTCT-3′ | 5′-RACE of lsrB |
| P37 | 5′-AAGGTCGGCTCGGTTTCGAT-3′ | 5′-RACE of trxB |
| P38 | 5′-GGTTACGAGGTCGTTGAC-3′ | 5′-RACE of trxB |
| P39 | 5′-AAAGTCCGATTCCCCGGA-3′ | 5′-RACE of lrp3 |
| P40 | 5′-TTGAGATTGGCGTCGGACTG-3′ | 5′-RACE of lrp3 |
| P41 | 5′-CGATCTCGTCTGATGCTGC-3′ | 5′-RACE of lpsC |
| P42 | 5′-TACATGAAACGGATGGGCCA-3′ | 5′-RACE of lpsC |
| P43 | 5′-TGGGACAAACTGCGCATTT-3′ | RT-PCR of lsrB |
| P44 | 5′-GCTTTCGAGCTTCATCAGCAC-3′ | RT-PCR of lsrB |
| P45 | 5′-ACATCCGACATGGCAATCG-3′ | RT-PCR of trxB |
| P46 | 5′-AGGTATTTCTCCGCTTCGAGC-3′ | RT-PCR of trxB |
| P47 | 5′-ATCGTGCGCTTCTGAATGC-3′ | RT-PCR of lrp3 |
| P48 | 5′-TCCTGAAAGCTCGCAAGGTT-3′ | RT-PCR of lrp3 |
| P49 | 5′-GCCTATCTCGGCAAGTGCAT-3′ | RT-PCR of lpsC |
| P50 | 5′-GATGTTGAAGCACCAGGGCT-3′ | RT-PCR of lpsC |
| P51 | 5′-CATATTCACTTCGGCACGGAG-3′ | RT-PCR of lpsD |
| P52 | 5′-GCAACTGCCAGGCCATTATG-3′ | RT-PCR of lpsD |
| P53 | 5′-CAAGGTTTTTGTGCATGCAACTGC-3′ | EMSA |
| P54 | 5′-GTTCCAAAAACACGTACGTTGACG-3′ | EMSA |
Italics represent restriction endonuclease sites.
LPS assay.
S. meliloti cells were collected from LB/MC cultures (optical density at 600 nm [OD600], ≈0.8). The crude LPS was extracted with both hot phenol and boiled methods and then treated by RNase, DNase I, and protease K (4, 26). The sulfate-anthrone method was performed to quantify the total sugar content of dialyzed LPS extract (27). The crude extract (with 0.5 μg of total protein) was used for DOC-PAGE (deoxycholate polyacrylamide gel electrophoresis) analysis (11).
5′-RACE.
In order to determine the transcriptional start sites of the lsrB, trxB, lrp3, and lpsC genes, RNA samples were isolated from S. meliloti 1021 using a TransZol plant kit (Transgen, Beijing, China). Random amplification of cDNA ends was performed using a 5′ full rapid amplification of cDNA ends (RACE) kit (TaKaRa, Dalian, China). The cDNA then was amplified by PCR using a 5′-RACE anchor outer primer (OutRAP) together with the nested outer primer of one target gene (outlsrB). Subsequently, a nested PCR was performed using the PCR products diluted 20-fold, obtained as described above with the inner primers of one target gene (inlsrB) and a 5′-RACE anchor inner primer (InRAP) (Table 2). The PCR amplification cycling protocol consisted of a predenaturing step at 95°C for 5 min and 35 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 1 min, followed by a final extension step at 72°C for 10 min. The corresponding P33 to P42 primers are listed in Table 2. The resulting PCR products were sequenced. The transcriptional start sites were determined by DNA sequence analysis.
RNA extraction and RT-PCR.
Total RNA for reverse transcription was isolated from cells grown to log phase (OD600, ∼0.8) in LB/MC medium using a TransZol plant kit (Transgen, Beijing, China). RNA quality was assayed by 1% agarose gel electrophoresis, and the RNA then was treated with RNase-free DNase (TakaRa, Dalian, China) for 30 min at 37°C to digest the remaining genomic DNA, which then was analyzed by PCR using multiple pairs of primers. The RNA preparations were used to synthesize cDNAs with a Primerscript RT Master Mix Perfect real-time kit (TakaRa, Dalian, China), and the first-strand cDNA was used as a template for PCR amplification. Real-time quantitative RT-PCR (qRT-PCR) was performed using SYBR green supermix (Toyobo, Osaka, Japan). The quantitative PCR system (20 μl) consisted of the following components: 10 μl of SYBR green real-time PCR master mix, 0.5 μl of each primer, 1 μl of the diluted cDNA sample, and 9 μl of sterile water. All readings were performed in triplicate using a real-time PCR system (Bio-Rad, Hercules, CA). The program consisted of a denaturing cycle at 95°C for 3 min; 40 cycles comprising 95°C for 10 s, 62°C for 30 s, and 72°C for 30 s; and a final step in which the temperature was elevated on a gradient from 65°C to 95°C to dissociate the double-stranded DNA products. The primers P19 to P32 were used for amplification of lsrB, lpsB, lpsC, lpsE, lrp3, trxB, and rpsF fragments (listed in Table 2). The transcripts of trxB, lsrB, lrp3, lpsC, and lpsD were amplified with the primers P43 to P52 (Table 2).
Construction of promoter-GUS fusion and GUS activity assay.
DNA fragments of approximately 300 bp, containing the lsrB, trxB, lrp3, and lpsC promoter regions, were amplified with the P7-P8, P9-P10, P11-P12, and P13-P14 primer pairs, respectively (Table 2). The amplicons were digested with BamHI, SmaI, or PstI and cloned into pRG960 (24), yielding the recombinant plasmids pLMG3, pLMG4, pLMG5, and pLMG6, respectively (Table 1). To assay β-glucuronidase activity, a rhizobial strain carrying an empty or a recombinant plasmid was grown in LB/MC medium to an OD600 of ∼0.8, and cells were collected by centrifugation at 6,000 × g for 2 min (4°C). The cells were crushed with a pestle fitted to an electric drill (J1Z-GL-10; 220 V, 50 Hz, 400 W; Modong Company, Shanghai, China) for 30 s three times on ice. The cell lysate was retained on ice to analyze β-glucuronidase activity with the substrate of p-nitrophenyl-β-d-glucuronide (Sigma, USA) as described by Jefferson et al. (28).
Expression, purification of LsrB protein, preparation of anti-LsrB antibodies, and immunoblotting.
The full-length lsrB ORF was amplified with the P17 and P18 primers (Table 2), purified, and ligated into pMD-18T (TaKaRa, Dalian, China). The foreign DNA was sequenced (Invitrogen, Shanghai, China). The correct plasmid was digested by SalI and NotI, and the released 900-bp DNA fragment was recycled and cloned into pET28b (+) (Novagen, Darmstadt, Germany) to construct a plasmid, pLMG8 (Table 1), which was used to transform competent cells of E. coli BL21(DE3)pLysS. This strain was cultured in 5 ml LB broth with kanamycin at 37°C overnight and then subcultured in 100 ml LB-kanamycin broth with 4 mg/ml isopropyl-β-d-thiogalactopyranoside (IPTG) at 28°C for 12 h. The bacterial cells were collected by centrifugation at 6,000 × g for 10 min (4°C) and washed once with 50 ml phosphate-buffered saline (PBS). The bacterial cells then were frozen with liquid nitrogen, ground to a powder, and resuspended in 10 ml PBS. The suspension was sonicated with a TY92-II sonifier (Scientz, Hangzhou, China) at a power of 400 W for 30 min in 10-s pulses. The supernatant was collected by centrifugation at 10,000 × g for 10 min and then loaded on a nickel-nitrilotriacetic acid (Ni-NTA) column at 4°C according to the Ni-NTA purification system procedure (Invitrogen, Shanghai, China). The eluted protein was analyzed using SDS-PAGE. The purified LsrB protein was diluted with 20% glycerol and stored at −80°C. The protein then was used to prepare anti-LsrB rabbit antibodies (Willget, Shanghai, China). Western blotting was carried out as previously described (29). The total cellular proteins were separated by SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane (Pharmacia, Stockholm, Sweden). LsrB protein was detected with the prepared anti-LsrB specific polyclonal primary antibodies and the secondary antibody, goat anti-rabbit IgG-HRP (horseradish peroxidase), from Abmart (Shanghai, China). The blots were stained with a 3,3′,5,5′-tetramethylbenzidine (TMB)-stabilized substrate for HRP (Promega, Madison, WI).
ChIP.
Chromatin immunoprecipitation (ChIP) was performed as described by Grainger et al. (30). In brief, 150 μl of formaldehyde was added to 5-ml volumes of fresh cultures (OD600, ∼0.8) of S. meliloti 1021 and the lsrB1-2 mutant. Cells were shaken for 10 min at 28°C. Cells were pelleted from 1.5 ml of culture, washed twice with 1.5 ml of PBS (pH 7.4), frozen in liquid nitrogen, and then crushed with a pestle attached to the electric drill for 30 s, three times, on ice. The cell lysate was suspended in 5 ml of IP buffer (50 mM Tris-HCl, pH 7.0, 150 mM NaCl, 1% Triton X-100) containing 1 mM phenylmethylsulfonyl fluoride (PMSF) and then sonicated with a TY92-II sonifier at a power of 300 W for 20 min in 5-s pulses. After centrifugation at 4°C for 10 min, 100 μl of supernatant was stored at −20°C as a positive control for PCR. A 1-μl volume of anti-LsrB antibody was added to 4.9 ml of sonicated supernatant and the mixture shaken gently at room temperature for 1 h, and then 25 μl of IgA-agarose beads (Abmart, Shanghai, China) was added. Samples were shaken gently at room temperature for 1 h. Beads were collected by centrifugation at 13,000 × g for 1 min and washed five times with IP buffer and then twice with TE buffer (10 mM Tris-HCl, pH 8.0, 0.1 mM EDTA). Agarose beads were suspended in 95 μl of TE buffer and 5 μl of 2% SDS and then incubated at 65°C overnight for reverse cross-linking. The sample was used as a PCR template to amplify the S. meliloti lrp3 promoter and rpsF (encoding a ribosome protein as a negative control) DNA fragments, using the primers P11 and P12 as well as P31 and P32 (Table 2), respectively.
Electrophoretic motility shift assay (EMSA).
The oligonucleotides containing TN11A boxes (P53 and P54) (Table 2) of lrp3 were synthesized by Invitrogen (Shanghai, China) and 3′ labeled using a DIG (digoxigenin) gel shift kit (Roche, Rotkreuz, Switzerland). The gel shift test of the LsrB-TN11A box was performed according to the manufacturer's protocol for the kit as well.
DNA sequence analysis.
The deduced protein sequence of LsrB was downloaded from the S. meliloti genome site (http://iant.toulouse.inra.fr/bacteria/annotation/cgi/rhime.cgi). Homologs of LsrB were aligned and downloaded from the NCBI homepage using the BLAST microbial genome program (http://www.ncbi.nlm.nih.gov/sutils/genom_table.cgi). The putative promoter DNA sequences were downloaded from the S. meliloti genome server, and potential promoters were predicted using the BDGP program (http://www.fruitfly.org/seq_tools/promoter.html). The BLAST nucleotide program from NCBI was used to analyze the DNA sequences.
RESULTS
Construction of an S. meliloti lsrB deletion mutant.
In our previous work, a plasmid insertion mutant of the S. meliloti lsrB (lsrB1) strain showed apparent symbiotic deficiency (19). To exclude a polar effect of lsrB1, an in-frame deletion mutant was constructed and named the lsrB1-2 mutant (Fig. 1A). Sinorhizobium genomic DNA was extracted, and PCR showed that a small DNA fragment (1.5 kb) was specifically amplified using the lsrB1-2 mutant genomic DNA as a template, but a large DNA fragment (2.4 kb) was detected using genomic DNA from S. meliloti 1021, the parent strain (Fig. 1B). Sequencing both DNA fragments showed the absence of the lsrB open reading frame in the PCR fragment obtained from the lsrB1-2 mutant. This result was confirmed by RT-PCR experiments (Fig. 1C). Primers P1 and P4 allowed the amplification of an internal fragment of the lsrB ORF in both the wild-type and the lsrB1-2 complemented strain but not in the lsrB1-2 mutant strain.
FIG 1.
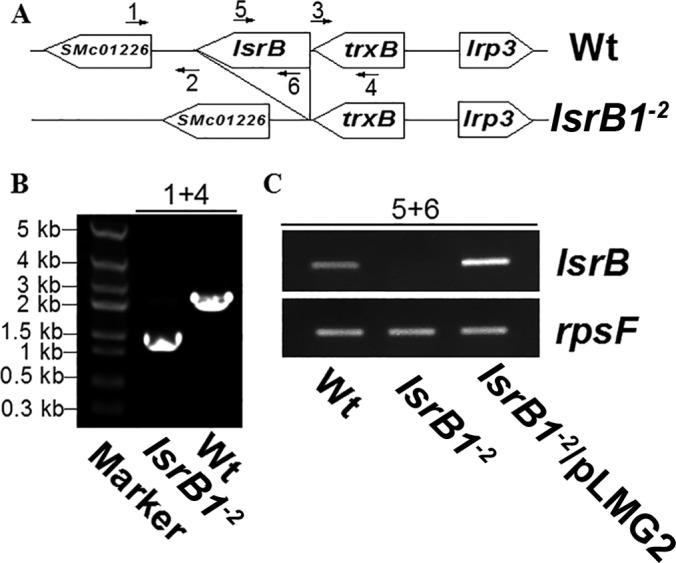
Verification of the constructed S. meliloti lsrB deletion mutant (lsrB1-2 mutant). (A) Genomic organization of the wild type (Wt; S. meliloti 1021) and the lsrB1-2 mutant. (B and C) The lsrB1-2 mutant was confirmed using PCR (B) and RT-PCR (C). The numbers 1 to 6 indicate primers used for PCR.
The S. meliloti lsrB1-2 mutant was sensitive to acidic pH and SDS.
The growth of the lsrB1-2 mutant was analyzed under different conditions. First, this mutant grew more slowly than the wild-type strain in a complex medium (LB/MC, pH 7.0) (Fig. 2A). Second, LB/MC medium at different pHs was used to assay sensitivity to acidic pH. The growth curve showed that the cell density of the lsrB1-2 mutant was clearly decreased in the medium at pH 6.0 and pH 6.5 compared to the density at pH 7.0 (Fig. 2A), indicating that this mutant was more sensitive to acidic pH. Third, when 0.1% SDS was added to the medium, the growth of lsrB2 was inhibited (Fig. 2B), suggesting that the mutant was sensitive to detergents. The expression of the lsrB gene from the complementation plasmid, pLMG2, rescued the growth deficiency of the mutant under different conditions (Fig. 2A and B). These results suggested that the envelope of the lsrB1-2 mutant was defective.
FIG 2.
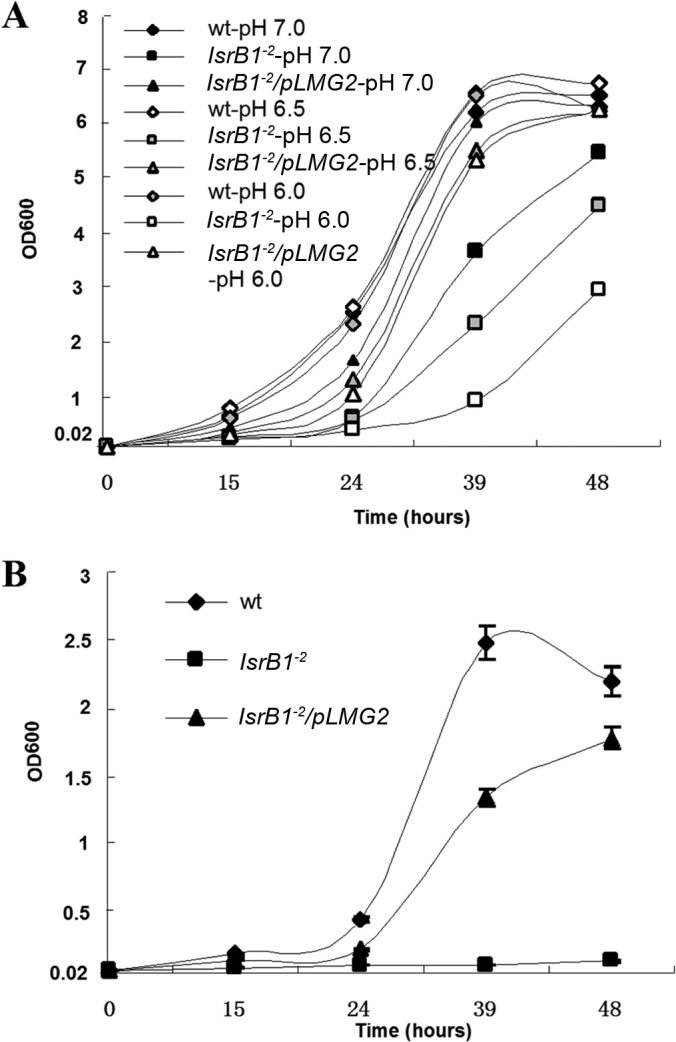
Growth of S. meliloti lsrB1-2 mutant under different stress conditions. (A) Sensitivity of the lsrB1-2 mutant to low pH in LB/MC broth; (B) sensitivity of the lsrB1-2 mutant to 0.1% SDS in LB/MC broth. All data were derived from three independent experiments and are expressed as means ± standard deviations (SD).
The S. meliloti lsrB1-2 mutant produced slightly less LPS.
Crude LPS from the S. meliloti lsrB1-2 mutant, the complemented mutant, and the parent strain was extracted with both the hot phenol and boiling methods. Both LPS extraction methods gave similar results. Total sugar in the LPS extracts was quantified using the sulfate-anthrone method. The results showed that the level of LPS from the lsrB1-2 mutant had a decreasing trend compared to the wild-type strain and the complemented mutant, although the difference was not very significant according to a Student t test. When DOC-PAGE was used to further analyze the LPS constituents, the lsrB1-2 mutant showed the same types of smooth and rough LPSs, but decreased levels were observed compared to those of the other two strains (Fig. 3). Taken together, these results indicated that LPS production was slightly reduced in the lsrB1-2 mutant and suggested that the S. meliloti LsrB protein was a positive regulator of LPS biosynthesis.
FIG 3.
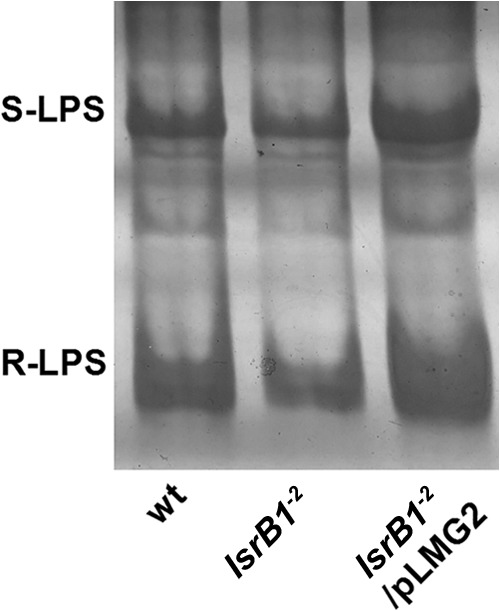
LPS produced by the S. meliloti lsrB1-2 mutant. LPS was assayed by DOC-PAGE. LPS was extracted from different S. meliloti strains that were grown in LB/MC broth (OD600, ∼0.8) with the boiled method. S-LPS and R-LPS represent smooth-LPS and rough-LPS, respectively.
Identification of lrp3-lpsCDE and trxB-lsrB operons.
Since a typical lysR gene often is located divergent from its target genes (16), the location and organization of S. meliloti lsrB and its adjacent genes were analyzed. In the genome of S. meliloti 1021, lsrB is adjacent to trxB, which is located divergent from lrp3 and lpsCDE. To test the possibility that lrp3-lpsC and trxB-lsrB genes consist of two operons, the transcriptional start sites of lsrB, trxB, lrp3, lpsC, and lpsD were mapped using 5′-RACE. Each of these genes (except lpsD) is transcribed from its own transcriptional start site (Fig. 4A), indicating that each gene has its own promoter. To determine whether they are cotranscribed from one operon, their transcripts were analyzed by RT-PCR. The results showed that transcripts containing both lrp3 and lpsCD were detected, just as trxB and lsrB were (Fig. 4B). These results suggest that lrp3 and lpsCDE constitute one operon but trxB and lsrB constitute another.
FIG 4.
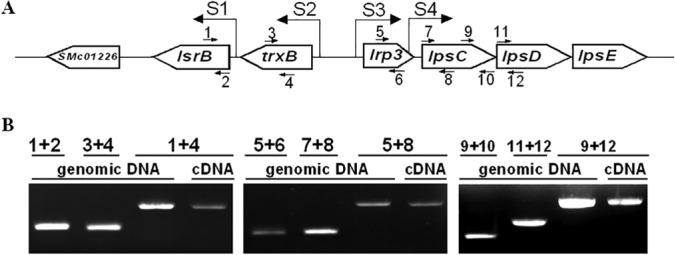
Transcripts and transcriptional start sites of lsrB, trxB, lrp3, lpsC, and lpsD. (A) Transcriptional start sites (S1 to S4) of lsrB, trxB, lrp3, and lpsC mapped by 5′-RACE are located at the nucleotide positions of 1700152, 1701200, 1701425, and 1701947 on the S. meliloti genome. TSSs of lsrB and trxB are almost consistent with published data (35). (B) The transcripts of the genes were determined by RT-PCR using the primers 1 to 12 and the template of genomic DNA and cDNA.
The expression of lpsCDE was positively regulated by S. meliloti LsrB.
Transcription of trxB, lrp3, lsrB, and the LPS core biosynthesis genes (lpsB, lpsC, and lpsE) was evaluated by qRT-PCR. The results showed that the transcript levels for lrp3, lpsC, and lpsE were significantly decreased in the lsrB1-2 mutant (only 1/53, 1/13, and 1/12 of the level for the wild type), whereas those of trxB and lpsB remained unchanged from those of the wild-type strain. A ribosome gene, rpsF, was used as an internal standard (Fig. 5A). These results suggested that LsrB regulates the expression of lrp3, lpsC, and lpsE. To confirm this, four promoter DNA fragments were predicted by the BDGP program together with the determined transcriptional start sites (Fig. 5A), fused to GUS, and introduced into S. meliloti strains (Fig. 5B). The GUS activities of these strains showed that the activities of the lsrB and lrp3 promoters decreased 175- and 273-fold in the lsrB1-2 mutant, but expression of the trxB and lpsC genes, in contrast to that determined by qRT-PCR, showed only 1.3- and 1.6-fold reductions (Fig. 5C). These results indicated that LsrB positively regulates the expression of lrp3 (together with lpsCDE) and lsrB at the transcriptional level.
FIG 5.
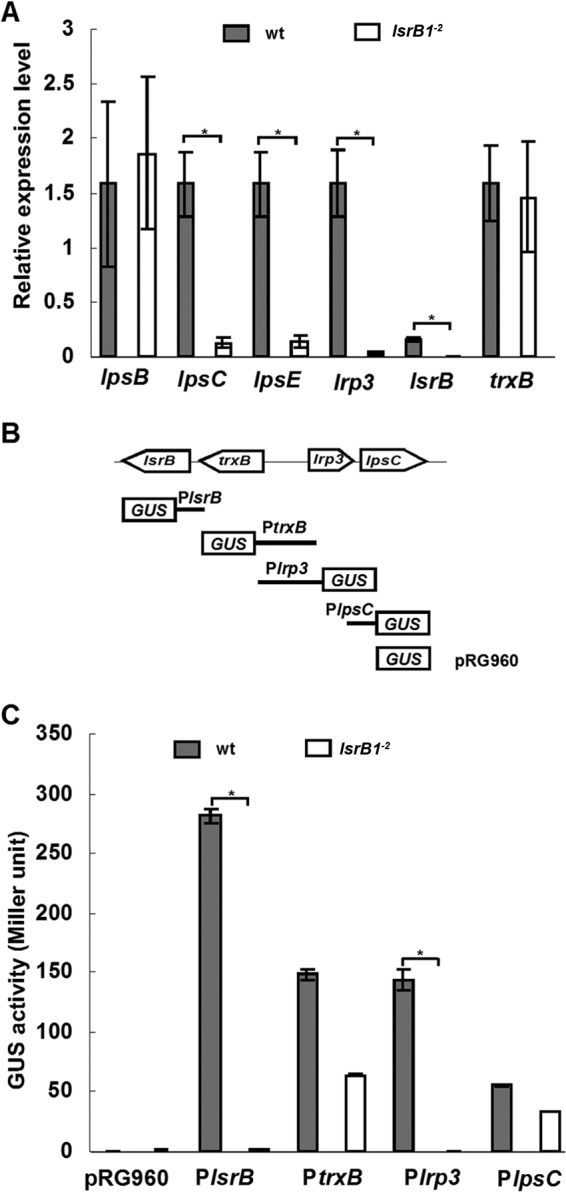
Expression of lpsC, lpr3, trxB, and lsrB genes in S. meliloti under free-living conditions. (A) Expression of LPS core biosynthesis genes (lpsB, lpsC, and lpsE) detected by qRT-PCR, with the internal standard of rpsF; (B) construction of promoter-GUS fusions; (C) β-glucuronidase activity of promoter-GUS fusions under free-living conditions. All data were derived from three independent experiments and are expressed as means ± SD. The S. meliloti strains were grown in LB/MC broth (OD600, ∼0.8).
LsrB binds to the TN11A box on the lrp3-lpsCDE promoter.
To determine whether LsrB regulates the transcription of the lrp3-lpsCDE operon by binding to the promoter DNA, we expressed the recombinant His-tagged LsrB protein in E. coli, and the purified protein was used to raise antibodies in two rabbits. The polyclonal antibodies then were applied to analyze the LsrB protein derived from S. meliloti strains. Immunoblotting confirmed the loss of LsrB protein in the lsrB1-2 mutant, whereas one specific band (at about 35 kDa) was detected in the lysates of the parent strain and the complemented mutant (Fig. 6A). Therefore, this anti-LsrB antibody was used in a ChIP assay to determine whether LsrB binds to specific genomic DNA in vivo. The ChIP data indicated that the DNA fragment derived from the lrp3 promoter was specifically enriched by the anti-LsrB antibodies in vivo (Fig. 6B). Hence, LsrB positively regulates the expression of the lrp3 and lpsCDE genes by binding to the promoter DNA of the operon.
FIG 6.
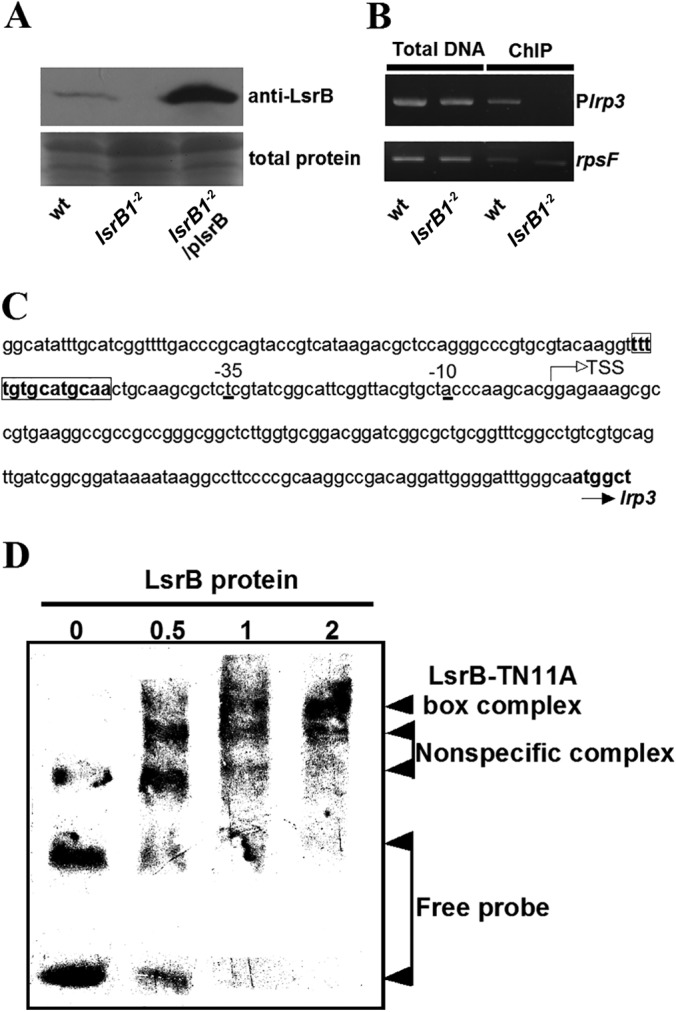
LsrB protein binds to the promoter of the lrp3-lpsCDE operon. (A) Immunoblot analysis of LsrB from the indicated S. meliloti strains. (B) ChIP assay of LsrB protein binding to the lrp3 promoter in vivo. Total DNA from the sonicated samples; wt, S. meliloti 1021; Plrp3, lrp3 promoter; rpsF, a ribosome protein gene used as a negative control. (C) The lrp3 promoter sequence. TSS, transcriptional start sites mapped by 5′-RACE. Box, TN11A box. (D) LsrB protein binding to the TN11A box of the lrp3 promoter in vitro. The amounts of loaded LsrB protein were 0, 0.5, 1, and 2 μg, respectively.
The target promoters of LysR family transcriptional factors usually contain at least one palindromic TN11A motif (17, 31). The putative overlapping TN11A boxes (TtttgtgcatgcA and TttgtgcatgcaA [uppercase letters represent the conserved nucleotides in a TN11A motif]) were found at position −47 from the transcriptional start site (Fig. 6C). Twenty-four-base-pair oligonucleotides containing the TN11A boxes were synthesized and used in an EMSA. The data showed that the recombinant LsrB protein bound directly to the TN11A boxes of the lrp3 promoter in vitro (Fig. 6D). These results indicate that LsrB regulates the expression of the lpsCDE genes by specifically binding to the TN11A box on the lrp3 promoter.
DISCUSSION
To our knowledge, there are a few regulators involved in LPS biosynthesis in rhizobia, although some S. meliloti gene mutants produce defective LPS (11). SyrA in S. meliloti and TtsI in S. fredii NGR234 contribute to the regulation of LPS O-antigen synthesis (15, 16). It also has been reported that the transcription elongation factor GreA participates in LPS core biosynthesis in S. meliloti 1021 and S. fredii HH103 (7). However, regulation of the genes for the LPS core is not well understood. In the present study, we found that LsrB, the S. meliloti transcriptional factor from the LysR family, positively regulated transcription of the LPS core biosynthetic genes, lpsCDE, by binding to the TN11A box on the lrp3-lpsC operon promoter. Therefore, LsrB is a new LysR family regulator that is associated with LPS production. Moreover, LsrB appears to modify only the amount of LPS, not its constituents, as is the case with SyrA and TtsI (15, 16).
The defective growth of the S. meliloti lsrB mutant could result from modification of the cell envelope or the redox status. At first, we observed that the lsrB1-2 mutant grew slowly on LB/MC or M9 agar plates. This result was consistent with the growth curve of the mutant in LB/MC broth (Fig. 2A). Importantly, this defective growth became more severe under stressful conditions, such as low pH and in the presence of a detergent (Fig. 2B), suggesting that the cell envelope of the mutant could be defective. This possibility was supported by LPS assays of S. meliloti cultures (Fig. 3). Additionally, the defective growth of the lsrB mutant could be associated with the cellular redox status. In our previously published data, the oxidative burst (reactive oxygen species accumulation) appeared in the lsrB-deleted cells because the catalase KatA was significantly increased and glutathione was somewhat decreased (32). Therefore, the growth defect of the lsrB mutant probably was caused by several factors.
The LPS decrease did not completely fit the expression of lpsCDE. We noticed from DOC-PAGE analysis that LPS production in the lsrB1-2 mutant was slightly reduced (Fig. 3), and this was consistent with downregulation of the lrp3-lpsCDE operon (Fig. 5). The quantification of total sugar revealed a trend of decreasing LPS in the lsrB1-2 mutant, but it was not very significant in a statistical analysis. It may be due to the upregulation of other polysaccharides (such as exopolysaccharide and cyclic-β-glucan) in the mutant. Importantly, the trend of decreasing LPS in the mutant was verified by another method, DOC-PAGE (Fig. 3). However, the extent of the LPS decrease in the lsrB mutant did not completely correspond with the amount of lpsCDE mRNA (Fig. 3 and 5). One possibility is that only a few LpsCDE proteins are required for LPS synthesis and that the expression of LpsCDE is regulated at the posttranscriptional level. The other is that other proteins play roles similar to those of LpsCDE in LPS core biosynthesis. These possibilities should be further verified.
Regulation of lpsCDE expression was complex under different conditions. The lsrB, trxB, lrp3, and lpsCDE genes can be organized into two different operons, but these genes are controlled by both their own promoters and the operon promoter (Fig. 4). However, the expression data suggested that only two promoters (those for lsrB and lrp3) could be regulated by LsrB (Fig. 5). The expression of lpsCDE also is controlled by two promoters (Plrp3 and PlpsC). Unlike Plrp3, PlpsC was not regulated by LsrB under free-living conditions, so the activity of the promoter-GUS fusion was not significantly altered between the wild type and the lsrB deletion mutant (Fig. 5C). This was supported by the EMSA data, as the PlpsC DNA was not able to bind to LsrB in vitro (data not shown), in contrast to Plrp3 (Fig. 6). We also detected the expression of promoter-GUS fusions in bacteroids from alfalfa nodules (31). Compared to their activity under free-living conditions, the activity of all four promoters was decreased in alfalfa nodules induced by the lsrB1-2 mutant (33), suggesting that the promoter activity for lpsC and trxB is regulated differently under free-living and symbiotic conditions.
The LsrB binding sites could be atypical. Two LsrB binding sites on the lrp3 promoter were predicted from the presence of TN11A motifs typical of LysR family regulators, but a palindromic sequence has not been found in these motifs (Fig. 6C). Some LysR family regulators, such as OxyR, do not have the binding sites containing a palindromic sequence (34). We also analyzed the LsrB binding sites on the lsrB promoter, and a putative TN11A motif was found. However, EMSAs showed that LsrB weakly bound to the DNA fragment containing this motif (data not shown). We did not exclude that other LsrB binding sites could exist on the promoter region. In time, it is likely that the conserved binding sites of LsrB on the S. meliloti genome will be identified using ChIP sequencing.
Interestingly, regulation of LPS biosynthesis mediated by LsrB could be conserved in some Rhizobium species, because the organization of lsrB, trxB, lrp3, and lpsC is conserved in S. meliloti 1021, S. medicae WSM419, S. fredii NGR234/HH103, Agrobacterium radiobacter K84, and most Mesorhizobium species, as revealed by genomic DNA analysis (Fig. 7).
FIG 7.
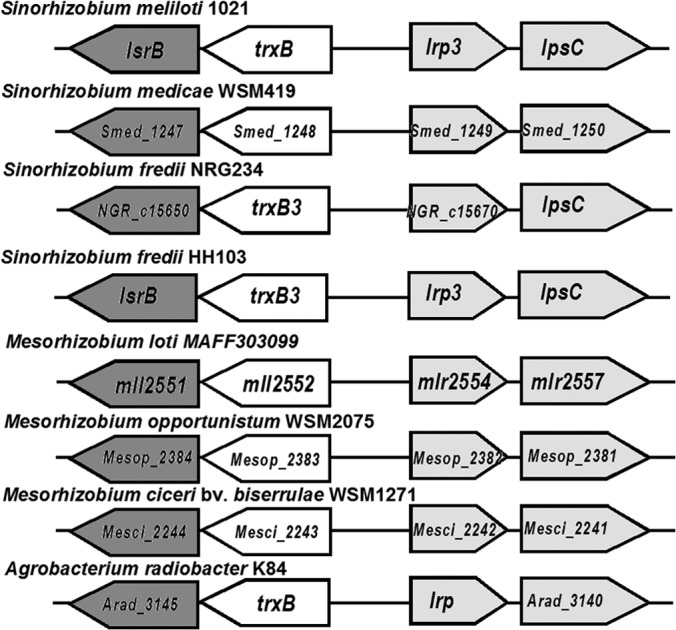
Organization of homologous genes to lsrB in various rhizobial genomes. Black boxes, lsrB homologs; gray boxes, the lrp-lpsC operon.
LPS production regulated by LsrB could be associated with alfalfa nodule development. In our previous work, the lsrB1-2 and lsrB1 (plasmid insertion) mutants induced heterogeneous, ineffective nodules on alfalfa (19, 33). Premature senescence appeared in these nodules, and abnormal bacteroid differentiation was found (33). Correspondingly, oxidative bursts were observed in most defective nodules (33). Therefore, it is possible that LsrB is involved in suppression of the host defense response. It has been reported that purified S. meliloti LPS can suppress the oxidative burst of Medicago cell cultures induced by invertase (8), and the null mutant of S. meliloti 1021 LPS core biosynthesis genes (lpsB and lpsCDE) induces deficient nodules with premature senescence on some ecotypes of alfalfa (4, 11). These data, together with our results, supported the possibility that LsrB can positively regulate LPS biosynthesis to suppress host defense responses that contribute to nodule premature senescence.
ACKNOWLEDGMENTS
We thank Christian Stahelin, Anke Becker, and Haiping Cheng for critical review of the manuscript.
This work was supported by the National Key Program for Basic Research (2010CB126501 and 2011CB100702) and the Natural Science Foundation of China (31070218 and 31370277) (L.L.).
Footnotes
Published ahead of print 20 June 2014
REFERENCES
- 1.Carlson RW, Hollingsworth RL, Dazzo FB. 1988. A core oligosaccharide component from the lipopolysaccharide of Rhizobium trifolii ANU843. Carbohydr. Res. 176:127–135. 10.1016/0008-6215(88)84064-3 [DOI] [PubMed] [Google Scholar]
- 2.Dazzo FB, Truchet GL, Hollingsworth RI, Hrabak EM, Pankratz HS, Philip-Hollingsworth S, Salzwedel JL, Chapman K, Appenzeller L, Squartini A. 1991. Rhizobium lipopolysaccharide modulates infection thread development in white clover root hairs. J. Bacteriol. 173:5371–5384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Maagd RA, Rao AS, Mulders IH, Goosen-de Roo L, van Loosdrecht MC, Wijffelman CA, Lugtenberg BJ. 1989. Isolation and characterization of mutants of Rhizobium leguminosarum bv. viciae 248 with altered lipopolysaccharides: possible role of surface charge or hydrophobicity in bacterial release from the infection thread. J. Bacteriol. 171:1143–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell GR, Reuhs BL, Walker GC. 2002. Chronic intracellular infection of alfalfa nodules by Sinorhizobium meliloti requires correct lipopolysaccharide core. Proc. Natl. Acad. Sci. U. S. A. 99:3938–3943. 10.1073/pnas.062425699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stacey G, So JS, Roth LE, Lakshmi SB, Carlson RW. 1991. A lipopolysaccharide mutant of Bradyrhizobium japonicum that uncouples plant from bacterial differentiation. Mol. Plant Microbe Interact. 4:332–340. 10.1094/MPMI-4-332 [DOI] [PubMed] [Google Scholar]
- 6.Reuhs BL, Geller DP, Kim JS, Fox JE, Kolli VS, Pueppke SG. 1998. Sinorhizobium fredii and Sinorhizobium meliloti produce structurally conserved lipopolysaccharides and strain-specific K antigens. Appl. Environ. Microbiol. 64:4930–4938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Margaret I, Lucas MM, Acosta-Jurado S, Buendia-Claveria AM, Fedorova E, Hidalgo A, Rodriguez-Carvajal MA, Rodriguez-Navarro DN, Ruiz-Sainz JE, Vinardell JM. 2013. The Sinorhizobium fredii HH103 lipopolysaccharide is not only relevant at early soybean nodulation stages but also for symbiosome stability in mature nodules. PLoS One 8:e74717. 10.1371/journal.pone.0074717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tellstrom V, Usadel B, Thimm O, Stitt M, Kuster H, Niehaus K. 2007. The lipopolysaccharide of Sinorhizobium meliloti suppresses defense-associated gene expression in cell cultures of the host plant Medicago truncatula. Plant Physiol. 143:825–837. 10.1104/pp.106.090985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheidle H, Gross A, Niehaus K. 2005. The lipid A substructure of the Sinorhizobium meliloti lipopolysaccharides is sufficient to suppress the oxidative burst in host plants. New Phytol. 165:559–565. 10.1111/j.1469-8137.2004.01214.x [DOI] [PubMed] [Google Scholar]
- 10.Lagares A, Hozbor DF, Niehaus K, Otero AJ, Lorenzen J, Arnold W, Puhler A. 2001. Genetic characterization of a Sinorhizobium meliloti chromosomal region in lipopolysaccharide biosynthesis. J. Bacteriol. 183:1248–1258. 10.1128/JB.183.4.1248-1258.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell GR, Sharypova LA, Scheidle H, Jones KM, Niehaus K, Becker A, Walker GC. 2003. Striking complexity of lipopolysaccharide defects in a collection of Sinorhizobium meliloti mutants. J. Bacteriol. 185:3853–3862. 10.1128/JB.185.13.3853-3862.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keating DH, Willits MG, Long SR. 2002. A Sinorhizobium meliloti lipopolysaccharide mutant altered in cell surface sulfation. J. Bacteriol. 184:6681–6689. 10.1128/JB.184.23.6681-6689.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferguson GP, Roop RM, Jr, Walker GC. 2002. Deficiency of a Sinorhizobium meliloti BacA mutant in alfalfa symbiosis correlates with alteration of the cell envelope. J. Bacteriol. 184:5625–5632. 10.1128/JB.184.20.5625-5632.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharypova LA, Niehaus K, Scheidle H, Holst O, Becker A. 2003. Sinorhizobium meliloti acpXL mutant lacks the C28 hydroxylated fatty acid moiety of lipid A and does not express a slow migrating form of lipopolysaccharide. J. Biol. Chem. 278:12946–12954. 10.1074/jbc.M209389200 [DOI] [PubMed] [Google Scholar]
- 15.Keating DH. 2007. Sinorhizobium meliloti SyrA mediates the transcriptional regulation of genes involved in lipopolysaccharide sulfation and exopolysaccharide biosynthesis. J. Bacteriol. 189:2510–2520. 10.1128/JB.01803-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marie C, Deakin WJ, Ojanen-Reuhs T, Diallo E, Reuhs B, Broughton WJ, Perret X. 2004. TtsI, a key regulator of Rhizobium species NGR234 is required for type III-dependent protein secretion and synthesis of rhamnose-rich polysaccharides. Mol. Plant Microbe Interact. 17:958–966. 10.1094/MPMI.2004.17.9.958 [DOI] [PubMed] [Google Scholar]
- 17.Schell MA. 1993. Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol. 47:597–626. 10.1146/annurev.mi.47.100193.003121 [DOI] [PubMed] [Google Scholar]
- 18.Galibert F, Finan TM, Long SR, Puhler A, Abola P, Ampe F, Barloy-Hubler F, Barnett MJ, Becker A, Boistard P, Bothe G, Boutry M, Bowser L, Buhrmester J, Cadieu E, Capela D, Chain P, Cowie A, Davis RW, Dreano S, Federspiel NA, Fisher RF, Gloux S, Godrie T, Goffeau A, Golding B, Gouzy J, Gurjal M, Hernandez-Lucas I, Hong A, Huizar L, Hyman RW, Jones T, Kahn D, Kahn ML, Kalman S, Keating DH, Kiss E, Komp C, Lelaure V, Masuy D, Palm C, Peck MC, Pohl TM, Portetelle D, Purnelle B, Ramsperger U, Surzycki R, Thebault P, Vandenbol M, et al. 2001. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 293:668–672. 10.1126/science.1060966 [DOI] [PubMed] [Google Scholar]
- 19.Luo L, Yao SY, Becker A, Ruberg S, Yu GQ, Zhu JB, Cheng HP. 2005. Two new Sinorhizobium meliloti LysR-type transcriptional regulators required for nodulation. J. Bacteriol. 187:4562–4572. 10.1128/JB.187.13.4562-4572.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finan TM, Kunkel B, De Vos GF, Signer ER. 1986. Second symbiotic megaplasmid in Rhizobium meliloti carrying exopolysaccharide and thiamine synthesis genes. J. Bacteriol. 167:66–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meade HM, Long SR, Ruvkun GB, Brown SE, Ausubel FM. 1982. Physical and genetic characterization of symbiotic and auxotrophic mutants of Rhizobium meliloti induced by transposon Tn5 mutagenesis. J. Bacteriol. 149:114–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schafer A, Tauch A, Jager W, Kalinowski J, Thierbach G, Puhler A. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73. 10.1016/0378-1119(94)90324-7 [DOI] [PubMed] [Google Scholar]
- 23.Khan SR, Gaines J, Roop RM, Jr, Farrand SK. 2008. Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl. Environ. Microbiol. 74:5053–5062. 10.1128/AEM.01098-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van den Eede G, Deblaere R, Goethals K, Van Montagu M, Holsters M. 1992. Broad host range and promoter selection vectors for bacteria that interact with plants. Mol. Plant Microbe Interact. 5:228–234. 10.1094/MPMI-5-228 [DOI] [PubMed] [Google Scholar]
- 25.Leigh JA, Signer ER, Walker GC. 1985. Exopolysaccharide-deficient mutants of Rhizobium meliloti that form ineffective nodules. Proc. Natl. Acad. Sci. U. S. A. 82:6231–6235. 10.1073/pnas.82.18.6231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buendia-Claveria AM, Moussaid A, Ollero FJ, Vinardell JM, Torres A, Moreno J, Gil-Serrano AM, Rodriguez-Carvajal MA, Tejero-Mateo P, Peart JL, Brewin NJ, Ruiz-Sainz JE. 2003. A purL mutant of Sinorhizobium fredii HH103 is symbiotically defective and altered in its lipopolysaccharide. Microbiology 149:1807–1818. 10.1099/mic.0.26099-0 [DOI] [PubMed] [Google Scholar]
- 27.Morris DL. 1948. Quantitative determination of carbohydrates with Dreywood's anthrone reagent. Science 107:254–255. 10.1126/science.107.2775.254 [DOI] [PubMed] [Google Scholar]
- 28.Jefferson RA, Burgess SM, Hirsh D. 1986. Beta-glucuronidase from Escherichia coli as a gene-fusion marker. Proc. Natl. Acad. Sci. U. S. A. 83:8447–8451. 10.1073/pnas.83.22.8447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu HY, Luo L, Yang MH, Cheng HP. 2012. Sinorhizobium meliloti ExoR is the target of periplasmic proteolysis. J. Bacteriol. 194:4029–4040. 10.1128/JB.00313-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grainger DC, Overton TW, Reppas N, Wade JT, Tamai E, Hobman JL, Constantinidou C, Struhl K, Church G, Busby SJ. 2004. Genomic studies with Escherichia coli MelR protein: applications of chromatin immunoprecipitation and microarrays. J. Bacteriol. 186:6938–6943. 10.1128/JB.186.20.6938-6943.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maddocks SE, Oyston PC. 2008. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154:3609–3623. 10.1099/mic.0.2008/022772-0 [DOI] [PubMed] [Google Scholar]
- 32.Lu D, Tang G, Wang D, Luo L. 2013. The Sinorhizobium meliloti LysR family transcriptional factor LsrB is involved in regulation of glutathione biosynthesis. Acta Biochim. Biophys. Sin. (Shanghai) 45:882–888. 10.1093/abbs/gmt083 [DOI] [PubMed] [Google Scholar]
- 33.Tang GR, Lu DW, Wang D, Luo L. 2013. Sinorhizobium meliloti lsrB is involved in alfalfa root nodule development and nitrogen-fixing bacteroid differentiation. Chin. Sci. Bull. 58:4077–4083. 10.1007/s11434-013-5960-6 [DOI] [Google Scholar]
- 34.Zheng M, Wang X, Doan B, Lewis KA, Schneider TD, Storz G. 2001. Computation-directed identification of OxyR DNA binding sites in Escherichia coli. J. Bacteriol. 183:4571–4579. 10.1128/JB.183.15.4571-4579.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlüter JP, Reinkensmeier J, Barnett MJ, Lang C, Krol E, Giegerich R, Long SR, Becker A. 2013. Global mapping of transcription start sites and promoter motifs in the symbiotic α-proteobacterium Sinorhizobium meliloti 1021. BMC Genomics 14:156. 10.1186/1471-2164-14-156 [DOI] [PMC free article] [PubMed] [Google Scholar]