

Abstract
Fresh pork sausage is produced without a microbial kill step and therefore chilled or frozen to control microbial growth. In this report, the microbiota in a chilled fresh pork sausage model produced with or without an antimicrobial combination of sodium lactate and sodium diacetate was studied using a combination of traditional microbiological methods and deep pyrosequencing of 16S rRNA gene amplicons. In the untreated system, microbial populations rose from 102 to 106 CFU/g within 15 days of storage at 4°C, peaking at nearly 108 CFU/g by day 30. Pyrosequencing revealed a complex community at day 0, with taxa belonging to the Bacilli, Gammaproteobacteria, Betaproteobacteria, Actinobacteria, Bacteroidetes, and Clostridia. During storage at 4°C, the untreated system displayed a complex succession, with species of Weissella and Leuconostoc that dominate the product at day 0 being displaced by species of Pseudomonas (P. lini and P. psychrophila) within 15 days. By day 30, a second wave of taxa (Lactobacillus graminis, Carnobacterium divergens, Buttiauxella brennerae, Yersinia mollaretti, and a taxon of Serratia) dominated the population, and this succession coincided with significant chemical changes in the matrix. Treatment with lactate-diacetate altered the dynamics dramatically, yielding a monophasic growth curve of a single species of Lactobacillus (L. graminis), followed by a uniform selective die-off of the majority of species in the population. Of the six species of Lactobacillus that were routinely detected, L. graminis became the dominant member in all samples, and its origins were traced to the spice blend used in the formulation.
INTRODUCTION
Fresh pork sausages are produced from a combination of pork meat, pork fat, water, and various spices. Unlike fermented sausages, these products are raw and therefore must be cooked thoroughly prior to consumption. Fresh pork sausages are preserved by refrigeration, typically being shipped frozen from the producer and subsequently maintained at refrigeration temperatures during retail sale and storage by the consumer. Remarkably, these products have a shelf life that can be as long as 55 to 60 days at refrigeration temperature, in contrast to a shelf life of only days for other fresh cuts of meat and fresh ground beef or ground pork. The explanation for such dramatic differences in shelf life is not completely understood but is due in part to the antimicrobial effects of the spice blends.
The shelf life of fresh sausage and other minced meat products at refrigeration is based largely on sensory characteristics and correlates with color and odor that can be perceived by the consumer. Color is influenced most strongly by the oxidation state of myoglobin in the tissue while odors are due to a complex combination of microbial products, remaining host enzymes, and physiochemical properties at the surface and within the meat matrix (1, 2). Traditional studies of the microbial populations found on meats and in meat production environments have associated spoilage of meats with broad groups of organisms, including members of the Proteobacteria (taxa belonging to the genera Actinobacter, Pseudomonas, Moraxella, Psychrobacter, Shewanella, and several members of the Enterobacteriaceae), the Bacilli (species of Bacillus, Brocothrix, Carnobacterium, Kurthia, Lactobacillus, Lactococcus, Leuconostoc, Pediococcus, Staphylococcus, and Weissella), and the Bacteroidetes (Bacteroides and Flavobacterium) (2, 3). Although these organisms occupy diverse phylogenetic space, they share in common the capacity for growth at refrigeration temperatures (psychrotrophic growth) and heterotrophic lifestyles on glucose, lipids, and amino acids, which are abundant in blended fresh ground meat products. These organisms have collectively been categorized as the spoilage-specific organisms (SSO), with a subset of SSO called the ephemeral spoilage organisms (ESO) (4) based on the ability of the latter to bloom transiently under conditions of storage or transport. From an ecological perspective, both the SSO and the ESO groups would be considered allochthonous species because both groups are acquired from other sources (e.g., hide, entrails, other ingredients, and the production environment) into the initially pristine muscle tissue after harvest.
Substrates for growth of the microbial population in meat products are abundant and diverse, ranging from glucose and simple carbohydrates, amino acids and protein, and readily available lipids from the fat that is blended into these products. Temporal analysis of some fresh sausages using culture-based approaches have shown that highly oxidative species of pseudomonads are detectable early, likely utilizing the available glucose and initiating proteolysis and lipid oxidation, followed by facultative/fermentative organisms such as Gammaproteobacteria and the lactic acid bacteria (LAB) (5, 6). The succession of microbial growth presumably leads to accumulation of metabolites that cause off-odors and off-flavors, discoloration, and gas production that are associated with sensory spoilage.
The microbial population that colonizes and ultimately spoils fresh sausage is likely to be highly variable, depending on which groups of microbial taxa the product has been exposed to and perhaps even the order in which they are encountered. The muscle tissue in situ is essentially devoid of microorganisms, with the exception of low levels of organisms that have been detected in lymph nodes. After hide removal, the once pristine tissue is rapidly colonized once exposed to the environment. Organisms are introduced onto the carcass surface through cross-contamination from hides, entrails, and the production environment, and organisms from spices can be introduced into the blended product. Fresh pork sausages can be produced from meats derived from hot-boned or cold-boned processes, referring specifically to prerigor processing that is done on unchilled carcasses or postrigor processing that is done on chilled carcasses. This choice of production system has a significant impact on the microbiota. For example, the hot-boning process can introduce organisms onto the carcass surface during dehiding and further downstream processing (e.g., entrails). Cold-boning, however, relies on hot water scalding of the skin surface, making it more likely that contamination will occur during evisceration or from the environment. In addition to differences in source of contamination, chilled postrigor processing exposes the incoming microflora to extremes of temperature. In contrast, hot-boned meat is often treated with a lactic acid wash immediately after dehiding, ultimately selecting for early colonization of the carcass by acid-tolerant organisms.
Given the rich history of study surrounding the microbiology of pork sausages, and the remaining gaps in our understanding of the actual growth and metabolic characteristics of the microbial population during spoilage, these meats were chosen as a model to examine the potential for next-generation DNA sequencing (NGS) methods to elucidate the detailed dynamics of microbial populations during spoilage. To this end, a combination of NGS-based analytical methods and traditional microbiological methods were used to quantify the microbiota of a model pork sausage.
MATERIALS AND METHODS
Sausage process/production.
The model samples for the present study were produced at a commercial manufacturing plant with a capacity to handle 500+ head per day. The raw material for making fresh pork sausage model was spent sows weighing about 500 lbs. The hogs received were stunned, followed by exsanguination of blood. This was a hot-boned or fresh-boned process. In the conventional processing of meats, “cold-boning” carcasses are chilled after slaughter for 24 h or more and processed in the chilled state (postrigor). In hot-boned process the meat was harvested, ground, and packaged immediately after slaughter (prerigor) at ambient temperature without chilling. The manufacturing process did not include an in-process and/or postprocess kill step in production of these fresh or raw or hot-boned pork sausage. However, the carcasses in this plant were treated sequentially with hot water spray and lactic acid shower for the reduction of microbial load and Gram-negative pathogens.
Fresh sausage was made from prerigor meat, trims, and spice blend. Meat, spice, and a small amount of added water was comminuted in a grinder, followed by blending in a blender to adjust for fat levels, and then packaged as samples. The proximate composition was 50% water plus additional 3% water as needed, 30% ± 3% fat and 40% meat. The samples used in the present study were made with mild sausage seasoning consisting of salt, sage, red pepper, black pepper, and minor amounts of additional spices and spice extractives (SpiceTec, Cranbury, NJ). Ground meat (45 lbs) was mixed with water, an antimicrobial system, and preseasoned mild sausage blend in a ribbon blender for 90 s and packaged as 1-lb chubs into coextruded packaging with ethylene vinyl alcohol (EVOH) as an oxygen and odor barrier and polyethylene as a moisture barrier on the food contact surface. The initial temperature of the ground chub was 33°C, and the final temperature after blending was 10°C. Samples were stuffed in the packaging material with appropriate code for each of the variables. Film was crimped with a poly clip and placed on a rack. Racks were transferred into a blast freezer at −20°C for 24 h and then transferred to a regular freezer. The model sausage chubs were then shipped and handled frozen. The total time was 30 to 45 min from live hogs to lactic acid shower and another 30 to 45 min from lactic acid shower to the kettle/grinding/packaging process. Thus, the “conversion” process of hogs to samples took place in <2 h. To simulate typical cold-chain management at retail, the frozen product was thawed in the refrigerator (slacking) for 2 days and subsequently stored at 4°C for the duration of the study. Day 0 began after the 2-day slacking period.
Antimicrobials.
The antimicrobial used in the present study is GRAS (generally recognized as safe) approved and is commercially available as a sodium lactate-sodium diacetate (Ultralac SL 564 [Hawkins, Minneapolis, MN]; i.e., sodium lactate [CAS 72-17-3] and sodium diacetate [CAS 126-96-5]). This is a clear, colorless to water white liquid, composed of 55 to 57% (wt/vol) sodium lactate, 3.5 to 4.5% (wt/vol) sodium diacetate, and 40% (wt/vol) water. Antimicrobials were added at 3, 4, 5, and 6% levels. The batch formula was adjusted for additional water (1.2, 1.6, 1.8, and 2.2%) from the antimicrobial liquid system. Also, untreated negative-control samples lacking the sodium lactate-sodium diacetate (LD) antimicrobial system were produced under the same production conditions. The untreated and treated model sausages were made in a single batch, and multiple chubs from each treatment were prepared and frozen to accommodate two technical replicates for plating and an additional replicate for NGS. Equal portions of chubs from each treatment were then shipped frozen to Deibel Laboratories for microbiological analysis and to the University of Nebraska Core for Applied Genomics and Ecology (CAGE) for microbiological analysis and NGS.
Sample preparation for traditional microbial plating and enumeration.
Chubs were stored in a frozen state until initiation of study. Two days prior to study initiation, all sausage samples were transferred from the freezer to a 4°C refrigerator. Samples thawed for 2 days before the day 0 pull. For the remainder of the study, all samples were stored at 4°C. On day 0, two 1-lb chubs of each sausage formulation were aseptically opened, and each was placed into a sterile blender bottle. The entire 1-lb chub was used for sampling to approximate the standard 325-g samples that are used for microbiological testing. A small portion of each sausage formulation was reserved for testing of pH, water activity (aw), sensory evaluation, and photographs. Each sausage sample was diluted with sterile Butterfield's phosphate buffer at a 1:5 dilution. A 1:5 dilution was used because the blender bottles were not big enough to accommodate a 1-lb sample diluted to 1:10. Each diluted sample was blended thoroughly until sample was homogenized in the buffer solution. From the 1:5 sample blender, 0.5 ml was removed and plated onto the respective media below for each analysis. This plating represented a 10−1 sample dilution. To make the first serial dilution, 0.5 ml from the blender was dispensed into 9.5 ml of sterile 0.1% peptone and vortexed thoroughly to yield a 10−2 sample dilution. For every serial dilution after the 10−2 dilution, 1 ml of peptone-diluted sample was dispensed into a 9-ml tube of 0.1% peptone water and vortexed. This was done to create dilutions of 10−3 to 10−6 as needed. The remaining sausage samples were stored under refrigeration (4°C) and sampled, as described above, on days 15, 30, 45, 60, and 80.
Incubation and enumeration.
Buffered and diluted samples were plated in duplicate onto the following media for each analysis. Standard methods agar incubated for 48 h at 35°C for the mesophilic aerobic plate count. Standard methods agar was incubated for 10 days at 4°C for the psychrotrophic aerobic plate count. Violet red bile glucose agar was incubated for 24 h at 35°C for Enterobacteriaceae. 3M Escherichia coli/coliform Petrifilm was incubated for 48 h at 35°C for coliforms and E. coli. APT agar with sucrose and bromcresol purple was incubated for 3 days at 25°C for lactic acid bacteria (LAB).
The log10 values for each test count were calculated and averaged for the two replicates of a given sample and for the two samples for each sausage formulation and pull day. Enumeration values of <10 CFU/g (nondetection) were assigned a log value of 1.00. Sample averages and standard deviations were calculated for each test, formulation, and pull day. The statistical difference between control and treatment variables and in between treatment variables was assessed at a 95% confidence level.
Sensory examination and electronic nose estimates.
The sensory characteristics were described for each sample by three individuals at the Deibel laboratory facility (including a meat scientist) and one individual at the CAGE laboratory. The individuals used a common range of categorical descriptors to describe odors, with increasing intensities from sour to very sour to rancid to very rancid to spoiled. The descriptors were reported and considered as categorical data, with consensus among the evaluations being required to mark the earliest time point for detectable sensory changes. The chemical composition of each sausage sample was also measured by an array of 10 metal oxide gas sensors in an Air-Sense Pen-3 E-Nose (electronic nose [eNose]). Samples archived at −80°C from each time point across treated and untreated model sausage were subsampled into 50-ml Falcon tubes and maintained at −20°C until the following day. While samples were still frozen, they were weighed (1.000 ± 0.050 g) in duplicate into 20-ml screw-cap headspace vials with polytetrafluoroethylene septa. The samples were immediately placed at 4°C until analysis. Immediately prior to analysis, the samples were equilibrated to room temperature for 15 min. A blank consisting of an empty vial was analyzed before each batch of samples. The run parameters on the eNose were as follows: flush time = 40 s, zero point trim = 10 s, presampling time = 5 s, measurement time = 60 s, chamber flow = 400 ml/min, injection flow = 400 ml/min, and dilution = 200 ml/min. Samples were simultaneously injected with two 18-gauge needles at the start of data collection, an inlet needle leading to room air and an outlet needle connected to the sample transfer line of the eNose. Since injection was performed manually, there was a ±1-s delay between injection and the start of data collection. Raw data from the eNose was exported in ASCII format. Mean responses from the sensors were used for subsequent data analyses. Responses from the individual sensors showing significant changes over time for individual treatments were first identified by analysis of variance (ANOVA). Candidate taxa associated with changes in the responses of individual sensors were identified by correlation analysis and linear regression with the relative abundances of individual taxa for each corresponding time point.
Next-generation sequencing.
Bacteria were removed from 10 g of the sausage by soaking in (Tris-EDTA [TE] buffer with 0.1% Triton X-100) and harvested by centrifugation at 10,000 × g. The pellet was resuspended in TE buffer, and the suspension was layered onto a cushion of 36% sucrose and centrifuged for 15 min at 2,000 × g. Bacterial cells from the resulting pellet were resuspended in ASL buffer, and DNA extractions were performed using the QIAamp stool kit (Qiagen) on a Biosprint 96 automated extraction system essentially as previously described (7, 8). The composition of the microbiota was assessed by deep pyrosequencing of PCR products originating from the V1-V2 region of the 16S rRNA gene with bar-coded fusion primers containing Roche-454 A or B titanium sequencing, followed by a unique eight-base barcode sequence (indicated by “B” in the following sequences) and the 5′ ends of primer A-8FM (5′-CCATCTCATCCCTGCGTGTCTCCGACTCAGBBBBBBBBAGAGTTTGATCMTGGCTCAG-3′) and primer B-357R (5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAGBBBBBBBBCTGCTGCCTYCCGTA-3′). All PCRs were quality controlled for amplicon saturation by quantifying and comparing band intensities of the PCR products after gel electrophoresis with standards using GeneTools software (Syngene). Amplicons from 48 individual samples were pooled in equal amounts, gel purified, quantified by Pico Green analysis, and used for emulsion PCR (emPCR). After recovery and enrichment for DNA-containing beads, the emPCR products from the 48-sample pools were sequenced on individual regions of two-region Picotitre plates on a Roche-GS-FLX machine using titanium sequencing chemistry.
Data processing.
Raw data from the Roche-454 GS-FLX machine was first processed through specialized scripts that filtered the data on the basis of the following criteria, with sequences not meeting these criteria being removed from further analysis: (i) a complete forward primer sequence and barcode; (ii) ≤2 “N” in a sequence read, where N is equivalent to an interrupted and resumed sequencing signal from sequential flows; (iii) a sequence of >200 and <500 nucleotides; and (iv) an average quality score of ≥20 across the entire length of the sequence.
After a filtering step, reads were trimmed to remove 5′ and 3′ adapter and primer sequences, parsed by barcode into corresponding sample files, automatically associated with a matching “.QUAL” file containing the quality scores, uploaded into a MySQL database, and associated with sample information. The MySQL database tables are stored on a database server and available online (http://cage.unl.edu).
To help normalize taxonomic assignment and phylogenetic distance estimates of individual sequence reads, the entire data set was initially processed through the Multi-CLASSIFIER algorithm, which assigns hierarchical taxonomic status to each sequence read based on a covariance model developed from a training set (9, 10). This algorithm is capable of processing very large data sets and was recently shown to provide adequate taxonomic assignments to pyrosequencing data (11). After being processed through the Multi-CLASSIFIER, the sequences were parsed into “classified” and “unclassified” sets based on meeting threshold limits of 0.8 at the genus level against the Multi-CLASSIFIER model.
Classified reads were assigned species-level status using a BLAST pipeline that associated the reads with species-level taxonomic assignment using a curated database developed from RDP and SILVA databases of curated 16S rRNA sequences (9, 12). Sequences were considered a species match if they achieved 97% identity with a reference sequence over a minimum of 200 bases of contiguous BLAST alignment. Sequence reads that failed to meet the 0.8 scoring threshold at the genus level from the Multi-CLASSIFIER algorithm (“unclassified” reads) were further processed into Operational taxonomic units (OTU) using CD-Hit to estimate the phylogenetic distances and cluster at a 97% cutoff (13). The taxonomic status of these OTU was approximated by BLAST against the curated database. Reads from each bin from the combined “classified” and “unclassified” portions of the pipeline were then normalized relative to the total number of reads for each sample (relative abundance). For statistical analyses, this relative abundance value was subjected to log10 transformation to reduce the effects of extensive variation in values across multiple samples. Alpha and beta diversity estimates were made with OTU developed from parsing our filtered data through the Pyrosequencing Pipeline of the Ribosomal Database Project using the ALIGNER and Complete linkage clustering programs at a 97% threshold to define the OTU.
Isolation of Lactobacillus species from model sausage and spice mix.
Samples (∼1 g) of the model sausage from archived samples of various time points and a 1-g sample of an archived sample of the spice blend were diluted in 0.1% peptone, and dilutions were plated onto APT or MRS agar (Neogen). A total of 96 colonies were randomly selected from dilutions, giving rise to well-isolated colonies across all of the samples. After being streaked onto MRS for purification, DNA was extracted from individual isolates and the 16S rRNA gene amplified by using the primers 8F (AGAGTTTGATCMTGGCTCAG) and 16SU2 (ATCGGYTACCTTGTTACGACTT) with PCR conditions of 95°C for 2 min, followed by 30 cycles of 95°C for 30 s, 55°C for 45 s, and 72°C for 45 s, with a final 5-min extension at 72°C on the last cycle. The resulting amplicons were confirmed by agarose gel electrophoresis, followed by Sanger DNA sequencing (Michigan State University). Taxonomic assignments were made by alignment to 16S rRNA sequences from type strains of Lactobacillus species by using cmALIGN and complete linkage clustering with distance cutoffs ranging from 0 to 3%. The genetic relationships of the isolates from the spice blend and from the model sausage were further examined by using RAPD [random(ly) amplified polymorphic DNA] with a single primer CCCGTCAGCA and PCR conditions of 95°C for 2 min, followed by 45 cycles of 95°C for 30 s, 32.5°C for 45 s, and 72°C for 45 s, with a final extension of 5 min at 75°C at the end of the 45 cycles. RAPD products were separated by agarose gel electrophoresis and visualized by ethidium bromide staining.
RESULTS
Viable cell counts from untreated model pork sausage.
To systematically evaluate the dynamics of the microbial populations in the product, we used a combination of traditional microbiological plating methods and 16S rRNA gene-based pyrosequencing. Aerobic plate counts (APCs) from untreated or samples at the initiation of the experiment (day 0) contained 103 mesophilic organisms (Fig. 1). When the plates were incubated at a low temperature, 10% of these day 0 mesophilic APCs were psychrotrophic and capable of growth at 4°C. Enumeration of LAB, Enterobacteriaceae, and coliforms at day 0 showed that each comprised 1 to 2 logs of CFU or ca. 10% of the total APC. After freezing and during the thaw and storage period at 4°C, the population showed a sharp rise in APC (Fig. 1), reaching 106 by day 15 and peaking at 108 by day 45. Not surprisingly, the psychrotrophic APC (Fig. 1) showed an identical profile, reaching 106 on day 15 and stabilizing at 107 by day 30. The LAB (Fig. 1) comprised the biggest proportion of the total and psychrotrophic APCs, rising to 107 by day 30 and remaining in this range for the duration of the experiment. The Enterobacteriaceae increased to 106 by day 30 and also remained constant thereafter. A similar profile was exhibited by the coliform population, which increased to 104 by day 15 and remained constant throughout the experiment.
FIG 1.

Enumeration of microorganisms from untreated pork sausage product stored at 4°C. Total mesophilic APCs, psychrotrophic APCs, Enterobacteriaceae, coliforms, generic E. coli, and LAB were determined. The averages of two replicate platings are plotted, with the standard deviations indicated by the whiskers.
Microbiota analysis and reproducibility.
To gain a more detailed view of the microbial population in the untreated product, deep pyrosequencing of 16S rRNA gene amplicons was performed across samples from the same batch of product used for traditional microbiological analysis above. Phylogenetic analysis of the pyrosequencing data from the time zero and endpoint (day 80) samples was first used to estimate alpha and beta diversity of the populations. A total of 904 OTU from 15,983 sequences were identified from the day 0 sample and 1,309 OTU from 23,229 sequences obtained from the untreated sample stored at 4°C for 80 days. Rarefaction and Shannon diversity estimates (Fig. 2) both showed a trend toward decreased diversity after 80 days of storage at 4°C, signifying that the total number of taxa changes only moderately, whereas the evenness of the taxonomic distributions was more susceptible to change over time at 4°C.
FIG 2.

Rarefaction and Shannon diversity estimates of 16S rRNA gene sequences from untreated pork sausage stored at 4°C. Phylogeny-based analysis was used to develop OTU from pyrosequencing data of the day 0 and day 80 samples of the untreated product using the cmALIGN and complete linkage clustering at 97% identity (RDP Pyrosequencing Pipeline). The data were rarefied using the RDP pipeline rarefaction application (y axis on left), while the Shannon diversity was estimated using the standard equation Σprop1 × ln prop1, where prop1 is the proportion of each individual taxon of the total sequences from a sample (y axis on the right).
With complex populations of microbes present at a range of abundances, sample error can be an issue that influences data structures. The threshold of relative abundance that was subject to significant sample error was therefore estimated by comparing the relative abundances of a range of taxa across the day 0 samples from untreated and treated samples. Because all of the treatment groups originated from the same batch of model sausage, samples from day 0 of each treatment group were essentially technical repeats. The top 20 most abundant taxa that were found across at least four of the five day 0 samples were used as a basis for measuring repeatability. Linear regression was performed on all pairwise comparisons of the abundances of these 20 taxa from each treatment (see Table SA1 and Fig. SA1 in the supplemental material), giving an overall correlation coefficient (R2 = 0.6483), with most dispersion in the data occurring with data points below a relative abundance of 0.001. This result is similar to what we have reported previously for fecal samples (7). Taxa consistently having abundances below this threshold or which were sparsely distributed were therefore avoided in subsequent analyses.
Microbiota analysis and a detailed view of microbial population dynamics.
To visualize the dynamic behavior of the complex microbial population over the course of the 80-day refrigerated storage, taxonomy-based analysis was used to compare taxonomic distributions across samples from 15-day intervals between day 0 and the endpoint at day 80 (Fig. 3). Plots of the relative abundances of the dominant taxa over time showed a bimodal dynamic brought about by dramatic changes in the relative abundances of dominant taxa. At time zero, the dominant members of the population belong to the classes Bacillus and Gammaproteobacteria, with smaller proportions of Actinobacteria, Clostridia, Bacteroidetes, and Flavobacteria also present.
FIG 3.

Dynamic changes in relative abundances of taxa over time in untreated pork sausage during refrigeration at 4°C. The relative abundances of dominant taxa identified by pyrosequencing of 16S rRNA gene amplicons are plotted from samples of the untreated pork sausage after thawing and storage at refrigeration over 80 days. Graphs were plotted with each of the 15-day time points over the 80-day storage period. Taxa are colored by phyla, with Firmicutes in shades of blue-purple, Proteobacteria in shades of green, Bacteroidetes in yellow, and Actinobacteria in red.
Members of the Bacilli dominate the population, with the family Leuconostocaceae being the most abundant taxon detected, and the species Weissella confusa and Leuconostoc citreum comprising 50 and 20%, respectively, of the total population. These two taxa, along with Lactobacillus gasseri (present at 1%), likely account for the enumerable LAB detected by plating, since other LAB species belonging to the genera Lactobacillus, Streptococcus, and Lactococcus were found in much smaller proportions ranging from 0.1 to 0.01%). From day 0 to day 15, the first phase (phase I) of population change occurs, with the majority of the taxa declining to 0.1% or lower in relative abundance, including Weissella confusa and Leuconostoc citreum. These taxa are displaced by a dramatic rise in the abundance of two dominant species of Pseudomonas, with P. lini comprising nearly 80% of the sequences and P. psychrophila comprising 6.6%. The rise of these two taxa corresponds with the dramatic >1,000-fold rise in the APC and the psychrotrophic APC (Fig. 4), and these two taxa alone most likely account for this increase. Also occurring during this first phase is the initial 500-fold decline in relative abundance of the LAB species Lactobacillus gasseri, which was the dominant LAB present at day 0. This decline is offset by a by a >500-fold increase in the abundance of Carnobacterium divergens, and collectively these two species likely account for the viable LAB enumerated by plating (Fig. 4). Three additional species that were undetectable at day 0 also become detectable by day 15: Yersinia mollaretti at nearly 1%, an OTU of Serratia sp. at 0.2%, and Buttiauxella brennerae at 0.1%. The rapidly increasing abundance of these species corresponds to the >100,000-fold increase in increase in the counts of Enterobacteriaceae from day 0 to day 15 observed by plating (Fig. 4).
FIG 4.

High-resolution snapshot of microbial population dynamics in untreated pork sausage. (A) Total mesophilic APCs, psychrotrophic APCs, and LAB, Enterobacteriaceae, coliforms, and generic E. coli were enumerated from untreated pork sausage after thawing and storage at refrigeration temperature for 80 days. The log-transformed data from each treatment group is plotted from each of the 15-day time points taken over the 80-day refrigeration period, and the means and standard deviations of the log-transformed data are plotted at each of the 15-day time points. The pH of the samples is plotted on the second y axis on the right. The light red arrow indicates onset of sour odors, and the red arrow at day 45 marks the onset of a sour, putrid, or spoiled odor that would yield the product objectionable. (B) Relative abundances of taxa of interest from the pyrosequencing data derived from paired untreated pork sausage after thawing and storage at refrigeration temperatures for the same 80-day period.
By day 30 the population again underwent a dramatic shift to phase II, with the dominant pseudomonads now displaced by a sharp 300-fold increase in the relative abundance of Lactobacillus graminis (to 36% of the total population) and nearly 10-fold increases in Carnobacterium divergens (now 5.7%), Yersinia mollaretti (now 5.6%), the OTU of Serratia (now 3%), and Buttiauxella brennerae (now 5.6%). In addition to the prominent change in dominant microbial taxa, consensus changes in the sensory characteristics occur with the onset of detectable sour odors (Fig. 4A), suggesting that the change in dominant species at day 30 could be a hallmark of sensory degradation and may be a signature of the onset of events leading to sensory spoilage. Electronic nose measurements of subsamples from these time points are also consistent with the onset of significant changes in the chemical content of the matrix (see below). The striking changes in species composition and onset of sensory changes was coincident with a small, but noticeable change in the apparent growth rate of the populations enumerated by plating (Fig. 3 and Fig. 4A), a result that would be expected with sharp changes in abundances of dominant (growing) species. The succession of members of the Enterobacteriaceae and the dominant LAB species Carnobacterium divergens and Lactobacillus graminis also converts the dominant population from oxidative metabolism to organisms that can respire on alternative terminal electron acceptors (Enterobacteriaceae) or are fermentative (Enterobacteriaceae and LAB) and which can utilize a wide variety of amino acid and lipid substrates Whether the population shift is actually driven by changes in substrate availability, the accumulation of metabolic end products, and/or competition for niches remains to be determined.
By day 45, Lactobacillus graminis, Yersinia mollaretti, Buttiauxella brennerae, and Carnobacterium divergens remain dominant and are steadily detected as the most abundant taxa throughout the shelf life. Lactobacillus graminis abundance peaked at day 30, declined slightly at day 45, and remained relative constant through the remainder of the incubation period, paralleling the behavior of the LAB counts detected by plating. In contrast, Yersinia mollaretti continued to increase until day 45, suggesting that this organism accounts for the continued increase in APC as determined by plating between day 30 and day 45 sampling. Importantly, day 45 also marks the emergence of objectionable sensory characteristics, with sour, putrid, or spoiled odors now becoming prominent. An interesting feature of the pyrosequencing data is that other taxa detected at day 0 declined until day 45 but abruptly increased in abundance at day 60 and day 80 (Fig. 3 and 4B). Because the pyrosequencing data reflect the relative abundances of taxa and not their absolute abundance, it is unclear whether this increase reflects the growth of the minor taxa or instead reflects a decrease in the dominant organisms of the population. The phylogenetic and physiological diversity of organisms whose relative abundance increases suggests that their increase is only apparent and due to decline in the dominant taxa.
Effects of LD treatment.
In raw products, antimicrobial compounds can be used to extend shelf life and provide a product with more uniform/predictable patterns of spoilage. Because compounds such as lactate-diacetate (LD) are known to influence the growth of microorganisms in fresh pork sausage and in processed meats (14–17), we introduced LD into the model sausage matrix at a range of concentrations from 3 to 6%. Across all LD treatments, the microbiota of the model sausage showed a dramatic response, with most groups of organisms remaining static or showing significantly reduced growth (Fig. 5A). The LD effect could be observed even in the mesophilic APCs, where the samples only rose to 103 by day 15 (Fig. 5A), whereas the untreated was already at 106 (Fig. 4A). On day 30, when the untreated sample had 107 CFU/g (Fig. 4A), the 3% LD rose to only 104 (Fig. 5A), whereas the 4 to 6% LD samples remained at 103 logs and hovered there for the duration of the experiment, with a trend toward a 0.5- to 1-log reduction at 60 to 80 days. Psychrotrophic APCs (Fig. 4A and 5A) showed a very similar response to the LD treatments, where the rapid >100,000-fold rise to 106 CFU/g observed by day 15 in untreated samples (Fig. 4A) was now limited to only the 102-to-103 range (Fig. 5A). Even the lowest level of LD (3%) limited the psychrotrophic population to only 103 by ca. 30, whereas other LD concentrations showed slight reductions in the population. Overall, the psychrotrophic APC data showed a pattern similar to that seen in the (mesophilic) APC testing: the control sample showed a rapid increase early in the study and remained high; the 3% LD showed a slight increase starting on day 30; and the 4, 5, and 6% LD samples remained relatively constant through 80 days, with a slight decrease at the end of the study.
FIG 5.

High-resolution snapshots of microbial population dynamics in untreated and treated pork sausage. (A) The total mesophilic APCs, psychrotrophic APCs, and lactic acid bacteria, and Enterobacteriaceae were enumerated from pork sausage containing 0, 3, 4, 5, and 6% LD. The log-transformed data from each treatment group are plotted from each of the 15-day time points taken over the 80-day refrigeration period, along with the standard deviations of the log-transformed data (whiskers). (B) The relative abundances of dominant taxa identified by pyrosequencing of 16S rRNA gene amplicons are plotted from paired samples taken at 15-day time points over the course of the 80-day shelf life (x axis) for the 3, 4, 5, and 6% LD treatments.
The effects of LD treatment on LAB (Fig. 5A) were also similar to that of mesophilic and psychrotrophic APCs, with the 3% LD treatment reducing LAB counts from 106 in untreated to 103 in treated samples on day 15, and the population reached a maximum of 104 by day 30, where it remained for the duration. Increasing amounts of LD appeared to have little effect as the remaining three treatments (4, 5, and 6%) rose to 103 on day 15 and remained at that level through the rest of the study. Although LD treatment lowered the growth rate of the mesophilic and psychrotrophic APCs and LAB, it completely suppressed growth of Enterobacteriaceae, coliforms, and E. coli, which remained at 10 CFU/g or lower in all treated samples.
As expected from the impact on the microbiota, the LD-treated sausage also displayed much more limited changes in sensory characteristics (Fig. 5A). In all treatments, consensus sensory changes were not perceptible until day 45. In the 3% LD treatment the rate of sensory deterioration was much slower than in the untreated samples, yielding objectionable putrid odors only at the endpoint. Within the limits of descriptive olfactory sensing, there was some degree of dose-response relationship, with the higher concentrations yielding only slightly rancid odors at the endpoint.
Pyrosequencing data from the LD-treated samples revealed just how dramatically the LD affects dynamics of the populations (Fig. 5B). Noticeably, LD completely suppressed the bimodal successions with multiple taxa that were observed in the untreated samples, yielding instead a single modality where the relative abundance of Lactobacillus graminis increased in all LD treatments and became the dominant member of the population. Lactobacillus graminis consistently became dominant at day 30 in the 3 to 5% LD treatments but was delayed until day 45 in the 6% treatment. Across all treatments, the increase in Lactobacillus graminis to dominance was also concurrent with decreased abundances of Leuconostoc citreum, Lactobacillus gasseri, and Weisella confusa, which were generally abundant in the samples at day 0 but decreased precipitously over time. The erratic behavior of Weisella confusa is a curiosity. Although this organism was dominant at day 0 of the untreated sample, it was undetected in day 0 samples of treated sausage but was detectable at relatively high levels from day 15 to day 30 in the 3, 5, and 6% samples, followed by a substantial drop in all except the 6% treatment. In contrast, many other taxa that were present at lower abundance (and thus more susceptible to sample error) were consistently detected and showed similar trending.
The behavior of L. graminis as detected by pyrosequencing is similar in untreated and LD treated sausage and essentially mirrors viable LAB plate counts from these same samples (Fig. 5A and B), with growth occurring during the first 30 to 45 days leading to a relatively stable population thereafter. In contrast, the majority of other taxa in the microbiota remain relatively stable for the first 30 to 45 days, followed by a remarkably uniform decline. Because the LAB abundances detected beyond day 30 to 45 are essentially flat, the apparent differential between the abundance of L. graminis and the remaining taxa at these later time points must therefore reflect both inactivation of the remaining taxa and degradation of their DNA. This result is consistent with the L. graminis population surviving a striking selective “die-off” that occurs among most of the other taxa in the microbiota in the LD-treated samples. This “die-off” is unique to the LD treatment, since many of these same organisms underwent a nearly synchronous increase in relative abundances at day 60 in the untreated samples.
Unique behavior of L. graminis compared to other Lactobacillus species.
Given the opposing behavior of L. gasseri and L. graminis, we subsequently compared the behavior of the different Lactobacillus species observed in the sausage samples. Six different species, including L. amylovorus, L. apodemi, L. curvatus, L. gasseri, L. graminis, and L. sakei could be assigned with a high degree of confidence (Fig. 6) (although there were other Lactobacillus OTU but with low-confidence species placements). An important observation was that the initial abundances of the Lactobacillus species do not account for their behavior: L. gasseri was actually more abundant than L. graminis, and though its abundance increased slightly in untreated samples, it fell precipitously in all treated samples. Only L. sakei, L. curvatus, and L. graminis showed any propensity for growth or stability in treated or untreated samples. L. graminis was the dominant taxon (Fig. 6B) and likely accounts for the LAB detected by plating (Fig. 6A). L. curvatus and L. apodemi were low-abundance taxa that showed some increase in the 3% LD samples, but the remaining treatment samples showed a sparse distribution of these organisms that is characteristic of sample error with low-abundance taxa.
FIG 6.

Dynamic behavior of different Lactobacillus species in untreated and LD-treated pork sausage. (A) The total mesophilic APCs, psychrotrophic APCs, and LAB, and Enterobacteriaceae were enumerated from pork sausage containing 0, 3, 4, 5, and 6% LD. The log-transformed data from each treatment group are plotted from each of the 15-day time points taken over the 80-day refrigeration period. (B) The relative abundances of the six different species of Lactobacillus that were detectable in the pyrosequencing data are plotted over time across the untreated and treated samples.
Signatures of spoilage in untreated and treated sausage.
A disparity in the dynamic behavior of phylogenetically related taxa such as Leuconostoc citreum and L. graminis suggests the existence of interactions between groups of organisms. To define the groups of taxa potentially showing interactions, we used correlation analysis to group the dominant taxa into five different groups that show strong correlation within group (A to E) (Fig. 7). The three Lactobacillus species L. graminis, L. curvatus, and L. apodemi comprise one of the most highly correlated groups (group A) and which shows strong negative correlation with nearly all taxa. A second group of correlated taxa (group B) is formed by Y. mollaretti, B. brennerae, C. divergens, and the Serratia OTU4, all of which increase in abundance during the second stage of the succession in untreated sausage and all of which also remain only as low abundance taxa in treated samples. Like the Lactobacillus graminis group, this group is negatively correlated with most organisms, but negative interactions of this group manifest only in untreated sausage, and this is reflected in their weak negative correlations with other taxa. A third group (group C) of taxa is made up of Pseudomonas lini and P. psychrophila, which comprise the taxa showing dramatic increases in relative abundance during the first stage of succession in untreated sausage. This group also shows only weak negative interactions with other taxa, reflecting the inability of this group to grow in any of the treated samples. Fourth (group D) is a group of highly correlated taxa that that show weak negative interactions with groups A to C. Group D taxa largely comprise organisms typically found in the gastrointestinal tract (e.g., Prevotella, Bacteroides, Ruminococcus, Blautia, and Roseburia). It seems unlikely that any of these taxa would grow well, if at all, in the sausage matrix, even at later stages of storage when the environment becomes much more reducing. However, they are consistently detected at low levels, indicating that the cells (or their genomic content) are stable at storage and remain detectable. Lastly, group E comprises a large number of taxa that show a high degree of negative interaction with group A and, to a lesser degree, group B. Several members of this group are species of Streptococcus, Acinetobacter, and Weissella, along with Leuconostoc citreum. When the abundances of these taxa are plotted along with Lactobacillus graminis (Fig. 8), it is clear that this core set of group E taxa are indeed negatively associated with L. graminis across all samples and time points. Even more striking is the location of the inflection point between L. graminis and group E taxa (best observed as the crossover between L. graminis and L. citreum). Remarkably, this inflection point in untreated and treated samples corresponds to the time at which sensory characteristics of the product begin to change and in treated samples also corresponds to the time at which the selective die-off initiates. Collectively, the behavior of group E taxa and L. graminis suggests that these organisms may represent a “microbial signature” of the onset of physiochemical changes that are associated with spoilage and the initial relative abundances of these taxa could serve as a predictor of shelf life.
FIG 7.

Correlation analysis of detectable taxa in the microbiota of the pork sausage. Correlations were calculated between all pairwise combinations of taxa for each time point in treated and untreated samples. The correlation matrix was sorted to group samples together with similar behavior. The scale for the R values is shown at the bottom left of the figure, with dark blue corresponding to positive R values and bright red corresponding to negative R values. Groups A to E show taxa that share similar dynamics.
FIG 8.

Dynamic behavior of correlation group E taxa in untreated and treated pork sausage. (A) Total mesophilic APCs, psychrotrophic APCs, and LAB, and Enterobacteriaceae were enumerated from pork sausage containing 0, 3, 4, 5, and 6% LD. The log-transformed data from each treatment group are plotted from each of the 15-day time points taken over the 80-day refrigeration period. (B) Relative abundances of group E taxa from the pyrosequencing data are at the 15-day sampling intervals during the 80-day storage at refrigeration. The pH of the samples is plotted in royal blue.
Microbial successions and chemical changes in the model sausage matrix.
To systematically test for associations between changes in the microbiota and sensory characteristics, an array of 10 chemical sensors on an Air-Sense Pen-3 E-Nose (eNose) was used to measure broad chemical compositional features of the matrix from each of the treatments and time points. The responses of each of the 10 individual sensors in the array were analyzed individually by ANOVA for significant differences at the different time points within each treatment. Several of the sensors showed significant changes (P < 0.05) in the untreated sausage, and box-whisker plots of data from those sensors (W1C, W1S, W2S, W3C, W5C, and W5S) are shown in Fig. 9. With the exception of W5S, these sensors each showed stepwise changes in their response patterns in the untreated samples, with significant step-ups or step-downs in responses occurring at day 30 and again at day 60. These same time points (Fig. 5) coincide with the onset of detectable sensory degradation (day 30) and objectionable sensory characteristics (day 60) in the untreated matrix. With respect to the microbiota, these time points are also coincident with onset of the phase II succession at day 30 and the notable decline in ratio of group A and B taxa to other taxa that occurs abruptly at day 60 (Fig. 5). It is notable that dominant populations of Pseudomonas that peak at day 15 result in little chemical change detectable by Pen3 eNose sensors at that time point. Consistent with the traditional microbiological analysis and the pyrosequencing data, the eNose sensors showed only muted responses over time in the LD-treated samples, with trends toward increases by day 60 to day 80 (Fig. 9).
FIG 9.

Box-and-whisker plots of responses from individual eNose sensors showing statistically significant responses. Each plot shows the responses of a single sensor from the eNose array for each individual sausage sample. Boxes represent the 95% confidence intervals, and whiskers depict maximum and minimum values for sensor responses over the 60-s measurements. The time points (days) are indicated above each individual sample, and boxes are color-coded as depicted at the bottom. Individual sensors detect aromatics (W1C), ammonia (W3C), aromatic-aliphatics (W5C), broad-range alcohols (W2S), and broad range with sensitivity to nitrogen oxides (W5S). The values for days 30 to 80 were significant (P < 0.05) as determined by ANOVA for each of the sensors shown, with the exception of the W5S sensor, where only the day 60 and day 80 untreated samples were significant.
The broad ranges of compounds that can be detected by the eNose sensors confounds inference to the exact chemical nature of the changes that are detected. However, the similarities in the pattern of reactivity of the W1C (Aromatics), W3C (ammonia), W5C (aromatic-aliphatics), W2S (broad-range alcohols), and W5S (broad range with sensitivity to nitrogen oxides), coupled with the physiological characteristics of dominant organisms at these time points, suggest a combination of by-products from lipid oxidation, mixed acid carbohydrate fermentations, and amino acid metabolism. To further define potential cause-effect relationships between taxa within the microbiota and chemical-sensory changes of the meat matrix, we next used correlation analysis and linear regression to identify groups of taxa and individual taxa that showed strong correlation with behavior of the eNose sensors. Correlation analysis (Fig. 10A) initially defined two broad groups of taxa based on the polarity of association with the sensors. Linear regression analysis (see Table SA2 in the supplemental material) was used as a fine-grained estimate of organisms that are likely to be directly associated with the chemical changes of the matrix. Individually, Lactobacillus graminis, Carnobacterium divergens, and Serratia OTU4 consistently had significant slopes (P < 0.05) and displayed the highest R2 values across most of the sensors. Dot plots of their relative abundances against the sensor response (Fig. 10B) showed remarkable dose-response relationships. Among the taxa that could be assigned to a genus by Multi-CLASSIFIER analysis, these organisms are likely to be among the best candidates for cause-effect relationships in the eNose arrays and, similarly, are likely to be candidates for cause-effect relationships in the sensory characteristics.
FIG 10.

Association of eNose sensor responses and relative of individual taxa. (A) Correlation matrix (R values) of the relative abundances of microbial taxa measured by pyrosequencing and the responses of individual eNose sensors. The color scale on the left and the positions of the eNose rows and rows of taxa are indicated on the right. (B) Dot plots of the relative abundances of specific taxa and responses of individual eNose sensors in untreated sausage samples. The correlation coefficients (R2 values) are highlighted by color-coded boxes that correspond to regression lines and data points from each sensor.
Spice blend as a source of the dominant Lactobacillus graminis.
With the high-resolution snapshots of the microbial population dynamics in the sausage and the ability to use simple data-mining techniques to sort taxa into groups with similar behavior, we next sought to determine whether the pyrosequencing data could be used to define the source of the organisms in the sausage matrix. To accomplish this, we extracted total DNA from a sample of the spice blend used to make the model sausage and subjected it to 16S rRNA gene amplicon pyrosequencing. Sequences from this sample and from the time zero sample of the untreated model sausage (which represents the meat plus spice blend) were then used for phylogenetic analysis. After alignment (cmALIGN) and cluster analysis, we selected the top 100 most abundant taxa (using 97% cutoff) and conducted phylogenetic analyses, keeping track of the relative number of reads from each sample (i.e., “Spice” versus “Meat + Spice”) that belonged to each taxon. Figure 11 depicts the dendrogram showing the phylogenetic relationships of these abundant taxa. Of the 83 taxa detected in this analysis, 44 were detected in both the spice sample and the meat-and-spice sample, implying that the spice blend may be responsible for introducing a significant portion of the microbiota into the sausage matrix. The large group of taxa found in the spice blend included the three phylogenetically related species of Lactobacillus (L. graminis, L. sakei, and L. curvatus) that are readily isolated from plant material (18), as well as a large number of Streptococcus, Lactococcus, and Enterococcus species.
FIG 11.

Source-tracking taxa from the pork sausage and its spice blend ingredient. The distribution of taxa is illustrated from the day 0 sample of the untreated pork sausage and from a sample of the spice blend mix that was used to develop the model pork sausage. The dendrogram was developed from representative sequences derived from each of the 82 different genera that were detected in the samples. Representative sequences were developed from each of the genus-level Multi-CLASSIFIER taxonomic bins by first collapsing to 97% identity using cmALIGN and then aligning it to a phylogenetic framework established from 16S reference sequences. The individual pie charts depict species-level OTU for each of the 82 genera and the relative abundance of each OTU in the spice blend (dark blue) or the meat-spice blend (light blue) is illustrated by the proportion in each pie.
To further examine the source-tracking pattern inferred from the pyrosequencing data, archived samples from the spice blend and days 15, 30, and 45 of the untreated and treated sausage were diluted and plated onto APT or MRS agar to determine whether the same species of Lactobacillus could indeed be isolated. Sanger-based DNA sequence analysis of 16S rRNA gene amplicons derived from individual isolates revealed many of the same or related taxa that were inferred by pyrosequencing, including species of Weissella, Staphylococcus, Lactococcus, Enterococcus, Carnobacterium, and Lactobacillus, as well as several species of Bacillus. Among the Lactobacillus species, isolates of L. sakei, L. curvatus, and L. graminis were all recovered from the sausage samples. Most isolates from the spice blend were species of Bacillus or Carnobacterium, but a single isolate of Lactobacillus was recovered, and its 16S rRNA gene sequence aligned with several of the L. graminis isolates in our database. If the L. graminis isolates recovered from the sausage indeed originated from the spice blend, the isolates from the sausage and from the spice blend would likely share very recent common ancestry and should be genetically related. Genetic relationships estimated by RAPD analysis of the L. graminis and L. sakei isolates (Fig. 12) showed the presence of two different populations of L. graminis in the sausage, one of which had a RAPD pattern that was indistinguishable from that of the spice isolate. This result is consistent with at least one of the two L. graminis populations being introduced into the model sausage from the spice blend. Of course, it is possible that either population of L. graminis was introduced into the spice blend and the sausage from the environment. The relatively low efficiency of recovery from the spice blend (a single Lactobacillus species recovered among the 25 isolates sequenced) suggests that sample depth and sampling error both could have contributed to our inability to recover both subtypes if present in the spice.
FIG 12.
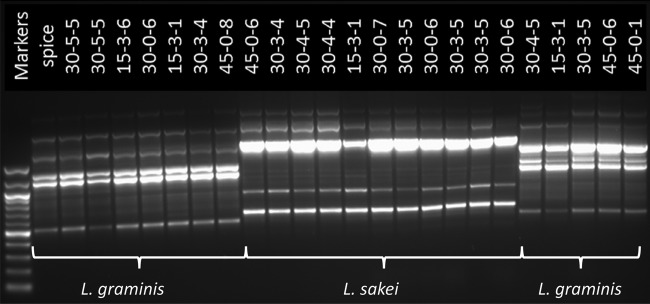
Agarose gel electrophoresis of RAPD products from Lactobacillus graminis and Lactobacillus sakei isolates. Isolates from individual sausage samples or the spice blend used to prepare the sausage were recovered from diluted samples plated onto APT or MRS agar. The species status was assigned on the basis of Sanger sequencing of 16S rRNA gene amplicons and is shown at the bottom of the gel. The origin of the isolate is indicated at the top of each lane (numbers and dashes indicate “day-treatment-isolate number”). Size markers (1-kb ladder) are indicated on the left side of the gel.
DISCUSSION
Complex combinations of physiochemical factors and microbial factors interact during the course of meat spoilage. The microbial populations found on meat products have largely been understood on the basis of standard microbiological methods that enumerate large groups of organisms (e.g., APCs), organisms that share physiological and/or phylogenetic relationships (e.g., LAB or coliforms), and, in a few instances, specific taxa that are associated with spoilage (e.g., Pseudomonas and Brochothrix). The development of non-culture-based denaturing gradient gel electrophoresis (DGGE) methodology (19) initially paved the way for semiquantitative analysis of microbial populations in foods, including a recent study in fresh pork sausages (20). When combined with culture-based methods, culture-independent methods have provided substantial insights into the microbiota of fresh meats (reviewed recently by Doulgeraki et al. [21]). Through this window, our understanding of the taxonomic composition of the microbiota from fresh meats and sausages has now progressed from low-resolution views of the alpha diversity to higher-resolution views that have begun to define population dynamics and beta diversity. By integrating high-resolution NGS-based pyrosequencing of 16S rRNA gene amplicons with traditional microbiological analysis, we have now extended this view to a highly quantitative characterization of alpha diversity, dynamic changes that occur during refrigerated storage, and the impact of antimicrobials on the population dynamics. Our studies have also provided methodology and approaches that can be built upon to define associations and cause-effect relationships between individual taxa and chemical characteristics of the meat matrix. Lastly, our work has established pyrosequencing as a tool to aid source-tracking organisms found in food matrices.
Population dynamics: successions of taxa in untreated pork sausage model.
Microbial successions with early phases of dominance by pseudomonads, followed by the dominance by combinations of Enterobacteriaceae and LAB, have been observed in refrigerated fresh meats with methodologies such as DGGE-PCR, T-RFLP, and RAPD (5, 21–24). Our application of 16S rRNA gene-based pyrosequencing has now extended our view of the dynamic behavior of complex microbial populations in fresh sausage, revealing the quantitative displacement of taxa that occur during microbial successions and the correlation of these events with changes in the chemical and sensory properties of the meat matrix. Two very distinct successions were observed in these studies, neither of which was evident from the plating data. The initial phase I was dominated by two species of pseudomonas (group C taxa), which displaced the species of Leuconostoc and Weissella (group E) that were dominant at time zero. A second succession in phase II (day 15 to day 30) led to the displacement of the pseudomonads by a complex of group A and group B species, including members of the Enterobacteriaceae (group B; Y. mollaretti, B. brennerae, and Serratia) and LAB (group A; C. divergens and Lactobacillus graminis) outranking the pseudomonas species by nearly 100-fold. Collectively, the group A and B taxa of L. graminis, Y. mollaretti, and C. divergens and the OTU4 taxon of Serratia remained the dominant taxa for the duration of the 80-day study. This two-phase dynamic likely reflects a combination of changes in redox potential and growth substrates, with aerobic growth of the pseudomonads depleting the free glucose, reducing the environment, and likely initiating lipid oxidation and proteolysis. The major population shift occurring at the onset of phase II leads to microbial populations of >109/g, and yet it occurs with only a modest shift in pH (pH change from 6.4 to 5.3).
In addition to the successions, the pyrosequencing data also revealed fundamental features of the population dynamics. First, the species that dominate the population at time zero are not the species that ultimately dominate the population during refrigerated storage. In fact, the most abundant species at time zero fell nearly 100-fold (Leuconostoc) or became undetectable (Weissella) by day 30, the onset of the second growth phase. Other species that were less abundant at day 0 (from 0.1 to 5% of the population) also showed this same dynamic, including several species of Streptococcus. The corollary of this concept is that the species that came to dominate the population during phase I or phase II were a small proportion of the population at time zero (Pseudomonas, Lactobacillus, and Carnobacterium) or were undetectable (Yersinia, Buttiauxella, and Serratia). Thus, initial relative abundances alone would not predict the final outcome of the population during refrigerated storage. Along with successions, we also observed abrupt changes in the relative abundances of less dominant taxa, most notably the abrupt increase in relative abundance of many taxa that occurs at days 45 and 60 in untreated sausage (Fig. 5B). Some of the species change in relative abundance by nearly 100-fold at these time points, implying that a significant event has occurred, and such events would not be detected by traditional methods. The nature of this event is not yet entirely clear, but we do note that the abrupt change of the lower abundance taxa seems to be associated with modest declines in the populations of Yersinia and Buttiauxella rather than a decline in the population of L. graminis (Fig. 4 and 5).
Undoubtedly, additional studies are needed to measure the degree of variation in the microbiota that occurs due to different processing methods, such as cold-boning versus hot-boning, and due to different production environments, as well as the impact of such variation on dynamic behavior of the populations. Studies are already under way to capture and estimate such variation and to determine whether models can be developed to predict outcome. Such data sets could usher in a new generation of modeling to optimize formulations or operations. Although we expect variation, we would also predict that the two distinct successions will be a conserved feature in fresh meats derived from hot-boning and packaged at ambient atmosphere and that they would be brought about by initial blooms of pseudomonads or other highly aerobic taxa, followed by the emergence of facultative members of the Enterobacteriaceae and LAB. In addition, we would also predict that the onset of phase II is the immediate antecedent of significant sensory changes.
Psychrotrophic growth characteristics of group A, B, and C species.
Although multiple species of Pseudomonas (P. poae, P. azotoformans, P. stutzeri, P. syringae, P. lini, and P. psychrophila) were present at low levels (∼0.005 to 0.01%) at time zero, only P. lini and P. psychrophila showed a dramatic increase in abundance during phase I. Strains belonging to each of these species are known to be psychrotrophic, and we would therefore have expected all six of these species to grow at refrigeration temperatures (25–29), leaving open the question as to why only P. lini and P. psychrophila showed such a dramatic growth in the sausage. It is possible that P. poae, P. azotoformans, and P. stutzeri are selectively inhibited by the antimicrobial characteristics of the spice blend. Alternatively, psychrotrophic growth traits may vary widely between subtypes of these species.
During phase II growth in untreated sausage, the pseudomonads were displaced by a complex combination of members of the Enterobacteriaceae and LAB (taxa Y. mollaretti, B. brennerae, Serratia spp., C. divergens, and L. graminis). It is interesting that only a single member of the Enterobacteriaceae (Citrobacter braakii) was even detectable at time zero, whereas the trio of Y. mollaretti, B. brennerae, and Serratia sp. was undetectable. All three of the latter organisms are known to be psychrotrophs and have been isolated from meats (22, 30). Psychrotrophic species of Yersinia and Serratia have further been associated with production of biogenic amines putrescine and cadaverine in fresh pork sausage (30). Similarly, C. divergens is a common member of the LAB found in refrigerated fresh meats and is a known producer of the biogenic amine tyramine (30). L. graminis, a dominant species of psychrotrophic LAB detected in our study, has previously been detected in fresh refrigerated meats (5), although little is known about its physiology. L. graminis was one of six different species of Lactobacillus that was detected during the course of the shelf life and, despite its very low initial abundances, it became the single most abundant species of LAB during refrigeration. The species is commonly found in wheat (18) and is often a member of the consortia found in buckwheat sourdoughs (31). Phylogenetically, L. graminis is very closely related to L. sakei and L. curvatus, which were also found in our samples at much lower abundances (Fig. 6A). L. sakei is commonly a dominant Lactobacillus species in spontaneously fermented sausages (32, 33), and the genome of this organism appears to have some unique adaptations that permit growth in the meat matrix, including multiple arginine deaminases that presumably promote utilization of exogenous arginine and arginyl-peptides from degraded meat proteins to generate energy via the arginine deaminase pathway (34). Whether this pathway is present in L. graminis and contributes to its ability to dominate fresh sausage remains to be determined.
With the unique behavior of L. graminis in the sausage matrix, the source of this organism becomes an increasingly important question. Since it is most associated with plants and plant-based products, it is not surprising to find that pyrosequencing identified this organism (along with L. curvatus and L. sakei) in the spice blend (Fig. 11). If L. graminis does have a unique propensity for growth in refrigerated fresh sausage, testing batches of spice mixture for the relative abundance of L. graminis could provide at least one biomarker for grading these ingredients into categories that could yield predictable shelf-lives (e.g., short, medium, or long).
Effects of the LD treatments.
At all four dosages, the LD treatments showed very similar effects, limiting the growth of the APC to a maximum of 4 logs by day 30 in 3% LD and 3 logs by day 30 in the remaining treatments. In all cases, the LAB and APCs were essentially equivalent. From the pyrosequencing data, the effect of LD eliminated the phase I succession and significantly altered the dynamics of phase II. The monophasic change in population composition was heralded by the dramatic increase in relative abundance of L. graminis and the corresponding drop in the abundances of nearly all other taxa. The inhibitory properties of LD against many non-LAB Firmicutes and Proteobacteria (including species of Yersinia and Serratia) are known from studies in pork sausage and in vitro cultures (14–16, 35). Somewhat surprising, however, was the selective survival of L. graminis among other LAB found in the treated samples. LD typically has an inhibitory activity against LAB such as L. sakei, and species of Carnobacterium and Weissella (17, 36), but the selective survival of L. graminis among a complex microbiota has not previously been observed. L. sakei did show a >100-fold increase in relative abundance in the 3% LD treatment, but it was otherwise sparsely detected in the remaining treatments. Even L. gasseri, which accounted for 2.5 to 10% of the population at day 0 in the treated samples, decreased by day 30 to day 45.
Perhaps the most remarkable feature of the LD treatment is the dynamics of the population die-off that occurs. The onset of the die-off begins at day 30 (3 and 4% LD) or day 45 (5 and 6% LD), and these same time points are coincident with the cessation of detectable LAB growth by plating. At these time points, an abrupt and surprisingly uniform decline begins in the relative amounts of DNA from nearly all taxa except L. graminis. In the absence of detectable growth by L. graminis, the continued decline in relative abundances of the remaining taxa must include not just a loss of viability of species in the microbiota but also degradation of their DNA, thereby precluding the amplification of 16S genes from dead cells. Degradation of nucleic acids is a common element of the major programmed cell death pathways that are found widely among prokaryotes (37). Given the abruptness at which the onset of the population die-off occurs and the quite uniform rates of death among widely different taxa, it is tempting to speculate that LD somehow brings about a type of programmed cell death that L. graminis is uniquely resistant to. Clearly, additional studies are necessary to better characterize this phenomenon.
Population dynamics and associations with chemical and sensory characteristics.
Our initial observation of the biphasic successions in untreated sausage and the relationship of the onset of phase II with sensory degradation suggests that specific taxa such as L. graminis and C. divergens and the group B taxa that dominate phase II could be more reliable microbial estimators of spoilage than previously believed (38). Association studies integrating quantitative data from individual taxa with measurements of broad chemical features from eNose sensor arrays (Fig. 10) also support the potential value of individual taxa, with strong correlations between individual taxa and chemical sensors suggestive of cause-effect (e.g., dose-effect) relationships. Linear regression (Fig. 10B) defined three main species—L. graminis, C. divergens, and Serratia OTU4—as having the most significant dose-response relationships with the eNose sensors, elevating these organisms as likely candidates. Surprisingly, there was no significant relationship between responses of the chemical sensors and phase I of the succession, despite substantial growth of two species of Pseudomonas. We also note that sensory characteristics remained intact at day 15 and did not appear to undergo degradation until day 30, when significant changes in the eNose sensors became detectable.
Despite the cause-effect relationships implied by strong associations with specific taxa, other studies have reported that significant batch-batch variation in microbiota and chemical composition confounds use of individual species or molecules as spoilage indicators (38). Such variation will undoubtedly need to be examined and carefully modeled. However, even if species-specific signatures are confounded by batch-batch variation, it seems likely that machine learning algorithms can be trained on data sets that allow predictive models to be developed that base prediction of the evolutionary relationships of organisms that can grow in phase I or phase II. In addition, prediction could also be based on the functional content, which can be inferred to some degree of accuracy from the taxonomic content (39) but are best measured directly from shotgun metagenomic sequencing. In this case, the relative abundances of genes involved in biosynthesis of spoilage-associated metabolic products can be quantified independently from the organisms from which they originate.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by a contract from ConAgra to the University of Nebraska.
J.R.D.D., S.E.G, G.S., C.E.D., and I.S. have equity interests in ConAgra Foods, Inc. A.K.B. is a technical consultant for ConAgra Foods, Inc.
Footnotes
Published ahead of print 13 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00774-14.
REFERENCES
- 1.Mancini RA, Hunt MC. 2005. Current research in meat color. Meat Sci. 71:100–121. 10.1016/j.meatsci.2005.03.003 [DOI] [PubMed] [Google Scholar]
- 2.Nychas G-JE, Marshall D, Sofos J. 2007. Meat, poultry, and seafood, p 105–140 In Doyle MP, Beuchat LR, Montville TJ. (ed), Food microbiology fundamentals and frontiers. ASM Press, Washington, DC [Google Scholar]
- 3.Stanbridge LH, ARD. 1998. The microbiology of chill-stored meat, p 174–219 In Davis AR, Board RG. (ed), The microbiology of meat and poultry. Blackie Academic and Professional, London, United Kingdom [Google Scholar]
- 4.Nychas G-JE, Skandamis PN, Tassou CC, Koutsoumanis KP. 2008. Meat spoilage during distribution. Meat Sci. 78:77–89. 10.1016/j.meatsci.2007.06.020 [DOI] [PubMed] [Google Scholar]
- 5.Ercolini D, Russo F, Torrieri E, Masi P, Villani F. 2006. Changes in the spoilage-related microbiota of beef during refrigerated storage under different packaging conditions. Appl. Environ. Microbiol. 72:4663–4671. 10.1128/AEM.00468-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janssens M, Myter N, De Vuyst L, Leroy F. 2012. Species diversity and metabolic impact of the microbiota are low in spontaneously acidified Belgian sausages with an added starter culture of Staphylococcus carnosus. Food Microbiol. 29:167–177. 10.1016/j.fm.2011.07.005 [DOI] [PubMed] [Google Scholar]
- 7.Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, Kachman SD, Moriyama EN, Walter J, Peterson DA, Pomp D. 2010. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. U. S. A. 107:18933–18938. 10.1073/pnas.1007028107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez I, Wallace G, Zhang C, Legge R, Benson AK, Carr TP, Moriyama EN, Walter J. 2009. Diet-induced metabolic improvements in a hamster model of hypercholesterolemia are strongly linked to alterations of the gut microbiota. Appl. Environ. Microbiol. 75:4175–4184. 10.1128/AEM.00380-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–D145. 10.1093/nar/gkn879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261–5267. 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Z, DeSantis TZ, Andersen GL, Knight R. 2008. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res. 36:e120. 10.1093/nar/gkn491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W., Jr 2007. SILVA: a comprehensive online resource for quality checked and aligned rRNA sequence data compatible with ARB. Nucleic Acids Res. 35:7188–7196. 10.1093/nar/gkm864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659. 10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- 14.Bradford DD, Huffman DL, Egbert WR, Jones WR. 1993. Low-fat fresh pork sausage patty stability in refrigerated storage with potassium lactate. J. Food Sci. 58:488–491. 10.1111/j.1365-2621.1993.tb04307.x [DOI] [Google Scholar]
- 15.Brewer MS, McKeith F, Martin SE, Dallmier AW, Meyer J. 1991. Sodium lactate effects on shelf-life, sensory, and physical characteristics of fresh pork sausage. J. Food Sci. 56:1176–1178. 10.1111/j.1365-2621.1991.tb04727.x [DOI] [Google Scholar]
- 16.Lamkey JW, Leak FW, Tuley WB, Johnson DD, West RL. 1991. Assessment of sodium lactate addition to fresh pork sausage. J. Food Sci. 56:220–223. 10.1111/j.1365-2621.1991.tb08015.x [DOI] [Google Scholar]
- 17.Peirson MD, Guan TY, Holley RA. 2003. Thermal resistances and lactate and diacetate sensitivities of bacteria causing bologna discoloration. Int. J. Food Microbiol. 86:223–230. 10.1016/S0168-1605(02)00548-2 [DOI] [PubMed] [Google Scholar]
- 18.Corsetti A, Settanni L, Chaves López C, Felis GE, Mastrangelo M, Suzzi G. 2007. A taxonomic survey of lactic acid bacteria isolated from wheat (Triticum durum) kernels and non-conventional flours. Syst. Appl. Microbiol. 30:561–571. 10.1016/j.syapm.2007.07.001 [DOI] [PubMed] [Google Scholar]
- 19.Muyzer G, Smalla K. 1998. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Van Leeuwenhoek 73:127–141. 10.1023/A:1000669317571 [DOI] [PubMed] [Google Scholar]
- 20.Cocolin L, Rantsiou K, Iacumin L, Urso R, Cantoni C, Comi G. 2004. Study of the ecology of fresh sausages and characterization of populations of lactic acid bacteria by molecular methods. Appl. Environ. Microbiol. 70:1883–1894. 10.1128/AEM.70.4.1883-1894.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doulgeraki AI, Ercolini D, Villani F, Nychas G-JE. 2012. Spoilage microbiota associated to the storage of raw meat in different conditions. Int. J. Food Microbiol. 157:130–141. 10.1016/j.ijfoodmicro.2012.05.020 [DOI] [PubMed] [Google Scholar]
- 22.Ercolini D, Russo F, Nasi A, Ferranti P, Villani F. 2009. Mesophilic and psychrotrophic bacteria from meat and their spoilage potential in vitro and in beef. Appl. Environ. Microbiol. 75:1990–2001. 10.1128/AEM.02762-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nieminen TT, Vihavainen E, Paloranta A, Lehto J, Paulin L, Auvinen P, Solismaa M, Björkroth KJ. 2011. Characterization of psychrotrophic bacterial communities in modified atmosphere-packed meat with terminal restriction fragment length polymorphism. Intl. J. Food Microbiol. 144:360–366. 10.1016/j.ijfoodmicro.2010.10.018 [DOI] [PubMed] [Google Scholar]
- 24.Pennacchia C, Ercolini D, Villani F. 2011. Spoilage-related microbiota associated with chilled beef stored in air or vacuum pack. Food Microbiol. 28:84–93. 10.1016/j.fm.2010.08.010 [DOI] [PubMed] [Google Scholar]
- 25.Behrendt U, Ulrich A, Schumann P. 2003. Fluorescent pseudomonads associated with the phyllosphere of grasses; Pseudomonas trivialis sp. nov., Pseudomonas poae sp. nov., and Pseudomonas congelans sp. nov. Intl. J. Syst. Evol. Microbiol. 53:1461–1469. 10.1099/ijs.0.02567-0 [DOI] [PubMed] [Google Scholar]
- 26.Delorme S, Lemanceau P, Christen R, Corberand T, Meyer J-M, Gardan L. 2002. Pseudomonas lini sp. nov., a novel species from bulk and rhizospheric soils. Intl. J. Syst. Evol. Microbiol. 52:513–523 [DOI] [PubMed] [Google Scholar]
- 27.Lo Giudice A, Brilli M, Bruni V, De Domenico M, Fani R, Michaud L. 2007. Bacterium-bacterium inhibitory interactions among psychrotrophic bacteria isolated from Antarctic seawater (Terra Nova Bay, Ross Sea). FEMS Microbiol. Ecol. 60:383–396. 10.1111/j.1574-6941.2007.00300.x [DOI] [PubMed] [Google Scholar]
- 28.Sahu B, Ray MK. 2008. Auxotrophy in natural isolate: minimal requirements for growth of the Antarctic psychrotrophic bacterium Pseudomonas syringae Lz4W. J. Basic Microbiol. 48:38–47. 10.1002/jobm.200700185 [DOI] [PubMed] [Google Scholar]
- 29.Yumoto I, Kusano T, Shingyo T, Nodasaka Y, Matsuyama H, Okuyama H. 2001. Assignment of Pseudomonas sp. strain E-3 to Pseudomonas psychrophila sp. nov., a new facultatively psychrophilic bacterium. Extremophiles 5:343–349. 10.1007/s007920100199 [DOI] [PubMed] [Google Scholar]
- 30.Curiel JA, Ruiz-Capillas C, de Las Rivas B, Carrascosa AV, Jimenez-Colmenero F, Munoz R. 2011. Production of biogenic amines by lactic acid bacteria and enterobacteria isolated from fresh pork sausages packaged in different atmospheres and kept under refrigeration. Meat Sci. 88:368–373. 10.1016/j.meatsci.2011.01.011 [DOI] [PubMed] [Google Scholar]
- 31.Moroni AV, Arendt EK, Bello FD. 2011. Biodiversity of lactic acid bacteria and yeasts in spontaneously fermented buckwheat and teff sourdoughs. Food Microbiol. 28:497–502. 10.1016/j.fm.2010.10.016 [DOI] [PubMed] [Google Scholar]
- 32.Aymerich T, Martín B, Garriga M, Hugas M. 2003. Microbial quality and direct PCR identification of lactic acid bacteria and nonpathogenic staphylococci from artisanal low-acid sausages. Appl. Environ. Microbiol. 69:4583–4594. 10.1128/AEM.69.8.4583-4594.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rantsiou K, Urso R, Iacumin L, Cantoni C, Cattaneo P, Comi G, Cocolin L. 2005. Culture-dependent and -independent methods to investigate the microbial ecology of Italian fermented sausages. Appl. Environ. Microbiol. 71:1977–1986. 10.1128/AEM.71.4.1977-1986.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaillou S, Champomier-Verges MC, Cornet M, Crutz-Le Coq AM, Dudez AM, Martin V, Beaufils S, Darbon-Rongere E, Bossy R, Loux V, Zagorec M. 2005. The complete genome sequence of the meat-borne lactic acid bacterium Lactobacillus sakei 23K. Nat. Biotechnol. 23:1527–1533. 10.1038/nbt1160 [DOI] [PubMed] [Google Scholar]
- 35.Grau FH. 1981. Role of pH, lactate, and anaerobiosis in controlling the growth of some fermentative Gram-negative bacteria on beef. Appl. Environ. Microbiol. 42:1043–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stekelenburg FK, Kant-Muermans MLT. 2001. Effects of sodium lactate and other additives in a cooked ham product on sensory quality and development of a strain of Lactobacillus curvatus and Listeria monocytogenes. Intl. J. Food Microbiol. 66:197–203. 10.1016/S0168-1605(00)00521-3 [DOI] [PubMed] [Google Scholar]
- 37.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810. 10.1016/j.cell.2007.06.049 [DOI] [PubMed] [Google Scholar]
- 38.Laursen BG, Byrne DV, Kirkegaard JB, Leisner JJ. 2009. Lactic acid bacteria associated with a heat-processed pork product and sources of variation affecting chemical indices of spoilage and sensory characteristics. J. Appl. Microbiol. 106:543–553. 10.1111/j.1365-2672.2008.04045.x [DOI] [PubMed] [Google Scholar]
- 39.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31:814–821. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.