

Abstract
Chromium pollution is potentially detrimental to bacterial soil communities, compromising carbon and nitrogen cycles that are essential for life on earth. It has been proposed that intracellular reduction of hexavalent chromium [Cr(VI)] to trivalent chromium [Cr(III)] may cause bacterial death by a mechanism that involves reactive oxygen species (ROS)-induced DNA damage; the molecular basis of the phenomenon was investigated in this work. Here, we report that Bacillus subtilis cells lacking a functional error prevention oxidized guanine (GO) system were significantly more sensitive to Cr(VI) treatment than cells of the wild-type (WT) strain, suggesting that oxidative damage to DNA is involved in the deleterious effects of the oxyanion. In agreement with this suggestion, Cr(VI) dramatically increased the ROS concentration and induced mutagenesis in a GO-deficient B. subtilis strain. Alkaline gel electrophoresis (AGE) analysis of chromosomal DNA of WT and ΔGO mutant strains subjected to Cr(VI) treatment revealed that the DNA of the ΔGO strain was more susceptible to DNA glycosylase Fpg attack, suggesting that chromium genotoxicity is associated with 7,8-dihydro-8-oxodeoxyguanosine (8-oxo-G) lesions. In support of this notion, specific monoclonal antibodies detected the accumulation of 8-oxo-G lesions in the chromosomes of B. subtilis cells subjected to Cr(VI) treatment. We conclude that Cr(VI) promotes mutagenesis and cell death in B. subtilis by a mechanism that involves radical oxygen attack of DNA, generating 8-oxo-G, and that such effects are counteracted by the prevention and repair GO system.
INTRODUCTION
Chromium, a common environmental pollutant, is present in divalent [Cr(II)], trivalent [Cr(III)], and hexavalent [Cr(VI)] oxidation states; the most stable and common forms in the environment are the hexavalent Cr(VI) and the trivalent Cr(III) species. The biological effects of the metal are highly dependent on its oxidation state. Compounds of Cr(VI) in the form of oxides, chromates, and dichromates have been widely recognized as toxic substances due to their high solubility (1). This property and its similarity to sulfate promote the active transport of chromate across biological membranes, and once internalized by cells, Cr(VI) exhibits a variety of genotoxic, mutagenic, and carcinogenic effects for all forms of life. In contrast, Cr(III) is considered less toxic than Cr(VI) because of its tendency to form insoluble complexes that are impeded in crossing cell membranes (2, 3). It has been proposed that the deleterious effects of Cr(VI) are a consequence of its intracellular reduction to Cr(III), leading to increased formation of reactive oxygen species (ROS), such as superoxide (O2·−), hydrogen peroxide (H2O2), and hydroxyl radicals (OH·) via a Fenton-like reaction between Cr(V) and H2O2 (4–6). Furthermore, results from in vitro studies have shown that Cr(VI) promotes a variety of DNA lesions, such as 7,8-dihydro-8-oxodeoxyguanosine (8-oxo-G), strand breaks, apurinic/apyrimidinic (AP) sites, and chromium-DNA adducts, among other modifications (7–9). The deleterious effects of Cr(VI) have also been associated with damage to the cellular envelopes. In rats, it has been proposed that hexavalent chromium, by altering the proportions of cholesterol and phospholipid, may promote damage to the cell membrane structure (10). In prokaryotes, exposure of Escherichia coli and Shewanella oneidensis MR-1 to Cr(VI) induced severe morphological changes, including formation of aseptated long filaments, cell aggregation, and damage to cell walls (11, 12).
Analysis of global responses to chromium exposure in some bacterial species has shown that Cr(VI) induces the synthesis of proteins with antioxidant functions, including catalase, superoxide dismutase, thioredoxin, and components of the SOS regulon (12–14). Moreover, analysis of the in vivo effects of Cr(VI) in the yeast Saccharomyces cerevisiae revealed that the main mechanism of toxicity of the oxyanion is exerted through oxidation of proteins, specifically glycolytic enzymes and heat shock proteins (15).
The ability of Cr(VI) to promote the synthesis of 8-oxo-G lesions in isolated calf thymus DNA has been demonstrated in cell-free systems composed of a chromate salt and hydrogen peroxide (16). The synthesis of this oxidized base was also detected in single- and double-stranded oligonucleotides that were incubated with Cr(V) complexes [N,N′-ethylenebis(salicylidene-animato)oxochromium(V)] (17). The 8-oxo-G lesion is mutagenic, as it promotes GC→TA and AT→CG transversions. In several microorganisms, the mutagenic effects of 8-oxo-G are counteracted by the oxidized guanine (GO) DNA repair system (18–20). This system is composed of the proteins MutM, MutY, and MutT; the first two are responsible for avoiding the mutagenic effects of 8-oxo-G once installed on DNA, whereas the last protein has a “sanitizing” role, hydrolyzing oxidized DNA and RNA precursors from the nucleotide pools (21). Bacillus subtilis possesses a complete GO system; in addition to YtkD and MutT, orthologs of the nucleotide diphosphohydrolase MutT of E. coli (22, 23), its genome contains genes encoding the MutM and MutY proteins (24). Recent studies have revealed that adaptive mutagenesis is strongly potentiated in starved B. subtilis cells lacking a functional GO system and have suggested that oxidative stress is an important component in the generation of genetic diversity (25).
To contend with the cytotoxic effects of Cr(VI), bacteria have evolved different strategies, including biosorption, catalytic reduction of the oxyanion to Cr(III), and extrusion of chromate ions by an energy-dependent efflux transporter termed ChrA (reviewed in reference 1). ywrA and ywrB, encoding putative short-chain monodomain versions of ChrA, have been described in B. subtilis. Although the role of YwrA/YwrB in the microorganism is currently unknown, a recent study revealed that the simultaneous, but not the single, expression of its encoding genes conferred chromate resistance on E. coli (26). Aside from this report, few studies have been conducted on the mechanisms involved in preventing and/or eliminating the in vivo genotoxicity of Cr(VI) in Bacillus species. Therefore, in this work, we took advantage of the well-characterized Gram-positive bacterium B. subtilis to investigate the type of DNA damage promoted by hexavalent chromium, the physiological consequences of such damage, and the mechanisms involved in counteracting those effects.
MATERIALS AND METHODS
Bacterial strains, culture conditions, and reagents.
All B. subtilis strains used in this work were derived from strain 168 and are listed in Table 1. The growth medium used routinely was Luria-Bertani (LB) medium. When required, neomycin (Neo) (10 μg ml−1), tetracycline (Tet) (10 μg ml−1), spectinomycin (Sp) (100 μg ml−1), chloramphenicol (Cm) (5 μg ml−1), kanamycin (Kan) (25 μg ml−1), erythromycin (Ery) (5 μg ml−1), or rifampin (Rif) (10 μg ml−1) was added to the medium. Liquid cultures were incubated with vigorous aeration (shaking at 250 rpm) at 37°C. Cultures on solid media were grown at 37°C. The optical density at 600 nm (OD600) of liquid cultures was monitored with a Pharmacia Ultrospec 2000 spectrophotometer.
TABLE 1.
B. subtilis strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| 168 | trpC2 | Laboratory stock |
| PERM431 | ΔkatA::Cm; Cmr | 28 |
| PERM432 | ΔkatB::Cm; Cmr | 28 |
| PERM434 | ΔsodA::Cm; Cmr | 28 |
| PERM1275 | ΔkatA::Ery ΔkatB::Cm; Eryr Cmr | This study |
| PERM1274 | ΔsodA::Cm ΔkatA::Ery; Cmr Eryr | This study |
| PERM679 | ΔperR::kan; Kanr | 29 |
| PERM342 | ΔsigB::Cm; Cmr | Laboratory stock |
| PERM603 | ΔytkD::Neo ΔmutM::Tc ΔmutY::Sp; Neor Tcr Spr | 25 |
| PERM882 | ΔkatA::Ery; Eryr | Laboratory stock |
| PERM1178 | amyE::(PkatA-GFP spc) trpC2 pheA1; Spr | 32 |
To determine intracellular ROS, a 10 mM stock solution of 2′,7′-dichlorofluorescein diacetate (DCFH-DA) (≥97% purity; Sigma, St. Louis, MO) was prepared in absolute ethanol and kept at −20°C in the dark for further use. To measure Cr(VI) susceptibility, a stock solution of potassium dichromate (K2Cr2O) (99.97% purity; JT Baker, Phillipsburg, NJ) at 10,000 ppm was prepared in sterile Milli-Q water. Working concentrations were prepared by diluting the stock with sterile Milli-Q water.
Strain construction.
To obtain KatA-deficient strains, we constructed a plasmid that integrates by Campbell-type recombination into the katA locus of B. subtilis. To this end, a 422-bp DNA fragment from nucleotides (nt) 123 to 544 of the katA open reading frame (ORF) was PCR amplified from chromosomal DNA of B. subtilis strain 168 using the oligonucleotide primers 5′-AAGCTTCCATTTCAACCGAGAACG-3′ (forward) and 5′-GGATCCGGTCAGACATCAGGATTG-3′ (reverse). The primers were designed to insert HindIII and BamHI sites, respectively (underlined). Amplification was performed with Vent DNA polymerase (New England BioLabs, Beverly, MA), and the PCR product was cloned into the HindIII/BamHI sites of the pMUTIN4 vector (27) to obtain pPERM879. This plasmid was used to transform competent cells of B. subtilis 168, thus generating B. subtilis strain PERM882 (katA; Eryr). To generate the katB katA and sodA katA mutants, the previously reported strains B. subtilis sodA (Cmr) and katB (Cmr) (kindly provided by Peter Setlow [28]) were used as recipients to interrupt the katA gene. To accomplish this, plasmid pPERM879 was used to transform competent cells of B. subtilis PERM432 (katB) and PERM434 (sodA), generating strains PERM1275 (katB katA; Cmr Eryr) and PERM1274 (sodA katA; Cmr Eryr), respectively (Table 1). B. subtilis strains PERM882, PERM1274, and PERM1275 were first characterized by analyzing their susceptibilities to hydrogen peroxide using assays of zones of growth inhibition (29) (Fig. 1). Furthermore, the recombination event that interrupted katA in these strains was confirmed by PCR using specific oligonucleotide primers that generated a PCR product of ∼1,050 bp extending from the interrupted katA gene through the lacZ gene of pMUTIN4, as shown in Fig. 1.
FIG 1.

Construction and molecular characterization of B. subtilis ΔkatA, ΔkatB katA, and ΔsodA katA strains. (A) The integrative plasmid pPERM879 (see Materials and Methods) was used to transform competent cells of B. subtilis strains 168 (WT), PERM432 (ΔkatB), and PERM434 (ΔsodA) to generate ΔkatA, ΔkatB katA, and ΔsodA katA strains, respectively. The Campbell-type recombination event leading to inactivation of katA in the three genetic backgrounds was confirmed by amplification of an ∼1,050-bp PCR product using specific oligonucleotide primers, as described in Materials and Methods. The PCR products from the three strains were separated in an agarose gel that was stained with ethidium bromide and are shown at the bottom: lane 1, ΔkatA; lane 2, ΔkatB katA; lane 3, ΔsodA katA. (B) The mutants obtained were characterized by their sensitivity to 100 mM H2O2 using a zone of inhibition assay, as described in Materials and Methods.
Determination of Cr(VI) toxicity.
To determine the dose-response curve of the survival of B. subtilis cells following Cr(VI) exposure, wild-type (WT) and mutant strains deficient in protection and DNA repair systems were grown at 37°C in LB medium to an OD600 of 1.0 (0.3 × 108 to 0.5 × 108 cells/ml). At that point, cultures that were free of spores as assessed by phase-contrast microscopy were treated with different concentrations of Cr(VI) (0, 12, 24, 48, or 80 μg ml−1) and incubated for 3 h at 37°C with shaking. After treatment, bacterial viability was estimated by counting the CFU on LB-agarose plates. To this end, samples of the bacterial suspensions were collected and serially diluted in 1× phosphate-buffered saline (PBS) (NaCl, 137 mM; KCl, 2.7 mM; Na2HPO4, 10 mM; and KH2PO4, 1.8 mM), and aliquots were plated on LB-agarose plates. Colonies were counted after overnight incubation at 37°C, and CFU were determined in appropriately diluted plated samples (30 to 300 CFU). Data were reported as 90% lethal dose (LD90) values, namely, the concentration of Cr(VI) that kills 90% of the bacterial population. The concentration of residual chromium(VI) remaining in the medium after the indicated incubation period was determined spectrophotometrically at 540 nm, using 1,5-diphenylcarbazide (DPC) as the complexing agent, as previously described (11, 30). The total Cr incorporated in the B. subtilis biomass was determined by electrothermal atomic adsorption spectroscopy (ETAA-AS) according to the procedure described by Muñoz et al. (31).
Adaptive response to Cr(VI).
To obtain unchallenged, challenged nonadapted, and challenged preadapted cultures (12), a single colony of B. subtilis strain 168 was inoculated into 5 ml of LB medium and grown with shaking at 37°C until the stationary phase of growth (OD600, 2.5). One aliquot of this culture was used to inoculate fresh LB medium, and the culture was grown with aeration at 37°C to an OD600 of 0.1. At that point, when the cells were in exponential growth, the culture was split into two subcultures; one subculture was left untreated, and the other was supplemented with an LD10 (3 ppm) of Cr(VI). These subcultures were shaken at 250 rpm and incubated at 37°C until the stationary phase of growth (OD600, 2.1 to 2.5). Then, the untreated culture was used to inoculate both the unchallenged and the challenged nonadapted LB cultures, and the treated culture was used to obtain the challenged preadapted LB culture. The unchallenged, challenged nonadapted, and challenged preadapted cultures were grown with aeration at 37°C to an OD600 of 0.1; at that point, challenged nonadapted and challenged preadapted cultures were supplemented with a Cr(VI) concentration equivalent to a LD20 (6 ppm), shaken at 250 rpm, and incubated at 37°C. The growth kinetics of the cultures were monitored with a Pharmacia Ultrospec 2000 spectrophotometer set at 600 nm every 30 min, and samples of the cell suspensions (0.1 ml) were collected at 0, 60, 120, 180, and 240 min; serially diluted into 1× PBS; and plated onto solid LB medium to determine viable counts.
Induction experiments and microscopy analysis.
B. subtilis cells carrying a translational PkatA-green fluorescent protein (GFP) fusion (32) were grown in LB medium to an OD600 of 0.5; at that point, the culture of exponentially growing cells was split into three subcultures of equal volumes. One subculture was left untreated, another was supplemented with the dose of Cr(VI) necessary to reach the LD50 (22 ppm), and the third was treated with an LD50 of H2O2 (i.e., 1.3 mM). These subcultures were shaken at 250 rpm and incubated at 37°C for 30 min. Subsequently, cells collected by centrifugation (15,000 × g for 10 min) were washed twice with 1× PBS and resuspended in 1 ml of this solution. Samples of the cell suspensions (0.1 ml) were collected, serially diluted into 1× PBS, and plated onto solid LB medium to determine viable counts. Finally, the fluorescence emitted by each sample was quantified with a Perkin-Elmer LS-55 fluorescence spectrometer (Perkin-Elmer, Waltham, MA) set at excitation and emission wavelengths of 488 nm and 515 nm, respectively. Emission values were reported as the number of fluorescence units/105 viable cells.
Cell samples (0.5 ml) removed from the cultures described above were centrifuged briefly (15,000 × g [20°C]); the cell pellets obtained were washed twice with 1× PBS and suspended in 50 μl of this solution. Samples spotted onto glass slides were mixed with a poly-l-lysine solution (Sigma) and analyzed by fluorescence microscopy with a Zeiss Axioscope A1 microscope equipped with an AxioCam ICc1 camera. Fluorescence and phase-contrast images were acquired by using AxioVision v. 4.8.2 software and adjusted only for brightness and contrast. The excitation and emission wavelengths employed were 490 and 510 nm for GFP.
Analysis of mutagenesis induced by Cr(VI).
Mutations to rifampin resistance (Rifr) of B. subtilis strains in the absence or presence of Cr(VI), added to reach the LD50, were determined as follows. Overnight LB cultures of each strain were inoculated into flasks containing fresh LB medium and grown to an OD600 of 1.0; each culture was then split into two subcultures, which were transferred into different flasks. One of the subcultures was untreated, and the other was treated with a Cr(VI) concentration necessary to reach the LD50: WT, 22 ppm, and ΔGO, 17 ppm. The untreated and Cr(VI)-treated cultures were shaken at 37°C for 16 h. Mutation frequencies were determined by plating aliquots of each culture onto six LB plates containing 10 μg ml−1 rifampin, as well as plating aliquots of appropriate dilutions onto LB plates without rifampin. Rifr colonies were counted after 24 h of incubation at 37°C.
Determination of intracellular ROS.
To measure intracellular ROS, the fluorescent probe DCFH-DA was used (33, 34). Briefly, B. subtilis cells were grown aerobically in LB medium to an OD600 of 1.0, at which point the cells were exposed to a Cr(VI) dose necessary to reach the LD50 and incubated for 3 h at 37°C with shaking. Subsequently, cells collected from culture flasks by centrifugation at 5,000 × g for 10 min were washed twice with 10 mM HEPES, and the cell pellets were suspended in 1 ml of 10 mM HEPES (pH 7.2) supplemented with 110 mM glucose (35). Then, the cell suspension was mixed with a final concentration of 10 mM DCFH-DA and incubated with shaking in the dark for 30 min at 37°C. Cells collected by centrifugation (5,000 × g; 10 min) were washed twice with 10 mM HEPES (pH 7.2). Samples of the bacterial suspensions (100 μl) were serially diluted in 1× PBS and plated to determine CFU. Finally, 100 μl of the resulting cell suspensions were mixed with 900 μl of the same buffer, and the fluorescence intensity of DCF was measured with a fluorescence spectrometer (LS-55; Perkin-Elmer, Sparta, NJ). Emission values were standardized by viable-cell counts (CFU).
Detection and quantitation of oxidative-stress-induced DNA damage.
In order to detect oxidative DNA damage, B. subtilis strains were grown in LB medium to an OD600 of 1.0 and exposed to an LD90 Cr(VI) dose for 3 h at 37°C with shaking. The cells were then lysed, and chromosomal DNA was extracted and purified as previously described (36). DNA was quantified using the GeneQuant Pro program (Amersham Biosciences, Pittsburg, PA). To detect the formation of 8-oxo-G, 5 μg of DNA was digested with 14 units of formamidopyrimidine (Fapy)-DNA glycosylase (Fpg) (New England, Biolabs, Ontario, Canada); this enzyme, in addition to its DNA glycosylase activity, possesses AP-lyase activity that generates single-strand breaks following 8-oxo-G attack (37). Subsequently, the DNA was maintained in a denatured state and electrophoresed through 0.8% alkaline agarose gels with buffer recirculation, as described previously (38).
Immunological quantitation of 8-oxo-G.
Quantitation of 8-oxo-G lesions present in samples of genomic DNA was performed with an HT 8-oxo-dG ELISA kit (Trevigen, Gaithersburg, MD). To this end, samples of DNA (30 μg) were added in duplicate to the wells of an enzyme-linked immunosorbent assay (ELISA) plate, along with different amounts of a standard 8-oxo-G solution to obtain a standard curve, following the manufacturer's instructions. Then, an 8-oxo-G monoclonal antibody that binds competitively to 8-oxo-G in the standard samples or to wells precoated with genomic DNA was added; the excess antibody was washed with PBST (1× PBS containing 0.1% Tween 20). The concentration of 8-oxo-G was determined based on the antibody retained in the wells using a horseradish peroxidase (HRP)-conjugated secondary antibody (anti-mouse IgG-HRP) and a colorimetric substrate (TACS-Sapphire [Trevigen]). The formation of yellow product indicated the presence of 8-oxo-G. Absorbance was measured in a microplate reader (Varioskan Flash Multimode Reader; Thermo Scientific, Pittsburgh, PA) at 450 nm, and quantitation of 8-oxo-G was calculated from the standard curve.
Statistical analysis.
The results obtained in the present work are represented as means ± standard errors of the mean (SEM), and a Wilcoxon signed-rank test was utilized to compare differences between wild-type and mutant strains. This nonparametric test was utilized because Box-Cox transformation was not enough to obtain data with a normal distribution (using the Shapiro-Wilk test). All tests were performed with the program Statistica v. 12, and differences were considered statistically significant at a P value of <0.05.
RESULTS
Roles of antioxidant enzymes in protecting B. subtilis from the cytotoxic effects of Cr(VI).
Previous studies with different biological systems exposed to hexavalent chromium have proposed that the oxyanion exerts its toxic effects by inducing the production of ROS that may impact different cell targets (5, 16, 39). Therefore, we first investigated whether the absence of the antioxidant enzymes KatA, KatB, and/or SodA increases the susceptibility of B. subtilis cells to Cr(VI). Accordingly, we initially determined the survival of the wild-type parental strain B. subtilis 168 following a 3-h exposure to a wide range of Cr(VI) concentrations, observing a dose-dependent decrease in survival and obtaining an LD90 of 48 ± 0.52 ppm (Fig. 2A). Following the same approach, we calculated the LD90 for each derived B. subtilis mutant and compared them with that of the wild-type parental strain. For the wild-type strain, we found that after the 3-h period of exposure to an LD50 (22 ppm) of Cr(VI), an amount equivalent to 6 ± 0.19 ppm of total chromium was incorporated into the cells. Moreover, we found no evidence of abiotic Cr(VI) reduction in an uninoculated LB culture supplemented with the same amount of the oxyanion (namely, 22 ppm) during the 3 h of incubation (data not shown).
FIG 2.

(A) Roles of KatA, KatB, SodA, PerR, and SigB in survival of B. subtilis cells exposed to hexavalent chromium. B. subtilis WT, PERM431 (ΔkatA), PERM432 (ΔkatB), PERM434 (ΔsodA), PERM1275 (ΔkatA katB), PERM1274 (ΔsodA katA), PERM679 (ΔperR), and PERM342 (ΔsigB) strains were grown in LB medium at 37°C to an OD600 of 1.0. At that point, the strains were treated with different concentrations of Cr(VI), and the cultures were grown for 3 h at 37°C with shaking. The LD90 was calculated for each strain from a dose-response curve. (B) B. subtilis strain PERM1178 (katA-GFP) was grown at 37°C in LB medium to an OD600 of 0.5; at that point, the culture was divided into three subcultures. One of the the subcultures was left as an untreated control (U), whereas the other two were supplemented with LD90s of Cr(VI) or H2O2, respectively, and shaken for an additional period of 30 min at 37°C. Samples collected from the control and treated cultures were washed and suspended in 1× PBS, and the samples' GFP fluorescence was measured as described in Materials and Methods. In panels A and B, each bar represents the mean of data collected from three independent experiments done in triplicate, and the error bars represent SEM. The asterisks indicate values that were significantly different. (C to H) Samples collected from the untreated culture (C and D) or from those treated with Cr(VI) (E and F) or H2O2 (G and H) were processed and photographed as described in Materials and Methods. (C, E, and G) Bright field; (D, F, and H) GFP channel. Scale bar, 2 μm.
Analyses revealed that cells deficient for KatA had significantly increased susceptibility to Cr(VI) (P < 0.05); however, such an effect was not observed in strains harboring single mutations on katB or sodA (Fig. 2A). Compelling evidence has shown that KatA is the main protein with catalase activity involved in preventing the toxicity of hydrogen peroxide in growing vegetative cells of B. subtilis (40); therefore, we investigated how the genetic inactivation of this function in the mutant strains deficient for KatB or SodA affected the susceptibility of B. subtilis to Cr(VI). The katA katB and katA sodA strains showed LD90 values for Cr(VI) treatment of 8 ± 0.71 ppm and 6 ± 0.34 ppm, respectively; therefore, these strains were statistically more susceptible to hexavalent chromium treatment than strains bearing single mutations on katA, katB, or sodA (P = 0.0076) (Fig. 2A). Together, these results suggest that the toxic effects of chromium are due to oxidative damage and that the main antioxidant enzymes provide protection from such toxicity to B. subtilis cells.
Several intra- and extracellular harmful factors, including oxidative stress, may activate transcriptional responses involved in maintaining the cell integrity of B. subtilis under stressful conditions (41–43). Therefore, we investigated whether Cr(VI) affects the survival of mutant strains lacking the transcriptional regulator PerR or the sigma factor σB, as both are involved in controlling the expression of genes encoding proteins with antioxidant activity (41). PerR acts as a transcriptional repressor that controls the peroxide regulon (29), whereas σB is responsible for controlling the expression of genes that belong to the general stress response (44). Accordingly, the LD90s of Cr(VI) were determined in B. subtilis strains lacking PerR or σB and were compared with that obtained for the wild-type strain. The results revealed that PerR-deficient cells, which constitutively express antioxidant genes, including katA and mrgA, were significantly more resistant (1.3-fold; P = 0.0076) to Cr(VI) than cells of the wild-type parental strain (Fig. 2A). In contrast, compared with the wild-type strain, the survival of the σB mutant was diminished ∼5-fold by Cr(VI), exhibiting an LD90 value of 9 ± 0.21 ppm (Fig. 2A). The data indicate that the components of the PerR and σB regulons contribute to survival in response to this stress, but it is not known if this is due to protection, DNA repair, or another mechanism. We further investigated this notion by analyzing whether hexavalent chromium had the ability to induce the expression of a translational PkatA-GFP fusion integrated in the amyE locus of B. subtilis strain PERM1178. As expected, with respect to an untreated control, the transcription of this fusion was increased ∼12-fold by hydrogen peroxide (Fig. 2B); of note, Cr(VI) was also able to increase the fluorescence levels of the katA-GFP fusion ∼5-fold. In line with these results, fluorescence microscopy analysis of samples obtained from cultures of the katA-GFP strain confirmed that H2O2 and Cr(VI) were able to activate synthesis of the KatA-Gfp fusion protein (Fig. 2C to H).
Cr(VI) induces an adaptive response in B. subtilis.
Since global stress responses are presumably involved in conferring protection from hexavalent chromium toxicity on B. subtilis, we next investigated whether Cr(VI) elicits an adaptive response in the microorganism. To this end, overnight B. subtilis cultures grown in the absence or presence of an LD10 (3 ppm) of hexavalent chromium were used to inoculate fresh LB medium supplemented with a higher dose (6 ppm) of the oxyanion, and the growth kinetics of these cultures, together with a control culture that was not challenged with Cr(VI), were monitored using the A600 and quantified by cell viability. Our results showed that the preadapted culture of B. subtilis was able to grow and survive better when challenged with a higher dose of hexavalent chromium than a culture of the same strain that was not previously grown in the presence of the oxyanion (Fig. 3).
FIG 3.
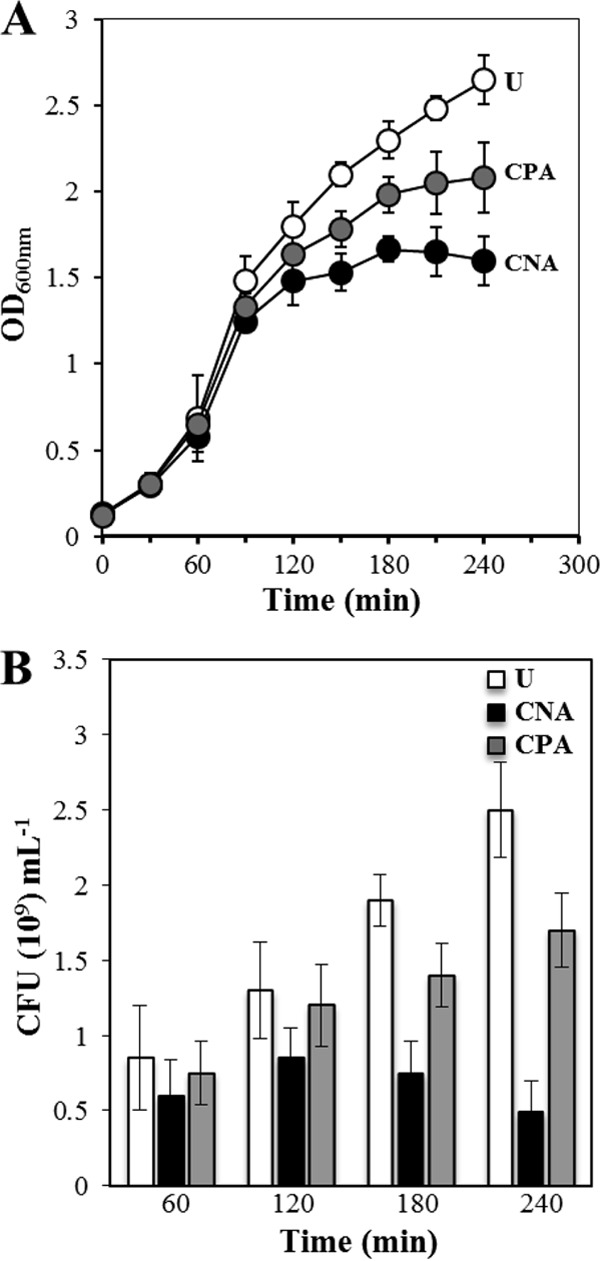
Growth kinetics (OD600) (A) and viable-cell counts (CFU) (B) for unchallenged (U), challenged nonadapted (CNA), and challenged preadapted (CPA) B. subtilis cells inoculated at an initial OD600 of 0.1 into LB cultures supplemented or not with 6 ppm of Cr(VI), as described in Materials and Methods. The values shown represent the means and standard deviations of three independent experiments done in triplicate.
Cr(VI) generates intracellular oxidative stress.
It has been proposed that the intracellular reduction of Cr(VI) to Cr(III) promotes oxidative stress, increasing the intracellular concentration of hydrogen peroxide, as well as superoxide and hydroxyl radicals that may cause oxidative damage to distinct intracellular targets (6, 45). Based on this notion, we used the probe DCFH-DA to determine the in vivo intracellular levels of ROS (33, 34) in wild-type B. subtilis cells that were grown in the presence or in the absence of hexavalent chromium. As shown in Fig. 4, the probe detected a very low level of intracellular ROS in untreated cells; however, the intracellular concentration of these toxic species dramatically increased (∼866- ± 4.85-fold; P = 0.0076) in the cells that were incubated with Cr(VI) (Fig. 4). Consequently, these results suggest that chromium affects cell viability by promoting the intracellular synthesis of oxygen radicals.
FIG 4.

Effect of Cr(VI) on the production of ROS in growing cells of different B. subtilis strains. B. subtilis WT and PERM603 (ΔGO) strains were grown at 37°C in LB medium to an OD600 of 1.0 and then exposed (gray bars) or not (white bars) for 3 h to an LD50 of Cr(VI). Cell samples of the different strains collected by centrifugation were washed with PBS and mixed with the fluorescent compound DCHFA-DA. After an incubation period of 30 min in darkness, the cells were processed to quantify the fluorescence intensity, as described in Materials and Methods. Each bar represents the mean of data collected from three independent experiments done in triplicate, and the error bars represent SEM. The asterisks indicate values that were significantly different.
Chromium kills B. subtilis cells by oxidative-stress-induced DNA damage.
As noted above, the absence of catalase and superoxide dismutase activities increased the susceptibility of B. subtilis cells to Cr(VI) (Fig. 2A); moreover, as shown above, Cr(VI) increased the intracellular concentration of ROS (Fig. 4). Several lines of evidence have revealed that one of the main cellular targets of ROS is nucleic acids (20, 46, 47); therefore, we investigated whether the oxidative stress generated by Cr(VI) modifies the genetic material of B. subtilis. To this end, chromosomal DNA samples, purified from wild-type B. subtilis cells that were treated or not with an LD90 of hexavalent chromium, were incubated with Fpg, a DNA glycosylase that specifically recognizes and hydrolyzes DNA containing the lesion 8-oxo-G (37), and the products of these reactions were analyzed by alkaline gel electrophoresis (AGE). The results showed that Fpg was able to generate slight smearing below the chromosomal DNA band isolated from cells that were not incubated with Cr(VI), indicating the presence of 8-oxo-Gs in these samples (Fig. 5A, lane 2, and Table 2). On the other hand the genomic-DNA sample that was obtained from cells treated with Cr(VI) showed significantly higher susceptibility to the attack of Fpg (Fig. 5A, lane 4, and Table 2), which is indicative of a major amount of oxidized guanines in these samples. To confirm these results, the genomic-DNA samples isolated from cultures of wild-type cells, treated or not with Cr(VI), were processed to quantify the oxidized base 8-oxo-G by ELISA using an antibody that specifically recognizes the lesion (48). The results in Fig. 5B revealed the existence of a significantly higher concentration of 8-oxo-Gs in the genomes of cells that were treated with hexavalent chromium than in those of cells that were not incubated with the oxyanion (Wilcoxon test; P = 0.027). Together, these results support the suggestion that oxidative stress promoted by chromium modifies the genetic material of B. subtilis, inducing the formation of the highly mutagenic 8-oxo-G lesion. In support of this concept, the frequency of mutation to Rifr of B. subtilis cells cultivated in the presence of Cr(VI) increased around 2.5-fold (Wilcoxon test; P = 0.011) with respect to the value calculated for B. subtilis cells grown in the absence of the oxyanion (Fig. 6B).
FIG 5.

Detection and quantification of 8-oxo-G in genomic-DNA samples isolated from different B. subtilis strains grown in the presence of Cr(VI). (A) Chromosomal DNA was purified from cell cultures of B. subtilis WT (lanes 1 to 4) and ΔGO (lanes 5 to 8) strains that were grown in the absence (lanes 1, 2, 5, and 6) or presence (lanes 3, 4, 7, and 8) of a Cr (VI) concentration equivalent to the LD90 for each strain. DNA samples (∼5 μg) were treated (lanes 2, 4, 6, and 8) or not (lanes 1, 3, 5, and 7) with 14 units of the enzyme Fpg, as described in Materials and Methods. Reaction mixture samples were electrophoresed on a 1% alkaline agarose gel that was then stained with ethidium bromide, as described in Materials and Methods. The results shown are representative of two experiments that yielded essentially similar results. CD, high-molecular-weight chromosomal DNA. (B) Chromosomal DNA was purified from cell cultures of B. subtilis WT and ΔGO strains grown in the absence (white bars) or presence (gray bars) of a concentration of Cr(VI) equivalent to the LD90 for each strain. DNA samples (30 μg) from each strain and experimental condition were used to quantify the levels of 8-oxo-G, using a specific anti-8-oxo-G antibody with an ELISA kit, as described in Materials and Methods. The values represent the average and SEM of two independent experiments done in triplicate for each sample. The asterisks indicate values that were significantly different.
TABLE 2.
Quantification of Fpg degradation of chromosomal DNA isolated from different B. subtilis strains treated or not with Cr(VI)a
| Strain | Avg (±SD) % band intensity remaining after enzyme (Fpg) treatmentb |
|
|---|---|---|
| −Cr(VI) | +Cr(VI) | |
| WT | 73 ± 2.7 | 54 ± 1.6 |
| ΔGO | 21 ± 3.5 | 14 ± 2.8 |
Samples of chromosomal DNA isolated from cell cultures of WT and ΔGO strains grown in the presence (+) or absence (−) of Cr(VI) were treated or not with Fpg, and the reaction mixtures were electrophoresed on a 1% alkaline agarose gel and stained with ethidium bromide (EtBr), as described in Materials and Methods.
For each reaction, the intensity of the chromosomal DNA band remaining in the gel well after Fpg treatment was quantified by densitometry, using the public domain ImageJ program. The reference was the chromosomal DNA band in the gel well from an identical amount of total chromosomal DNA in an untreated control reaction that was run in a lane adjacent to the lane with the same amount of total Fpg-treated chromosomal DNA, as shown in Fig. 5A, lanes 1 to 8. The values are averages ± standard deviations for duplicate determinations in three separate experiments.
FIG 6.

Effect of Cr(VI) on the resistance properties (A) and mutation frequencies (B) of different B. subtilis strains. (A) B. subtilis WT and PERM603 (ΔGO) strains were grown in LB medium at 37°C to an OD600 of 1.0; at that point, the cultures were treated with different concentrations of Cr(VI) and grown for 3 h at 37°C with shaking. The LD90 was calculated from a dose-response curve for each strain. (B) B. subtilis WT and PERM603 (ΔGO) strains were grown at 37°C in LB medium to an OD600 of 1.0 and then divided into two Erlenmeyer flasks; one of the flasks was left as an untreated control (white bars), and the other was supplemented with an LD50 of Cr(VI) (gray bars). The cultures were shaken for an additional period of 16 h at 37°C and processed to calculate the frequencies of mutation to Rifr, as described in Materials and Methods. Each bar represents the mean of data collected from at least two independent experiments for each sample, and the error bars represent SEM. The asterisks indicate values that were significantly different.
The GO system counteracts the genotoxic effects promoted by chromium in B. subtilis.
In organisms from the three domains of life, the genotoxic effects of the oxidized base 8-oxo-G are mainly counteracted by a set of three proteins collectively termed the GO system (49–51). The system is constituted of the DNA glycosylases MutM and MutY and the Nudix hydrolase MutT (18). Several experiments support the idea that a functional GO system exists in B. subtilis cells (23–25). Therefore, we investigated whether these repair proteins are involved in protecting B. subtilis cells from the noxious effects promoted by hexavalent chromium. To this end, an isogenic strain deficient in the DNA repair proteins YtkD (MutTA), MutM, and MutY was treated with different amounts of Cr(VI), and the LD90 of the mutant was compared with that calculated for the wild-type strain. As shown in Fig. 6A, absence of the full GO system markedly increased the susceptibility of B. subtilis to chromium compared to the wild-type parental strain containing the functional proteins. We further inquired whether the major susceptibility of the GO-deficient strain to Cr(VI) treatment is associated with elevated concentrations of intracellular ROS. As shown in Fig. 4, the presence of chromium dramatically increased the amount of ROS in the GO-deficient strain; in fact, the concentration of oxygen radicals was around 17 times greater in the GO mutant than in the wild-type strain (Fig. 4). To further corroborate that chromium-promoted oxidative stress induces DNA damage and that such damage is increased in the ΔGO mutant, we determined the frequency of mutation to Rifr in the presence or absence of Cr(VI). The results showed that although the ΔGO strain exhibited a hypermutagenic Rifr phenotype with a calculated spontaneous mutation frequency of ∼600 per 109 cells, the value was increased around 3-fold (1,787 ± 23; P = 0.011) by the presence of chromium in the culture medium (Fig. 6B). These results strongly suggest that one of the mechanisms employed by chromium to kill B. subtilis cells involves the production of the genotoxic 8-oxo-G lesion promoted by ROS and that the DNA repair GO system is involved in counteracting such effects. In support of these hypotheses, AGE analysis showed that in comparison with purified DNA of the wild-type strain, a significantly large amount of 8-oxo-G was detected by the Fpg probe in genomic-DNA samples isolated from the ΔGO strain, even in the absence of the metal (Fig. 5A, compare lanes 5 and 6, and Table 2). Notably, the AGE analysis also revealed that Fpg-promoted degradation dramatically increased in the DNA sample of the GO-deficient strain that was treated with Cr(VI), as revealed by the production of major smearing of these genomic-DNA samples (Fig. 5A, compare lanes 7 and 8, and Table 2). Therefore, in the absence of a functional GO system, the genome of B. subtilis seems to accumulate a large amount of 8-oxo-G; however, the presence of chromium increased the amount of oxidized guanine in the strain even more. The ELISA employed in this work to specifically quantify the concentration of 8-oxo-Gs confirmed these observations, as it revealed that with respect to wild-type B. subtilis cells, a substantial amount of the lesion was detected in the DNA purified from B. subtilis cells lacking a functional GO repair system (Fig. 5B). However, as shown in Fig. 5B, a significantly large concentration of the lesion was detected in cells of the mutTA mutM mutY triple-knockout strain that were treated with Cr(VI) (P = 0.027).
DISCUSSION
Due to their ability to proliferate in distinct environments, Bacillus species have the potential to be confronted with the toxic oxyanion Cr(VI), which can be found in either natural or polluted settings. In this work, we used B. subtilis as a model (52, 53) to explore the consequences of the soil microorganism's exposure to hexavalent chromium. In several biological systems, the toxicity of Cr(VI) has been associated with ROS-promoted damage of several intracellular targets, including nucleic acids (8, 17, 39). To contend with the genotoxic effects of oxygen radicals, cells employ three general types of strategies: (i) preventing, (ii) eliminating, or (iii) tolerating the damage (54–56). Analyses of global responses to Cr(VI) exposure by different microorganisms have revealed a common trend, i.e., the transcriptional activation of catalase and superoxide dismutase genes and additional genes whose encoded products are involved in counteracting the toxic effects of oxidative stress (12–14). Our results showed that the preventive catalase/superoxide dismutase system existing in vegetative cells, composed of KatA, KatB, and SodA (40, 57, 58), plays a prominent role in protecting B. subtilis against the toxicity of Cr(VI), as revealed by the higher susceptibility to the oxyanion exhibited by katA katB and katA sodA mutants relative to the wild-type strain. From these results, it was inferred that the functions of KatA and KatB are redundant, as the absence of KatB alone did not sensitize B. subtilis cells to chromium treatment whereas the KatA-deficient strain was only slightly more sensitive than the wild-type strain. A similar redundant function in the protection conferred by catalases to Cr(VI) was reported in E. coli, where a single katE, but not a katG, mutant was more susceptible than the parental wild-type strain to the deleterious effects of the metal (12). In B. subtilis, katA is mainly expressed in growing cells, and its transcription can be stimulated by hydrogen peroxide (40). On the other hand, katB transcription takes place during the transition growth phase as part of the σB regulon; of note, in addition to heat, ethanol, and hyperosmotic stresses, the expression of this regulon can also be stimulated by oxidative-stress inducers, including hydrogen peroxide (41, 59). On the basis of these observations, we can speculate that Cr(VI) promotes oxidative stress in vegetative B. subtilis cells and that such an event activates the PerR and σB transcriptional responses to counteract the deleterious effects of the oxyanion. In support of this contention, we found that the loss of PerR, which constitutively keeps the expression of katA, mrgA, and other members of the transcriptional regulon active (29), significantly increased the resistance of the bacterium to hexavalent chromium. In contrast, the resistance to Cr(VI) of a σB mutant that is impaired in activating the general stress response (59) was severely affected. Notably, this transcriptional circuitry controls the expression of katB, sodA, and other genes with antioxidant properties, such as dps (60, 61). In marked contrast, in E. coli cells, chromate was incapable of activating the general stress response controlled by the RpoS (σS) factor, as evidenced by the lack of induction of a pexB::lacZ transcriptional fusion that has been used as a bona fide measure of σS levels (12). The adaptive effect observed after preculturing B. subtilis with a sublethal dose of Cr(VI) also supports the notion that global transcriptional responses are involved in counteracting the toxic effects of Cr(VI) in the microorganism. Our results are not unprecedented, as similar adaptive responses elicited by Cr(VI) were also reported in S. oneidensis and E. coli (11, 12).
A wide range of enzymes and cellular metabolites have the ability to reduce Cr(VI) to Cr(III), producing short-lived highly reactive species, such as Cr(V), which in the presence of H2O2 generates hydroxyl radicals (OH·). In this process, part of the Cr(V) is oxidized back to Cr(VI), generating repeating redox cycles that could produce large amounts of OH·, subjecting the cells to severe oxidative stress (4, 5). Our results supported this mechanism, since B. subtilis cells cultured in the presence of Cr(VI), but not an untreated control, showed the presence of high levels of intracellular ROS, as detected by the DCFH-DA probe, a chemical compound whose fluorescence is activated inside the cells only after reacting with oxygen radicals (33, 34). Additional support for this mechanism came from a recent report that employed the ROS-activated green fluorescent dye 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) to demonstrate the presence of high levels of intracellular ROS in growing cells of E. coli after chromium treatment (12).
The results from previous reports have revealed that ROS, in particular the hydroxyl radical, can react with nucleic acids affecting essential cellular processes, including replication and transcription, which may promote mutagenesis and cell death (62, 63). Despite the fact that in S. cerevisiae the lethal effects of Cr(VI) have been associated with damage to proteins (15), our results revealed that in B. subtilis the noxious effects of the oxyanion could be exerted through oxidative DNA damage, specifically through the formation of 8-oxo-G. Indeed our results revealed that a B. subtilis knockout strain lacking a functional GO system was significantly more susceptible to Cr(VI) treatment than its wild-type parental strain. Of note, the levels of ROS and the amounts of 8-oxo-G lesions were higher in the mutant than in the wild-type strain in the presence of Cr(VI). We hypothesized that such effects could be attributed to the inability of the ΔGO mutant to repair GO lesions promoted by hexavalent chromium. These results strongly suggest that the higher susceptibility to hexavalent chromium of the GO-deficient strain is associated with the formation of 8-oxo-G in DNA and that the GO repair system plays a pivotal role in counteracting such effects.
It must be pointed out that the pleiotropic effects leading to major production of ROS in the GO-deficient strain are unknown; however, experiments to elucidate this phenotype using a proteomic approach are under way. Our results are consistent with a previous study (64) that employed a lymphoblastoid cell line (TK6) treated with Cr(VI), resulting in the generation of oxidative-stress-associated DNA lesions, including 8-oxo-G, which was detected by Fpg on a comet assay. Accordingly, the results of this report confirmed that production of oxidized bases is a mechanism by which Cr(VI) exerts its noxious effects in prokaryotic and eukaryotic cells (64). It has been reported that, in addition to 8-oxo-G, Fpg is also capable of processing 4,6-diamino-5-formamidopyrimidine (FapyAde) or 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua), two DNA lesions that are also generated by oxidative stress (65). Therefore, in addition to 8-oxo-G, treatment of B. subtilis cells with hexavalent chromium may result in the production of these and other types of DNA lesions, including single-strand breaks that arise during repair of oxidized bases by DNA glycosylases with associated lyase activity (39, 66) or as by-products resulting from attack of the deoxyribose sugar by oxygen radicals (62). The results from our AGE experiment support these concepts, as slight smearing of fragmented DNA was detected below the chromosomal bands isolated from the wild-type and GO-deficient strains grown in the presence of Cr(VI) (Fig. 5A, lanes 4 and 8). However, the repertoire of genetic lesions promoted by Cr(VI) could be even wider and may include bulky lesions that are substrates for the nucleotide excision and/or homologous-recombination repair pathways. In support of this contention, we found that addition of chromium to exponentially growing B. subtilis cells activated the SOS response, as demonstrated by the ability of the oxyanion to induce the expression of a recA-lacZ transcriptional fusion (data not shown). The bacterial SOS response activation property of chromium was previously observed in E. coli and other microorganisms (13, 14, 67) and may be associated with the promotion by the metal of the formation of Cr-DNA monoadducts, DNA interstrand cross-links (ICLs), and DNA-Cr-protein cross-links (DPCs) (8) that may represent blocks for replication and/or transcription. These lesions have been shown to be substrates in vitro for the UvrABC machinery of the thermoresistant bacterium Bacillus caldotenax (68). Furthermore, in B. subtilis, the translation synthesis (TLS)-encoding yqjW gene is also induced as part of the transcriptional SOS response (69), which may be in part responsible for the mutagenic effects promoted by Cr(VI) in the microorganism. Therefore, additional work will be required to better define other types of genetic injuries generated by Cr(VI) in soil microorganisms, as well as the pathways responsible for its repair.
ACKNOWLEDGMENTS
This work was supported by the University of Guanajuato (grant DAIP-324-2013) and by the Consejo Nacional de Ciencia y Tecnología (CONACYT) (grants 205744 and 221231) of México. F.S.-E. was supported by scholarships from CONACYT and the University of Guanajuato.
We are indebted to Norma Ramírez and Fernando Ramírez for invaluable technical assistance and to Jorge A. Contreras-Garduño for support with statistical analysis.
Footnotes
Published ahead of print 27 June 2014
REFERENCES
- 1.Cervantes C, Campos-García J, Devars S, Gutiérrez-Corona F, Loza-Tavera H, Torres-Guzmán JC, Moreno-Sánchez R. 2001. Interactions of chromium with microorganisms and plants. FEMS Microbiol. Rev. 25:335–347. 10.1111/j.1574-6976.2001.tb00581.x [DOI] [PubMed] [Google Scholar]
- 2.Wong P. 1988. Mutagenicity of heavy metals. Bull. Environ. Contam. Toxicol. 40:597–603. 10.1007/BF01688386 [DOI] [PubMed] [Google Scholar]
- 3.Quievryn G, Peterson E, Messer J, Zhitkovich A. 2003. Genotoxicity and mutagenicity of chromium(VI)/ascorbate-generated DNA adducts in human and bacterial cells. Biochemistry 42:1062–1070. 10.1021/bi0271547 [DOI] [PubMed] [Google Scholar]
- 4.Shi X, Dalal N. 1994. Generation of hydroxyl radical by chromate in biologically relevant systems: role of Cr(V) complexes versus tetraperoxochromate(V). Environ. Health Perspect. 102:231–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu KJ, Shi X. 2001. In vivo reduction of chromium(VI) and its related free radical generation. Mol. Cell. Biochem. 222:41–47. 10.1023/A:1017994720562 [DOI] [PubMed] [Google Scholar]
- 6.Itoh M, Nakamura M, Suzuki T, Kawai K, Horitsu H, Takamizawa K. 1995. Mechanism of chromium(VI) toxicity in Escherichia coli: is hydrogen peroxide essential in Cr(VI) toxicity? J. Biochem. 117:780–786 [DOI] [PubMed] [Google Scholar]
- 7.Qi W, Reiter R, Tan D, Garcia J, Manchester L, Karbownik M, Calvo J. 2000. Chromium(III)-induced 8-hydroxydeoxyguanosine in DNA and its reduction by antioxidants: comparative effects of melatonin, ascorbate, and vitamin E. Environ. Health Perspect. 108:399–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhitkovich A. 2005. Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium(VI). Chem. Res. Toxicol. 18:3–11. 10.1021/tx049774+ [DOI] [PubMed] [Google Scholar]
- 9.Salnikow K, Zhitkovich A. 2008. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic and chromium. Chem. Res. Toxicol. 21:28–44. 10.1021/tx700198a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dey S, Nayak P, Roy S. 2001. Chromium-induced membrane damage: protective role of ascorbic acid. J. Environ. Sci. (China) 13:272–275 [PubMed] [Google Scholar]
- 11.Chourey K, Thompson M, Morrell-Falvey J, VerBerkmoes N, Brown S, Shah M, Zhou J, Doktycz M, Hettich R, Thompson D. 2006. Global molecular and morphological effects of 24-hour chromium(VI) exposure on Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 72:6331–6344. 10.1128/AEM.00813-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ackerley D, Barak Y, Lynch S, Curtin J, Matin A. 2006. Effect of chromate stress on Escherichia coli K-12. J. Bacteriol. 188:3371–3381. 10.1128/JB.188.9.3371-3381.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu P, Brodie E, Suzuki Y, McAdams H, Andersen G. 2005. Whole-genome transcriptional analysis of heavy metal stresses in Caulobacter crescentus. J. Bacteriol. 187:8437–8449. 10.1128/JB.187.24.8437-8449.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown S, Thompson M, VerBerkmoes N, Chourey K, Shah M, Zhou J, Hettich R, Thompson D. 2006. Molecular dynamics of the Shewanella oneidensis response to chromate stress. Mol. Cell. Proteomics 5:1054–1071. 10.1074/mcp.M500394-MCP200 [DOI] [PubMed] [Google Scholar]
- 15.Sumner E, Shanmuganathan A, Sideri T, Willetts S, Houghton J, Avery S. 2005. Oxidative protein damage causes chromium toxicity in yeast. Microbiology 151:1939–1948. 10.1099/mic.0.27945-0 [DOI] [PubMed] [Google Scholar]
- 16.Aiyar J, Berkovits H, Floyd R, Wetterhahn K. 1991. Reaction of chromium(VI) with glutathione or with hydrogen peroxide: identification of reactive intermediates and their role in chromium(VI)-induced DNA damage. Environ. Health Perspect. 92:53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugden K, Campo C, Martin B. 2001. Direct oxidation of guanine and 7,8-dihydro-8-oxoguanine in DNA by a high-valent chromium complex: a possible mechanism for chromate genotoxicity. Chem. Res. Toxicol. 14:1315–1322. 10.1021/tx010088+ [DOI] [PubMed] [Google Scholar]
- 18.Michaels M, Miller J. 1992. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J. Bacteriol. 174:6321–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruner S, Nash H, Lane W, Verdine G. 1998. Repair of oxidatively damaged guanine in Saccharomyces cerevisiae by an alternative pathway. Curr. Biol. 8:393–403 [DOI] [PubMed] [Google Scholar]
- 20.Sekiguchi M, Tsuzuki T. 2002. Oxidative nucleotide damage: consequences and prevention. Oncogene 21:8895–8904. 10.1038/sj.onc.1206023 [DOI] [PubMed] [Google Scholar]
- 21.Tajiri T, Maki H, Sekiguchi M. 1995. Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat. Res. 336:257–267. 10.1016/0921-8777(94)00062-B [DOI] [PubMed] [Google Scholar]
- 22.Ramírez M, Castellanos-Juárez F, Yasbin R, Pedraza-Reyes M. 2004. The ytkD (mutTA) gene of Bacillus subtilis encodes a functional antimutator 8-oxo-(dGTP/GTP)ase and is under dual control of sigma A and sigma F RNA polymerases. J. Bacteriol. 186:1050–1059. 10.1128/JB.186.4.1050-1059.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castellanos-Juárez F, Alvarez-Alvarez C, Yasbin R, Setlow B, Setlow P, Pedraza-Reyes M. 2006. YtkD and MutT protect vegetative cells but not spores of Bacillus subtilis from oxidative stress. J. Bacteriol. 188:2285–2289. 10.1128/JB.188.6.2285-2289.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasaki M, Yonemura Y, Kurusu Y. 2000. Genetic analysis of Bacillus subtilis mutator genes. J. Gen. Appl. Microbiol. 46:183–187. 10.2323/jgam.46.183 [DOI] [PubMed] [Google Scholar]
- 25.Vidales L, Cárdenas L, Robleto E, Yasbin R, Pedraza-Reyes M. 2009. Defects in the error prevention oxidized guanine system potentiate stationary-phase mutagenesis in Bacillus subtilis. J. Bacteriol. 191:506–513. 10.1128/JB.01210-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Díaz-Magaña A, Aguilar-Barajas E, Moreno-Sánchez R, Ramírez-Díaz M, Riveros-Rosas H, Vargas E, Cervantes C. 2009. Short-chain chromate ion transporter proteins from Bacillus subtilis confer chromate resistance in Escherichia coli. J. Bacteriol. 191:5441–5445. 10.1128/JB.00625-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vagner V, Dervyn E, Ehrlich S. 1998. A vector for systematic gene inactivation in Bacillus subtilis. Microbiology 144:3097–3104. 10.1099/00221287-144-11-3097 [DOI] [PubMed] [Google Scholar]
- 28.Casillas-Martinez L, Setlow P. 1997. Alkyl hydroperoxide reductase, catalase, MrgA, and superoxide dismutase are not involved in resistance of Bacillus subtilis spores to heat or oxidizing agents. J. Bacteriol. 179:7420–7425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuangthong M, Herbig A, Bsat N, Helmann J. 2002. Regulation of the Bacillus subtilis fur and perR genes by PerR: not all members of the PerR regulon are peroxide inducible. J. Bacteriol. 184:3276–3286. 10.1128/JB.184.12.3276-3286.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Urone P. 1955. Stability of colorimetric reagent for chromium, s-diphenylcarbazide, in various solvents. Anal. Chem. 27:1354–1355. 10.1021/ac60104a048 [DOI] [Google Scholar]
- 31.Muñoz A, Corona F, Wrobel K, Soto G, Wrobel K. 2005. Subcellular distribution of aluminum, bismuth, cadmium, chromium, copper, iron, manganese, nickel, and lead in cultivated mushrooms (Agaricus bisporus and Pleurotus ostreatus). Biol. Trace Elem. Res. 106:265–277. 10.1385/BTER:106:3:265 [DOI] [PubMed] [Google Scholar]
- 32.Hoover S, Xu W, Xiao W, Burkholder W. 2010. Changes in DnaA-dependent gene expression contribute to the transcriptional and developmental response of Bacillus subtilis to manganese limitation in Luria-Bertani medium. J. Bacteriol. 192:3915–3924. 10.1128/JB.00210-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Setsukinai K, Urano Y, Kakinuma K, Majima H, Nagano T. 2003. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J. Biol. Chem. 278:3170–3175. 10.1074/jbc.M209264200 [DOI] [PubMed] [Google Scholar]
- 34.LeBel C, Ischiropoulos H, Bondy S. 1992. Evaluation of the probe 2′,7′-dichiorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem. Res. Toxicol. 5:227–231. 10.1021/tx00026a012 [DOI] [PubMed] [Google Scholar]
- 35.Campos S, Ibarra-Rodriguez J, Barajas-Ornelas R, Ramírez-Guadiana F, Obregón-Herrera A, Setlow P, Pedraza-Reyes M. 2014. Interaction of apurinic/apyrimidinic endonucleases Nfo and ExoA with the DNA integrity scanning protein DisA in the processing of oxidative DNA damage during Bacillus subtilis spore outgrowth. J. Bacteriol. 196:568–578. 10.1128/JB.01259-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cutting S, Vander-Horn P. 1990. Genetic analysis, p 27–74 In Harwood C, Cuttin S. (ed), Molecular biological methods for Bacillus. John Wiley and Sons Ltd, West Sussex, England [Google Scholar]
- 37.Tchou J, Bodepudi V, Shibutani S, Antoshechkin I, Miller J, Grollman A, Johnson F. 1994. Substrate specificity of Fpg protein: recognition and cleavage of oxidatively damaged DNA. J. Biol. Chem. 269:15318–15324 [PubMed] [Google Scholar]
- 38.Sambrook J, Russell D. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 39.da Casadevalla M, Andreas CPK. 1999. Chromium(VI)-mediated DNA damage: oxidative pathways resulting in the formation of DNA breaks and abasic sites. Chem. Biol. Interact. 123:117–132 [DOI] [PubMed] [Google Scholar]
- 40.Bol D, Yasbin R. 1991. The isolation, cloning and identification of a vegetative catalase gene from Bacillus subtilis. Gene 109:31–37. 10.1016/0378-1119(91)90585-Y [DOI] [PubMed] [Google Scholar]
- 41.Helmann J, Wu M, Gaballa A, Kobel P, Morshedi M, Fawcett P, Paddon C. 2003. The global transcriptional response of Bacillus subtilis to peroxide stress is coordinated by three transcription factors. J. Bacteriol. 185:243–253. 10.1128/JB.185.1.243-253.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mostertz J, Scharf C, Hecker M, Homuth G. 2004. Transcriptome and proteome analysis of Bacillus subtilis gene expression in response to superoxide and peroxide stress. Microbiology 150:497–512. 10.1099/mic.0.26665-0 [DOI] [PubMed] [Google Scholar]
- 43.Zuber P. 2009. Management of oxidative stress in Bacillus. Annu. Rev. Microbiol. 63:575–597. 10.1146/annurev.micro.091208.073241 [DOI] [PubMed] [Google Scholar]
- 44.Haldenwang W. 1995. The sigma factors of Bacillus subtilis. Microbiol. Rev. 59:1–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu K, Shi X, Dalal N. 1997. Synthesis of Cr(IV)-GSH, its identification and its free hydroxyl radical generation: a model compound for Cr(VI) carcinogenicity. Biochem. Biophys. Res. Commun. 235:54–58. 10.1006/bbrc.1997.6277 [DOI] [PubMed] [Google Scholar]
- 46.Cooke M, Olinski R, Loft S. 2008. Measurement and meaning of oxidatively modified DNA lesions in urine. Cancer Epidemiol. Biomarkers Prev. 17:3–14. 10.1158/1055-9965.EPI-07-0751 [DOI] [PubMed] [Google Scholar]
- 47.Kasai H. 2002. Chemistry-based studies on oxidative DNA damage: formation, repair, and mutagenesis. Free Radic. Biol. Med. 33:450–456. 10.1016/S0891-5849(02)00818-3 [DOI] [PubMed] [Google Scholar]
- 48.Yin B, Whyatt R, Perera F, Randall M, Cooper T, Santella R. 1995. Determination of 8-hydroxydeoxyguanosine by an immunoaffinity chromatography-monoclonal antibody-based ELISA. Free Radic. Biol. Med. 18:1023–1032. 10.1016/0891-5849(95)00003-G [DOI] [PubMed] [Google Scholar]
- 49.Tsuzuki T, Nakatsu Y, Nakabeppu Y. 2007. Significance of error-avoiding mechanisms for oxidative DNA damage in carcinogenesis. Cancer Sci. 98:465–470. 10.1111/j.1349-7006.2007.00409.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oliver A, Sánchez J, Blázquez J. 2002. Characterization of the GO system of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 217:31–35. 10.1111/j.1574-6968.2002.tb11452.x [DOI] [PubMed] [Google Scholar]
- 51.Lu A, Li X, Gu Y, Wright P, Chang D. 2001. Repair of oxidative DNA damage: mechanisms and functions. Cell Biochem. Biophys. 35:141–170. 10.1385/CBB:35:2:141 [DOI] [PubMed] [Google Scholar]
- 52.Earl A, Losick R, Kolter R. 2008. Ecology and genomics of Bacillus subtilis. Trends Microbiol. 16:269–275. 10.1016/j.tim.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessiéres P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell SC, Bron S, Brouillet S, Bruschi CV, Caldwell B, Capuano V, Carter NM, Choi SK, Codani JJ, Connerton IF, Cummings NJ, Daniel RA, Denizot F, Devine KM, Düsterhöft A, Erlich SD, Emmerson PT, Entian KD, Errington J, Fabret C, Ferrari E, Foulger D, Fritz C, Fujita M, Fujita Y, Fuma S, Galizzi A, Galleron N, Ghim SY, Glaser P, Goffeau A, Golightly EJ, Grandi G, Guiseppi G, Guy BJ, Haga K, et al. 1997. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390:249–256. 10.1038/36786 [DOI] [PubMed] [Google Scholar]
- 54.Imlay J. 2008. Cellular defenses against superoxide and hydrogen peroxide. Annu. Rev. Biochem. 77:755–776. 10.1146/annurev.biochem.77.061606.161055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lushchak V. 2001. Oxidative stress and mechanisms of protection against it in bacteria. Biochemistry (Moscow) 66:476–489 [DOI] [PubMed] [Google Scholar]
- 56.Sale J, Lehmann A, Woodgate R. 2012. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 13:141–152. 10.1038/nrm3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Inaoka T, Matsumura Y, Tsuchido T. 1998. Molecular cloning and nucleotide sequence of the superoxide dismutase gene and characterization of its product from Bacillus subtilis. J. Bacteriol. 180:3697–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Engelmann S, Lindner C, Hecker M. 1995. Cloning, nucleotide sequence, and regulation of katE encoding a σB-dependent catalase in Bacillus subtilis. J. Bacteriol. 177:5598–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reder A, Höper D, Gerth U, Hecker M. 2012. Contributions of individual σB-dependent general stress genes to oxidative stress resistance of Bacillus subtilis. J. Bacteriol. 194:3601–3610. 10.1128/JB.00528-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petersohn A, Brigulla M, Haas S, Hoheisel J, Völker U, Hecker M. 2001. Global analysis of the general stress response of Bacillus subtilis. J. Bacteriol. 183:5617–5631. 10.1128/JB.183.19.5617-5631.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Höper D, Völker U, Hecker M. 2005. Comprehensive characterization of the contribution of individual SigB-dependent general stress genes to stress resistance of Bacillus subtilis. J. Bacteriol. 187:2810–2826. 10.1128/JB.187.8.2810-2826.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Balasubramanian B, Wendy K, Thomas D. 1998. DNA strand breaking by the hydroxyl radical is governed by the accessible surface areas of the hydrogen atoms of the DNA backbone. Proc. Natl. Acad. Sci. U. S. A. 95:9738–9743. 10.1073/pnas.95.17.9738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Avery S. 2011. Molecular targets of oxidative stress. Biochem. J. 434:201–210. 10.1042/BJ20101695 [DOI] [PubMed] [Google Scholar]
- 64.El-Yamani N, Zúñiga L, Stoyanova E, Creus A, Marcos R. 2011. Chromium-induced genotoxicity and interference in human lymphoblastoid cell (TK6) repair processes. J. Toxicol. Environ. Health A 74:1030–1039. 10.1080/15287394.2011.582282 [DOI] [PubMed] [Google Scholar]
- 65.Karakaya A, Jaruga P, Bohr V, Grollman A, Dizdaroglu M. 1997. Kinetics of excision of purine lesions from DNA by Escherichia coli Fpg protein. Nucleic Acids Res. 25:474–479. 10.1093/nar/25.3.474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stearns D, Kennedy L, Courtney K, Giangrande P, Phieffer L, Wetterhahn K. 1995. Reduction of chromium(VI) by ascorbate leads to chromium-DNA binding and DNA strand breaks in vitro. Biochemistry 34:910–919. 10.1021/bi00003a025 [DOI] [PubMed] [Google Scholar]
- 67.Llagostera M, Garrido S, Guerrero R, Barbé J. 1986. Induction of SOS genes of Escherichia coli by chromium compounds. Environ. Mutagen. 8:571–577. 10.1002/em.2860080408 [DOI] [PubMed] [Google Scholar]
- 68.O'Brien T, Jiang G, Chun G, Mandel H, Westphal C, Kahen K, Montaser A, States J, Patierno S. 2006. Incision of trivalent chromium [Cr(III)]-induced DNA damage by Bacillus caldotenax UvrABC endonuclease. Mutat. Res. 610:85–92. 10.1016/j.mrgentox.2006.06.015 [DOI] [PubMed] [Google Scholar]
- 69.Duigou S, Ehrlich S, Noirot P, Noirot-Gros M. 2004. Distinctive genetic features exhibited by the Y-family DNA polymerases in Bacillus subtilis. Mol. Microbiol. 54:439–451. 10.1111/j.1365-2958.2004.04259.x [DOI] [PubMed] [Google Scholar]