

Abstract
Insects are the most abundant animals on Earth, and the microbiota within their guts play important roles by engaging in beneficial and pathological interactions with these hosts. In this study, we comprehensively characterized insect-associated gut bacteria of 305 individuals belonging to 218 species in 21 taxonomic orders, using 454 pyrosequencing of 16S rRNA genes. In total, 174,374 sequence reads were obtained, identifying 9,301 bacterial operational taxonomic units (OTUs) at the 3% distance level from all samples, with an average of 84.3 (±97.7) OTUs per sample. The insect gut microbiota were dominated by Proteobacteria (62.1% of the total reads, including 14.1% Wolbachia sequences) and Firmicutes (20.7%). Significant differences were found in the relative abundances of anaerobes in insects and were classified according to the criteria of host environmental habitat, diet, developmental stage, and phylogeny. Gut bacterial diversity was significantly higher in omnivorous insects than in stenophagous (carnivorous and herbivorous) insects. This insect-order-spanning investigation of the gut microbiota provides insights into the relationships between insects and their gut bacterial communities.
INTRODUCTION
Many symbiotic associations of bacteria have developed within the insect gut (1). Bacterial mutualists in insect guts play an important role in regulating the host's metabolism, and they also promote efficient digestion for extraction of maximum energy from ingested foods (2) and protect the host from other, potentially harmful microbes (3). In addition to beneficial functions, the insect gut microbiota may also engage in opportunistically harmful interactions with the host (4–6). Our previous study on Drosophila flies revealed that alterations in the gut microbiota induced by the host's deregulated immune genotype led to host mortality (7).
The microbiota of the insect gut has been analyzed using both culture-dependent (8–10) and culture-independent (11–15) methods. However, culture-dependent methods often produce biased results, depending upon the conditions and techniques used. Culture-independent molecular ecological approaches based on analysis of the 16S rRNA gene have yielded a better and more comprehensive picture of bacterial communities and have resulted in a dramatic improvement in our understanding of the microbes living within the guts of insects. The use of recent advances in molecular biology and the application of high-throughput next-generation sequencing technologies to microbial ecology have shown that the diversity in microbial populations is significantly higher than previously estimated by traditional culture-based and conventional molecular methods and that “rare biospheres” may be masked by dominant microorganisms (16). Comprehensive analysis of the bacterial diversity within a host species is a prerequisite in both insect physiology and microbial ecology to allow a better understanding of the ecological roles of insect symbionts and interactions with their insect hosts. However, most studies on gut bacterial diversity have been taxon specific, focusing on insects such as termites (10, 17, 18), ants (19–21), fire bugs (22–24), fruit flies (13, 15, 25), beetles (9, 26, 27), and bees (28–31), leaving a need for broader and systematic characterization and comparison across all insects.
To date, there are no reports regarding the general pattern and composition of gut microbes in diverse orders of Insecta, even though previous research on bacterial diversity in insects has focused mostly on a phylogenetically restricted group of insect taxa. We now need to confirm the symbiotic relationships between hosts and their gut bacteria for diverse insects; therefore, the present study aimed to make a comprehensive, insect-order-spanning investigation of the gut microbiota by use of an extensive sampling strategy and deep-sequencing technology. In particular, this work focused on the detailed bacterial profiles within the insect gut and the effects of the environmental habitat, diet, developmental stage, and phylogenetic position of insect hosts on the composition of the insect gut microbiota.
MATERIALS AND METHODS
Insect sampling.
A total of 305 insect specimens representing 218 species were collected from 15 sites in South Korea between April 2010 and March 2012 (see Table S1 and Fig. S1 in the supplemental material) and were transferred immediately to the laboratory for DNA extraction. The collected insects belonged to 21 different orders of Insecta: Archaeognatha, Blattaria, Coleoptera, Dermaptera, Diptera, Ephemeroptera, Hemiptera, Hymenoptera, Isoptera, Lepidoptera, Mantodea, Mecoptera, Megaloptera, Neuroptera, Odonata, Orthoptera, Phasmatodea, Plecoptera, Thysanoptera, Thysanura, and Trichoptera. To investigate the effects of geographical location and insect developmental stage on the gut microbiota, Apis mellifera (honeybee) specimens were collected from five different locations. Specimens of Lycorma delicatula (planthopper) and Illeis koebelei (yellow ladybird) were collected at different developmental stages (larva and adult) to determine their gut microbiota. Collected specimens were grouped into four habitat types in a given developmental stage, as follows: “sky” insects that primarily use flight to search for food or a mate, “underground” insects that spend most of their lifetime underground (although they might sometimes be exposed to search for food), “aquatic” insects that are primarily aquatic (including those that feed or mate in water), and “ground” insects that are not categorized in the other habitat types. A host-associated insect sample was excluded from the analysis of the habitat-dependent microbiota.
DNA extraction.
Gut samples obtained from the crop to the hindgut of adults and larvae/nymphs were removed using sterile forceps and homogenized by shaking in a sterile tube containing glass beads (0.5-mm diameter) and 0.5 ml STES buffer (1% SDS, 0.2 M Tris-HCl [pH 7.6], 10 mM EDTA, 0.5 M NaCl) for 15 min, using a CM-1000 mixer (Eyela, Tokyo, Japan). For very small insects (i.e., Isoptera sp., Thrips palmi, and Bemisia tabaci), DNAs were extracted from whole bodies after washing twice in 100% ethanol. Genomic DNA was extracted using standard phenol-chloroform and ethanol precipitation methods (32). To identify the phylogenetic status of the collected insects, host DNA was also extracted from the legs or bodies by using the same methodology.
Determination of insect 18S rRNA gene sequences and phylogenetic analysis of insects.
Insects were initially named on the basis of morphological characteristics before their identity was resolved using 18S rRNA gene sequencing. The 18S rRNA genes were amplified from insect DNA by using Maxime PCR PreMix (i-Taq) (iNtRON Biotechnology, South Korea) and the primer pair 18S-1L (5′-TACCTGGTTGATCCTGCCAGT-3′) and 18S-3R (5′-CCTACGGAAACCTTGTTACG-3′) or 18S-1.2F (5′-TGCTTGTCTCAAAGATTAAGC-3′) and 18S-7R (5′-GCATCACAGACCTGTTATTGC-3′). Conditions for PCR were initial denaturation for 10 min at 95°C, followed by 30 cycles of 95°C for 30 s, 58°C for 45 s, and 72°C for 1 min 20 s and a final extension step of 10 min at 72°C. PCR products were sequenced using an ABI Prism 3730XL DNA analyzer (Applied Biosystems). Amplified 18S rRNA gene fragments were assembled using SeqMan, and the sequences (approximately 1,000 bp) were then compared with others deposited in the nucleotide collection (nr/nt) in GenBank, using BLASTN searches. Alignment of 18S rRNA gene sequences was performed using ClustalW, and a phylogenetic tree was constructed based on the neighbor-joining algorithm, using MEGA5. The 18S rRNA gene sequence of the hard tick Ixodes ricinus (accession no. GU074706) was used as the outgroup.
Pyrosequencing of bacterial 16S rRNA genes.
The bacterial compositions of insect guts were determined by PCR amplification of purified genomic DNA by use of Maxime PCR PreMix (i-Taq) (iNtRON Biotechnology, South Korea). PCR amplification of the V1-V2 region of the 16S rRNA gene was performed using the following primers, which contain linker sequences (TC or CA) and eight-base, sample-specific bar-coded sequences (designated X): 8F (5′-X-TC-AGAGTTTGATCCTGGCTCAG-3′) and 338R (5′-X-CA-TGCTGCCTCCCGTAGGAGT-3′). Each sample was replicated three times (technical replicates). The conditions for PCR were as follows: initial denaturation at 94°C for 2 min, followed by 30 cycles of denaturation at 94°C for 1 min, annealing at 60°C for 30 s, and extension at 72°C for 1 min and a final extension step of 10 min at 72°C. Potential contamination of buffers and primer sets was checked by PCRs using DNA-free samples. PCR products were pooled and then purified. Equimolar amounts of amplicons were combined, and the resultant DNA quality was evaluated on a Bioanalyzer 2100 instrument, using a DNA1000 lab chip (Agilent). Pooled DNA samples were then amplified by emulsion PCR before 454 pyrosequencing was performed by a sequencing provider (Macrogen, South Korea), using a GS FLX Titanium system according to the manufacturer's instructions (Roche 454 Life Sciences).
Bacterial 16S rRNA gene sequence analysis.
Low-quality sequences that were shorter than 250 nucleotides (nt) or contained more than one ambiguous base and sequences of the 16S rRNA gene primers and bar codes were removed using the trim.seqs script in mothur v.1.30.2 (33). High-quality sequences were aligned using the Infernal aligner at the Ribosomal Database Project (RDP) website (34) and were trimmed to represent the V1-V2 region by using the BioEdit program for accurate analysis with the same regions and an increase of the alignment speed. Sequences corresponding to chloroplast or eukaryotic genes, which may have been from undigested food particles or host tissues, were removed on the basis of the RDP classifier outcome (threshold bootstrap value of 80%). The remaining 222,465 sequences were aligned against the SILVA alignment database (http://www.mothur.org/w/images/9/98/Silva.bacteria.zip). The pre.cluster scripts in mothur were used to denoise sequences, and the screen.seq, filter.seq, and chimera.slayer scripts in mothur were used to screen for high-quality sequences. Operational taxonomic units (OTUs) were determined using the cluster script with the furthest-neighbor algorithm, based on a 3% distance level, as previously described by Nam et al. (35). After further processing, 209,535 sequences were used for SILVA-based classification, using the classify.seqs script with the k-nearest-neighbor algorithm, with a bootstrap confidence score of 100 and a bootstrap value of 80 for taxonomic assignment. The closest bacterial relatives were assigned according to the best match in the database. Sequences that were identified as chloroplast (10,402 sequences) and singleton (24,577 sequences) sequences and had bootstrap values of <80 (182) were removed based on the SILVA-based classification. The remaining 174,374 sequences were used for bacterial community analysis. The mothur package was used to calculate Good's coverage, the abundance-based coverage estimator (ACE), the bias-corrected Chao1 richness estimator, the jackknife estimator of species richness, and the Shannon-Weaver and Simpson diversity indices, and the results (means ± standard deviations [SD]) are presented in Table S2 in the supplemental material. To determine the bacterial communities at the level of the insect order, the 16S rRNA gene sequences were compared with those in the CAMERA Prokaryotic Nucleotide Sequence Database, with E values of <10−10 for calculation of percent identities of 16S rRNA gene sequences. The acquired percent identities were used for column statistics analysis, using GraphPad Prism 5.0 for Windows (GraphPad Software).
Statistical comparison of bacterial communities based on habitat, diet, developmental stage, and phylogeny.
Oxygen requirements for the growth of insect gut bacteria comprising more than 1% abundance in this study were defined as aerobes, microaerobes, facultative anaerobes, and anaerobes, according to genera reported in previous studies (see Table S3 in the supplemental material). The aerobe and microaerobe groups were collectively considered aerobes for this analysis. The classified groups “fungivore” and “no-feed” (according to diet type) and insect orders containing fewer than five samples (phylogenetic position of insect hosts) were not included for the statistical analyses due to low sampling depth. For statistical analyses shown in Fig. 4 and 5, the two-tailed unpaired t test was performed to compare the two different groups of developmental stages. The Kruskal-Wallis test was used to analyze differences between the relative abundances of aerobes, facultative anaerobes, and anaerobes in insects classified by environmental habitat, diet, and phylogeny, and also between species richness and the Shannon indices of the classified insects. P values for pairwise comparisons were corrected with Dunn's posttest, using GraphPad Prism 5.0.
FIG 4.

Relative abundances of insect gut bacterial groups, categorized by their oxygen requirements, according to the hosts' environmental habitat, diet, developmental stage, and phylogenetic position. White, gray, and black bars represent aerobes, facultative anaerobes, and anaerobes, respectively. The bacteria were characterized based on previous descriptions of the phenotypic features of each of the bacterial genera, as shown in Table S3 in the supplemental material. Asterisks indicate statistically significant differences among all pairs of relative abundances (*, P < 0.05; **, P < 0.01).
FIG 5.

Bacterial diversity in insect gut microbiota with respect to the hosts' environmental habitat, diet, developmental stage, and phylogeny. The amount of bacterial diversity was determined by comparing the numbers of OTUs (top) and the Shannon diversity indices (bottom). Asterisks indicate statistically significant differences among all pairs of values (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
To determine the relationships between bacterial communities and the different factors (environmental habitat, diet, developmental stage, and host phylogeny) shown in Table S4 in the supplemental material, similarities of microbiota composition or structure among groups were qualified and quantified using a metric based on OTU richness (Jaccard) or abundance (Thetayc or Bray-Curtis dissimilarity). Significant effects of group types were tested by analysis of molecular variance (AMOVA), analysis of similarity (ANOSIM), and UniFrac analysis (unweighted and weighted), based on a PHYLIP-formatted distance matrix, using Jclass and Thetayc calculators of community membership and structure similarity, respectively, in mothur. The generated PHYLIP-formatted distance matrix based on Jaccard values was used for principal coordinate analysis (PCoA) to determine microbial community differences across the range of insects classified by environmental factors.
To identify shared OTUs among the gut bacteria of Apis mellifera, Lycorma delicatula, and Illeis koebelei according to geographical location and developmental stage, the make.shared and get.shared.seqs scripts in mothur were used. A comparison of the bacterial diversities in the guts of bees was made using 16S rRNA gene sequences from previous studies (28–30). For principal component analysis (PCA), sequences showing abundances of >1% at the level of bacterial family in each insect sample were used. Profile comparisons were performed using Pearson's correlation (36, 37) with 0.1% optimization, and a dendrogram was generated using the unweighted-pair group method using average linkages (UPGMA) with BioNumerics software (Applied Maths, Belgium).
Comparison of insect and mammalian gut microbiota.
Bacterial 16S rRNA gene sequences from mammalian feces were obtained from the published data set (total of 25,851 sequences) reported by Ley et al. (38). Sequences shorter than 250 nt were removed, and those remaining were trimmed to maintain the V1-V2 region. A total of 24,445 sequences were analyzed by PCoA. The sequences that originated from mammals were analyzed by use of mothur as described above for insect gut bacteria.
Nucleotide sequence accession numbers.
The 16S rRNA gene sequences identified in the insect gut microbiota and the 18S rRNA gene sequences of insect samples reported in this study were submitted to NCBI under accession numbers SRA061337 and KC413646 to KC413939, respectively.
RESULTS
Analysis of bacterial 16S rRNA gene sequences.
A total of 174,374 high-quality sequence reads were identified as belonging to the domain Bacteria. Each insect sample had an average of 571.7 (±544.4) reads (see Table S2 in the supplemental material). The total and average numbers of OTUs for each insect were 9,301 and 84.3 (±97.7), respectively. Good's coverage, which estimates the percentage of OTUs represented in an insect sample, averaged 92% (±0.07%), suggesting that the majority of bacterial phylotypes in the insect gut were included in this study.
Taxonomic classification of bacteria in the insect gut.
A total of 18 bacterial phyla and unclassified bacteria were detected across 21 orders (Fig. 1). The majority of sequences were those of the Proteobacteria (62.1% of the classified sequences) and Firmicutes (20.7%), followed by Bacteroidetes (6.4%), Actinobacteria (4.8%), Tenericutes (1.9%), and unclassified bacteria (3.0%) (see Fig. S2 in the supplemental material). At the bacterial class level, 34.1%, 7.5%, and 19.6% of the total sequences represented the Alpha-, Beta-, and Gammaproteobacteria, respectively. Bacilli and Clostridia (belonging to the phylum Firmicutes) represented 18.0% and 2.3% of the sequences, respectively, followed by 4.8% Actinobacteria, 3.1% Bacteroidia, 2.1% Flavobacteria (Bacteroidetes), and 1.9% Mollicutes (Tenericutes). At the family level, the Anaplasmataceae (14.1%) and Enterobacteriaceae (12.0%) were the most dominant. At the genus level, the Wolbachia group (14.1%) was most prevalent. To determine the novelty of the bacterial communities in the insect guts, the 16S rRNA gene sequences reported here were compared with those in the CAMERA database. The mean values for percent sequence identity ranged from 93.1% to 99.2% (average, 97.1% ± 0.10%) (Fig. 2). Relatively low sequence similarity values were obtained for the orders Megaloptera (mean value, 92.1%), Mecoptera (93.5%), Blattaria (94.7%), Archaeognatha (96.1%), and Plecoptera (96.1%). This indicates that a large number of novel candidate bacterial groups are present in insect guts. On the other hand, sequences assigned to the genus Wolbachia were commonly found in 17 insect orders (Wolbachia was the dominant species in Ephemeroptera, Megaloptera, Diptera, Lepidoptera, Hymenoptera, Orthoptera, Neuroptera, and Dermaptera) but were not present in the 13 insects belonging to the orders Mantodea, Blattaria, Isoptera, and Phasmatodea (Fig. 3a). Sequences of Rickettsia were present in 19 insect orders, and sequences of Spiroplasma were present in 17 insect orders. “Candidatus Cardinium” sequences were present in only 1 insect order. Although the presence of heritable symbionts varied between individual insects, a large number of specimens harbored these bacteria from the Wolbachia, Rickettsia, and Spiroplasma genera. This indicates that possible bacterial symbionts might be distributed more widely across insect taxa than previously thought.
FIG 1.

Abundance and composition of gut microbiota in 218 insect species. 18S rRNA gene sequences were aligned using the ClustalW program, and a phylogenetic tree was constructed based on the neighbor-joining algorithm, using the MEGA5 program. (Left) The 21 insect orders in the phylogenetic tree are represented by colored lines. (Right) The compositions of the gut microbiota at the phylum level, based on 16S rRNA gene sequences, are shown in the bar graph. The insect species in the phylogenetic tree are described in Table S1 in the supplemental material.
FIG 2.
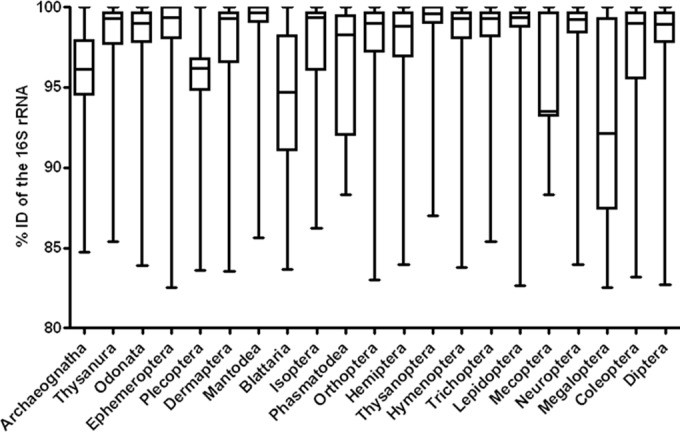
Similarities of bacterial 16S rRNA gene sequences in different orders of insects. % ID indicates % identity.
FIG 3.
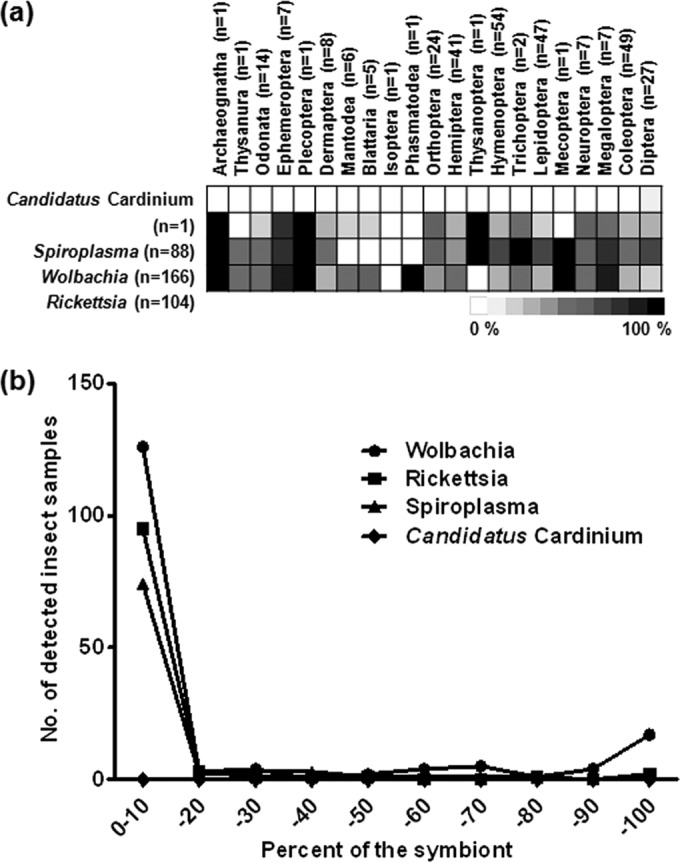
Distribution of 4 bacterial symbionts in the insect gut. The relative abundances of the symbionts in 21 insect orders (a) and their infection frequencies (b) are shown.
Diversity in the gut microbiota is associated with the environmental habitat, diet, developmental stage, and phylogeny of the host.
The relative abundances of bacteria based on their oxygen demands were determined with respect to environmental habitat, diet, developmental stage, and phylogenetic position of insect hosts. From a total of 303 bacterial genera, 184 were assigned as aerobes (60.7%), 48 as facultative anaerobes (15.8%), and 71 as anaerobes (23.4%), based on the literature. The environmental habitats occupied by the 305 insects were categorized as follows: sky (43.3%), ground (47.9%), underground (4.9%), and aquatic (3.9%) (see Table S1 in the supplemental material). In determining the relationship between gut microbiota and host diet, insects were classified into the following five groups according to feeding preference: herbivore (n = 183), carnivore (n = 62), omnivore (n = 27), scavenger (n = 14), fungivore (n = 10), and “no-feed” (n = 9) (see Table S1). There were significant differences in the relative abundances of anaerobes in insects categorized by environmental habitat (Kruskal-Wallis test; P = 0.0006), diet (P = 0.0099), developmental stage (t test; P = 0.0034), and phylogeny (Kruskal-Wallis test; P < 0.0001). However, the relative abundances of aerobes and facultative anaerobes showed no significant differences between insects, regardless of the categorization factors (Fig. 4). Aerobes were more abundant in the guts of terrestrial insects (sky, ground, and underground) than in those of the aquatic group of insects. The abundance of anaerobes was significantly higher in the guts of aquatic insects than in the guts of terrestrial insects (P = 0.0006). When bacterial abundances in different developmental stages were compared, anaerobe abundances were significantly higher in the larval stages than in adults (P = 0.0034). Additionally, anaerobes were more abundant in the guts of omnivorous and Blattaria insects, which is supported by a previous report showing that dictyopteran insects, such as cockroaches and termites, have larger proportions of anaerobic Bacteroidia and Clostridia species (39).
The analysis of bacterial diversity associated with the different host factors (Fig. 5) revealed significant differences in the numbers of OTUs between insects with different diets (P = 0.0106) and those at different developmental stages (P = 0.0178). These results were supported by analyses of Shannon diversity indices (P = 0.0020 and P < 0.0001, respectively). Bacterial diversity was significantly greater in the guts of insects characterized as omnivores than in those of carnivores or herbivores. Analysis revealed significant differences in the numbers of OTUs (P = 0.0178) and the Shannon diversity indices (P < 0.0001) between insects at different developmental stages. However, no significant differences were observed in either the number of OTUs or the Shannon diversity index between insects categorized by habitat or phylogenetic position.
Bacterial community composition in relation to geographical location and developmental stage of hosts.
To investigate the impact of geography on the bacterial community composition of insect guts, honeybees were collected from five different locations (see Fig. S1 in the supplemental material). Knowing that honeybee colonies forage within several kilometers of their nests (an average of 1.5 to 1.7 km) (40, 41), a sufficient distance between the five sampling sites (105.8 ± 14.7 km) was ensured. Figure 6a compares the bacterial diversities in the guts of A. mellifera bees obtained here and in previous studies (28–30). Principal component analysis showed that the gut microbiota of honeybees clustered separately from those of other insects (Fig. 6b). To determine which of the commonly detected OTUs identified in replicates were shared, the 16S rRNA gene sequences were compared with previously published sequences (30), based on a 3% distance level. From a total of 532 OTUs (11,204 sequences) identified, only 14 (4,017 sequences) were common among the honeybees from the five regions in our study and two bees from Arizona (30), and these were assigned to the Lactobacillus Firm-4, Lactobacillus Firm-5, Snodgrassella alvi (Beta), and Gilliamella apicola (Gamma-1) groups. OTUs belonging to Alpha-1, Alpha-2, the Frischella perrara (Gamma-2) group, and Lactococcus spp. were commonly found in most of the bees.
FIG 6.

Diversity of gut microbiota of Apis mellifera, Illeis koebelei, and Lycorma delicatula. The gut microbiota of honeybees (Apis mellifera) were collected from separate locations (CG [site 12 in Fig. S1 in the supplemental material], Chilgok-Gun; JG [site 13], Jinan-Gun; WS [site 5], Wonju-Si; SS [site 6], Seosan-Si; and GG [site 10], Goesan-Gun) or reported in previous studies. (a) Compositions of gut microbiota at the phylum level. (b) PCA showing the gut microbiota of A. mellifera collected from separate locations or reported in previous studies. Bacterial community analysis and PCA were performed using data generated in this study and from previous studies. The bacterial distributions of gut microbiota in I. koebelei (planthopper) (c) and L. delicatula (yellow ladybird) (d) collected at different insect developmental stages are shown. (e) Numbers of shared OTUs of the bacterial community at the different developmental stages of I. koebelei and L. delicatula. Shared OTUs in L. delicatula nymphs were not observed. The insect samples of I. koebelei were collected from the same region, except for the “Adult 4” sample. (f) PCoA showing the gut microbiota of I. koebelei and L. delicatula collected at different insect developmental stages.
Comparisons were made between the compositions of bacterial communities in the guts of planthoppers and yellow ladybirds collected at different developmental stages (Fig. 6c to f). OTUs shared between both developmental stages were identified in each replicate of a larva or adult by analysis of the common OTUs. In the guts of Illeis koebelei insects, 29 of 357 OTUs were shared at the larval stage, and 3 of the 248 OTUs were common at the adult stage. Only 2 OTUs (183 sequences) were shared between the different developmental stages, and these were assigned to Ralstonia and Frateuria species. In the guts of Lycorma delicatula insects, 2 of the 526 OTUs were assigned to Imtechium and were shared only in the adult stage (Fig. 6e). PCoA revealed that the bacterial compositions of the gut at different developmental stages of Illeis koebelei and Lycorma delicatula were not tightly clustered (Fig. 6f). The results indicated that among all the bacteria, only Ralstonia and Frateuria species (Proteobacteria) are commonly found in the gut microbiota of Illeis koebelei.
DISCUSSION
This large-scale study comprehensively examined the insect gut microbiota by using next-generation sequencing. A previous study of 106 individual mammals showed that gut microbiota are dominated by the Firmicutes and Bacteroidetes (82% of classified sequences) (38). Our study showed that the predominant phyla in the guts of the insects examined were Proteobacteria and Firmicutes, representing 82.8% of the total sequences. This result is supported by a previous study (12) showing that Proteobacteria and Firmicutes were the predominant phyla in 81 insect gut samples, comprising 57.4% and 21.7% of sequences, respectively. Forty-six percent of bacterial OTUs were observed in individual insects only (see Fig. S3 in the supplemental material). This distribution pattern is similar to the results of a previous study reporting that most bacterial OTUs were confined to a single environmental sample and that a single OTU was not observed in the clone libraries of more than 6 of the 14 habitat types (42). In this study, PCoA revealed a distinct difference in the compositions of bacterial communities in the guts of insects and mammals, showing that the variation in gut microbiota among insects is much larger than that in mammals (Fig. 7). Most of the vertebrate gut microbiota, including those of mammals, consisted of Bacteroidetes and Firmicutes as the most dominant phyla (43). On the other hand, although insect gut microbiota were generally dominated by Proteobacteria and Firmicutes, there were dominant phyla of Bacteroidetes, Actinobacteria, and Tenericutes in some insect samples (Fig. 1). The dynamic variation in insect gut microbiota can be determined by gut morphology and physicochemical conditions, such as pH and oxygen availability in the insect gut (44, 45). The insect gut as a bacterial habitat shows different morphologies, generally depending on the insect order (45); furthermore, the gut morphology is sharply changed by metamorphosis, according to the life cycle of the insect (46). Additionally, the oxygen availability can be influenced by gut shape (47), metabolism of colonizing bacteria (48, 49), and partial pressure of oxygen from the outside environment (50), and the pH, from acidic to extremely alkaline conditions, is determined by different gut compartments in diverse insect individuals (51, 52). These diverse gut conditions may cause the variation in host-specific gut microbiota in insects.
FIG 7.

Composition of the insect gut microbiota compared with that of the mammalian gut microbiota from the study of Ley et al. (38). Principle coordinate analysis was performed using a PHYLIP-formatted distance matrix based on Jaccard values.
In a recent substantial analysis of the bacteria within whole insect bodies, Jones et al. (11) reported a low bacterial diversity, with fewer than eight phylotypes in each insect sample. This is not consistent with the data presented in this study (84.3 OTUs per sample), where only insect guts harboring most OTUs found in only one sample were analyzed. Jones et al. excluded phylotypes representing less than 1% of the bacterial community in individual samples. The purpose of this study was to characterize bacterial populations within the guts of insects by using a deep-sequencing approach where all 16S rRNA gene sequences were analyzed, apart from singletons. This approach of analyzing rare sequences may produce inconsistent results on the diversity of insect-associated bacteria. The unexpectedly higher community diversity in insect guts may be supported by the fact that most OTUs were found in only one sample (see Fig. S3 in the supplemental material) and that the insect microbiota, including environmental bacteria, can be highly variable (45). In comparing the bacterial 16S rRNA gene sequences from this study with those reported previously, bacteria with relatively low percent identities (e.g., Clostridiales bacterium canine oral taxon 141 from Ephemeroptera had 82.5% identity, Desulfovibrio sp. ABHU2SB from Megaloptera had 82.6% identity, and Desulfuromonadales bacterium Tc37 from Diptera had 82.7% identity) were recovered from insect guts (Fig. 2), suggesting a remarkably diverse population of novel bacterial taxa with low 16S rRNA gene sequence similarity. These newly identified members of the insect gut microbiota may enable us to discover new bacterial lineages with more extensive sequencing and cultivation efforts. The 16S rRNA gene sequences with relatively low similarities found in the orders Megaloptera and Mecoptera are of particular interest for understanding the origins and functions of these bacteria, because the adult insects do not feed and their juvenile guts are shed when they metamorphose into adults.
Members of the genus Wolbachia infect many members of the Arthropoda (53, 54) and were predominant among the heritable symbionts identified. Our study showed that Wolbachia sequences were found in 166 of the 305 insect samples (54.4%); this is higher than levels reported by previous studies (17 to 35%) for diverse insect species, based on the PCR detection approach (55, 56), and higher than a recent estimation of Wolbachia prevalence (40%) (57). The distribution of the insect hosts harboring Wolbachia in the present study was much wider than previously reported. Studies by Jeyaprakash and Hoy (58) and Russell et al. (59) showed that Wolbachia strains were found in 18 different insect orders (Blattaria, Coleoptera, Dermaptera, Diptera, Hemiptera, Homoptera, Hymenoptera, Isoptera, Lepidoptera, Neuroptera, Odonata, Orthoptera, Phthiraptera, Plecoptera, Siphonaptera, Strepsiptera, Thysanoptera, and Thysanura); however, we identified Wolbachia in five additional insect orders (Arcaeognatha, Ephemeroptera, Mecoptera, Megaloptera, and Tricoptera), suggesting that Wolbachia strains may be more widespread throughout the insects. We also found that the frequency of Wolbachia sequences within a species indicates a “most-or-few” infection pattern, showing a very high (>90%) or low (<10%) infection frequency (Fig. 3b), in accordance with a statistical analysis reported by Hilgenboecker et al. (54). Despite several reports about the distribution of intracellular symbionts that regulate host reproduction, such as Wolbachia, Spiroplasma, and “Candidatus Cardinium” (53–56, 58–60), there are few studies addressing the broad distribution of host-specific bacteria from a wide range of insect hosts.
Relative bacterial abundances in the gut varied according to the environmental habitats of the insects and were most likely associated with the levels of oxygen. Insects adapt to different environmental habitats, in which oxygen availability is variable (61). Previous studies of wood-feeding termites reported that bacteria contribute to the hypoxic environment in the peripheral compartment of the hindgut, with their respiratory activities consuming oxygen that penetrates into the hindgut, resulting in a significant oxygen sink (10, 48). This hypoxic environment can be disturbed when the levels of diffused oxygen exceed the oxygen-consuming capability of these hindgut bacteria (62). An increased partial pressure of oxygen induces oxic conditions in the hindgut of the wood-feeding lower termite (50). In the present study, we found significant differences in the relative abundance of anaerobes depending on the environmental habitat occupied by the host (Fig. 4). The host habitat may influence the community composition of the insect gut microbiota.
Diet is a controlling factor for microbial diversity. In the mammalian gut, the diversity of bacteria increases according to host diet, from carnivores to omnivores to herbivores (38). The intestinal bacteria of insects such as termites and aphids produce compounds that are essential and otherwise inaccessible for the host (e.g., detritus, phloem, sap, wood, and xylem) (28, 63, 64). Most studies on the relationships between insect diet and the distribution of gut microbiota have tended to focus on specific microbes in a single insect group; however, a recent, wider-ranging study based on previously published and newly generated 16S rRNA gene sequences from 62 insect species from seven orders reported that both host diet and taxonomy affect insect gut bacterial communities (12). In our study of phylogenetically diverse insects from a range of habitats and environments, we found that gut bacterial diversity (in terms of both species richness and the Shannon index) was significantly higher in omnivorous insects than in stenophagous (carnivore and herbivore) ones (Fig. 5). This pattern could be explained by the assumption that omnivores would be expected to consume more different foods, including diverse bacterial species, than carnivores and herbivores; therefore, the higher level of bacterial diversity may be related to the consumed food types (65, 66). In addition, detailed statistical comparisons of bacterial communities to determine the effects of different factors by using AMOVA, ANOSIM, and UniFrac analysis (see Table S4 in the supplemental material) showed that there were significant differences within each major factor but that each difference between groups was variable. In particular, the R values from ANOSIM among the four groups classified by diet were negative, indicating a high level of within-group variability. The gut microbiota of honeybees were clustered separately from those of other insects (Fig. 6b) but were not separated in relation to host diet, habitat, phylogenetic position, or developmental stage (see Fig. S4). This supports the findings of a previous study (12) showing that host diet appears to affect the composition of the gut microbiota in some, but not all, groups of insects. As a general conclusion, our findings suggest that some groups within the microbiota and bacterial richness in the insect gut may be affected by host diet, habitat, and developmental stage.
Despite some profound differences in the factors that could influence bacterial composition in the guts of the honeybee (collected from five locations), planthopper, and yellow ladybird, which consume the same diet despite insect metamorphosis (67, 68) and were collected at different developmental stages, there were still some shared sequences that may represent members of the core microbiota in their guts. Microbiomes such as the human gut (69), the human oral cavity (70), and zebrafish guts (71) have also been investigated using a deep-sequencing approach. In the present study, Lactobacillus and Ralstonia species were recorded in the guts of Apis mellifera and Illeis koebelei, respectively, and this was consistent regardless of locality and developmental stage. Although the factors that influenced the development of particular shared OTUs in the insect gut remain unresolved, the identification of phylotypes comprising the core microbiota may provide fundamental information to enable diagnosis of physiological states of insects, as demonstrated in the study of the core human gut microbiome (69). Further studies might be needed to understand the coevolution mechanism of host and core microorganisms.
The present study outlines a detailed investigation of the composition and diversity of the gut microbiota of 305 individual insects from 218 species belonging to 21 taxonomic orders of Insecta, using 454 pyrosequencing of the 16S rRNA gene. The current study shows a higher bacterial diversity in insect guts, a remarkably diverse population of novel bacterial taxa, and a broad distribution of the host-specific bacteria from a wide range of insect hosts. We also found significant differences in the relative abundances of anaerobes in insects, regardless of the categorization factors and of the gut bacterial diversity according to host diet and developmental stage, as well as the possible core microbiota in a few insect groups. Although there are limitations of this study, such as the sequencing depth per sample being insufficient to reach saturation for some of the samples and the lack of consideration of age, sex, and biological replicates of the samples, this study presents basic information on both the microbial diversity in the guts of insects and the associations of microbes and their hosts. This order-spanning investigation of gut microbiota could be of value and interest in invertebrate microbiology, providing insights into the relationships between insects and their gut bacterial communities.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Institute of Biological Resources (NIBR) (grant 2013-02-001) and the Mid-Career Researcher Program (grant 2011-0028854), through the National Research Foundation of Korea (NRF).
We thank Young-Seuk Park and Nak-Soon Yoon for providing the insect samples and Jong-Chul Jeong for identifying insect morphologies.
Footnotes
Published ahead of print 13 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01226-14.
REFERENCES
- 1.Hooper LV, Gordon JI. 2001. Commensal host-bacterial relationships in the gut. Science 292:1115–1118. 10.1126/science.1058709 [DOI] [PubMed] [Google Scholar]
- 2.Kaufman MG, Klug MJ. 1991. The contribution of hindgut bacteria to dietary carbohydrate utilization by crickets (Orthoptera, Gryllidae). Comp. Biochem. Physiol. 98:117–123. 10.1016/0300-9629(91)90588-4 [DOI] [Google Scholar]
- 3.Dillon R, Charnley K. 2002. Mutualism between the desert locust Schistocerca gregaria and its gut microbiota. Res. Microbiol. 153:503–509. 10.1016/S0923-2508(02)01361-X [DOI] [PubMed] [Google Scholar]
- 4.Bourtzis K, Miller T. 2003. Insect symbiosis. CRC Press, Boca Raton, FL [Google Scholar]
- 5.Broderick NA, Raffa KF, Handelsman J. 2006. Midgut bacteria required for Bacillus thuringiensis insecticidal activity. Proc. Natl. Acad. Sci. U. S. A. 103:15196–15199. 10.1073/pnas.0604865103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hurst GD, Jiggins FM. 2000. Male-killing bacteria in insects: mechanisms, incidence, and implications. Emerg. Infect. Dis. 6:329–336. 10.3201/eid0604.000402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryu JH, Kim SH, Lee HY, Bai JY, Nam YD, Bae JW, Lee DG, Shin SC, Ha EM, Lee WJ. 2008. Innate immune homeostasis by the homeobox gene Caudal and commensal-gut mutualism in Drosophila. Science 319:777–782. 10.1126/science.1149357 [DOI] [PubMed] [Google Scholar]
- 8.Apte-Deshpande A, Paingankar M, Gokhale MD, Deobagkar DN. 2012. Serratia odorifera a midgut inhabitant of Aedes aegypti mosquito enhances its susceptibility to dengue-2 virus. PLoS One 7:e40401. 10.1371/journal.pone.0040401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arias-Cordero E, Ping L, Reichwald K, Delb H, Platzer M, Boland W. 2012. Comparative evaluation of the gut microbiota associated with the below- and above-ground life stages (larvae and beetles) of the forest cockchafer, Melolontha hippocastani. PLoS One 7:e51557. 10.1371/journal.pone.0051557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tholen A, Schink B, Brune A. 1997. The gut microflora of Reticulitermes flavipes, its relation to oxygen, and evidence for oxygen dependent acetogenesis by the most abundant Enterococcus sp. FEMS Microbiol. Ecol. 24:137–149. 10.1016/S0168-6496(97)00053-6 [DOI] [Google Scholar]
- 11.Jones RT, Sanchez LG, Fierer N. 2013. A cross-taxon analysis of insect-associated bacterial diversity. PLoS One 8:e61218. 10.1371/journal.pone.0061218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colman DR, Toolson EC, Takacs-Vesbach CD. 2012. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 21:5124–5137. 10.1111/j.1365-294X.2012.05752.x [DOI] [PubMed] [Google Scholar]
- 13.Chandler JA, Lang JM, Bhatnagar S, Eisen JA, Kopp A. 2011. Bacterial communities of diverse Drosophila species: ecological context of a host-microbe model system. PLoS Genet. 7:e1002272. 10.1371/journal.pgen.1002272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toju H, Fukatsu T. 2011. Diversity and infection prevalence of endosymbionts in natural populations of the chestnut weevil: relevance of local climate and host plants. Mol. Ecol. 20:853–868. 10.1111/j.1365-294X.2010.04980.x [DOI] [PubMed] [Google Scholar]
- 15.Wong CN, Ng P, Douglas AE. 2011. Low-diversity bacterial community in the gut of the fruitfly Drosophila melanogaster. Environ. Microbiol. 13:1889–1900. 10.1111/j.1462-2920.2011.02511.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc. Natl. Acad. Sci. U. S. A. 103:12115–12120. 10.1073/pnas.0605127103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boucias DG, Cai Y, Sun Y, Lietze VU, Sen R, Raychoudhury R, Scharf ME. 2013. The hindgut lumen prokaryotic microbiota of the termite Reticulitermes flavipes and its responses to dietary lignocellulose composition. Mol. Ecol. 22:1836–1853. 10.1111/mec.12230 [DOI] [PubMed] [Google Scholar]
- 18.Kohler T, Dietrich C, Scheffrahn RH, Brune A. 2012. High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation of the gut in wood-feeding higher termites (Nasutitermes spp.). Appl. Environ. Microbiol. 78:4691–4701. 10.1128/AEM.00683-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poulsen M, Sapountzis P. 2012. Behind every great ant, there is a great gut. Mol. Ecol. 21:2054–2057. 10.1111/j.1365-294X.2012.05510.x [DOI] [PubMed] [Google Scholar]
- 20.Funaro CF, Kronauer DJ, Moreau CS, Goldman-Huertas B, Pierce NE, Russell JA. 2011. Army ants harbor a host-specific clade of Entomoplasmatales bacteria. Appl. Environ. Microbiol. 77:346–350. 10.1128/AEM.01896-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russell JA, Moreau CS, Goldman-Huertas B, Fujiwara M, Lohman DJ, Pierce NE. 2009. Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. Proc. Natl. Acad. Sci. U. S. A. 106:21236–21241. 10.1073/pnas.0907926106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sudakaran S, Salem H, Kost C, Kaltenpoth M. 2012. Geographical and ecological stability of the symbiotic mid-gut microbiota in European firebugs, Pyrrhocoris apterus (Hemiptera, Pyrrhocoridae). Mol. Ecol. 21:6134–6151. 10.1111/mec.12027 [DOI] [PubMed] [Google Scholar]
- 23.Salem H, Kreutzer E, Sudakaran S, Kaltenpoth M. 2013. Actinobacteria as essential symbionts in firebugs and cotton stainers (Hemiptera, Pyrrhocoridae). Environ. Microbiol. 15:1956–1968. 10.1111/1462-2920.12001 [DOI] [PubMed] [Google Scholar]
- 24.Kaltenpoth M, Winter SA, Kleinhammer A. 2009. Localization and transmission route of Coriobacterium glomerans, the endosymbiont of pyrrhocorid bugs. FEMS Microbiol. Ecol. 69:373–383. 10.1111/j.1574-6941.2009.00722.x [DOI] [PubMed] [Google Scholar]
- 25.Roh SW, Nam YD, Chang HW, Kim KH, Kim MS, Ryu JH, Kim SH, Lee WJ, Bae JW. 2008. Phylogenetic characterization of two novel commensal bacteria involved with innate immune homeostasis in Drosophila melanogaster. Appl. Environ. Microbiol. 74:6171–6177. 10.1128/AEM.00301-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reid NM, Addison SL, Macdonald LJ, Lloyd-Jones G. 2011. Biodiversity of active and inactive bacteria in the gut flora of wood-feeding huhu beetle larvae (Prionoplus reticularis). Appl. Environ. Microbiol. 77:7000–7006. 10.1128/AEM.05609-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hulcr J, Rountree NR, Diamond SE, Stelinski LL, Fierer N, Dunn RR. 2012. Mycangia of ambrosia beetles host communities of bacteria. Microb. Ecol. 64:784–793. 10.1007/s00248-012-0055-5 [DOI] [PubMed] [Google Scholar]
- 28.Engel P, Martinson VG, Moran NA. 2012. Functional diversity within the simple gut microbiota of the honey bee. Proc. Natl. Acad. Sci. U. S. A. 109:11002–11007. 10.1073/pnas.1202970109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohr KI, Tebbe CC. 2006. Diversity and phylotype consistency of bacteria in the guts of three bee species (Apoidea) at an oilseed rape field. Environ. Microbiol. 8:258–272. 10.1111/j.1462-2920.2005.00893.x [DOI] [PubMed] [Google Scholar]
- 30.Martinson VG, Danforth BN, Minckley RL, Rueppell O, Tingek S, Moran NA. 2011. A simple and distinctive microbiota associated with honey bees and bumble bees. Mol. Ecol. 20:619–628. 10.1111/j.1365-294X.2010.04959.x [DOI] [PubMed] [Google Scholar]
- 31.Cox-Foster DL, Conlan S, Holmes EC, Palacios G, Evans JD, Moran NA, Quan PL, Briese T, Hornig M, Geiser DM, Martinson V, vanEngelsdorp D, Kalkstein AL, Drysdale A, Hui J, Zhai J, Cui L, Hutchison SK, Simons JF, Egholm M, Pettis JS, Lipkin WI. 2007. A metagenomic survey of microbes in honey bee colony collapse disorder. Science 318:283–287. 10.1126/science.1146498 [DOI] [PubMed] [Google Scholar]
- 32.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 33.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–D145. 10.1093/nar/gkn879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nam YD, Jung MJ, Roh SW, Kim MS, Bae JW. 2011. Comparative analysis of Korean human gut microbiota by barcoded pyrosequencing. PLoS One 6:e22109. 10.1371/journal.pone.0022109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neyrinck AM, Possemiers S, Verstraete W, De Backer F, Cani PD, Delzenne NM. 2012. Dietary modulation of clostridial cluster XIVa gut bacteria (Roseburia spp.) by chitin-glucan fiber improves host metabolic alterations induced by high-fat diet in mice. J. Nutr. Biochem. 23:51–59. 10.1016/j.jnutbio.2010.10.008 [DOI] [PubMed] [Google Scholar]
- 37.Dieckmann R, Helmuth R, Erhard M, Malorny B. 2008. Rapid classification and identification of salmonellae at the species and subspecies levels by whole-cell matrix-assisted laser desorption ionization–time of flight mass spectrometry. Appl. Environ. Microbiol. 74:7767–7778. 10.1128/AEM.01402-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI. 2008. Evolution of mammals and their gut microbes. Science 320:1647–1651. 10.1126/science.1155725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sabree ZL, Moran NA. 2014. Host-specific assemblages typify gut microbial communities of related insect species. SpringerPlus 3:138. 10.1186/2193-1801-3-138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Visscher PK, Seeley TD. 1982. Foraging strategy of honeybee colonies in a temperate deciduous forest. Ecology 63:1790–1801. 10.2307/1940121 [DOI] [Google Scholar]
- 41.Steffan-Dewenter I, Kuhn A. 2003. Honeybee foraging in differentially structured landscapes. Proc. R. Soc. B Biol. Sci. 270:569–575. 10.1098/rspb.2002.2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nemergut DR, Costello EK, Hamady M, Lozupone C, Jiang L, Schmidt SK, Fierer N, Townsend AR, Cleveland CC, Stanish L, Knight R. 2011. Global patterns in the biogeography of bacterial taxa. Environ. Microbiol. 13:135–144. 10.1111/j.1462-2920.2010.02315.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. 2008. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6:776–788. 10.1038/nrmicro1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dillon RJ, Dillon VM. 2004. The gut bacteria of insects: nonpathogenic interactions. Annu. Rev. Entomol. 49:71–92. 10.1146/annurev.ento.49.061802.123416 [DOI] [PubMed] [Google Scholar]
- 45.Engel P, Moran NA. 2013. The gut microbiota of insects—diversity in structure and function. FEMS Microbiol. Rev. 37:699–735. 10.1111/1574-6976.12025 [DOI] [PubMed] [Google Scholar]
- 46.Moll RM, Romoser WS, Modrzakowski MC, Moncayo AC, Lerdthusnee K. 2001. Meconial peritrophic membranes and the fate of midgut bacteria during mosquito (Diptera: Culicidae) metamorphosis. J. Med. Entomol. 38:29–32. 10.1603/0022-2585-38.1.29 [DOI] [PubMed] [Google Scholar]
- 47.Ke J, Sun JZ, Nguyen HD, Singh D, Lee KC, Beyenal H, Chen S-L. 2010. In-situ oxygen profiling and lignin modification in guts of wood-feeding termites. Insect Sci. 17:277–290. 10.1111/j.1744-7917.2010.01336.x [DOI] [Google Scholar]
- 48.Brune A, Emerson D, Breznak JA. 1995. The termite gut microflora as an oxygen sink: microelectrode determination of oxygen and pH gradients in guts of lower and higher termites. Appl. Environ. Microbiol. 61:2681–2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brune A, Miambi E, Breznak JA. 1995. Roles of oxygen and the intestinal microflora in the metabolism of lignin-derived phenylpropanoids and other monoaromatic compounds by termites. Appl. Environ. Microbiol. 61:2688–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ebert A, Brune A. 1997. Hydrogen concentration profiles at the oxic-anoxic interface: a microsensor study of the hindgut of the wood-feeding lower termite Reticulitermes flavipes (Kollar). Appl. Environ. Microbiol. 63:4039–4046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kappler A, Brune A. 1999. Influence of gut alkalinity and oxygen status on mobilization and size class distribution of humic acids in the hindgut of soil-feeding termites. Appl. Soil Ecol. 13:219–229. 10.1016/S0929-1393(99)00035-9 [DOI] [Google Scholar]
- 52.Gross EM, Brune A, Walenciak O. 2008. Gut pH, redox conditions and oxygen levels in an aquatic caterpillar: potential effects on the fate of ingested tannins. J. Insect Physiol. 54:462–471. 10.1016/j.jinsphys.2007.11.005 [DOI] [PubMed] [Google Scholar]
- 53.Werren JH, Windsor DM. 2000. Wolbachia infection frequencies in insects: evidence of a global equilibrium? Proc. Biol. Sci. 267:1277–1285. 10.1098/rspb.2000.1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hilgenboecker K, Hammerstein P, Schlattmann P, Telschow A, Werren JH. 2008. How many species are infected with Wolbachia?—a statistical analysis of current data. FEMS Microbiol. Lett. 281:215–220. 10.1111/j.1574-6968.2008.01110.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Werren JH, Windsor D, Guo L. 1995. Distribution of Wolbachia among neotropical arthropods. Proc. Biol. Sci. 262:197–204. 10.1098/rspb.1995.0196 [DOI] [Google Scholar]
- 56.Kikuchi Y, Fukatsu T. 2003. Diversity of Wolbachia endosymbionts in heteropteran bugs. Appl. Environ. Microbiol. 69:6082–6090. 10.1128/AEM.69.10.6082-6090.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zug R, Hammerstein P. 2012. Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS One 7:e38544. 10.1371/journal.pone.0038544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jeyaprakash A, Hoy MA. 2000. Long PCR improves Wolbachia DNA amplification: wsp sequences found in 76% of sixty-three arthropod species. Insect Mol. Biol. 9:393–405. 10.1046/j.1365-2583.2000.00203.x [DOI] [PubMed] [Google Scholar]
- 59.Russell JA, Funaro CF, Giraldo YM, Goldman-Huertas B, Suh D, Kronauer DJ, Moreau CS, Pierce NE. 2012. A veritable menagerie of heritable bacteria from ants, butterflies, and beyond: broad molecular surveys and a systematic review. PLoS One 7:e51027. 10.1371/journal.pone.0051027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zchori-Fein E, Perlman SJ. 2004. Distribution of the bacterial symbiont Cardinium in arthropods. Mol. Ecol. 13:2009–2016. 10.1111/j.1365-294X.2004.02203.x [DOI] [PubMed] [Google Scholar]
- 61.Hoback WW, Stanley DW. 2001. Insects in hypoxia. J. Insect Physiol. 47:533–542. 10.1016/S0022-1910(00)00153-0 [DOI] [PubMed] [Google Scholar]
- 62.Brune A, Frenzel P, Cypionka H. 2000. Life at the oxic-anoxic interface: microbial activities and adaptations. FEMS Microbiol. Rev. 24:691–710. 10.1111/j.1574-6976.2000.tb00567.x [DOI] [PubMed] [Google Scholar]
- 63.Moran NA, McCutcheon JP, Nakabachi A. 2008. Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 42:165–190. 10.1146/annurev.genet.41.110306.130119 [DOI] [PubMed] [Google Scholar]
- 64.Tartar A, Wheeler MM, Zhou X, Coy MR, Boucias DG, Scharf ME. 2009. Parallel metatranscriptome analyses of host and symbiont gene expression in the gut of the termite Reticulitermes flavipes. Biotechnol. Biofuels 2:25. 10.1186/1754-6834-2-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anderson KE, Sheehan TH, Mott BM, Maes P, Snyder L, Schwan MR, Walton A, Jones BM, Corby-Harris V. 2013. Microbial ecology of the hive and pollination landscape: bacterial associates from floral nectar, the alimentary tract and stored food of honey bees (Apis mellifera). PLoS One 8:e83125. 10.1371/journal.pone.0083125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Michener CD. 1974. The social behavior of the bees: a comparative study. Harvard University Press, Cambridge, MA [Google Scholar]
- 67.Kim JG, Lee EH, Seo YM, Kim NY. 2011. Cyclic behavior of Lycorma delicatula (Insecta: Hemiptera: Fulgoridae) on host plants. J. Insect Behav. 24:423–435. 10.1007/s10905-011-9266-8 [DOI] [Google Scholar]
- 68.Takeuchi M, Sasaki Y, Sato C, Iwakuma S, Isozaki A, Tamura M. 2000. Seasonal host utilization of mycophagous ladybird Illeis koebelei (Coccinellidae: Coleoptera). J. Appl. Entomol. Zool. (Jpn.) 44:89–94. 10.1303/jjaez.2000.89 [DOI] [Google Scholar]
- 69.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. 2009. A core gut microbiome in obese and lean twins. Nature 457:480–484. 10.1038/nature07540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zaura E, Keijser BJ, Huse SM, Crielaard W. 2009. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 9:259. 10.1186/1471-2180-9-259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roeselers G, Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM, Guillemin K, Rawls JF. 2011. Evidence for a core gut microbiota in the zebrafish. ISME J. 5:1595–1608. 10.1038/ismej.2011.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.