

Abstract
Gordonia polyisoprenivorans strain VH2, a potent rubber-degrading actinomycete, harbors two latex clearing proteins (Lcps), which are known to be essential for the microbial degradation of rubber. However, biochemical information on the exact role of this protein in the degradation of polyisoprene was lacking. In this study, the gene encoding Lcp1VH2 was heterologously expressed in strains of Escherichia coli, the corresponding protein was purified, and its role in rubber degradation was examined by measurement of oxygen consumption as well as by chromatographic and spectroscopic methods. It turned out that active Lcp1VH2 is a monomer and is responsible for the oxidative cleavage of poly(cis-1,4-isoprene) in synthetic as well as in natural rubber by the addition of oxygen (O2) to the cis double bonds. The resulting oligomers possess repetitive isoprene units with aldehyde (CHO-CH2—) and ketone (—CH2-CO-CH3) functional groups at the termini. Two fractions with average isoprene contents of 18 and 10, respectively, were isolated, thus indicating an endocleavage mechanism. The activity of Lcp1VH2 was determined by applying a polarographic assay. Alkenes, acyclic terpenes, or other rubber-like polymers, such as poly(cis-1,4-butadiene) or poly(trans-1,4-isoprene), are not oxidatively cleaved by Lcp1VH2. The pH and temperature optima of the enzyme are at pH 7 and 30°C, respectively. Furthermore, it was demonstrated that active Lcp1VH2 is a Cu(II)-containing oxygenase that exhibits a conserved domain of unknown function which cannot be detected in any other hitherto-characterized enzyme. The results presented here indicate that this domain might represent a new protein family of oxygenases.
INTRODUCTION
The high annual production and consumption rates of natural and synthetic poly(cis-1,4-isoprene) rubber and rubber-containing materials cause huge challenges in the disposal of this polymer. Most waste management strategies that are implemented at present, such as combustion or stockpiling in landfills, exhibit a serious hazard to the environment and to human health. Therefore, finding alternative processes for the recycling of polyisoprenoids is desirable (1). Processes involving complete or partial biodegradation could be an environment-friendly opportunity for reutilization of the polymer. For such a process, enzymes capable of the initial cleavage of the very inert polymer would be of great interest.
Until now, only one enzyme responsible for a primary attack of poly(cis-1,4-isoprene) has been characterized in detail. This rubber oxygenase A (RoxA) was first detected in Xanthomonas sp. strain 35Y, which secretes this heme-dependent dioxygenase into medium during growth on rubber (2, 3). Purified RoxA cleaves the double bonds of rubber by incorporation of both oxygen groups into the polymer, generating 12-oxo-4,8-dimethyltrideca-4,8-diene-1-al (ODTD) as the main product (4, 5). Recently, RoxA was crystallized, structural and functional analyses were carried out, and a first cleavage mechanism was proposed (6–9). However, RoxA was detected in only a few Gram-negative bacteria (2, 10). In Gram-positive bacteria, especially actinomycetes, in which the capability of rubber degradation seems to be quite widespread (11), no RoxA orthologs can be detected (10). In contrast, all rubber-degrading actinomycetes seem to have at least one copy of a gene encoding a latex clearing protein (Lcp). This enzyme is considered to catalyze the initial oxidative cleavage of rubber within this group of Gram-positive bacteria (10, 12–16) and does not possess any similarity to the amino acid sequence of RoxA. Lcp was first detected in Streptomyces sp. strain K30 by generating a chemically induced mutant that cannot form clear zones on latex overlay plates. Complementation experiments using lcp restored this capability (16). Homologs can also be detected in members of the non-clear-zone-forming group of actinomycetes, such as strains of Nocardia and Gordonia (12, 13). Representatives of this group grow adhesively on the polymer and appear to be more effective in disintegration of the rubber than strains of the clear-zone-forming group (17). Within the adhesively growing group, Gordonia polyisoprenivorans strain VH2 was selected as a model strain, inter alia, due to its fast metabolization of poly(cis-1,4-isoprene). On the basis of the whole genome sequence together with practical experiments such as transposon mutagenesis and generation of deletion mutants, a full rubber degradation pathway for strain VH2 was predicted. Within the genome of this strain, two Lcp-encoding genes were detected (lcp1VH2 and lcp2VH2) (14) and are thought to be responsible for the oxidative cleavage of the rubber polymer for several reasons: (i) when cells of strain VH2 grow on rubber, an increase of carbonyl functional groups as well as a decrease of the cis double bonds were observed (17); (ii) the deletion of both genes resulted in a non-rubber-degrading phenotype, whereas single-deletion mutants still grew on rubber (14); (iii) heterologous expression of lcp1VH2 in non-clear-zone-forming Streptomyces strain TK23 enabled this strain to form translucent halos and aldehydes on latex overlay plates; (iv) incubation of poly(cis-1,4-isoprene) with this recombinant strain resulted in the generation of fragments of lower molecular weight; and (v) lcp1VH2 transcripts could be detected by reverse transcription-PCR analysis only when cells were grown on rubber but not when cells were grown on, e.g., sodium acetate (13).
Nevertheless, biochemical information on how Lcp is enzymatically involved in the degradation of poly(cis-1,4-isoprene) and whether other enzymes are necessary for rubber disintegration by actinobacteria is missing. It was the aim of this study to unravel the reaction catalyzed by Lcp.
MATERIALS AND METHODS
Bacterial strains, media, growth conditions, and oligonucleotides.
Cells of Escherichia coli were cultivated aerobically at 37°C in lysogeny broth (LB) (18), whereas cells of Gordonia polyisoprenivorans strain VH2 (DSMZ 44266) were aerobically cultivated at 30°C in Standard-I (St-I) medium (Merck, Darmstadt, Germany). Liquid cultures were incubated in Erlenmeyer flasks on horizontal rotary shakers with agitation at 100 to 150 rpm. Preparation of solid medium was carried out by the addition of 1.5% (wt/vol) agar-agar. Antibiotics were added as described previously by Sambrook et al. (18).
Chemicals.
Chemicals were purchased from Roth (Karlsruhe, Germany) and Sigma-Aldrich (Steinheim, Germany). Synthetic poly(cis-1,4-isoprene) (Roth, Karlsruhe, Germany) was purified and grained to a defined size of 63 to 500 μm, as described previously (14). Natural rubber (CAS no. 9006-04-06) was received from Weber & Schaer GmbH & Co. KG (Hamburg, Germany) as latex milk (Neotex Latz). The stabilizing ammonia was removed via centrifugation (5 min at 10,000 × g). The top layer was removed and used for further experiments.
Bioinformatic analyses.
For prediction of cleavage sites, TatP 1.0 (19) was applied, using default parameters. Prediction and analyses of domains were carried out by using the Pfam database (Pfam A and B) (20). Alignments were performed by applying ClustalW with default parameters (21).
Isolation and modification of DNA.
Total DNA of G. polyisoprenivorans strain VH2 was isolated by using the DNeasy Blood & Tissue kit (Qiagen, Hilden, Germany). The gene encoding Lcp1VH2 was amplified from total DNA of G. polyisoprenivorans strain VH2 by using oligonucleotides lcp1_fw_oTAT_NdeI (5′-AAAACATATGCACCACCACCACCACCACAACTCGATCCCGGGTGTGGG-3′) and lcp_rv_Stop_BamHI (5′-AAAAGGATCCTCAGTTGTAGTTCGGGTTGTTGAAGTAGG-3′), synthesized by Eurofins MWG Operon (Ebersberg, Germany). Oligonucleotide lcp1_fw_oTAT_NdeI generates a hexahistidine tag at the N terminus. Amplification was carried out by using Phusion high-fidelity DNA polymerase (Thermo Fisher Scientific, Waltham, MA, USA). The amplified DNA fragment was purified by employing the peqGOLD gel extraction kit (Peqlab Biotechnologie GmbH, Erlangen, Germany) and cloned into the pJET 1.2 Blunt cloning vector (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol. The construct was transferred into chemically competent cells (14) of E. coli Mach1 T1R (Life Technologies, Carlsbad, CA, USA).
Plasmids were isolated from cells growing on medium containing an appropriate antibiotic by using a peqGOLD plasmid minikit (Peqlab Biotechnologie, Erlangen, Germany). The correctness of the DNA fragment was verified by sequencing at Seqlab (Sequence Laboratories Göttingen, Göttingen, Germany) using the pJET1.2 forward and reverse sequencing primers (Thermo Fisher Scientific, Waltham, MA, USA). The correct fragment was excised from pJET1.2::Hislcp1VH2 by using the restriction endonucleases BamHI and NdeI (Thermo Fisher Scientific, Waltham, MA, USA) and was purified (peqGOLD gel extraction kit; Peqlab Biotechnologie, Erlangen, Germany) according to the manufacturer's protocols. The fragment was then ligated into the BamHI- and NdeI-linearized expression vector pET23a(+) (Merck, Darmstadt, Germany), and the resulting plasmid was transformed into chemically competent cells (18) of E. coli Mach1 T1R. The correctness of the insertion and fragment was confirmed by plasmid isolation and subsequent restriction and sequencing analysis (using the T7 promoter and terminator primer), as described above.
Purification of Lcp1VH2.
Overexpression of lcp1VH2 was carried out with E. coli C41(DE3) containing pET23a(+)::Hislcp1VH2, as described in the pET system manual (Merck, Darmstadt, Germany), with the exception that cells were grown for 16 h at 20°C to 25°C after induction at an optical density at 600 nm (OD600) of 0.4 to 0.5. The cells were harvested by centrifugation (4°C at 3,500 × g) and stored at −20°C. Cell disruption was carried out after resuspension in 100 mM Tris-HCl buffer containing 40 mM imidazole and 500 mM NaCl (pH 7.4, 4°C) by a 5-fold passage through a cooled French press (Aminco, Silver Spring, MD, USA). The soluble fraction was obtained from the crude extract by 30 min of centrifugation at 21,000 × g (4°C).
All purification steps were carried out at 4°C. The soluble fraction was loaded (0.5 ml/min) onto a Ni-nitrilotriacetic acid (NTA) Sepharose column (HisTrap FF, 1 ml; GE Healthcare Lifescience, Uppsala, Sweden) that was equilibrated according to the manufacturer's instructions. For binding, a solution consisting of 100 mM Tris-HCl buffer containing 40 mM imidazole and 500 mM NaCl (pH 7.4) was used. For purification, the following program was chosen: the flow rate was 1 ml/min, the first washing step was carried out for 20 min with 40 mM imidazole, and the second washing step was done for 10 min with 60 mM imidazole in 100 mM Tris-HCl buffer containing 500 mM NaCl (pH 7.4). Elution was carried out for 10 min at a concentration of 500 mM imidazole in the latter buffer. The eluted fractions were pooled and then concentrated to approximately 1 ml by using Vivaspin ultrafiltration spin columns (molecular weight cutoff [MWCO] of 10,000; polyethersulfone [PES] membrane) by centrifugation (3,500 × g). Since the protein was not totally pure after immobilized metal affinity chromatography (IMAC) was performed, a further purification step was performed via size exclusion chromatography (SEC) using a Superdex 200 HP 16/600 column (GE Healthcare Lifescience, Uppsala, Sweden). This step also served for molecular mass detection of Lcp1VH2. Calibration was carried out by applying a high-molecular-mass gel filtration calibration kit (GE Healthcare Lifescience, Uppsala, Sweden), according to the manufacturer's protocol. The concentrated eluate was loaded onto the column, which was equilibrated with 20 mM sodium phosphate buffer containing 150 mM NaCl (pH 7.4). A flow rate of 1 ml/min was applied. Fractions were determined by monitoring the absorbance at 280 nm. The fractions were pooled and concentrated, and the buffer was changed to Bis-Tris buffer (200 mM, pH 7.0) by centrifugation (3,500 × g) using Vivaspin ultrafiltration spin columns.
Protein concentrations were determined according to the Bradford method (22). Proteins were separated, and purity was examined via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (23). Protein verification and control of purity were executed by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) at the Institut für Mikrobiologie, Ernst-Moritz-Arndt-Universität, Greifswald, Germany.
Oxygen consumption assay.
Oxygen consumption was determined by using the module-based microrespiration system and MicOx software from Unisense (Aarhus, Denmark). Bis-Tris buffer (200 mM, pH 7.0), 5 μg/ml purified protein, and 20 μl/ml of a 20% latex emulsion were mixed in Unisense microrespiration chambers (1 ml). Consumption of oxygen was determined at 23°C with a stirring rate of 200 rpm by using a standard glass sensor (OX-MR; Unisense, Aarhus, Denmark) that was prepolarized and calibrated according to the manufacturer's protocol. This assay was also applied for determination of the temperature optimum, which was performed within a heating cabinet at temperatures mentioned in the text. Determination of the pH optimum was achieved by using the buffers mentioned in the text. Furthermore, the substrate range of Lcp1VH2 was also tested via oxygen consumption using substrates other than latex (as specified). For this, 1% (vol/vol) of each compound was added to the assay mixture. In order to exclude inhibitory effects of the corresponding compound, 20 μl/ml of a 20% latex emulsion was additionally added, and the oxygen consumption rate was compared to that of the assay mixtures with latex emulsion only. The oxygen consumption assay was also used by default to check the activity of the purified protein as well as for the chelating experiments.
Isolation of degradation products and spectral analyses.
For spectral analyses, 50 μl/ml of a 20% (vol/vol) latex emulsion was mixed with 15 μg/ml of purified protein (negative control: enzyme heat inactivated at 98°C for 30 min) in Bis-Tris buffer (200 mM, pH 7.0) and inoculated into test tubes for 20 h at 30°C on a rotary shaker at 150 rpm (sample volume, 1 ml). Furthermore, 15 mg/ml of granules of synthetic rubber (in 200 mM Bis-Tris buffer, pH 7.0) was inoculated into test tubes for 5 days with a daily addition of 10 μg/ml of purified protein (negative control: heat-inactivated enzyme). The test tubes containing synthetic rubber were autoclaved before the enzyme was added for the first time. The mixtures were extracted by using diethyl ether. The resulting fractions were dried by using anhydrous MgSO4, filtered, and concentrated in a rotary evaporator at 40°C under reduced pressure. The degradation products were purified by chromatography on a silica gel 60 (0.040 to 0.063 mm; 230 to 400 mesh) column using dichloromethane as an eluent. Analysis of the fractions (each ca. 10 ml) with a thin-layer chromatography (TLC) plate using dichloromethane as an eluent was performed by using precoated plastic sheets with detection by UV (254 nm) and by coloration with a cerium molybdenum solution [phosphomolybdic acid (25 g), Ce(SO4)2 · H2O (10 g), concentrated H2SO4 (60 ml), H2O (940 ml)]. Two product-containing fractions (fraction 1 and fraction 2) were collected, and the solvent was removed in a rotary evaporator at 40°C under reduced pressure.
1H and 13C spectra of the extracts and isolated products were recorded at room temperature in CDCl3 on a spectrometer at 600 and 151 MHz, respectively. The chemical shifts are given in ppm relative to the internal standard tetramethylsilane (TMS) {1H, δ [Si(CH3)4] = 0.00 ppm} or relative to the resonance of the solvent (13C, δ [CDCl3] = 77.0 ppm). 1H and 13C signals were assigned by means of C, H, correlation spectroscopy (COSY), and heteronuclear single-quantum coherence (HSQC) spectroscopy. Infrared spectra were recorded by Fourier transform infrared spectroscopy (FTIR) (SpectrumOne; PerkinElmer, Waltham, MA, USA). The samples were measured directly on an attenuated total reflectance (ATR) sample head. The positions of the absorption bands are given in cm−1. The FTIR spectrum is given for the second fraction, with an average chain length, n, of 10 for FTIR (film) with 2,961, 2,918, 2,853, 1,722, 1,449, 1,376, 1,082, and 837 cm−1. Furthermore, nuclear magnetic resonance (NMR) spectroscopy data (1H and 13C) for both fractions are 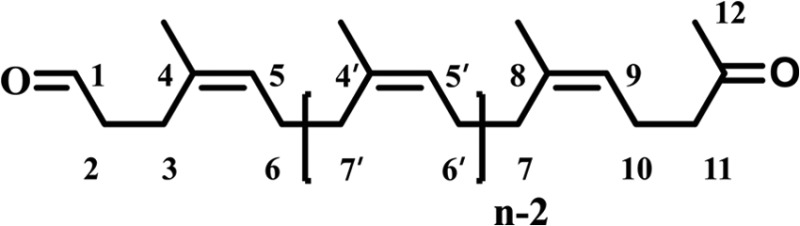
Fraction 1 1H NMR (600 MHz, CDCl3): δ = 1.68 (s, 53.3 H, 4-Me, 8-Me), 2.01 to 2.09 (m, 67.6 H, 6-H, 7-H), 2.13 (s, 3 H, 12-H), 2.26 (td, 3J10,11 = 7.7 Hz, 3J10,9 = 7.4 Hz, 2 H, 10-H), 2.35 (t, 3J3,2 = 7.8 Hz, 2 H, 3-H), 2.44 (t, 3J11,10 = 7.7 Hz, 2 H, 11-H), 2.49 (td, 3J2,3 = 7.8 Hz, 3J2,1 = 1.6 Hz, 2 H, 2-H), 5.08 (t, 3J9,10 = 7.4 Hz, 1 H, 9-H), 5.11 to 5.18 (m, 17.0 H, 5-H), 9.78 (t, 3J1,2 = 1.6 Hz, 1 H, 1-H) ppm.
Fraction 1 13C NMR (151 MHz, CDCl3): δ = 22.3 (C-10), 23.1 (4-Me), 23.4 (8-Me), 24.3 (C-3), 26.4 (C-6), 29.7 (C-12), 32.2 (C-7), 42.4 (C-2), 44.0 (C-11), 123.4 (C-9), 125.0 (C-5), 135.2 (C-8), 202.1 (C-1), 208.7 (C=O) ppm.
Fraction 2 1H NMR (600 MHz, CDCl3): δ = 1.68 (s, 29.7 H, 4-Me, 8-Me), 2.01 to 2.09 (m, 35.4 H, 6-H, 7-H), 2.13 (s, 3 H, 12-H), 2.26 (td, 3J10,11 = 7.7 Hz, 3J10,9 = 7.4 Hz, 2 H, 10-H), 2.35 (t, 3J3,2 = 7.8 Hz, 2 H, 3-H), 2.44 (t, 3J11,10 = 7.7 Hz, 2 H, 11-H), 2.49 (td, 3J2,3 = 7.8 Hz, 3J2,1 = 1.6 Hz 2 H, 2-H), 5.08 (t, 3J9,10 = 7.4 Hz, 1 H, 9-H), 5.11 to 5.18 (m, 9.0 H, 5-H), 9.78 (t, 3J1,2 = 1.6 Hz, 1 H, 1-H) ppm.
Fraction 2 13C NMR (151 MHz, CDCl3): δ = 22.3 (C-10), 23.1 (4-Me), 23.4 (8-Me), 24.3 (C-3), 26.4 (C-6), 29.7 (C-12), 32.2 (C-7), 42.4 (C-2), 44.0 (C-11), 123.4 (C-9), 125.0 (C-5), 135.2 (C-8), 202.1 (C-1), 208.7 (C=O) ppm.
Possible degradation of poly(trans-1,4-isoprene) and poly(cis-1,4-butadiene) using purified Lcp1VH2 was also determined by using 1H NMR analysis. For this, 1.5% (wt/vol) of each polymer was incubated in 200 mM Bis-Tris buffer (pH 7.0) in test tubes for 5 days with a daily addition of 10 μg/ml of purified protein (negative control: without enzyme) as described above for rubber. The test tubes were autoclaved before the test was started. The mixtures were extracted, dried, filtered, and concentrated as described above for rubber. 1H spectra of the extracts were recorded at room temperature in CDCl3 on a spectrometer at 600 MHz.
Identification and characterization of cofactors in purified Lcp1VH2.
Metal content analyses using inductively coupled plasma optical emission spectroscopy (ICP-OES) were carried out at the Chemical Analysis Lab/Center for Applied Isotope Studies at the University of Georgia.
The oxidation state of the transition metal within the active enzyme was determined by applying a spectroscopic assay with bathocuproine sulfonate (BCS), which specifically chelates Cu(I) (24). Samples contained 50 μM Lcp1VH2 in 20 mM sodium phosphate buffer containing 150 mM NaCl (pH 7.4) and 1% (wt/vol) SDS within a quartz cuvette. BCS (final concentration of 500 μM) was added to form yellow Cu(I)(BCS)2 complexes with Cu(I), which were detected spectroscopically at approximately 480 nm. Reduction of Cu(II) was performed by the addition of Na-ascorbate (final concentration of 500 μM). To chelate divalent ions, EDTA was added to a concentration of 20 mM. UV-visible (UV-Vis) spectra were recorded on a Jasco V-550 UV-Vis spectrophotometer (Jasco Corporation, Tokyo, Japan) or a Shimadzu UV-2600 UV-Vis spectrophotometer (Shimadzu, Kyoto, Japan).
RESULTS
Purification of Lcp1VH2.
Although functional expression of lcp1VH2 in an lcp double-deletion mutant of G. polyisoprenivorans strain VH2 (14) resulted in the complementation of the rubber-degrading activity, purification of the enzyme from this mutant was not successful. Furthermore, the protein of heterologously expressed lcp1VH2 was not processed and secreted in E. coli, although analysis of the protein sequence of Lcp1VH2 (GenBank accession no. YP_005285193) revealed the occurrence of a twin-arginine (TAT) signal peptide cleavage site between amino acids 36 and 37 (…WAPA-NSIP…). Instead, it remained in the cells mainly as inclusion bodies. Additionally, Lcp1VH2 could not be purified by IMAC when a hexahistidine tag was adhered C-terminally under nondenaturing conditions; purification was possible only under denaturing conditions (not shown).
For these reasons, we replaced the predicted TAT cleavage site with an N-terminal hexahistidine tag. The resulting protein (377 amino acids) exhibited a theoretical molecular mass of 42.59 kDa. The overproduced protein was purified to electrophoretic homogeneity (Fig. 1) in two subsequent steps: (i) via immobilized metal affinity chromatography and (ii) by size exclusion chromatography. The latter chromatographic step revealed an apparent molecular mass of 38.5 kDa (±1.5 kDa) for modified Lcp1VH2, which correlates with a monomeric protein. The average yield of purified Lcp1VH2 from 1,000 ml of culture volume was approximately 1.5 mg.
FIG 1.
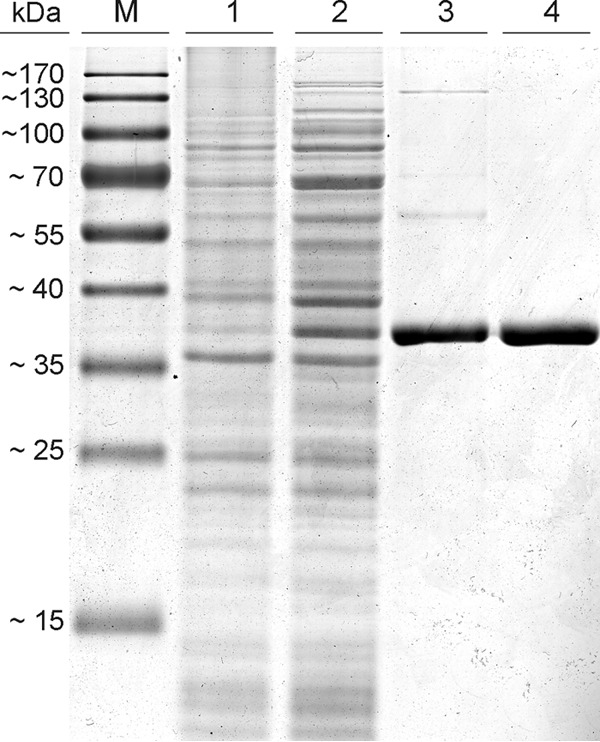
Purification of Lcp1VH2 by applying an immobilized metal affinity chromatography step (lane 3) and subsequent size exclusion chromatography (lane 4), as analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10 μg of protein was applied in lanes 3 and 4). Lanes 1 and 2 contain 40 μg of protein of the soluble fraction of the controls [E. coli C41(DE3) containing pET23a(+) (lane 1) as well as of E. coli C41(DE3) containing pET23a(+)::hislcp1VH2 (lane 2)], both induced with isopropyl β-d-1-thiogalactopyranoside at an OD600 of 0.4. M, marker proteins.
Oxygen consumption assay.
Since it was anticipated that Lcp is an oxygenase, measurements of oxygen consumption by Lcp1VH2 were carried out with the active enzyme as well as the heat-inactivated protein (98°C for 30 min) in the presence of latex by an enzyme assay, as described in Materials and Methods (Fig. 2). The assay clearly showed that oxygen consumption of Lcp1VH2 is latex dependent and does not occur in the absence of the polyisoprene substrate. In order to demonstrate that the consumption of oxygen is due to the active enzyme and not to the other components of the applied assay mixture, negative-control experiments were carried out. For example, the assay was performed with heat-inactivated protein, with buffer only, or with buffer together with latex but without enzyme. Since all these controls were negative, the oxygen consumption can be attributed to the enzyme activity. Furthermore, clearing of the samples with active enzyme was observed in these assays, whereas the control mixtures (use of denatured enzyme or latex only) remained milky. This clearly demonstrated that active Lcp1VH2 consumes oxygen to change the structure of molecules that are present in latex. An evaluation of oxygen consumption within the first 10 min (without a lag phase) revealed a specific activity of Lcp1VH2 of approximately 1.3 μmol/min per mg of purified protein.
FIG 2.
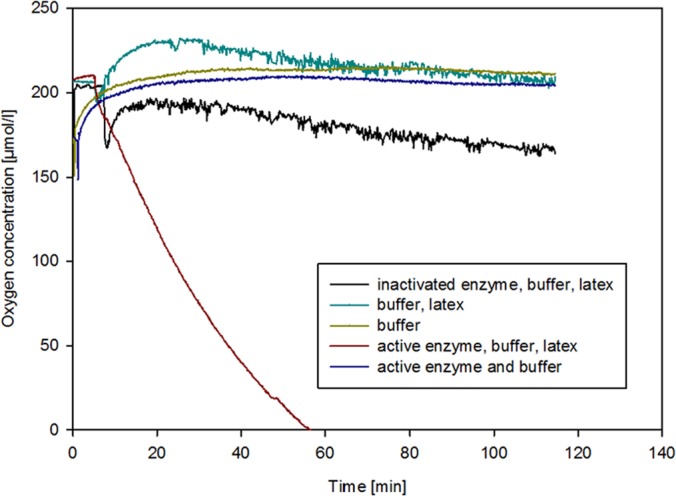
Measurements of Lcp1VH2-driven oxygen consumption. Analyses were carried out in the presence of 20 μl/ml of a latex suspension and 5 μg/ml of Lcp1VH2 in Bis-Tris buffer (200 mM, pH 7.0). All components for the negative controls were added as indicated in the key at the concentrations mentioned above. All measurements were carried out at 23°C at a stirring rate of 200 rpm. Lcp1VH2 was inactivated by heat.
Analysis of degradation products via spectral analyses.
After incubation of latex with active or heat-inactivated enzyme, the resulting degradation products were extracted, dried, filtered, and concentrated, as described in Materials and Methods. Thereafter, they were analyzed by FTIR and NMR spectroscopy. Furthermore, the degradation products were purified by using preparative column chromatography with silica gel. The products were eluted with dichloromethane, and two fractions were collected. The product fractions were also analyzed by means of FTIR and NMR spectroscopy.
Latex treated with active or heat-inactivated Lcp1VH2 gave FTIR spectra (Fig. 3) that were identical to that of poly(cis-1,4-isoprene) reported in the literature (25, 26). This includes the assignment of the C=C double bonds as well as of the aliphatic CHx groups, confirming that the extracts consisted of natural rubber from latex milk. However, the analysis of the product fractions, which could be isolated when the rubber was incubated with active Lcp1VH2, showed FTIR spectral bands at 1,722 cm−1, which clearly indicated the presence of carbonyl functional groups. Latex samples of the negative control incubated with denatured enzyme did not exhibit these groups. This finding is in good agreement with data from previous analyses of microbiologically deteriorated rubber analyzed via infrared spectroscopy (17, 27) and indicates a degradation mechanism involving the addition of oxygen to the polyisoprene molecules.
FIG 3.

FTIR absorbance spectrum of the second product fraction after incubation of latex milk with active Lcp1VH2 in comparison to the spectrum of latex of the negative control. From a latex emulsion containing 20% (vol/vol) latex, 50 μl/ml was mixed with 15 μg/ml of purified protein (negative control: heat-inactivated enzyme) in Bis-Tris buffer (200 mM, pH 7.0) and incubated for 20 h at 30°C on a rotary shaker (150 rpm). R′ to R′′′, alkyl residues.
Furthermore, the formation of aldehydes as well as ketones during the degradation of rubber was detected by 1H NMR spectroscopy of the two product fractions (Fig. 4; see Fig. S1 in the supplemental material for the full 1H NMR spectrum of the second fraction in comparison to the negative control). The triplet at 9.78 ppm, which was not detected in the negative control, clearly shows the appearance of aldehyde groups during the degradation process and therefore confirmed the findings made by FTIR analysis. Furthermore, an occurrence of a singlet at 2.13 ppm, which represents the protons of the methyl ketone, appeared only in samples that were treated with active Lcp1VH2 but not in those treated with denatured protein. The signals, which appeared in the sample with active enzyme at between 2.26 ppm and 2.49 ppm, are due to the formation of terminal alkyl chains during the Lcp1VH2-mediated degradation process. The position of the multiplet signals at 5.13 ppm and 2.04 ppm as well as the singlet signal at 1.68 ppm were detected in both samples and are to be assigned to the poly(cis-1,4-isoprene) structure, as presented at position 5 (olefinic protons), positions 6 and 7 (methylene protons), and positions 4 and 8 (methyl protons), respectively, in Fig. 4. All these spectra are in agreement with spectra that were reported previously for intact poly(cis-1,4-isoprene) as well as for the biodegradation products, like those isolated from a culture of Nocardia sp. strain 835A growing on rubber as the sole source of carbon and energy (3, 28–31). Furthermore, the occurrence of these products was confirmed by 13C NMR spectroscopy, and the data also corresponded to spectra for biodegraded rubber particles obtained previously (3, 29). The two isolated fractions differed from each other by the average chain length, n, which could be estimated by using the 1H NMR integrals by dividing the signal integrals by the number of protons of the corresponding groups (3). Here an average n of 17.9 for the first fraction and an average n of 10.0 for the second fraction were calculated (see Table S1 in the supplemental material for detailed calculations). All experiments were also carried out with synthetic poly(cis-1,4-isoprene) and led to identical results (not shown).
FIG 4.

1H NMR spectrum of the second fraction. For clarity, parts of the spectrum have been omitted. Symbols: s, singlet; d, doublet; t, triplet; td, triplet of doublet; m, multiplet; Me, methyl.
Biochemical characterization of Lcp1VH2.
The temperature and pH optima of purified Lcp1VH2 were elucidated by using different buffers and the oxygen consumption assay described above (Fig. 5). The temperature optimum was at around 30°C, that of the pH was at around 7. The activity of Lcp1VH2 at pH <5 could not be determined since the rubber of latex coagulates at low pH values. Furthermore, the substance range of the enzyme was evaluated, and it clearly appeared that alkanes and also alkenes were not oxidatively cleaved by Lcp1VH2, as revealed by the oxygen consumption assay. This assay also revealed that acyclic terpenes, such as squalene or squalane, and also unsaturated fatty acids, such as palmitoleic acid or linoleic acid, were also not cleaved. Furthermore, enzyme assays employing active Lcp1VH2 and subsequent NMR analyses were applied, as described in Materials and Methods. These analyses clearly revealed that the enzyme does not cleave the double bonds of polymers having a structure like poly(cis-1,4-isoprene), such as poly(trans-1,4-isoprene) and poly(cis-1,4-butadiene), since the spectra of polymers treated with active Lcp1VH2 exhibited no differences from those of the negative controls (in the absence of the enzyme) (data not shown).
FIG 5.
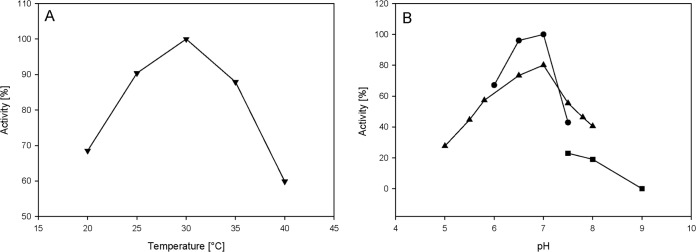
Temperature (A) and pH (B) optima of purified Lcp1VH2. A 20-μl/ml latex suspension was incubated with 5 μg/ml of purified Lcp1VH2, and oxygen consumption was measured. For determination of the temperature optimum, Bis-Tris buffer (200 mM, pH 7.0) was chosen. For evaluation of the pH optimum of the enzyme, Bis-Tris (▲), PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] (●), and Tris (◼) (each 200 mM) were applied at the corresponding pH values at 23°C.
Lcp1VH2 possesses no domain for which a function is known and also shows no similarity to an already characterized protein. Therefore, a distinct prediction of a putatively utilized transition metal cannot be done in silico. However, purified and concentrated Lcp1VH2 shows an orange color with a UV-visible spectrum illustrated in Fig. 6. This spectrum shows a broad band at around 420 nm, also typical for, e.g., copper-dependent “white” laccases (32, 33) and indicating a putatively bound transition factor. Therefore, the influence of different chelating agents on the enzyme activity was tested. EDTA and tiron (sodium 4,5-dihydroxybenzene-1,3-disulfonate) did not significantly inhibit the activity of the enzyme, even after an incubation time of 30 min, but 1,10-phenanthroline as well as 2,2-bipyridyl inhibited the activity at high concentrations (Table 1). The latter compounds are known not only to chelate Fe(II) but also to form complexes with, e.g., Cu(II) (34, 35). Therefore, ethylxanthate, highly specific for copper at low concentrations (36), was also applied. The latter clearly showed significant inhibition of the reaction at low concentrations of even 0.2 mM.
FIG 6.

UV-visible spectrum of purified Lcp1VH2 (A) and results of spectroscopic assays for determination of the oxidation state of copper within Lcp1VH2 by applying bathocuproine sulfonate (BCS) (B). For both panels, the buffer for all samples consisted of 20 mM sodium phosphate buffer containing 150 mM NaCl (pH 7.4). For panel B, All samples contained 50 μM Lcp1VH2 and 1% (wt/vol) SDS. The black solid line in panel B represents the absorption of all components, including 20 mM EDTA but excluding BCS. The light-gray broken line indicates a sample containing 500 μM BCS. The dark-gray broken line indicates a sample containing 500 μM BCS and 500 μM ascorbate. The light-gray solid line indicates a sample that contained 20 mM EDTA and 500 μM BCS.
TABLE 1.
Compounds tested for inhibition of Lcp1VH2
| Compound added | Concn (mM) | Relative activity (%) |
|---|---|---|
| Standard assay mixture without added compound | 0 | 100 |
| Chelators | ||
| EDTA | 10 | 108 |
| Sodium 4,5-dihydroxybenzene-1,3-disulfonate | 10 | 96 |
| 1,10-Phenanthroline | 1 | 53 |
| 10 | 0 | |
| 2,2-Bipyridyl | 10 | 65 |
| Ethylxanthate | 0.2 | 70 |
| 2 | 0 | |
| Alkylating reagent | ||
| N-Ethylmaleimide | 1 | 101 |
| 10 | 70 | |
| Reducing agent | ||
| 2-Mercaptoethanol | 5 | 17 |
In order to detect whether other metals could also be involved in the enzymatic activity, a metal content analysis of purified Lcp1VH2 was performed by using inductively coupled plasma optical emission spectroscopy (ICP-OES). This analysis clearly showed that the only metal present in the purified and active Lcp1VH2 is copper. As also described previously for other recombinant copper-dependent enzymes that were purified from cells of E. coli (37), active Lcp1VH2 had only a substoichiometric content of copper. In order to specify the oxidation state of the transition metal within the active enzyme, a spectroscopic assay applying bathocuproine sulfonate (BCS), specifically chelating Cu(I) (24), was employed (Fig. 6). When BCS was added to the enzyme, denatured with SDS, an increase of the absorbance at approximately 480 nm [associated with the formation of the Cu(I)(BCS)2 complex] indicated the presence of Cu(I). However, the reduction of possibly contained Cu(II) to Cu(I) by applying ascorbate resulted in a further increase of the absorbance, thus indicating that Cu(II) is also present. Since Cu(I) could also evolve by intrinsic reduction of Cu(II), e.g., by cysteine, tyrosine, or tryptophan (38) within this environment, the assay was performed with the addition of EDTA before the enzyme was denatured with SDS. After the subsequent addition of BCS, the absence of Cu(I)(BCS)2 complexes clearly indicated that there was no Cu(I) within the sample. Therefore, it was concluded that the purified and active Lcp1VH2 contains Cu(II).
DISCUSSION
Until now, several studies on the participation of Lcp in the degradation of poly(cis-1,4-isoprene) have been carried out (12, 13, 16), and the essentiality of Lcp for the degradation process was unequivocally demonstrated recently (14). However, concrete statements on how Lcp is involved in the degradation of rubber and if other coenzymes or cofactors are essential for the initial step of rubber disintegration by actinobacteria could not be made until now. In this study, we clearly show that Lcp1VH2 is responsible for this initial oxidative step by demonstrating the reaction in vitro employing the purified enzyme. Lcp1VH2 cleaves rubber at the cis double bonds of the polymer by incorporation of oxygen, which leads to the formation of oligomers containing terminal carbonyl functional groups. Furthermore, this process relies on an endocleavage mechanism, resulting in degradation products of different molecular weights.
The degradation products possess the structures shown in Fig. 4, which are in good agreement with the degradation products generated by actinomycetes growing on natural and synthetic as well as vulcanized rubber. However, the molecular weights of the fractions always differ from strain to strain (2 to 190 isoprene units) (3, 12, 13, 29). Since Lcp is present in all rubber-degrading actinomycetes (10, 12–16), Lcps from different strains obviously degrade the polymer to cleavage products of different average chain lengths by endocleavage. However, it should be pointed out that all the publications mentioned above depict a completely different experimental setup, since they were carried out with living cells and never with the purified enzyme. Therefore, an additional modification of the resulting degradation products or the rapid consumption of lower-molecular-weight products by the cells is conceivable. Furthermore, other factors in these experiments, such as the length of the incubation period or the ratio of enzyme to substrate, might also influence the average length of the identified products.
For purified RoxA of Xanthomonas sp. strain 35Y, the only rubber-cleaving oxygenase that was investigated in more detail until now, the formation of products of various sizes and consisting of repetitive isoprene units with carbonyl functional groups at the termini was also described. This enzyme also cleaves rubber at the double bonds upon the incorporation of oxygen outside the cell and without the utilization of cosubstrates such as NAD(P) (4, 5, 29). For RoxA, a degradation mechanism involving the oxidative cleavage of the polyisoprene molecule via the generation of an allylic hydroxyperoxide and the subsequent formation of a substrate-dioxetane intermediate was proposed (5, 9). Since Lcp also cleaves rubber by adding oxygen to the double bonds of the polymer outside the cell (13, 14, 16, 17, 39), and since in vitro experiments with Lcp1VH2 could also be performed without additional cosubstrates, parallels within the degradation mechanism are possible.
However, the amino acid sequences of latex clearing proteins exhibit no similarities to that of RoxA, and the characterization of Lcp1VH2 clearly revealed great differences between Lcp and RoxA: (i) the main average cleavage products of Lcp1VH2 contain five and nine times more isoprene units than RoxA; (ii) Lcp amino acid sequences contain a domain of unknown function (DUF2236; Pfam accession no. PF09995), whereas RoxA sequences possess conserved sequences that belong to the family of diheme cytochrome c peroxidases (Pfam accession no. PF03150); (iii) Lcp1VH2, in contrast to the heme-containing RoxA, is a Cu(II)-dependent oxygenase; (iv) BLAST analyses reveal that Lcp is highly distributed, whereas RoxA is detectable in only very few Gram-negative bacteria (4, 10); and (v) a comparison of the evaluated specific activities indicates that Lcp1VH2 has a higher specific activity (1.3 μmol/min per mg) than RoxA of Xanthomonas sp. strain 35Y (0.3 μmol/min per mg) (5). These activities are, however, comparable to those of other double-bond-cleaving oxygenases, such as benzene or toluene dioxygenases (activity of between 0.045 and 2.58 μmol/min per mg) (40, 41), but are clearly lower than those of, e.g., the secreted, copper-dependent quercetin 2,3-dioxygenases (up to 177 μmol/min per mg) (42).
Due to the inhibition of Lcp1VH2 activity by the utilized chelating agents with a simultaneous absence of metals other than copper and the results of the applied bathocuproine sulfonate-based spectroscopic assays, and since it is known from the literature that hexahistidine-tagged, purified, copper-binding enzymes do not contain significantly larger amounts of copper than the same proteins that were purified by applying streptavidin tags or for which the hexahistidine tag was removed (43, 44), it can be concluded that purified Lcp1VH2 possesses Cu(II) as a transition factor. The divalent copper ions are known to preferably complex with histidine, serine, threonine, or tyrosine amino acid residues (45). Furthermore, copper sites can be assigned to three groups due to their spectroscopic capabilities. Since the enzyme shows no strong absorption band at 600 nm (typical for type 1 copper centers) or 300 nm (typical for type 3 copper centers) (46, 47) but shows a UV-visible spectrum that is similar to that of a nontypical, single-copper-containing, laccase-related enzyme (33), it can be speculated that the Cu(II) center within Lcp1VH2 is of type 2. These centers are found mainly in oxygenases, Cu/Zn superoxide dismutases, and copper oxidases that depict no blue color and most commonly ligate Cu(II) by histidine (45, 48). A comparison of >50 Lcp-similar sequences taken from publicly accessible databases revealed that the amino acid sequences of Lcp are highly conserved, especially in the area presented in Fig. 7. Within this region, all Lcp sequences of adhesive growing strains but also of clear-zone-forming bacteria show three highly conserved histidine residues: H195, H200, and H228 (in Lcp1VH2). It might therefore be possible that these three histidine residues play a substantial role in the coordination of the copper ion, as also described for other copper-containing oxygenases, such as the quercetin 2,3-dioxygenase mentioned above (42). However, this assumption has to be verified by further investigations in the future.
FIG 7.

Alignment of the most conserved segment within Lcp sequences of actinomycetes showing adhesive growth on rubber (strains of Gordonia and Nocardia) as well as of strains forming clear zones on latex overlay plates (strains of Streptomyces). Highly conserved histidine amino acids are marked with arrows.
The amino acid sequences of both Lcps of G. polyisoprenivorans strain VH2 are very similar and show >60% identity. Both proteins, like all Lcp sequences, possess a twin-arginine signal peptide and the DUF2236 domain. Since a deletion of only one lcp gene within the genome of strain VH2 did not result in a mutant that lost its ability to grow on rubber as the sole source of carbon and energy, whereas a double-deletion mutant exhibited a total loss of this ability (14), it is likely that Lcp2VH2 also cleaves rubber by incorporation of oxygen in a similar manner.
The conserved region presented in Fig. 7 is located within the conserved domain of unknown function that is detectable in all Lcp sequences. In Lcp1VH2, this domain covers >50% of the amino acid sequence. However, not all DUF2236 domain-containing proteins can be annotated as Lcp by similarity, but the domain can be detected in 541 species of Eubacteria (mainly Actinobacteria and Proteobacteria) and 113 species of Eukaryota (mainly Ascomycota) (Pfam version 27.0 [20]). All these phyla are known for their high degradative capabilities, in particular for the degradation of hardly degradable polymers. Since lcp1VH2 encodes an oxygenase, this domain might be detectable in a new family of oxygenases. However, confirmation of this statement requires further experiments in the future.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge financial support by the Deutsche Forschungsgemeinschaft (project STE 386/10-1), the Fonds der Chemischen Industrie (scholarship to D.B.), the Heinrich Heine Universität Düsseldorf, and Forschungszentrum Jülich GmbH for research related to this publication.
We thank Weber & Schaer GmbH & Co. KG (Hamburg, Germany) for providing Neotex Latz. We also thank S. Fetzner, A. L. Drees, D. Nianios, and S. Thierbach at the Institut für Mikrobiologie und Biotechnologie (WWU Münster) for their help regarding cofactor determination.
Footnotes
Published ahead of print 13 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01502-14.
REFERENCES
- 1.Sienkiewicz M, Kucinska-Lipka J, Janik H, Balas A. 2012. Progress in used tyres management in the European Union: a review. Waste Manag. 32:1742–1751. 10.1016/j.wasman.2012.05.010 [DOI] [PubMed] [Google Scholar]
- 2.Jendrossek D, Reinhardt S. 2003. Sequence analysis of a gene product synthesized by Xanthomonas sp. during growth on natural rubber latex. FEMS Microbiol. Lett. 224:61–65. 10.1016/S0378-1097(03)00424-5 [DOI] [PubMed] [Google Scholar]
- 3.Tsuchii A, Suzuki T, Takeda K. 1985. Microbial degradation of natural rubber vulcanizates. Appl. Environ. Microbiol. 50:965–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braaz R, Fischer P, Jendrossek D. 2004. Novel type of heme-dependent oxygenase catalyzes oxidative cleavage of rubber (poly-cis-1,4-isoprene). Appl. Environ. Microbiol. 70:7388–7395. 10.1128/AEM.70.12.7388-7395.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braaz R, Armbruster W, Jendrossek D. 2005. Heme-dependent rubber oxygenase RoxA of Xanthomonas sp. cleaves the carbon backbone of poly(cis-1,4-isoprene) by a dioxygenase mechanism. Appl. Environ. Microbiol. 71:2473–2478. 10.1128/AEM.71.5.2473-2478.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birke J, Hambsch N, Schmitt G, Altenbuchner J, Jendrossek D. 2012. Phe317 is essential for rubber oxygenase RoxA activity. Appl. Environ. Microbiol. 78:7876–7883. 10.1128/AEM.02385-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitt G, Seiffert G, Kroneck PM, Braaz R, Jendrossek D. 2010. Spectroscopic properties of rubber oxygenase RoxA from Xanthomonas sp., a new type of dihaem dioxygenase. Microbiology 156:2537–2548. 10.1099/mic.0.038992-0 [DOI] [PubMed] [Google Scholar]
- 8.Hoffmann M, Braaz R, Jendrossek D, Einsle O. 2008. Crystallization of the extracellular rubber oxygenase RoxA from Xanthomonas sp. strain 35Y. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64:123–125. 10.1107/S1744309108001206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seidel J, Schmitt G, Hoffmann M, Jendrossek D, Einsle O. 2013. Structure of the processive rubber oxygenase RoxA from Xanthomonas sp. Proc. Natl. Acad. Sci. U. S. A. 110:13833–13838. 10.1073/pnas.1305560110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birke J, Röther W, Schmitt G, Jendrossek D. 2013. Functional identification of rubber oxygenase (RoxA) in soil and marine myxobacteria. Appl. Environ. Microbiol. 79:6391–6399. 10.1128/AEM.02194-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jendrossek D, Tomasi G, Kroppenstedt RM. 1997. Bacterial degradation of natural rubber: a privilege of actinomycetes? FEMS Microbiol. Lett. 150:179–188. 10.1016/S0378-1097(97)00072-4 [DOI] [PubMed] [Google Scholar]
- 12.Ibrahim EM, Arenskötter M, Luftmann H, Steinbüchel A. 2006. Identification of poly(cis-1,4-isoprene) degradation intermediates during growth of moderately thermophilic actinomycetes on rubber and cloning of a functional lcp homologue from Nocardia farcinica strain E1. Appl. Environ. Microbiol. 72:3375–3382. 10.1128/AEM.72.5.3375-3382.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bröker D, Dietz D, Arenskötter M, Steinbüchel A. 2008. The genomes of the non-clearing-zone-forming and natural rubber-degrading species Gordonia polyisoprenivorans and Gordonia westfalica harbor genes expressing Lcp activity in Streptomyces strains. Appl. Environ. Microbiol. 74:2288–2297. 10.1128/AEM.02145-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiessl S, Schuldes J, Thürmer A, Halbsguth T, Bröker D, Angelov A, Liebl W, Daniel R, Steinbüchel A. 2012. Involvement of two latex-clearing proteins during rubber degradation and insights into the subsequent degradation pathway revealed by the genome sequence of Gordonia polyisoprenivorans strain VH2. Appl. Environ. Microbiol. 78:2874–2887. 10.1128/AEM.07969-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imai S, Ichikawa K, Muramatsu Y, Kasai D, Masai E, Fukuda M. 2011. Isolation and characterization of Streptomyces, Actinoplanes, and Methylibium strains that are involved in degradation of natural rubber and synthetic poly(cis-1,4-isoprene). Enzyme Microb. Technol. 49:526–531. 10.1016/j.enzmictec.2011.05.014 [DOI] [PubMed] [Google Scholar]
- 16.Rose K, Tenberge KB, Steinbüchel A. 2005. Identification and characterization of genes from Streptomyces sp. strain K30 responsible for clear zone formation on natural rubber latex and poly(cis-1,4-isoprene) rubber degradation. Biomacromolecules 6:180–188. 10.1021/bm0496110 [DOI] [PubMed] [Google Scholar]
- 17.Linos A, Berekaa MM, Reichelt R, Keller U, Schmitt J, Flemming HC, Kroppenstedt RM, Steinbüchel A. 2000. Biodegradation of cis-1,4-polyisoprene rubbers by distinct actinomycetes: microbial strategies and detailed surface analysis. Appl. Environ. Microbiol. 66:1639–1645. 10.1128/AEM.66.4.1639-1645.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 19.Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S. 2005. Prediction of twin-arginine signal peptides. BMC Bioinformatics 6:167. 10.1186/1471-2105-6-167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. 2013. Pfam: the protein families database. Nucleic Acids Res. 42:D222–D230. 10.1093/nar/gkt1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 23.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 24.Blair D, Diehl H. 1961. Bathophenanthrolinedisulphonic acid and bathocuproinedisulphonic acid, water soluble reagents for iron and copper. Talanta 7:163–174. 10.1016/0039-9140(61)80006-4 [DOI] [Google Scholar]
- 25.Salomon G, van der Schee AC. 1954. Infrared analysis of isomerized, vulcanized and oxidized natural rubber. J. Polym. Sci. 14:181–192. 10.1002/pol.1954.120147405 [DOI] [Google Scholar]
- 26.Kuptsov AH, Zhizhin GN. 1998. Handbook of Fourier transform Raman and infrared spectra of polymers. Elsevier Science, Amsterdam, Netherlands [Google Scholar]
- 27.Cundell AM, Mulcock AP. 1975. The biodegradation of vulcanized rubber, p 89–96 In Underkofler LA. (ed), Developments in industrial microbiology, vol 16 Society of Industrial Microbiology, Arlington, VA [Google Scholar]
- 28.Tanaka A, Takeuchi Y, Kobayashi M, Tadokoro H. 1971. Characterization of diene polymers. I. Infrared and NMR studies: nonadditive behavior of characteristic infrared bands. J. Polym. Sci. A2 9:43–57 [Google Scholar]
- 29.Tsuchii A, Takeda K. 1990. Rubber-degrading enzyme from a bacterial culture. Appl. Environ. Microbiol. 56:269–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato H, Tanaka Y. 1979. 1H-NMR study of polyisoprenes. J. Polym. Sci. Polym. Chem. 17:3551–3558. 10.1002/pol.1979.170171113 [DOI] [Google Scholar]
- 31.Anachkov MP, Rakovski SK, Stefanova RV. 2000. Ozonolysis of 1,4-cis-polyisoprene and 1,4-trans-polyisoprene in solution. Polym. Degrad. Stabil. 67:355–363. 10.1016/S0141-3910(99)00137-8 [DOI] [Google Scholar]
- 32.Palmieri G, Giardina P, Bianco C, Scaloni A, Capasso A, Sannia G. 1997. A novel white laccase from Pleurotus ostreatus. J. Biol. Chem. 272:31301–31307. 10.1074/jbc.272.50.31301 [DOI] [PubMed] [Google Scholar]
- 33.Schuckel J, Matura A, van Pee KH. 2011. One-copper laccase-related enzyme from Marasmius sp.: purification, characterization and bleaching of textile dyes. Enzyme Microb. Technol. 48:278–284. 10.1016/j.enzmictec.2010.12.002 [DOI] [PubMed] [Google Scholar]
- 34.Moss ML, Mellon MG. 1942. Colorimetric determination of iron with 2,2′-bipyridyl and with 2,2′,2′-terpyridyl. Ind. Eng. Chem. Anal. Ed. 14:862–865. 10.1021/i560111a014 [DOI] [Google Scholar]
- 35.Brandt WW, Dwyer FP, Gyarfas ED. 1954. Chelate complexes of 1,10-phenanthroline and related compounds. Chem. Rev. 54:959–1017. 10.1021/cr60172a003 [DOI] [Google Scholar]
- 36.Oka T, Simpson FJ. 1971. Quercetinase, a dioxygenase containing copper. Biochem. Biophys. Res. Commun. 43:1–5. 10.1016/S0006-291X(71)80076-1 [DOI] [PubMed] [Google Scholar]
- 37.Martins LO, Soares CM, Pereira MM, Teixeira M, Costa T, Jones GH, Henriques AO. 2002. Molecular and biochemical characterization of a highly stable bacterial laccase that occurs as a structural component of the Bacillus subtilis endospore coat. J. Biol. Chem. 277:18849–18859. 10.1074/jbc.M200827200 [DOI] [PubMed] [Google Scholar]
- 38.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. 1985. Measurement of protein using bicinchoninic acid. Anal. Biochem. 150:76–85. 10.1016/0003-2697(85)90442-7 [DOI] [PubMed] [Google Scholar]
- 39.Yikmis M, Arenskötter M, Rose K, Lange N, Wernsmann H, Wiefel L, Steinbüchel A. 2008. Secretion and transcriptional regulation of the latex-clearing protein, Lcp, by the rubber-degrading bacterium Streptomyces sp. strain K30. Appl. Environ. Microbiol. 74:5373–5382. 10.1128/AEM.01001-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bagneris C, Cammack R, Mason JR. 2005. Subtle difference between benzene and toluene dioxygenases of Pseudomonas putida. Appl. Environ. Microbiol. 71:1570–1580. 10.1128/AEM.71.3.1570-1580.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lynch NA, Jiang H, Gibson DT. 1996. Rapid purification of the oxygenase component of toluene dioxygenase from a polyol-responsive monoclonal antibody. Appl. Environ. Microbiol. 62:2133–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fusetti F, Schroter KH, Steiner RA, van Noort PI, Pijning T, Rozeboom HJ, Kalk KH, Egmond MR, Dijkstra BW. 2002. Crystal structure of the copper-containing quercetin 2,3-dioxygenase from Aspergillus japonicus. Structure 10:259–268. 10.1016/S0969-2126(02)00704-9 [DOI] [PubMed] [Google Scholar]
- 43.Beloqui A, Pita M, Polaina J, Martínez-Arias A, Golyshina OV, Zumárraga M, Yakimov MM, García-Arellano H, Alcalde M, Fernández VM, Elborough K, Andreu JM, Ballesteros A, Plou FJ, Timmis KN, Ferrer M, Golyshin PN. 2006. Novel polyphenol oxidase mined from a metagenome expression library of bovine rumen: biochemical properties, structural analysis, and phylogenetic relationships. J. Biol. Chem. 281:22933–22942. 10.1074/jbc.M600577200 [DOI] [PubMed] [Google Scholar]
- 44.González-Guerrero M, Eren E, Rawat S, Stemmler TL, Argüello JM. 2008. Structure of the two transmembrane Cu+ transport sites of the Cu+-ATPases. J. Biol. Chem. 283:29753–29759. 10.1074/jbc.M803248200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karlin S, Zhu ZY, Karlin KD. 1997. The extended environment of mononuclear metal centers in protein structures. Proc. Natl. Acad. Sci. U. S. A. 94:14225–14230. 10.1073/pnas.94.26.14225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sakurai T, Kataoka K. 2007. Structure and function of type I copper in multicopper oxidases. Cell. Mol. Life Sci. 64:2642–2656. 10.1007/s00018-007-7183-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Solomon EI, Sundaram UM, Machonkin TE. 1996. Multicopper oxidases and oxygenases. Chem. Rev. 96:2563–2606. 10.1021/cr950046o [DOI] [PubMed] [Google Scholar]
- 48.MacPherson IS, Murphy ME. 2007. Type-2 copper-containing enzymes. Cell. Mol. Life Sci. 64:2887–2899. 10.1007/s00018-007-7310-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.