

Abstract
Methicillin-resistant Staphylococcus aureus (MRSA) infections pose a major challenge in health care, yet the limited heterogeneity within this group hinders molecular investigations of related outbreaks. Pulsed-field gel electrophoresis (PFGE) has been the gold standard approach but is impractical for many clinical laboratories and is often replaced with PCR-based methods. Regardless, both approaches can prove problematic for identifying subclonal outbreaks. Here, we explore the use of whole-genome sequencing for clinical laboratory investigations of MRSA molecular epidemiology. We examine the relationships of 44 MRSA isolates collected over a period of 3 years by using whole-genome sequencing and two PCR-based methods, multilocus variable-number tandem-repeat analysis (MLVA) and spa typing. We find that MLVA offers higher resolution than spa typing, as it resolved 17 versus 12 discrete isolate groups, respectively. In contrast, whole-genome sequencing reproducibly cataloged genomic variants (131,424 different single nucleotide polymorphisms and indels across the strain collection) that uniquely identified each MRSA clone, recapitulating those groups but enabling higher-resolution phylogenetic inferences of the epidemiological relationships. Importantly, whole-genome sequencing detected a significant number of variants, thereby distinguishing between groups that were considered identical by both spa typing (minimum, 1,124 polymorphisms) and MLVA (minimum, 193 polymorphisms); this suggests that these more conventional approaches can lead to false-positive identification of outbreaks due to inappropriate grouping of genetically distinct strains. An analysis of the distribution of variants across the MRSA genome reveals 47 mutational hot spots (comprising ∼2.5% of the genome) that account for 23.5% of the observed polymorphisms, and the use of this selected data set successfully recapitulates most epidemiological relationships in this pathogen group.
INTRODUCTION
The control of methicillin-resistant Staphylococcus aureus (MRSA) infections in hospitals has become a global challenge (1). The worldwide incidence of MRSA outbreaks has been rising steadily since the first strain of this group was reported in 1961 (1, 2). MRSA has also emerged in the general community (community-acquired MRSA [CA-MRSA]) and is recognized as a source of infection in hospitalized patients (3). Because MRSA can be transmitted rapidly as a nosocomial pathogen, particularly in intensive care units, it is important for patient morbidity and mortality to quickly and accurately identify the spread of any singular strain so that effective steps can be taken to curb burgeoning outbreaks. However, molecular epidemiological investigations of MRSA have been complicated by the emergence and rapid dissemination of a single dominant MRSA clone, USA300 (sequence type 8 [ST8]-MRSA-IV), which is now the major community-acquired strain in the United States (4). This clonal dominance has led to challenges in differentiating true outbreak strains from genomically similar strains that have been independently acquired from external sources. The inherently high degree of relatedness among members of MRSA USA300 may consequently render molecular typing results uninterpretable in the event that multiple independent USA300 subclones are recovered during an investigation of a potential outbreak (5). The appearance of distinct and dominant MRSA clones in other nations has generated similar problems within other health care systems (6).
Many molecular methods are available to type MRSA strains, including pulsed-field gel electrophoresis (PFGE) (7), PCR assays for rapidly evolving repeat elements (8), randomly amplified polymorphic DNA (RAPD) analysis (9), multilocus sequence typing (MLST) (10), typing of the polymorphic spa gene (11–13), ribotyping (14), mecA:Tn554 probe typing (15), staphylococcal cassette chromosome mec element (SCCmec) typing (16), multilocus variable-number tandem-repeat analysis (MLVA) (17–19), and DNA macroarray (20). These approaches have individual strengths and weaknesses. Historically, PFGE has been considered a gold standard, although recent works have called this status into question (21–23). Regardless, the approach is labor-intensive, time-consuming, and technically challenging to implement for most multipurpose clinical laboratories (24); consequently, this sometimes leads to typing artifacts (21). Alternatively, methods utilizing PCR to interrogate hypervariable loci, including spa typing and MLVA, can be more easily executed than and have similar performance characteristics and resolving power as those of PFGE (13, 17, 18, 24), and they have been used as convenient alternatives to that approach. Even so, none of these approaches may be sufficient to resolve subclonal relationships in the background of a highly prevalent clone, as the amount of divergence detectable in such cases may be inadequate to resolve strains (6).
In recent years, next-generation DNA sequencing technologies have become increasingly available to clinical microbiology laboratories (25, 26). The use of these platforms allows the complete genome sequences of bacterial isolates to be determined within a matter of days, enabling discrimination among genomes at the theoretical resolution of a single nucleotide difference and consequently permitting the resolution of subclonal strain relationships. Such technologies have offered an unprecedented ability to differentiate MRSA strains isolated during outbreak investigations (27–31), although they have not yet been integrated into routine laboratory practice. Here, we investigate the use of conventional molecular typing and next-generation sequencing methods for clinical laboratory investigations of MRSA epidemiology, with a consideration of the subclonal strain relationships. We compare the performance of the whole-genome data against two well-established molecular typing strategies, spa typing and MLVA, using a panel of 44 MRSA isolates that were collected across the state of Washington and sent to our laboratory for typing.
MATERIALS AND METHODS
Strains and molecular typing.
The strains used, institution of origin codes, and dates of acquisition are indicated in Table 1. All MRSA isolates were received and maintained on blood agar. DNA was extracted from the clinical isolates using the UltraClean microbial DNA isolation kit (Mo Bio). spa typing and MLVA were performed on all isolates, as described elsewhere (32), except that the products were analyzed by QIAxcel (Qiagen) capillary electrophoresis using the DNA screening kit (33).
TABLE 1.
MRSA strains used in this study
| Strain no. | Institution code | Date isolated (mo/day/yr) |
|---|---|---|
| 1 | A | 4/23/2009 |
| 2 | A | 6/15/2009 |
| 3 | A | 6/18/2009 |
| 4 | B | 9/1/2009 |
| 5 | B | 9/2/2009 |
| 6 | B | 9/12/2009 |
| 7 | B | 10/2/2009 |
| 8 | B | 10/16/2009 |
| 9 | C | 12/30/2009 |
| 10 | C | 12/30/2009 |
| 11 | C | 12/30/2009 |
| 12 | C | 12/30/2009 |
| 13 | C | 12/30/2009 |
| 14 | A | 2/22/2010 |
| 15 | A | 3/8/2010 |
| 16 | A | 3/22/2010 |
| 17 | A | 3/22/2010 |
| 18 | D | 4/12/2010 |
| 19 | E | 4/22/2010 |
| 20 | D | 5/31/2010 |
| 21 | D | 8/6/2010 |
| 22 | D | 8/12/2010 |
| 23 | D | 8/19/2010 |
| 24 | F | 9/30/2010 |
| 25 | B | 10/24/2010 |
| 26 | B | 10/26/2010 |
| 27 | G | 12/11/2010 |
| 28 | G | 2/27/2011 |
| 29 | G | 2/27/2011 |
| 30 | G | 3/1/2011 |
| 31 | G | 3/1/2011 |
| 32 | E | 3/4/2011 |
| 33 | H | 3/8/2011 |
| 34 | G | 3/9/2011 |
| 35 | E | 3/10/2011 |
| 36 | E | 3/19/2011 |
| 37 | E | 4/21/2011 |
| 38 | E | 4/21/2011 |
| 39 | E | 4/21/2011 |
| 40 | E | 4/21/2011 |
| 41 | I | 4/25/2011 |
| 42 | E | 4/25/2011 |
| 43 | E | 4/26/2011 |
| Reference | NAa | 2006 |
NA, not applicable.
Whole-genome sequencing.
Oligonucleotides were synthesized by integrated DNA technology (IDT). One hundred nanograms each DNA was digested for 1 h at 37°C in a 10-μl volume using 0.3 μl NEBNext double-stranded DNA (dsDNA) Fragmentase (New England BioLabs). DNA was simultaneously end repaired and A tailed in a 40-μl reaction mixture containing 1× Rapid ligation buffer (Enzymatics), 0.1675 mM each deoxynucleoside triphosphate (dNTP) (New England BioLabs), 0.1 μl Escherichia coli DNA polymerase I (New England BioLabs), 0.5 μl T4 polynucleotide kinase (PNK) (New England BioLabs), and 0.02 μl Taq DNA polymerase (New England BioLabs); this was incubated at 37°C for 30 min and 72°C for 20 min. Annealed Y-adaptors (5′-PO4-GATCGGAAGAGCGGTTCAGCAGGAATGCCGAG-3′ and 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′) were added at a concentration of 0.2 μM and ligated at 25°C for 20 min using T4 DNA ligase in Rapid ligation buffer (Enzymatics). After purification with AMPure beads (Agencourt), the library was PCR amplified with Kapa HiFi HotStart ReadyMix using the primer PRECAP_FWD_AMP_COMMON (5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGC-3′) and sample-specific bar-coded primers (5′-CAAGCAGAAGACGGCATACGAGATNNNNNNNNCGGTCTCGGCATTCCTGCTGAACCG-3′, where N indicates the position of an 8-bp sample-specific index). The cycling conditions were 95°C for 3 min, 10 cycles of 98°C for 20 s, 65°C for 15 s, and 72°C for 1 min, followed by one cycle of 72°C for 5 min. The PCR product was purified with AMPure beads (Agencourt), pooled, and sequenced on a MiSeq (Illumina) using 250-bp paired-end reads, with a custom Index primer mix (5′AGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCG-3′), to an average genomic read depth of ∼62× per sample (range, 15.5 to 300× coverage), as estimated by Cortex version 1.0.5.17 (34).
Data analysis.
The adaptors were trimmed and PCR duplicates were removed using the program fastq-mcf, with skew filtering disabled and other parameters at their defaults. De novo sequence assembly and variant calling were performed using Cortex version 1.0.5.17 (34), which uses colored de Bruijn graphs to detect both simple and complex genetic variants, employing the independent workflow and the whole-genome sequence of S. aureus USA300 strain FPR3757 (35) (GenBank accession no. CP000255.1) as a reference. Rare sites covered by <5 reads were masked as unknown data using custom scripts. Variants were annotated using SnpEff version 3.3 (36). A neighbor-joining phylogenetic tree of whole-genome sequence variants was generated using SplitsTree4 (37), and bootstrap support values were calculated using 1,000 pseudoreplicates.
Mutational hot spots were identified by cataloging the number of unique variants (SNPs and indels) called within a 500-bp sliding window. The windows were considered hot spots if they exceeded a threshold of 150 variants.
Accession number.
Sequence reads for this project are available from the Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra), under study accession number SRP042340.
RESULTS
MRSA strain typing by spa typing and MLVA.
The strain collection considered in this study included 44 MRSA isolates that were previously sent to our laboratory for the purpose of strain typing from different facilities across Washington state (Table 1). The collection dates for this collection spanned a 2-year period, and the isolates included members of the now-dominant USA300 group (as typed by our laboratory using MLVA and spa typing), as well as alternative strains. We also included a typed reference strain from the USA300 group, MRSA FPR3757, whose whole-genome sequence was generated by conventional methods (35). For the ease of data interpretation, we coded these strains numerically by their dates of isolation.
We initially characterized MRSA isolates using MLVA and spa typing techniques (Fig. 1A). spa typing identified 12 different MRSA groups, whereas MLVA demonstrated a higher degree of resolving power and partitioned a total of 17 distinct groups. In general, there was concordance among the groups identified by these independent methods. One population identified by MLVA (group 15) was split into two subgroups by spa typing analysis (groups 10 and 11). Four groups classified by spa typing (groups 1, 5, 9, and 12) were divided into at least two subgroups each by MLVA typing (groups 1 and 2, groups 6, 7, and 8, groups 12, 13, and 14, and groups 16 and 17, respectively). spa typing clustered all members from the USA300 group into a single category, whereas MLVA partitioned the USA300 strains into 2 separate, albeit closely related, groups. One of the USA300 strain groups identified by MLVA typing (group 17) comprised five isolates (strains 9 to 13) obtained from different patients at the same institution and on the same date, which therefore is consistent with an outbreak.
FIG 1.

Whole-genome phylogenetic tree of 44 MRSA strains. (A) Relationships among strains as inferred by whole-genome sequencing (phylogeny), MLVA (electrophoresis patterns), and spa typing (repeat codes). The MLVA and spa typing groups are numbered on the right. USA300 strains are indicated by a square bracket. (B) Whole-genome phylogeny for USA300 strains only. The scale bars for phylogenies in panels A and B are expressed in the number of changes per site; note the difference in scale between the two panels.
MRSA strain typing by whole-genome sequencing.
We next performed whole-genome sequencing of each MRSA strain and reconstructed the relationships among those isolates using phylogenetic analysis (Fig. 1A). A total of 131,424 unique single nucleotide variants and indels were identified across the genomes of the 44 MRSA isolates in our panel, which were shared to different degrees among the various isolates. In contrast to the other molecular methods examined, whole-genome sequencing enabled each strain to be uniquely distinguished from all other strains: even the two most genetically similar isolates (strains 9 and 12) were discernible from one another by two variants. The bootstrap support values for the phylogenetic reconstruction were high (>70%) for the great majority of the branches (data not shown), providing confidence in the reliability of the inferred relationships.
Because the polymorphisms distinguishing the isolates were effectively quantified by whole-genome analysis, the relationships among the strains were represented more continuously than by spa typing and MLVA, which are capable only of assigning isolates to distinct groups. This difference was most pronounced in the USA300 group (Fig. 1B), where the genomic phylogeny was able to resolve the relationships among individual isolates having identical spa or MLVA types (Fig. 1A). The five isolates constituting a possible outbreak formed a distinct clade (Fig. 1B), supported by high bootstrap support values (100%; data not shown), and among these isolates, polymorphisms were identified that uniquely marked each strain. Even so, the phylogenetic clades of the MRSA strains inferred by whole-genome sequencing recapitulate the partitions identified through MLVA gel electrophoresis patterns and spa types, suggesting a general concordance in the relative genetic distance measured by spa typing, MLVA, and whole-genome sequence analysis.
Reproducibility of whole-genome sequencing.
In order to ascertain whether a whole-genome sequencing approach reproducibly identifies the genetic variants in the strains typed, we sequenced 5 MRSA isolates selected from throughout the inferred phylogeny (strains 41, 35, 26, 22, and 7) in duplicate. Paired libraries from these strains were prepared independently and can therefore be considered true technical replicates. Whole-genome analysis of the corresponding libraries generated from these strains identified no discordant variants in the genotypes of the technical replicates. These findings suggest that the data obtained from whole-genome sequencing by this approach are highly reproducible and that the number of variants that can be attributed to technical errors is negligible. We attribute this high degree of concordance among the technical replicates to robust library preparation techniques, robust methods of informatics analysis, and the high fidelity of Illumina sequencing (38).
Resolution of whole-genome sequencing for molecular epidemiology.
We compared the resolution of strain typing achievable by whole-genome analysis to that enabled by MLVA and spa typing by quantifying the average number of genomic variants that differentiated the groups identified by each method (as defined in Fig. 1A), as well as the average degree of genomic divergence among members of the same group.
The minimum average number of polymorphisms identified by whole-genome sequencing that separated any two spa types was 1,241 (Table 2, group 10 compared to group 11), with an average pairwise distance between the spa groups of 40,676 variants. In comparison, MLVA offered higher resolution, as we found it was able to distinguish two groups of USA300 strains separated by an average of only 193 variants (MLVA groups 16 and 17), although the overall average pairwise distance between discrete groups was comparable to that of the spa typing results at 39,569 variants (Table 3).
TABLE 2.
Intergroup and intragroup genomic distances by spa typing
| spa typing group | No. of Isolates | Intragroup genomic variants |
Avg intergroup genomic variants against spa typing group: |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Avg | SD | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | ||
| 1 | 2 | 1,592.00 | NAa | 3,022 | 40,439 | 56,279 | 64,747 | 60,574 | 58,949 | 58,285 | 58,637 | 59,413 | 57,756 | 61,169 | |
| 2 | 1 | NA | NA | 39,617 | 55,084 | 63,802 | 58,714 | 57,991 | 56,999 | 57,286 | 57,697 | 56,390 | 60,076 | ||
| 3 | 1 | NA | NA | 58,844 | 65,521 | 63,143 | 63,469 | 62,341 | 63,760 | 65,923 | 64,057 | 70,951 | |||
| 4 | 1 | NA | NA | 28,555 | 23,242 | 24,858 | 28,102 | 28,837 | 27,160 | 26,533 | 28,579 | ||||
| 5 | 5 | 3,426.33 | 2,107.37 | 29,372 | 28,944 | 35,426 | 34,799 | 32,172 | 30,993 | 38,447 | |||||
| 6 | 7 | 228.10 | 119.84 | 20,995 | 28,417 | 26,397 | 27,940 | 27,772 | 28,321 | ||||||
| 7 | 1 | NA | NA | 23,022 | 23,176 | 25,003 | 24,787 | 23,644 | |||||||
| 8 | 1 | NA | NA | 3,944 | 31,553 | 28,641 | 28,663 | ||||||||
| 9 | 7 | 3,525.81 | 1,750.80 | 28,679 | 27,854 | 28,376 | |||||||||
| 10 | 1 | NA | NA | 1,241 | 30,435 | ||||||||||
| 11 | 1 | NA | NA | 28,833 | |||||||||||
| 12 | 16 | 179.22 | 97.21 | ||||||||||||
NA, not applicable.
TABLE 3.
Intergroup and intragroup genomic distances by MLVA
| MLVA group | No. of Isolates | Intragroup genomic variants |
Avg no. of intergroup genomic variants against MLVA group: |
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Avg no. | SD | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | ||
| 1 | 1 | NAa | NA | 1,592 | 2,836 | 41,359 | 56,324 | 64,351 | 65,665 | 65,603 | 60,590 | 58,911 | 59,510 | 58,738 | 60,105 | 60,901 | 58,601 | 61,374 | 61,385 | |
| 2 | 1 | NA | NA | 3,207 | 39,518 | 56,234 | 64,113 | 65,436 | 65,371 | 60,558 | 58,987 | 57,060 | 56,292 | 57,667 | 58,451 | 58,568 | 60,953 | 60,965 | ||
| 3 | 1 | NA | NA | 39,617 | 55,084 | 63,724 | 63,891 | 63,870 | 58,714 | 57,991 | 56,999 | 56,399 | 58,739 | 57,392 | 57,044 | 60,087 | 60,062 | |||
| 4 | 1 | NA | NA | 58,844 | 64,239 | 66,832 | 66,735 | 63,143 | 63,469 | 62,341 | 62,844 | 62,463 | 64,860 | 64,990 | 70,934 | 70,987 | ||||
| 5 | 1 | NA | NA | 28,630 | 28,392 | 28,491 | 23,242 | 24,858 | 28,102 | 29,248 | 28,784 | 28,444 | 26,847 | 28,552 | 28,609 | |||||
| 6 | 3 | 589.00 | 123.04 | 4,883 | 4,826 | 31,303 | 29,012 | 33,302 | 33,549 | 34,808 | 36,534 | 32,999 | 38,635 | 38,711 | ||||||
| 7 | 1 | NA | NA | 678 | 27,405 | 28,861 | 37,509 | 36,317 | 35,884 | 32,363 | 30,174 | 38,149 | 38,239 | |||||||
| 8 | 1 | NA | NA | 27,477 | 28,890 | 37,592 | 36,351 | 35,918 | 32,454 | 30,159 | 38,223 | 38,292 | ||||||||
| 9 | 7 | 228.10 | 119.84 | 20,995 | 28,417 | 27,996 | 27,158 | 24,544 | 27,856 | 28,296 | 28,350 | |||||||||
| 10 | 1 | NA | NA | 23,022 | 23,322 | 23,192 | 23,024 | 24,895 | 23,605 | 23,688 | ||||||||||
| 11 | 1 | NA | NA | 2,545 | 3,055 | 5,640 | 30,097 | 28,659 | 28,668 | |||||||||||
| 12 | 3 | 1,258.00 | 497.11 | 3,218 | 5,148 | 29,874 | 28,464 | 28,469 | ||||||||||||
| 13 | 1 | NA | NA | 4,008 | 29,247 | 29,095 | 29,174 | |||||||||||||
| 14 | 3 | 973.33 | 392.01 | 26,868 | 28,067 | 28,058 | ||||||||||||||
| 15 | 2 | 1,241.00 | 0.00 | 29,601 | 29,671 | |||||||||||||||
| 16 | 11 | 206.00 | 96.16 | 193 | ||||||||||||||||
| 17 | 5 | 77.07 | 60.35 | |||||||||||||||||
NA, not applicable.
Whole-genome sequencing cataloged significant sequence heterogeneity within groups of MRSA isolates that were considered genetically identical by both spa typing and MLVA. Five of the 12 spa type groups comprised multiple isolates and could therefore be used to assess the degree of intragroup genomic heterogeneity. For the least genomically diverse spa type group (group 2), an average of 228 variants distinguished the isolates from one another, whereas this value increased to 3,526 variants in the most heterogeneous spa type group (Table 2, group 9). Mirroring the values of intergroup genomic distance, the groups identified by MLVA were on average less genomically heterogeneous than were the spa groups. For the 7 MLVA groups composed of more than one isolate, an average of 77 and 1,258 genomic variants distinguished among the populations of isolates comprising the least and most heterogeneous groups, respectively (Table 3).
We conclude that the MLVA and spa typing methods are comparatively insensitive to the underlying genomic polymorphisms that are identifiable by whole-genome sequencing.
Identification of mutational hot spots in the MRSA genome.
We next explored the allocation of mutations across the MRSA genome. We plotted the empirical cumulative distribution of each of the 131,424 unique polymorphisms detected in our panel of 44 MRSA isolates against their position within the MRSA genome (Fig. 2). If mutations were distributed uniformly across the genome, we expected that this plot would present a uniform diagonal line (39). Qualitatively, however, the distribution of variants deviated from uniformity at several intervals, indicating nonrandom clustering of mutations within several discrete genomic regions.
FIG 2.

Cumulative distribution of mutations across the MRSA genome. The total number of unique mutations detected in the panel of 44 MRSA isolates (y axis) is shown as a function of genomic position (x axis; numbering is for the S. aureus USA300 FPR3757 reference genome). The slope of the plot is proportional to the density of mutations at the corresponding genomic coordinates. Genomic windows of ≥500 bp containing ≥150 unique variants each are shaded in black.
To quantitatively define genomic mutational hot spots, we cataloged genomic windows of ≥500 bp in length that met or exceeded a threshold of 150 unique variants each. This cutoff identified a total of 47 separate genomic regions (Fig. 2 and Table 4) comprising 74,089 bp of the MRSA genome (2.54%) and 30,937 of the total variants detected across the full MRSA panel (23.5%). The majority of the variants in the mutational hot spots (29,774 variants [96.2%]) occurred in noncoding DNA (Table 5). However, of the remaining 1,163 mutations occurring in annotated genes, 1,017 (95.2%) were nonsynonymous. Several of the genes displaying high variability were located at recognized mutational hot spots that are currently used for S. aureus strain typing, specifically, agrB/agrC/agrD (40), coa (41), clfA (42), and fnbA/fnbB (43). Although 10 unique variants were detected in the spa gene (designated SAUSA300_0113 in MRSA FPR3757; 1,526 bp), it did not qualify for our designation as a mutational hot spot.
TABLE 4.
Genomic positions of mutational hot spots in the MRSA genome
| Start positiona | Stop position | Interval size (nt)b |
|---|---|---|
| 104664 | 108444 | 3,781 |
| 181345 | 185747 | 4,403 |
| 266489 | 267675 | 1,187 |
| 419896 | 421394 | 1,499 |
| 441398 | 444174 | 2,777 |
| 446530 | 447027 | 498 |
| 460363 | 463105 | 2,743 |
| 614228 | 616216 | 1,989 |
| 619660 | 620686 | 1,027 |
| 636752 | 637513 | 762 |
| 854788 | 856593 | 1,806 |
| 860116 | 860646 | 531 |
| 861196 | 863444 | 2,249 |
| 865148 | 865923 | 776 |
| 979757 | 985650 | 5,894 |
| 1151084 | 1151643 | 560 |
| 1212311 | 1212966 | 656 |
| 1328959 | 1329816 | 858 |
| 1331148 | 1331982 | 835 |
| 1399433 | 1400358 | 926 |
| 1482194 | 1482797 | 604 |
| 1533265 | 1534046 | 782 |
| 1883823 | 1884358 | 536 |
| 1906914 | 1907476 | 563 |
| 1944433 | 1945417 | 985 |
| 1959882 | 1960757 | 876 |
| 2090250 | 2091436 | 1,187 |
| 2092435 | 2092936 | 502 |
| 2096341 | 2098407 | 2,067 |
| 2121667 | 2125251 | 3,585 |
| 2125394 | 2126660 | 1,267 |
| 2132348 | 2134784 | 2,437 |
| 2146812 | 2148632 | 1,821 |
| 2265906 | 2267144 | 1,239 |
| 2267251 | 2268150 | 900 |
| 2346947 | 2352540 | 5,594 |
| 2600834 | 2602158 | 1,325 |
| 2614491 | 2618103 | 3,613 |
| 2630570 | 2631344 | 775 |
| 2633993 | 2635583 | 1,591 |
| 2643288 | 2644159 | 872 |
| 2644216 | 2644858 | 643 |
| 2647275 | 2649354 | 2,080 |
| 2684647 | 2685362 | 716 |
| 2760796 | 2761504 | 709 |
| 2818261 | 2818780 | 520 |
| 2854183 | 2854725 | 543 |
Numbering is for the S. aureus USA300 FPR3757 reference genome.
nt, nucleotides.
TABLE 5.
Hypermutable genes in MRSA strainsa
| Geneb | Total no. of mutations | No. of synonymous mutations |
|---|---|---|
| SAUSA300_0097 | 74 | |
| cap5H | 7 | |
| cap5I | 14 | |
| cap5J | 21 | |
| cap5K | 21 | |
| coa | 16 | |
| SAUSA300_0369 | 19 | |
| SAUSA300_0390 | 26 | |
| SAUSA300_0391 | 40 | |
| SAUSA300_0395 | 151 | 46 |
| SAUSA300_0407 | 32 | |
| SAUSA300_0408 | 31 | |
| sdrD | 18 | |
| sdrE | 2 | |
| SAUSA300_0767 | 10 | |
| SAUSA300_0768 | 3 | |
| clfA | 13 | |
| SAUSA300_0773 | 33 | |
| SAUSA300_0775 | 4 | |
| oppA | 23 | |
| oppF | 7 | |
| oppD | 4 | |
| oppB | 22 | |
| oppC | 8 | |
| SAUSA300_1053 | 4 | |
| SAUSA300_1106 | 4 | |
| SAUSA300_1207 | 4 | |
| SAUSA300_1208 | 4 | |
| SAUSA300_1212 | 1 | |
| SAUSA300_1327 | 177 | 101 |
| SAUSA300_1366 | 2 | |
| SAUSA300_1702 | 4 | |
| SAUSA300_1759 | 9 | |
| SAUSA300_1925 | 15 | |
| SAUSA300_1926 | 1 | |
| SAUSA300_1928 | 4 | |
| SAUSA300_1929 | 3 | |
| SAUSA300_1930 | 17 | |
| SAUSA300_1966 | 3 | |
| SAUSA300_1967 | 6 | |
| SAUSA300_1968 | 11 | |
| SAUSA300_1969 | 12 | |
| SAUSA300_1970 | 23 | |
| SAUSA300_1971 | 45 | |
| SAUSA300_1977 | 17 | |
| SAUSA300_1978 | 16 | |
| agrB | 4 | |
| agrD | 2 | |
| agrC | 4 | |
| SAUSA300_2101 | 30 | |
| SAUSA300_2167 | 17 | |
| SAUSA300_2168 | 8 | |
| SAUSA300_2169 | 14 | |
| rpsI | 2 | |
| SAUSA300_2415 | 4 | |
| SAUSA300_2431 | 23 | |
| SAUSA300_2432 | 1 | |
| fnbB | 5 | |
| fnbA | 23 | |
| SAUSA300_2451 | 34 | |
| SAUSA300_2452 | 12 |
Bold type indicates genes also differentiating USA300 strains.
Ordered by genomic position.
We also cataloged the unique variants that distinguished the USA300 strains from one another (Table 6). As with the variants identified in the mutational hot spots, the majority of these alterations (1,293 out of 1,331 [97.1%]) occurred in noncoding regions of the genome. Thirty-four of the 38 (89%) mutations occurring in the coding genes were nonsynonymous. Four genes identified in this analysis (SAUSA300_0407, clfA, SAUSA300_1327, and SAUSA300_1968) were recovered as highly mutated genes in the full MRSA data set (Table 5).
TABLE 6.
Gene mutations distinguishing MRSA USA300 strainsa
| Geneb | Total no. of mutations | No. of synonymous mutations |
|---|---|---|
| SAUSA300_0240 | 1 | |
| SAUSA300_0407 | 2 | |
| gltB | 1 | |
| clfA | 2 | |
| purE | 2 | 1 |
| SAUSA300_1327 | 20 | 3 |
| SAUSA300_1333 | 1 | |
| ebpS | 1 | |
| SAUSA300_1478 | 2 | |
| SAUSA300_1968 | 1 | |
| sdrH | 1 | |
| SAUSA300_2237 | 1 | |
| clfB | 1 | |
| SAUSA300_2581 | 1 | |
| SAUSA300_2589 | 1 |
Bold type indicates genes also identified as a mutational hot spot.
Ordered by genomic position.
Resolution of mutational hot spots for molecular epidemiology.
In order to ascertain the practical amount of information carried within the mutational hot spots, we lastly performed a phylogenetic reconstruction of the strain relationships using only the genomic variants identified within the boundaries of mutational hot spots (Fig. 3). Compared to the reconstruction based on whole-genome sequence data, most major divisions describing the relatedness of the isolates were recovered using the selected genomic hot spot data: only the topological order of two major branches, comprising 3 strains (strain 3, and strains 14 and 21), required whole-genome sequence data for correct placement within the phylogeny. Similarly, the finer branching order of only 5 strains within their respective clades (strains 31, 37, 43, 42, and the reference genome), required complete genome data for proper resolution. Importantly, the possible subclonal outbreak of USA300 isolates (strains 9 to 13) was resolved as a distinct group within the larger USA300 clade.
FIG 3.
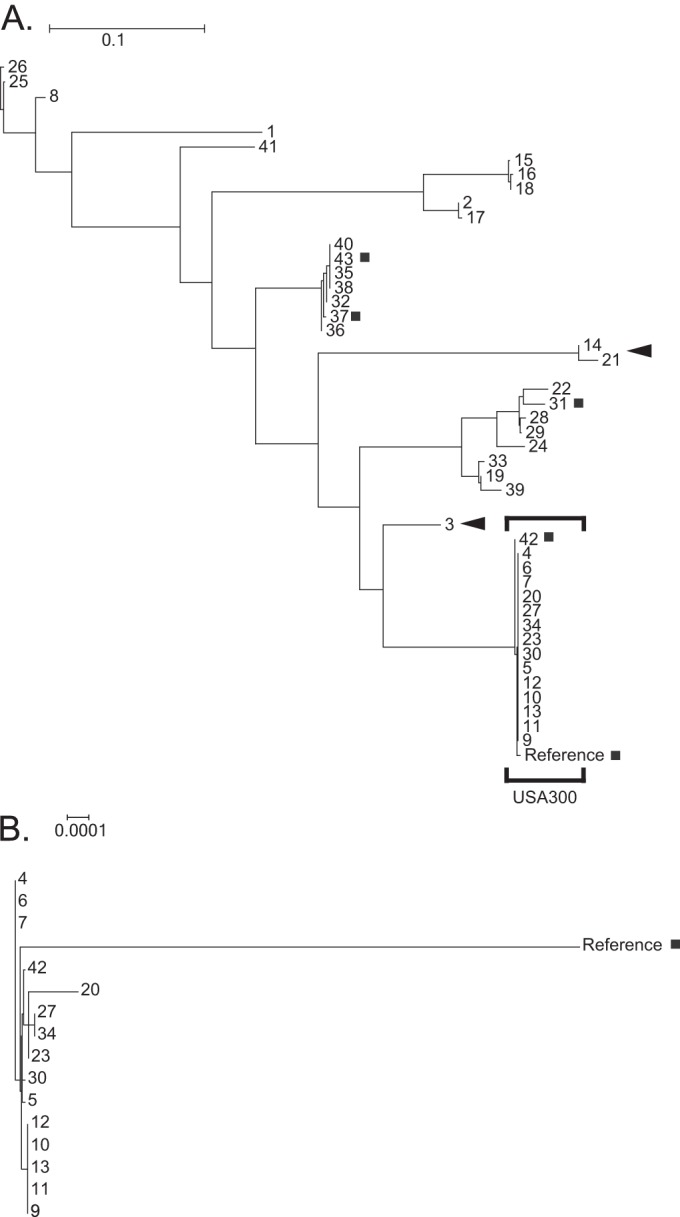
Phylogenetic tree based on mutational hot spots. Shown are the relationships among all 44 strains (A) and among USA300 strains only (B). Clades misplaced with respect to the genomic phylogeny are indicated by black arrowheads. Strains misplaced within their clades, compared to the genomic phylogeny, are marked with a gray square. The scale bars for the phylogenies in panels A and B are expressed in the number of changes per site; note the difference in scale between the two panels.
It was not possible to uniquely identify all strains using selected mutational hot spot information. The 5 possible outbreak strains, isolates 4, 6, and 7, and two pairs of isolates (27 and 34, and 35 and 38) required whole-genome sequence data in order to be distinguished from one another. Correspondingly, the bootstrap support values for this phylogenetic reconstruction were in general somewhat lower than those observed for the genomic tree (data not shown). However, high-confidence support values were obtained for the majority of the relationships (58 of 85 branches). We conclude that a selective analysis of these 47 mutational hot spot regions can be useful for initial laboratory screening of strains for their relatedness.
DISCUSSION
In addition to important functions in the prevention of nosocomial infection and the reconstruction of disease outbreaks (28), the ability to accurately and effectively assay the molecular signatures of MRSA strains has important implications for hospital resource allocation and reimbursement. Significant direct health care costs and efforts may be incurred by the care and treatment of nosocomial MRSA infections, while current guidelines from the Centers for Medicare and Medicaid Services (CMS) (44) prevent reimbursement for hospital-acquired conditions. Aside from the obvious implications for patient care, failures to properly resolve preadmission MRSA colonization from nosocomially acquired strains may additionally incur financial losses, both for the patients and the hospitals. For these reasons, it is necessary to identify outbreaks with both high sensitivity and high specificity.
PFGE was used as a reference method for strain discrimination in the pregenomic era. We did not perform PFGE in this study, as the level of technical skill and time required to implement this testing is impractical, or even infeasible, for many clinical laboratories that perform other diagnostic functions. At the same time, few reference laboratories currently offer PFGE services to outside clients. Aside from these practical considerations, mounting evidence suggests that PFGE results are not infallible (21). When experimental PFGE data have been compared to predicted banding patterns inferred from whole-genome sequence analysis, PFGE recovered only a fraction of the expected bands, and the results were found to correlate poorly with the relatedness of the strains (22). Further, the results obtained with different restriction enzymes often yield discordant interpretations, and genetic diversity in closely related outbreak strains may be insufficient to permit the resolution of their relationships (22). In some contexts, alternative molecular methods, such as limited sequence analysis by MLST, provide higher discriminatory power than does PFGE (23), while in other instances, the equivalency of PFGE with various molecular methods has been demonstrated (17, 18). MRSA whole-genome sequencing has been reported (27) to outperform PFGE in a research context.
Here, we explore the use of whole-genome sequencing, MLVA, and spa typing (which require similar protocols and levels of molecular biology bench skills to implement as those for whole-genome sequencing) as laboratory approaches for reconstructing the molecular epidemiology of MRSA strains, with special attention to their ability to discriminate subclonal strain outbreaks. MLVA of MRSA isolates demonstrated higher discriminatory power than did spa typing, as evidenced by its identification of 17 groups compared to the 12 groups resolved by spa typing (Fig. 1A). In contrast, each MRSA isolate was uniquely identified by whole-genome sequencing, and higher-resolution relationships among MRSA strains were ascertained (Fig. 1A and B). The quantitation of the underlying genomic variants that distinguish between populations identified as genetically identical by either spa typing or MLVA revealed that substantial numbers of genomic variants may be present among isolates within different groups, and surprisingly, among members of the same typed group (Tables 2 and 3). These values help define the number of absolute genomic differences that are reflected by the group partitions as defined by spa and MLVA typing. We conclude that MLVA and spa typing are relatively insensitive to underlying genomic polymorphisms and may therefore classify strains together when they are actually only indirectly related. As a consequence of this insufficiently high level of genetic resolution, false identification of outbreaks may occur, with the risk of unnecessary follow-up investigations and clinical responsive actions. As a case in point, MLVA was able in this study to resolve a potential USA300 outbreak group from other USA300 strains, whereas spa typing was not.
Whole-genome sequencing can more effectively facilitate the identification of true outbreak strains by allowing the genomic variants that differentiate isolates to be unambiguously and quantitatively cataloged. In this study, every MRSA isolate was found to have a unique genomic signature, allowing the hierarchy of relationships among strains to be inferred with a high resolution (Fig. 1A). The suspected outbreak strains were confidently separated from other USA300 isolates (Fig. 1B) and were distinguishable from one another. Two strains in the suspected MRSA outbreak differing by only 2 variants were successfully resolved, which is close to the theoretical detection limit of a single change. Given the high degree of reproducibility we observed for detected mutations, even small numbers of variants can be considered with confidence. Given the period of time occurring between the collection of any pairwise combination of strains and the number of genomic differences that distinguish them, it should be possible to calculate the probability that two strains derive from a recent common ancestry and then establish clonality. In long-term population-based studies, MRSA genomes have been estimated to accumulate approximately 1 mutation per 38-day period (27); however, scant data exist on the degree of genomic identity needed to define isolates as part of an outbreak, and there currently is no defined threshold for the number of genomic variants that define an isolate as unique. Dedicated studies of MRSA mutation frequencies occurring during large numbers of short-term transmission events will be required before such rules can be established. Although the dynamics of person-to-person MRSA transmission may be more complicated than was previously believed, including issues relating to a single individual being colonized by multiple genetically distinct strains (31, 45), the degree of resolution offered by whole-genome sequencing may be sufficiently high to permit an investigation of even short-term epidemiological events. This type of information offers obvious benefits for identifying strains that cause singular or repeated outbreaks within an institution, as well as for identifying the underlying origins. The identification and evaluation of mobile genetic elements, including transposons and plasmids, although not considered in this study, might further improve the resolution of whole-genome-based analysis.
We have found that not all portions of the MRSA genome are as epidemiologically informative as others, with mutations accumulating more frequently in particular regions (Fig. 2 and Table 4). The majority of the hypervariable regions are noncoding, suggesting that, by and large, these hot spots represent portions of the genome that are not under evolutionary constraints. Intriguingly, we found that annotated coding sequences falling within mutational hot spots contained almost entirely nonsynonymous variants (Table 5), suggesting that these sequences are under active positive selection. Several of the genes identified by this analysis are currently used in MRSA strain typing: the regulatory locus agr, for example, controls the balance of virulence factor expression in the colonization and invasion phases of infection by modulating adhesin- and toxin-producing genes (46), suggesting that it is a plausible target of evolutionary pressure. Regardless, the majority of the hypermutable sites are not explicitly targeted by existing assays and do not have obvious explanations for being under positive selection. There was limited overlap among the highly mutated genes identified through population-level analysis and the mutations differentiating the USA300 strains (Table 6), most likely due to insufficient divergence times among the USA300 strains for mutations to accumulate and become manifest. Regardless, these regions may prove informative in more targeted assays, in which targeting limited numbers of hypermutable markers for molecular epidemiological analysis is desirable. The dedicated study of these rapidly evolving genes may shed light on the pathways important to MRSA pathogenesis and virulence.
Surprisingly, the exclusive analysis of mutational hot spots was informative for the purpose of molecular epidemiology reconstruction (Fig. 3) and properly identified the subclonal outbreak present in our strain collection. Although this more limited approach did not provide the same level of resolution as the whole-genome data did, it was still possible to uniquely distinguish all but 10 strains from one another, offering significantly higher discriminatory power than that of MLVA. Thus, although there are clear advantages to the use of whole-genome sequencing data when performing high-resolution epidemiological analysis, a targeted sequencing strategy may be useful for approximating comprehensive genomic results, and it may offer practical benefits in terms of assay costs and turnaround times.
In summary, conventional strain typing, regardless of the method employed, relies on limited information parsed from a fraction of the genome, making it difficult to define precise epidemiological relationships (27, 47), especially those relationships occurring over short time periods. Much higher resolution is afforded by a whole-genome sequencing approach, enabling investigations of both historical relationships and short-term transmission events, as well as preventing artifactual calling of outbreaks due to insufficient genetic heterogeneity. Whole-genome sequencing using increasingly inexpensive benchtop genomic sequencers offers relatively simple workflow and clinically compatible turnaround times, providing whole-genome sequence data 2 to 3 days after isolating pure bacterial colonies. The major barrier to the implementation of a clinical whole-genome sequencing approach is bioinformatics analysis (26), as many programs used to analyze whole-genome sequencing data require computational infrastructure and knowledge of command-line interfaces. These requirements are prohibitive for many clinical laboratories; nevertheless, the quality and breadth of the data offered by whole-genome sequencing are sufficiently greater than those offered by conventional molecular methods and justify the additional efforts needed to implement these technologies.
ACKNOWLEDGMENTS
This work was supported in part by the National Center For Advancing Translational Sciences of the National Institutes of Health (grant UL1TR000423) and by developmental research project grant 5U54AI057141-08REV from the National Institute of Allergy and Infectious Diseases (NIAID)/Northwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (NWRCE).
Footnotes
Published ahead of print 21 May 2014
REFERENCES
- 1.Ayliffe GA. 1997. The progressive intercontinental spread of methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 24(Suppl 1):S74–S79 [DOI] [PubMed] [Google Scholar]
- 2.Parker MT, Jevons MP. 1964. A survey of methicillin resistance in Staphylococcus aureus. Postgrad. Med. J. 40(Suppl):170–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Brien FG, Pearman JW, Gracey M, Riley TV, Grubb WB. 1999. Community strain of methicillin-resistant Staphylococcus aureus involved in a hospital outbreak. J. Clin. Microbiol. 37:2858–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strommenger B, Bartels MD, Kurt K, Layer F, Rohde SM, Boye K, Westh H, Witte W, De Lencastre H, Nübel U. 2013. Evolution of methicillin-resistant Staphylococcus aureus towards increasing resistance. J. Antimicrob. Chemother. 69:616–622. 10.1093/jac/dkt413 [DOI] [PubMed] [Google Scholar]
- 5.Tenover FC, Goering RV. 2009. Methicillin-resistant Staphylococcus aureus strain USA300: origin and epidemiology. J. Antimicrob. Chemother. 64:441–446. 10.1093/jac/dkp241 [DOI] [PubMed] [Google Scholar]
- 6.Engelthaler DM, Kelley E, Driebe EM, Bowers J, Eberhard CF, Trujillo J, Decruyenaere F, Schupp JM, Mossong J, Keim P, Even J. 2013. Rapid and robust phylotyping of spa t003, a dominant MRSA clone in Luxembourg and other European countries. BMC Infect. Dis. 13:339. 10.1186/1471-2334-13-339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH, Swaminathan B. 1995. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J. Clin. Microbiol. 33:2233–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deplano A, Schuermans A, Van Eldere J, Witte W, Meugnier H, Etienne J, Grundmann H, Jonas D, Noordhoek GT, Dijkstra J, van Belkum A, van Leeuwen W, Tassios PT, Legakis NJ, van der Zee A, Bergmans A, Blanc DS, Tenover FC, Cookson BC, O'Neil G, Struelens MJ. 2000. Multicenter evaluation of epidemiological typing of methicillin-resistant Staphylococcus aureus strains by repetitive-element PCR analysis. The European Study Group on Epidemiological Markers of the ESCMID. J. Clin. Microbiol. 38:3527–3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saulnier P, Bourneix C, Prévost G, Andremont A. 1993. Random amplified polymorphic DNA assay is less discriminant than pulsed-field gel electrophoresis for typing strains of methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 31:982–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG. 2000. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 38:1008–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frénay HM, Bunschoten AE, Schouls LM, van Leeuwen WJ, Vandenbroucke-Grauls CM, Verhoef J, Mooi FR. 1996. Molecular typing of methicillin-resistant Staphylococcus aureus on the basis of protein A gene polymorphism. Eur. J. Clin. Microbiol. Infect. Dis. 15:60–64. 10.1007/BF01586186 [DOI] [PubMed] [Google Scholar]
- 12.Shopsin B, Gomez M, Montgomery SO, Smith DH, Waddington M, Dodge DE, Bost DA, Riehman M, Naidich S, Kreiswirth BN. 1999. Evaluation of protein A gene polymorphic region DNA sequencing for typing of Staphylococcus aureus strains. J. Clin. Microbiol. 37:3556–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harmsen D, Claus H, Witte W, Rothgänger J, Claus H, Turnwald D, Vogel U. 2003. Typing of methicillin-resistant Staphylococcus aureus in a university hospital setting by using novel software for spa repeat determination and database management. J. Clin. Microbiol. 41:5442–5448. 10.1128/JCM.41.12.5442-5448.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richardson JF, Aparicio P, Marples RR, Cookson BD. 1994. Ribotyping of Staphylococcus aureus: an assessment using well-defined strains. Epidemiol. Infect. 112:93–101. 10.1017/S0950268800057459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dominguez MA, de Lencastre H, Linares J, Tomasz A. 1994. Spread and maintenance of a dominant methicillin-resistant Staphylococcus aureus (MRSA) clone during an outbreak of MRSA disease in a Spanish hospital. J. Clin. Microbiol. 32:2081–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pérez-Roth E, Lorenzo-Díaz F, Batista N, Moreno A, Méndez-Alvarez S. 2004. Tracking methicillin-resistant Staphylococcus aureus clones during a 5-year period (1998 to 2002) in a Spanish hospital. J. Clin. Microbiol. 42:4649–4656. 10.1128/JCM.42.10.4649-4656.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabat A, Krzyszton-Russjan J, Strzalka W, Filipek R, Kosowska K, Hryniewicz W, Travis J, Potempa J. 2003. New method for typing Staphylococcus aureus strains: multiple-locus variable-number tandem repeat analysis of polymorphism and genetic relationships of clinical isolates. J. Clin. Microbiol. 41:1801–1804. 10.1128/JCM.41.4.1801-1804.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malachowa N, Sabat A, Gniadkowski M, Krzyszton-Russjan J, Empel J, Miedzobrodzki J, Kosowska-Shick K, Appelbaum PC, Hryniewicz W. 2005. Comparison of multiple-locus variable-number tandem-repeat analysis with pulsed-field gel electrophoresis, spa typing, and multilocus sequence typing for clonal characterization of Staphylococcus aureus isolates. J. Clin. Microbiol. 43:3095–3100. 10.1128/JCM.43.7.3095-3100.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Francois P, Huyghe A, Charbonnier Y, Bento M, Herzig S, Topolski I, Fleury B, Lew D, Vaudaux P, Harbarth S, van Leeuwen W, van Belkum A, Blanc DS, Pittet D, Schrenzel J. 2005. Use of an automated multiple-locus, variable-number tandem repeat-based method for rapid and high-throughput genotyping of Staphylococcus aureus isolates. J. Clin. Microbiol. 43:3346–3355. 10.1128/JCM.43.7.3346-3355.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trad S, Allignet J, Frangeul L, Davi M, Vergassola M, Couve E, Morvan A, Kechrid A, Buchrieser C, Glaser P, El-Solh N. 2004. DNA macroarray for identification and typing of Staphylococcus aureus isolates. J. Clin. Microbiol. 42:2054–2064. 10.1128/JCM.42.5.2054-2064.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung M, de Lencastre H, Matthews P, Tomasz A, Adamsson I, Aires de Sousa M, Camou T, Cocuzza C, Corso A, Couto I, Dominguez A, Gniadkowski M, Goering R, Gomes A, Kikuchi K, Marchese A, Mato R, Melter O, Oliveira D, Palacio R, Sá-Leão R, Santos Sanches I, Song JH, Tassios PT, Villari P, Multilaboratory Project Collaborators 2000. Molecular typing of methicillin-resistant Staphylococcus aureus by pulsed-field gel electrophoresis: comparison of results obtained in a multilaboratory effort using identical protocols and MRSA strains. Microb. Drug Resist. 6:189–198. 10.1089/mdr.2000.6.189 [DOI] [PubMed] [Google Scholar]
- 22.Davis MA, Hancock DD, Besser TE, Call DR. 2003. Evaluation of pulsed-field gel electrophoresis as a tool for determining the degree of genetic relatedness between strains of Escherichia coli O157:H7. J. Clin. Microbiol. 41:1843–1849. 10.1128/JCM.41.5.1843-1849.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nemoy LL, Kotetishvili M, Tigno J, Keefer-Norris A, Harris AD, Perencevich EN, Johnson JA, Torpey D, Sulakvelidze A, Morris JG, Jr, Stine OC. 2005. Multilocus sequence typing versus pulsed-field gel electrophoresis for characterization of extended-spectrum beta-lactamase-producing Escherichia coli isolates. J. Clin. Microbiol. 43:1776–1781. 10.1128/JCM.43.4.1776-1781.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strandén A, Frei R, Widmer AF. 2003. Molecular typing of methicillin-resistant Staphylococcus aureus: can PCR replace pulsed-field gel electrophoresis? J. Clin. Microbiol. 41:3181–3186. 10.1128/JCM.41.7.3181-3186.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Didelot X, Bowden R, Wilson DJ, Peto TE, Crook DW. 2012. Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 13:601–612. 10.1038/nrg3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fricke WF, Rasko DA. 2013. Bacterial genome sequencing in the clinic: bioinformatic challenges and solutions. Nat. Rev. Genet. 15:49–55. 10.1038/nrg3624 [DOI] [PubMed] [Google Scholar]
- 27.Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, Gardete S, Tavares A, Day N, Lindsay JA, Edgeworth JD, de Lencastre H, Parkhill J, Peacock SJ, Bentley SD. 2010. Evolution of MRSA during hospital transmission and intercontinental spread. Science 327:469–474. 10.1126/science.1182395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris SR, Cartwright EJ, Török ME, Holden MTG, Brown NM, Ogilvy-Stuart AL, Ellington MJ, Quail MA, Bentley SD, Parkhill J, Peacock SJ. 2013. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect. Dis. 13:130–136. 10.1016/S1473-3099(12)70268-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Köser CU, Holden MT, Ellington MJ, Cartwright EJ, Brown NM, Ogilvy-Stuart AL, Hsu LY, Chewapreecha C, Croucher NJ, Harris SR, Sanders M, Enright MC, Dougan G, Bentley SD, Parkhill J, Fraser LJ, Betley JR, Schulz-Trieglaff OB, Smith GP, Peacock SJ. 2012. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N. Engl. J. Med. 366:2267–2275. 10.1056/NEJMoa1109910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nübel U, Nachtnebel M, Falkenhorst G, Benzler J, Hecht J, Kube M, Bröcker F, Moelling K, Bührer C, Gastmeier P, Piening B, Behnke M, Dehnert M, Layer F, Witte W, Eckmanns T. 2013. MRSA transmission on a neonatal intensive care unit: epidemiological and genome-based phylogenetic analyses. PLoS One 8:e54898. 10.1371/journal.pone.0054898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindsay JA. 2013. Evolution of Staphylococcus aureus and MRSA during outbreaks. Infect. Genet. Evol. 21:548–553. 10.1016/j.meegid.2013.04.017 [DOI] [PubMed] [Google Scholar]
- 32.Woodward JF, Sengupta DJ, Cookson BT, Park JO, Dellinger EP. 2010. Disseminated community-acquired USA300 methicillin-resistant Staphylococcus aureus pyomyositis and septic pulmonary emboli in an immunocompetent adult. Surg. Infect. (Larchmt.) 11:59–63. 10.1089/sur.2009.015 [DOI] [PubMed] [Google Scholar]
- 33.Chia JH, Lai HC, Su LH, Kuo AJ, Wu TL. 2013. Molecular epidemiology of Clostridium difficile at a medical center in Taiwan: persistence of genetically clustering of A−B+ isolates and increase of A+B+ isolates. PLoS One 8:e75471. 10.1371/journal.pone.0075471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iqbal Z, Caccamo M, Turner I, Flicek P, McVean G. 2012. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat. Genet. 44:226–232. 10.1038/ng.1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367:731–739. 10.1016/S0140-6736(06)68231-7 [DOI] [PubMed] [Google Scholar]
- 36.Cingolani P, Platts A, Wang le L, Coon LM, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23:254–267. 10.1093/molbev/msj030 [DOI] [PubMed] [Google Scholar]
- 38.Lam HY, Clark MJ, Chen R, Chen R, Natsoulis G, O'Huallachain M, Dewey FE, Habegger L, Ashley EA, Gerstein MB, Butte AJ, Ji HP, Snyder M. 2012. Performance comparison of whole-genome sequencing platforms. Nat. Biotechnol. 30:78–82. 10.1038/nbt.2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seringhaus M, Kumar A, Hartigan J, Snyder M, Gerstein M. 2006. Genomic analysis of insertion behavior and target specificity of mini-Tn7 and Tn3 transposons in Saccharomyces cerevisiae. Nucleic Acids Res. 34:e57. 10.1093/nar/gkl184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geisinger E, Chen J, Novick RP. 2012. Allele-dependent differences in quorum-sensing dynamics result in variant expression of virulence genes in Staphylococcus aureus. J. Bacteriol. 194:2854–2864. 10.1128/JB.06685-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaida S, Miyata T, Yoshizawa Y, Kawabata S, Morita T, Igarashi H, Iwanaga S. 1987. Nucleotide sequence of the staphylocoagulase gene: its unique COOH-terminal 8 tandem repeats. J. Biochem. 102:1177–1186 [DOI] [PubMed] [Google Scholar]
- 42.Gomes AR, Vinga S, Zavolan M, de Lencastre H. 2005. Analysis of the genetic variability of virulence-related loci in epidemic clones of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 49:366–379. 10.1128/AAC.49.1.366-379.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rice K, Huesca M, Vaz D, McGavin MJ. 2001. Variance in fibronectin binding and fnb locus polymorphisms in Staphylococcus aureus: identification of antigenic variation in a fibronectin binding protein adhesin of the epidemic CMRSA-1 strain of methicillin-resistant S. aureus. Infect. Immun. 69:3791–3799. 10.1128/IAI.69.6.3791-3799.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.GPO. 2006. Deficit Reduction Act (DRA) of 2005. Public law 109-171–Feb. 8, 2006 U.S. Government Printing Office, Washington, DC: http://www.gpo.gov/fdsys/pkg/PLAW-109publ171/pdf/PLAW-109publ171.pdf [Google Scholar]
- 45.Price JR, Golubchik T, Cole K, Wilson DJ, Crook DW, Thwaites GE, Bowden R, Walker AS, Peto TE, Paul J, Llewelyn MJ. 2014. Whole-genome sequencing shows that patient-to-patient transmission rarely accounts for acquisition of Staphylococcus aureus in an intensive care unit. Clin. Infect. Dis. 58:609–618. 10.1093/cid/cit807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Papakyriacou H, Vaz D, Simor A, Louie M, McGavin MJ. 2000. Molecular analysis of the accessory gene regulator (agr) locus and balance of virulence factor expression in epidemic methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 181:990–1000. 10.1086/315342 [DOI] [PubMed] [Google Scholar]
- 47.Köser CU, Ellington MJ, Cartwright EJ, Gillespie SH, Brown NM, Farrington M, Holden MT, Dougan G, Bentley SD, Parkhill J, Peacock SJ. 2012. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. 8:e1002824. 10.1371/journal.ppat.1002824 [DOI] [PMC free article] [PubMed] [Google Scholar]