

Abstract
Mycobacterium avium subsp. paratuberculosis is the etiological agent of paratuberculosis, a granulomatous enteritis affecting a wide range of domestic and wild ruminants worldwide. A variety of molecular typing tools are used to distinguish M. avium subsp. paratuberculosis strains, contributing to a better understanding of M. avium subsp. paratuberculosis epidemiology. In the present study, PCR-based typing methods, including mycobacterial interspersed repetitive units/variable-number tandem repeats (MIRU-VNTR) and small sequence repeats (SSR) in addition to IS1311 PCR-restriction enzyme analysis (PCR-REA), were used to investigate the genetic heterogeneity of 200 M. avium subsp. paratuberculosis strains from dairy herds located in the province of Quebec, Canada. The majority of strains were of the “cattle type,” or type II, although 3 strains were of the “bison type.” A total of 38 genotypes, including a novel one, were identified using a combination of 17 genetic markers, which generated a Simpson's index of genetic diversity of 0.876. Additional analyses revealed no differences in genetic diversity between environmental and individual strains. Of note, a spatial and spatiotemporal cluster was evidenced regarding the distribution of one of the most common genotypes. The population had an overall homogeneous genetic structure, although a few strains stemmed out of the consensus cluster, including the bison-type strains. The genetic structure of M. avium subsp. paratuberculosis populations within most herds suggested intraherd dissemination and microevolution, although evidence of interherd contamination was also revealed. The level of genetic diversity obtained by combining MIRU-VNTR and SSR markers shows a promising avenue for molecular epidemiology investigations of M. avium subsp. paratuberculosis transmission patterns.
INTRODUCTION
Mycobacterium avium subsp. paratuberculosis is the etiological agent of Johne's disease, or paratuberculosis, a chronic granulomatous enteritis of domestic and wild ruminants, first described in 1895 (1). Paratuberculosis is a spectral disease characterized by an extended subclinical phase of several months or years, followed by an inevitable terminal clinical stage (2). Bovine paratuberculosis is endemic to most parts of the world, including Canada (3–5), and is considered one of the most costly infectious diseases of dairy cattle (6, 7). In Canada, paratuberculosis has also been detected in sheep (8), goats (9), and wild ruminant species (10–12). M. avium subsp. paratuberculosis is most likely introduced in ruminant herds through trading of subclinically infected animals, although wildlife reservoirs are also thought to play a role in spreading the bacteria to livestock (13). M. avium subsp. paratuberculosis is widespread in farm environments due to its excretion in high quantities in the feces of infected animals and to its environmental resilience. In addition, while M. avium subsp. paratuberculosis has been associated with human Crohn's disease, its role in causality remains to be demonstrated (14, 15). Nonetheless, M. avium subsp. paratuberculosis remains a public health concern.
Typing methods can be applied in molecular epidemiology studies to improve the understanding of transmission patterns of M. avium subsp. paratuberculosis and to investigate the role of wildlife reservoirs by tracking strains (16, 17). The knowledge thereby generated may improve the design of efficient control measures. Based on historical reasons, on the patterns obtained by pulsed-field gel electrophoresis (PFGE) analyses, and to a lesser extent, on growth characteristics and pigmentation, M. avium subsp. paratuberculosis strains have been divided into three major groups: the S (sheep) type (further segregated into subtypes I and III), the C (cattle) type (type II), and the B (bison) type (18, 19). However, the association of M. avium subsp. paratuberculosis types with either cattle, sheep, or bison is not absolute since strains from all lineages can cause diseases in these ruminants (19). IS900 restriction fragment length polymorphism (RFLP) has been traditionally used to investigate the genetic diversity and epidemiology of M. avium subsp. paratuberculosis strains. However, RFLP is technically demanding and generates limited discriminatory power. Typing techniques such as multilocus short sequence repeat typing (MLSSR) and variable-number tandem repeats of mycobacterial interspersed repetitive units (MIRU-VNTR) have gained interest mainly because of their ease of use and good discriminatory powers (17, 20). However, M. avium subsp. paratuberculosis is a highly monomorphic species, and using a combination of multilocus typing methods is necessary to increase their overall discriminatory power, as previously shown for the M. tuberculosis complex (21, 22) and M. avium subsp. paratuberculosis strains from different host species and different geographic locations (17, 23–26). A typing scheme based on 8 MIRU-VNTR loci (called INMV typing) has been previously proposed for the M. avium complex (26).
The goals of this study were (i) to investigate the genetic structure of bovine M. avium subsp. paratuberculosis populations originating from a total of 83 dairy herds, (ii) to describe the genetic diversity obtained by individual animal sampling versus environmental sampling, and (iii) to investigate the distribution of M. avium subsp. paratuberculosis genotypes in space and time. IS1311 PCR-restriction enzyme analysis (PCR-REA) was used as a primary typing tool in combination with 17 distinct genomic loci (MLSSR and MIRU-VNTR) to study the genetic diversity of M. avium subsp. paratuberculosis.
MATERIALS AND METHODS
Source of strains and primary isolation.
The present investigation was based on a collection of strains from 83 dairy herds located across the province of Quebec, Canada. These herds were sampled between one and five times between September 2007 and June 2011 in the context of different research projects, most of them including both environmental and individual sampling. There were no strict inclusion criteria applicable to all herds. Individual fecal samples from cows 3 years and older (maximum, 30 cows per herd), a single intestinal tissue sample, and pooled environmental manure samples were submitted to the Laboratoire d'épidémiosurveillance animale du Québec. Primary isolation of M. avium subsp. paratuberculosis strains was performed using 2 g of feces or pooled manure using the Bactec MGIT 960 mycobacterial detection system from Becton, Dickinson and Company. The protocol suggested by the manufacturer was strictly followed.
M. avium subsp. paratuberculosis subculture for molecular typing.
A total of 500 μl of growth medium from positive MGIT vials (231 samples in total) was diluted 1:5 in sterile phosphate-buffered saline (PBS). Aliquots of 100 μl of each dilution were then inoculated on 5 replicate slants of modified MiddleBrook 7H10 medium with mycobactin (27) and Herrold's egg yolk medium with mycobactin (28). Slants were incubated at 37°C and observed every 2 weeks for the appearance of colonies for up to 20 weeks.
Primary typing of M. avium subsp. paratuberculosis colonies (IS1311 PCR_REA).
Primary typing was performed using IS1311 PCR followed by PCR-REA directly from individual M. avium subsp. paratuberculosis colonies growing on slants. IS1311 PCRs were performed as described by Sevilla et al. (18) and Singh et al. (29) with slight modifications to adapt for colony PCR. Briefly, a single visible colony was harvested with a sterile toothpick and diluted in 10 μl of PCR grade water and heated at 95°C for 10 min. The mixture was then transferred to 40 μl of a PCR mixture consisting of 5 μl of 10× PCR buffer, 200 μM deoxynucleoside triphosphates (dNTPs), 2.5 mM MgCl2, 0.6 μM primers M-56 and M-119, and 2 U of Taq polymerase. Amplification was performed under the following conditions: one cycle of 3 min at 94°C and 37 cycles of 30 s at 94°C, 15 s at 62°C, and 1 min at 72°C. Amplification reactions were analyzed on a Qiaxcel capillary electrophoresis instrument (Qiagen), which has greater resolution (3 to 5 bp) over that of traditional gel electrophoresis. The amplicon size was 608 bp. REA reactions were performed as described previously (18, 29). Briefly, restriction reactions were carried out in a volume of 30 μl, containing 20 μl of positive IS1311 PCR, 3 μl of reaction 10× buffer, and 2 U of endonucleases HinfI and MseI. Reaction mixtures were incubated at 37°C for 2.5 h. Restriction reactions were analyzed using a Qiaxcel capillary electrophoresis instrument. Band patterns were interpreted following the recommendations of Sevilla et al. (18).
Genomic DNA extraction.
M. avium subsp. paratuberculosis colonies were harvested from slants and stored in mycobacterial storage medium (20% glycerol solution containing 3.0% [wt/vol] tryptone soya broth). A total of 200 μl of the storage medium containing M. avium subsp. paratuberculosis cells was used to isolate genomic DNA using the QIAamp DNA minikit according to the recommended protocol, with a slight modification that included an initial enzymatic lysis step using 200 μl of 20 mg/ml lysozyme, and incubated for 3 h at 37°C before proceeding with the manufacturer's protocol. DNA was stored at −20°C until needed.
Molecular typing.
MIRU-VNTR typing was carried out by PCR amplification on a total of 13 distinct loci: VNTR X3 (also known as MIRU-3), VNTR 3, VNTR 7, VNTR 10, VNTR 25, VNTR 32, VNTR 47, VNTR 292 (also known as MIRU-2), VNTR 259 (23), VNTR 1067 and VNTR 3527 (30), and MIRU-1 and MIRU-4 (31). For each marker, PCR conditions as described by the authors were strictly followed. Following PCR amplification, the reaction mixtures were analyzed on a Qiaxcel capillary electrophoresis instrument to determine the sizes of the amplicons at each locus. The number of repeats at each locus was determined according to the sizes of the amplicons using an allele-calling table as described by the authors of the above-cited works (23, 30, 31). The INRA (Institut National de Recherche Agronomique, France) MIRU-VNTR (INMV) nomenclature was used to analyze the results for MIRU-VNTR loci 292, X3, 25, 47, 3, 7, 10, and 32 as previously defined (26). For the remaining loci (MIRU 1, MIRU 4, VNTR 259, VNTR 1067, and VNTR 3527), a new nomenclature (MV typing) is proposed.
Multilocus short sequence repeat typing analysis was carried out by PCR amplification and sequencing of 4 short sequence repeat loci 1, 2, 8, and 9 (32). The primers and PCR conditions used were as described previously (32). All PCR amplicons were sequenced using BigDye v3.1 chemistry from Applied Biosystems on a 3730xl sequencer (Applied Biosystems). MLSSR types were expressed as the combination of number of repeats found in the amplified SSR loci. If the number of G repeats (stretches of guanosine residues) at loci 1 and 2 were equal to or greater than 11, G repeats for these loci were denoted as ≥11g. The allelic diversity at the different loci was calculated as previously described (33). This allelic diversity represents the probability that two alleles randomly selected from the population are different from each other.
Genetic diversity.
The genetic diversity was estimated using Simpson's index of diversity with a 95% confidence interval (CI) for the individual typing methods and their combinations (34). The following analyses were based on a combination of methods offering the largest diversity. Descriptive statistics were first used to present the number of different genotypes found among strains from all samples collected in the same herd at a given sampling point. Individual rarefaction curves with 95% confidence intervals were used to compare the genetic diversity according to the sampling method used (i.e., fecal samples from individual cases versus environmental manure samples), performed in Past software version 2.17c (35). In brief, rarefaction methods can be used to compare genetic diversity in samples of different sizes (36). Spatial, temporal, and spatiotemporal scan tests were performed to detect clusters in the distribution of the more common genotypes, i.e., those detected in at least 5 herds. Herds were geocoded at the centroid of the area covered by the 6-digit postal code of the farm, which corresponds approximately to the municipality in rural areas of Quebec. The analysis was limited to one sampling point in time per herd, with a random selection of the sampling point for herds sampled more than once. This was done to avoid the detection of a temporal cluster of strains due to potential repeated sampling of the same animals. A Bernoulli model was used with cases defined as herds with the specific complete genotypes and controls defined as all other herds. The maximal size of a cluster was set as including a maximum of 50% of herds. The statistical significance of clusters was determined using the Monte Carlo simulation with 999 permutations performed in SaTScan version 9.1.1 (37).
Genetic relationship analysis.
The genetic relationship between strains was investigated by creating minimum spanning trees (MST) using the BioNumerics software (Applied Maths, Austin, TX, USA) and a total of 17 combined VNTR and SSR markers. The MST is an undirected network that uses pairwise distances to describe the degree of dissimilarity between strains. The MST represents nodes (strains) linked together by the shortest possible distance.
RESULTS
Recovery of M. avium subsp. paratuberculosis strains in subculture.
A total of 231 samples originating from 83 herds (1 to 14 samples per herd) were cultured in the Bactec MGIT 960 system and confirmed by PCR [TaqMan MAP (Johne's) Reagents; Life Technologies]. From this total, 200 samples originating from 69 herds gave visible colonies after 6 to 20 weeks on solid medium and are referred to as strains henceforth (Table 1). Colonies were rough, whitish, and generally small, although some samples yielded larger colonies.
TABLE 1.
Distribution of M. avium subsp. paratuberculosis genotypes in 200 isolates from 69 dairy cattle herds of Quebec, Canada
| Farm ID | Yr-mo of collection | No. of isolates | Source of isolatesa | Main groupb | Genotype profile |
|||
|---|---|---|---|---|---|---|---|---|
| INMV | MV | MLSSR | Combined | |||||
| 1 | 2011-04 | 1 | FC | C | INMV 2 | MV 1 | S7 | MapGnt 10 |
| 2 | 2009-03 | 1 | EV | C | INMV 2 | MV 1 | S1 | MapGnt 5 |
| 2009-07 | 2 | FC | C | INMV 2 | MV 1 | S1 | MapGnt 5 | |
| 2009-10 | 2 | FC | C | INMV 2 | MV 1 | S1 | MapGnt 5 | |
| 1 | FC | C | INMV 3 | MV 1 | S1 | MapGnt 22 | ||
| 2010-03 | 2 | FC | C | INMV 2 | MV 1 | S1 | MapGnt 5 | |
| 2011-06 | 1 | FC | C | INMV 2 | MV 1 | S1 | MapGnt 5 | |
| 3 | 2007-09 | 1 | FC | C | —c | MV 1 | S13 | — |
| 1 | FC | C | INMV 8 | MV 3 | S6 | MapGnt 31 | ||
| 4 | 2007-10 | 1 | FC | C | INMV 3 | MV 1 | — | — |
| 2 | FC | C | INMV 3 | MV 1 | S3 | MapGnt 24 | ||
| 2007-11 | 1 | EV | C | INMV 3 | MV 1 | S3 | MapGnt 24 | |
| 5 | 2007-10 | 3 | FC | C | INMV 2 | MV 1 | S1 | MapGnt 5 |
| 6 | 2007-10 | 1 | FC | B | INMV X | MV 2 | S14 | MapGnt 35 |
| 1 | FC | C | INMV 1 | MV 1 | S11 | MapGnt 1 | ||
| 7 | 2007-11 | 1 | FC | C | INMV 2 | MV 1 | S13 | MapGnt 14 |
| 8 | 2008-01 | 1 | FC | B | INMV X | MV 2 | — | — |
| 1 | FC | B | INMV X | MV 2 | S20 | MapGnt 36 | ||
| 9 | 2008-04 | 1 | FC | C | INMV 1 | MV 1 | S12 | MapGnt 2 |
| 10 | 2008-04 | 1 | FC | C | — | MV 1 | S18 | — |
| 11 | 2008-06 | 1 | FC | C | INMV 2 | MV 1 | S1 | MapGnt 5 |
| 12 | 2008-09 | 1 | FC | C | INMV 3 | MV 1 | S6 | MapGnt 26 |
| 13 | 2009-03 | 1 | FC | C | INMV 2 | MV 1 | S6 | MapGnt 9 |
| 14 | 2009-04 | 1 | CO | C | INMV 3 | MV 1 | S15 | MapGnt 28 |
| 15 | 2009-04 | 1 | EV | C | INMV 2 | MV 1 | S2 | MapGnt 6 |
| 17 | 2009-04 | 1 | EV | C | INMV 2 | MV 1 | S1 | MapGnt 5 |
| 18 | 2009-04 | 1 | EV | C | INMV 3 | MV 1 | S5 | MapGnt 25 |
| 19 | 2009-05 | 1 | EV | C | INMV 2 | MV 1 | S21 | MapGnt 18 |
| 2010-04 | 3 | EV | C | INMV 2 | MV 1 | S21 | MapGnt 18 | |
| 1 | EV | C | INMV 2 | MV 1 | — | — | ||
| 2 | FC | C | INMV 2 | MV 1 | S21 | MapGnt 18 | ||
| 20 | 2009-05 | 2 | EV | C | INMV 2 | MV 1 | S2 | MapGnt 6 |
| 2010-04 | 3 | EV | C | INMV 2 | MV 1 | S2 | MapGnt 6 | |
| 6 | FC | C | INMV 2 | MV 1 | S1 | MapGnt 5 | ||
| 4 | FC | C | INMV 2 | MV 1 | S2 | MapGnt 6 | ||
| 1 | FC | C | INMV 2 | MV 1 | S8 | MapGnt 11 | ||
| 2010-05 | 1 | FC | C | INMV 2 | MV 1 | S2 | MapGnt 6 | |
| 23 | 2009-09 | 1 | FC | C | INMV 2 | MV 1 | S2 | MapGnt 6 |
| 24 | 2008-04 | 1 | EV | C | INMV 2 | MV 1 | S13 | MapGnt 14 |
| 1 | FC | C | INMV 2 | MV 3 | S13 | MapGnt 20 | ||
| 2009-09 | 1 | EV | C | INMV 2 | MV 1 | S18 | MapGnt 16 | |
| 1 | EV | C | INMV 3 | MV 1 | S18 | MapGnt 29 | ||
| 2010-06 | 1 | EV | C | INMV 2 | MV 1 | S13 | MapGnt 14 | |
| 2 | FC | C | INMV 2 | MV 1 | S13 | MapGnt 14 | ||
| 3 | FC | C | INMV 2 | MV 1 | S18 | MapGnt 16 | ||
| 25 | 2009-10 | 1 | EV | C | INMV 2 | MV 1 | S18 | MapGnt 16 |
| 26 | 2009-11 | 1 | EV | C | INMV 2 | MV 1 | S17 | MapGnt 15 |
| 1 | EV | C | INMV 2 | MV 1 | S21 | MapGnt 18 | ||
| 2010-03 | 2 | FC | C | INMV 2 | MV 1 | S21 | MapGnt 18 | |
| 27 | 2009-11 | 1 | FC | C | INMV 1 | MV 1 | S14 | MapGnt 3 |
| 28 | 2009-11 | 1 | FC | C | INMV 3 | MV 1 | S3 | MapGnt 24 |
| 29 | 2009-12 | 1 | EV | C | INMV 2 | MV 1 | S2 | MapGnt 6 |
| 2010-03 | 5 | FC | C | INMV 2 | MV 1 | S2 | MapGnt 6 | |
| 1 | FC | C | INMV 2 | MV 1 | S18 | MapGnt 16 | ||
| 31 | 2010-03 | 1 | EV | C | INMV 2 | MV 1 | S18 | MapGnt 16 |
| 33 | 2010-03 | 1 | FC | C | — | MV 3 | S2 | — |
| 34 | 2010-03 | 1 | FC | C | INMV 3 | MV 1 | S1 | MapGnt 22 |
| 2010-08 | 1 | FC | C | INMV 3 | MV 1 | — | — | |
| 35 | 2010-04 | 1 | FC | C | INMV 13 | MV 1 | S10 | MapGnt 33 |
| 36 | 2010-04 | 1 | FC | C | INMV 2 | MV 1 | S2 | MapGnt 6 |
| 37 | 2010-04 | 2 | EV | C | INMV 2 | MV 1 | S18 | MapGnt 16 |
| 38 | 2010-04 | 1 | EV | C | INMV 2 | MV 1 | S1 | MapGnt 5 |
| 1 | EV | C | INMV 2 | MV 1 | S2 | MapGnt 6 | ||
| 2010-07 | 1 | FC | C | INMV 2 | MV 1 | S2 | MapGnt 6 | |
| 39 | 2010-04 | 2 | EV | C | INMV 2 | MV 1 | S13 | MapGnt 14 |
| 40 | 2010-04 | 1 | EV | C | INMV 2 | MV 1 | S1 | MapGnt 5 |
| 2 | EV | C | INMV 2 | MV 1 | S2 | MapGnt 6 | ||
| 1 | EV | C | — | MV 1 | — | — | ||
| 2 | FC | C | INMV 1 | MV 1 | S14 | MapGnt 3 | ||
| 1 | FC | C | INMV 1 | MV 1 | S16 | MapGnt 4 | ||
| 7 | FC | C | INMV 2 | MV 1 | S1 | MapGnt 5 | ||
| 41 | 2010-05 | 1 | EV | C | INMV 2 | MV 1 | S21 | MapGnt 18 |
| 42 | 2010-05 | 1 | FC | C | INMV 3 | MV 1 | S2 | MapGnt 23 |
| 43 | 2010-08 | 1 | FC | C | INMV 2 | — | S18 | — |
| 44 | 2010-09 | 1 | FC | C | INMV 3 | MV 1 | S2 | MapGnt 23 |
| 47 | 2010-09 | 1 | EV | C | INMV 3 | MV 1 | S2 | MapGnt 23 |
| 1 | EV | C | INMV 3 | MV 1 | S18 | MapGnt 29 | ||
| 48 | 2010-10 | 1 | EV | C | INMV 2 | MV 1 | S9 | MapGnt 12 |
| 49 | 2010-10 | 2 | EV | C | INMV 2 | MV 1 | S17 | MapGnt 15 |
| 1 | EV | C | INMV 2 | MV 1 | — | — | ||
| 50 | 2010-10 | 2 | EV | C | INMV 2 | MV 1 | S2 | MapGnt 6 |
| 1 | EV | C | INMV 2 | MV 1 | — | — | ||
| 52 | 2010-11 | 1 | EV | C | INMV 3 | MV 1 | S1 | MapGnt 22 |
| 1 | EV | C | INMV 3 | MV 1 | — | — | ||
| 53 | 2010-11 | 1 | FC | C | INMV 2 | MV 1 | S6 | MapGnt 9 |
| 2 | FC | C | INMV 3 | MV 1 | S1 | MapGnt 22 | ||
| 2 | FC | C | INMV 3 | MV 1 | S2 | MapGnt 23 | ||
| 1 | FC | C | INMV 3 | MV 1 | — | — | ||
| 54 | 2010-11 | 3 | EV | C | INMV 3 | MV 1 | S8 | MapGnt 27 |
| 56 | 2011-03 | 1 | FC | C | INMV 2 | MV 1 | S3 | MapGnt 7 |
| 57 | 2011-03 | 1 | FC | C | INMV 3 | MV 1 | S1 | MapGnt 22 |
| 58 | 2011-04 | 1 | FC | C | INMV 2 | MV 1 | S1 | MapGnt 5 |
| 60 | 2011-04 | 1 | FC | C | INMV 2 | MV 1 | — | — |
| 62 | 2011-05 | 1 | FC | C | INMV 2 | MV 1 | S1 | MapGnt 5 |
| C | 2007-10 | 1 | FC | C | INMV 3 | MV 1 | S6 | MapGnt 26 |
| D | 2007-11 | 1 | FC | C | INMV 13 | MV 1 | S1 | MapGnt 32 |
| E | 2007-11 | 1 | EV | C | INMV 13 | MV 1 | S10 | MapGnt 33 |
| 1 | FC | C | INMV 13 | MV 1 | S10 | MapGnt 33 | ||
| G | 2007-11 | 1 | EV | C | INMV 2 | MV 1 | S1 | MapGnt 5 |
| 2 | EV | C | INMV 2 | MV 1 | S2 | MapGnt 6 | ||
| 1 | EV | C | INMV 2 | MV 1 | S4 | MapGnt 8 | ||
| 2 | FC | C | INMV 2 | MV 1 | S2 | MapGnt 6 | ||
| H | 2007-11 | 1 | EV | C | INMV 2 | MV 1 | S19 | MapGnt 17 |
| 3 | FC | C | INMV 2 | MV 1 | S19 | MapGnt 17 | ||
| I | 2007-11 | 2 | EV | C | INMV 2 | MV 1 | S2 | MapGnt 6 |
| 5 | FC | C | INMV 2 | MV 1 | S2 | MapGnt 6 | ||
| J | 2007-11 | 1 | EV | C | INMV 1 | MV 1 | S16 | MapGnt 4 |
| 1 | FC | C | INMV 2 | MV 1 | S2 | MapGnt 6 | ||
| 1 | FC | C | INMV 1 | MV 1 | S16 | MapGnt 4 | ||
| L | 2008-01 | 2 | EV | C | — | MV 1 | S1 | — |
| 2 | FC | C | — | MV 1 | S1 | — | ||
| M | 2008-01 | 4 | EV | C | INMV 2 | MV 1 | S1 | MapGnt 5 |
| 1 | FC | C | INMV 2 | MV 1 | S1 | MapGnt 5 | ||
| 1 | FC | C | INMV 2 | MV 1 | S6 | MapGnt 9 | ||
| N | 2008-02 | 1 | EV | C | INMV 2 | MV 1 | S2 | MapGnt 6 |
| 1 | EV | C | INMV 2 | MV 1 | S3 | MapGnt 7 | ||
| O | 2008-02 | 1 | EV | C | INMV 2 | MV 3 | S1 | MapGnt 19 |
| 1 | FC | C | INMV 2 | MV 3 | S1 | MapGnt 19 | ||
| P | 2008-03 | 1 | FC | C | INMV 2 | MV 1 | S13 | MapGnt 14 |
| R | 2008-04 | 1 | EV | C | INMV 2 | MV 1 | S1 | MapGnt 5 |
| 2 | EV | C | INMV 2 | MV 1 | S3 | MapGnt 7 | ||
| 1 | FC | C | INMV 2 | MV 1 | — | — | ||
| S | 2008-04 | 2 | FC | C | INMV 3 | MV 1 | S2 | MapGnt 23 |
| U | 2008-04 | 1 | FC | C | INMV 2 | MV 1 | S12 | MapGnt 13 |
| V | 2008-05 | 1 | FC | C | INMV 2 | MV 1 | S1 | MapGnt 5 |
| 7 | FC | C | INMV 2 | MV 1 | S2 | MapGnt 6 | ||
| W | 2008-05 | 1 | FC | C | INMV 2 | MV 1 | S2 | MapGnt 6 |
| X | 2008-06 | 2 | EV | C | INMV 2 | MV 1 | S13 | MapGnt 14 |
| 1 | EV | C | INMV 2 | MV 1 | S18 | MapGnt 16 | ||
| 1 | FC | C | INMV 2 | MV 1 | S1 | MapGnt 5 | ||
| 3 | FC | C | INMV 2 | MV 1 | S13 | MapGnt 14 | ||
| Total no. | 200 | 6d | 3d | 21d | 33d | |||
EV, environmental pooled sample; FC, individual fecal culture; CO, colon culture.
C, cattle; B, bison.
—, 8 isolates were not genotyped by INMV, 1 isolate was not genotyped by MV, and 11 isolates were not genotyped by MLSSR.
Number of different genotype profiles for the method.
Group typing (IS1311 PCR-REA).
Group typing of M. avium subsp. paratuberculosis strains revealed that the cattle type, or type II, was by far the most common group (Table 1). Of the 200 strains typed by IS1311 PCR-REA, 197 (98.5%) were of the cattle type and only 3 (1.5%) were of the bison type (Table 1). In herd 6, both cattle-type and bison-type strains were present. In herd 8, the bison type was the only type detected. All bison-type strains were recovered from individual samples. No sheep-type strain was identified in this population.
Typing results (MIRU-VNTR and MLSSR). (i) MIRU-VNTR.
Results from MIRU-VNTR typing are reported separately for INMV profiles (MIRU-VNTR loci 292, X3, 25, 47, 3, 7, 10, and 32) and MV profiles (MIRU-VNTR loci 1, 4, 259, 1067, and 3527) (Table 1).
A total of 6 distinct INMV profiles were observed, among which 1 had never been reported before and was therefore considered novel. The novel profile was given a tentative name (INMV X, profile 22532228). The new INMV X profile was specific to the bison-type strains. Table 1 shows the different INMV profiles found in this study: INMV 1 (n = 8), INMV 2 (n = 146), INMV 3 (n = 30), INMV 8 (n = 1), INMV 13 (n = 4), and INMV X (n = 3). INMV 2 (73% of strains and 64% of herds) and INMV 3 (15% of strains and 25% of herds) were the most frequent INMV profiles detected.
A total of three different MV profiles (MV 1, MV 2, and MV 3) were identified (Table 1). MV 1 was the most frequent profile observed (96% of strains in 94% of herds). The 3 strains with the MV 2 profile belonged to the bison-type group, whereas all strains with the MV 1 or MV 3 profile were from the cattle-type group.
(ii) MLSSR profiles.
MLSSR profiling was applicable to only 189 strains from a total of 68 herds. Amplification failure occurred for at least one of the SSR loci in the remaining 11 strains. MLSSR segregated the 189 strains into 21 distinct profiles (Table 1). Types S1 (28% of strains in 22% of herds) and S2 (29% of strains in 22% of herds) were the most frequent profiles observed (Table 1).
Allelic diversity.
The relative frequencies and diversity of the various alleles are shown in Table 2. INMV loci 3 and 32 and MV loci MIRU 1, MIRU 4, 259, and 3527 were all monomorphic. The highest allelic diversity (0.560) was observed for MLSSR locus 2.
TABLE 2.
Relative frequency of isolates according to the specific allele copy number and allelic diversity for each locus
| Locus | No. of isolates | Relative frequency (%) of isolates according to specific allele copy no.: |
Allelic diversity | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ≥11 | |||
| INMV | |||||||||||||
| 292 | 192 | 3.6 | 92.2 | 4.2 | 0.147 | ||||||||
| X3 | 192 | 100 | 0.000 | ||||||||||
| 25 | 192 | 98.4 | 1.6 | 0.031 | |||||||||
| 47 | 192 | 100 | 0.000 | ||||||||||
| 3 | 192 | 100 | 0.000 | ||||||||||
| 7 | 192 | 99.5 | 0.5 | 0.010 | |||||||||
| 10 | 192 | 15.6 | 84.4 | 0.264 | |||||||||
| 32 | 192 | 100 | 0.000 | ||||||||||
| MV | |||||||||||||
| MIRU 1 | 199 | 100 | 0.000 | ||||||||||
| MIRU 4 | 199 | 100 | 0.000 | ||||||||||
| 259 | 199 | 100 | 0.000 | ||||||||||
| 1067 | 199 | 2.5 | 96 | 1.5 | 0.078 | ||||||||
| 3527 | 199 | 100 | 0.000 | ||||||||||
| MLSSR | |||||||||||||
| 1 | 189 | 29.1 | 1.6 | 2.1 | 5.8 | 61.4 | 0.535 | ||||||
| 2 | 189 | 6.9 | 43.4 | 49.7 | 0.560 | ||||||||
| 8 | 189 | 5.8 | 87.3 | 6.9 | 0.230 | ||||||||
| 9 | 189 | 10.6 | 89.4 | 0.189 | |||||||||
Genetic diversity.
Simpson's index of diversity was calculated for a total of 181 strains having a combined MLSSR, MV, and INMV profile (Table 1). The genetic diversities revealed by MLSSR, MV, and INMV typing methods were statistically different from each other based on the comparison of confidence intervals. The MLSSR method used individually or in combination with others was associated with high indices of diversity ranging from 0.83 to 0.88. Although the inclusion of MV and/or INMV in addition to MLSSR was not associated with a large increase in genetic diversity, it allowed an important increase in the number of different genotypes recovered, from 21 to a maximum of 33 (Table 3). All further analyses were performed based on the genotypes identified by the combination of the three typing methods (MV, INMV, and MLSSR).
TABLE 3.
Simpson's index of diversity with 95% confidence intervals based on different typing methods used separately or in combinationa
| Typing method | No. of different profiles | Simpson's index (95% CI) |
|---|---|---|
| Individual typing method | ||
| MLSSR | 21 | 0.829 (0.794–0.865) |
| MV | 3 | 0.065 (0.014–0.115) |
| INMV | 6 | 0.381 (0.297–0.464) |
| Combination of two typing methods | ||
| MLSSR + MV | 25 | 0.836 (0.801–0.872) |
| MLSSR + INMV | 31 | 0.870 (0.839–0.902) |
| MV + INMV | 7 | 0.406 (0.321–0.490) |
| Combination of three typing methods | ||
| MLSS + MV + INMV | 33 | 0.876 (0.845–0.907) |
n = 181 isolates.
A total of 33 distinct clusters (genotypes) were defined for 181 strains originating from 64 herds by combining the typing results from the MV, INMV, and MLSSR loci (Table 1). A total of 19 strains could not be typed using either MV, INMV, or MLSSR and had therefore no genotype assigned. The cattle-type group of strains was divided into a total of 31 distinct genotypes. Two strains of the bison-type group were segregated into 2 distinct genotypes; one bison type strain failed to give an MLSSR profile and could not be assigned a genotype (Table 1). M. avium subsp. paratuberculosis genotype 5 (MapGnt 5) and MapGnt 6 were the most frequent genotypes observed among all strains (20% and 23% of strains, respectively) and herds (20% of herds each). These two genotypes differed only in their MLSSR profile (Table 1).
The number of strains with a complete genotype available for each sampling point (i.e., one herd sampled at one time) ranged from 1 to 14, with a maximum of 4 different genotypes found per sampling point (Table 4). Among the 181 strains with a complete genotype, 66 were isolated from pooled environmental samples, 111 were from individual fecal samples, and 1 was isolated from the colon of a suspected clinical case submitted to necropsy. According to the rarefaction curves, no difference in genetic diversity was observed between strains from environmental and individual fecal samples (Fig. 1).
TABLE 4.
Number of complete genotypes detected for each sampling point according to the number of isolates tested
| No. of isolates per sampling pointa | Frequency | No. of different genotypes |
||
|---|---|---|---|---|
| Mean | Minimum | Maximum | ||
| 1 | 43 | 1 | 1 | 1 |
| 2 | 18 | 1.4 | 1 | 2 |
| 3 | 5 | 1.6 | 1 | 2 |
| 4 | 1 | 1 | 1 | 1 |
| 5 | 2 | 2 | 1 | 3 |
| 6 | 4 | 2.3 | 2 | 3 |
| 7 | 2 | 2 | 1 | 3 |
| 8 | 1 | 2 | 2 | 2 |
| 13 | 1 | 4 | 4 | 4 |
| 14 | 1 | 3 | 3 | 3 |
A sampling point represents all isolates collected from a single farm at the same time.
FIG 1.

Rarefaction curves with 95% confidence intervals comparing the numbers of genotypes observed in 66 strains (isolates) from environmental sampling and 114 from individual case sampling.
The spatiotemporal distribution of the 6 most common genotypes was analyzed (MapGnt 5, 6, 14, 16, 22, and 23 genotypes). For MapGnt 16, statistically significant spatial (P = 0.01) and space-time (P = 0.04) clusters were observed (mapped in Fig. 2). The space-time cluster comprised 4 herds with genotype 16 sampled between June 2009 and May 2010. No other clusters in space and/or time were detected (for all, P ≥ 0.07).
FIG 2.

Geographical distribution of the 64 herds included in the study. Point size is proportional to the number of herds (1 to 4) located in the postal code area. Distribution of MapGnt 16 is illustrated in blue, as are the distributions of the two most common genotypes (MapGnt 5 and MapGnt 6, in red and green, respectively). The large blue dotted line circle represents the significant spatial cluster of MapGnt 16, and the small dotted line blue circle represent herds included in the significant spatiotemporal cluster of MapGnt 16.
To analyze the genetic relationships of the population, a minimum spanning tree that included a total of 181 strains having a combined VNTR and SSR profile (17 loci) was constructed (Fig. 3). The predominance of genotypes 5 and 6 (red and green, respectively) in the population is illustrated by the sizes of the nodes. Node sizes are proportional to the number of strains with the corresponding genotype. Both genotypes 5 and 6 appeared as consensus sequences to which most alternate genotypes in the population were related (Fig. 3). The population was generally homogeneous, with most genotypes closely related to the 2 principal nodes, differing by a maximum of 3 loci and more generally by only 1. However, a cluster of strains comprised of genotypes 1, 3, 4, 35, and 36, the latter two being from the bison-type strains, were more distantly related to the main cluster.
FIG 3.
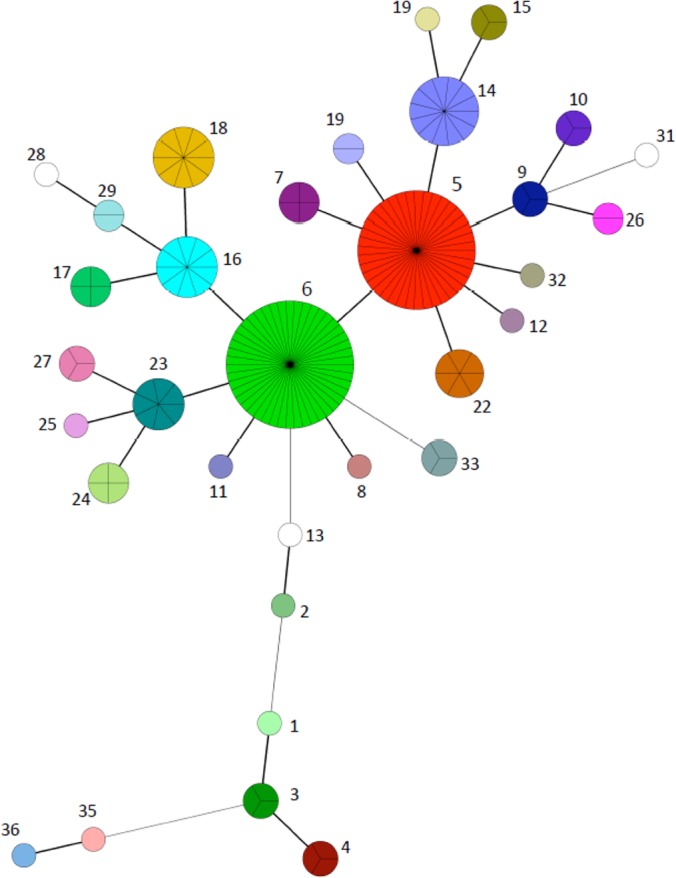
Minimum spanning tree based on combined VNTR and SSR profiles of a set of 17 loci from 181 M. avium subsp. paratuberculosis strains. Node sizes are proportional to the number of strains sharing a given genotype (cluster). Each cluster is represented as a uniquely colored and numbered pie chart, where the number of subdivisions illustrates the number of strains. The number of loci differing between the nodes is represented by the style of the connecting lines: thick and short, 1 difference; thin and intermediate length, 2 differences; thin and long, 3 differences.
A second MST was generated to investigate the intra- and interherd genetic diversity using strains from all herds from which more than one strain was recovered. Figure 4 shows the strains grouped by genotypes (nodes) and the corresponding herds from which they were recovered (represented as distinct colors subdividing the nodes and their corresponding number or letter placed outside the nodes). Most herds harbored strains with identical or closely related genotypes (e.g., herd 2 in beige and herd 20 in light green). However, other herds harbored strains with more distantly related genotypes (e.g., herd 53 in dark brown and herd 40 in red). Interestingly, herds 40 and J (red and deep purple, respectively) both harbored strains belonging to two genetically distant genotypes (4 and 6). Also of note, herd 24 (purple) harbored strains belonging to a total of 4 closely related genotypes.
FIG 4.
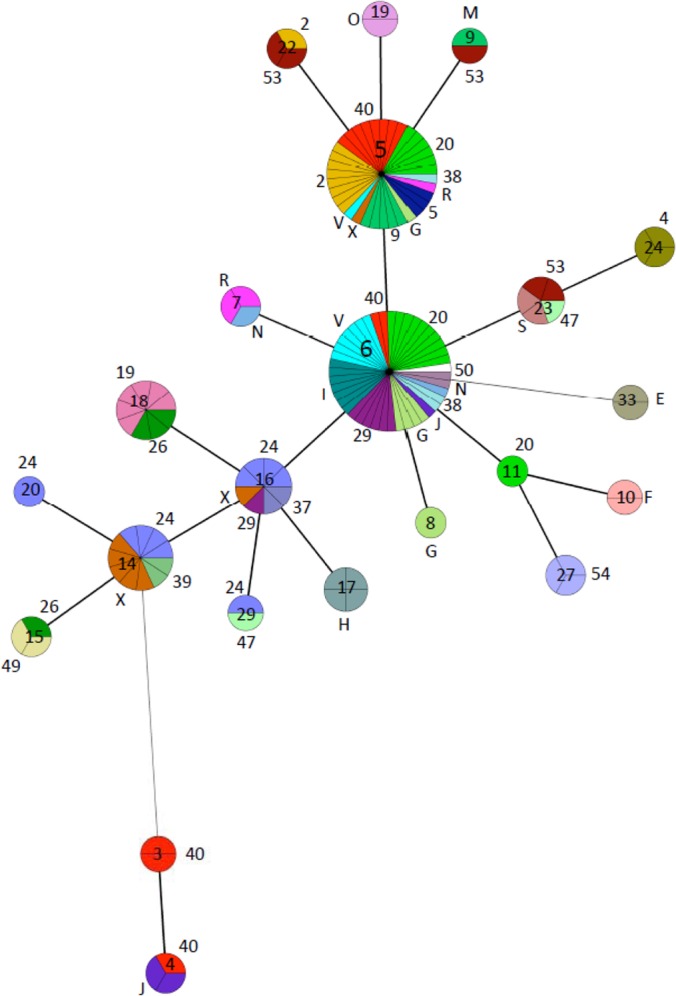
Minimum spanning tree based on combined VNTR and SSR profiles of M. avium subsp. paratuberculosis strains originating from herds with more than one strain. Node sizes are proportional to the number of strains sharing a given genotype (cluster). A distinct color and its corresponding letter or number placed outside the node represent each herd within a cluster. For each cluster, the number of subdivisions represents the number of strains. The corresponding genotype is indicated inside the node. The number of loci differing between the nodes is represented by the style of the connecting lines: thick and short, 1 difference; thin and intermediate length, 2 differences; thin and long, ≥3 differences.
DISCUSSION
This study aimed at investigating the genetic diversity and population structure of M. avium subsp. paratuberculosis strains isolated from cattle herds in a defined geographical region of Canada (Quebec). A combination of PCR-based typing methods was used to discriminate between M. avium subsp. paratuberculosis strains. In line with current knowledge, the cattle or C type was the dominant group detected among cattle strains, although three strains of the bison type originating from 2 different herds were also isolated. These two herds were located more than 200 km from each other, and insufficient epidemiological data were available to establish a link between them. To our knowledge, this is the first report of bison-type M. avium subsp. paratuberculosis strains isolated from domestic species on the American continent. Bison-type strains were first reported from wild animal species (bison) in Montana, USA (38). These strains were subsequently isolated in India and Korea from different domestic and wild host species and identified as carrying the major infecting genotype in some domestic animals (39–41). More recently, bison-type strains have been reported to infect domestic species in Africa (42). The presence of bison-type strains in domestic cattle of Quebec could suggest potential spillover events between unknown reservoirs and dairy herds. Quebec has a considerable population of wild deer, which may serve as a reservoir of infection of bison-type strains for domestic ruminants as previously suggested (13). There are also a small number of bison herds in the province, which could act as a source for these atypical strains, although an epidemiological link remains to be established. Bison-type strains have also been isolated from patients with Crohn's disease, which underscores their potential to colonize the human intestine (43). Collectively, these results underpin the need to better understand the role of these potential reservoirs not only as sources of M. avium subsp. paratuberculosis strains for domestic cattle, but also as potential sources for human infection.
The relative frequencies of the three main INMV types (INMV1, INMV2, and INMV3) are in slight contrast with other studies from Europe and South America, where INMV 1 and INMV 2 were identified as the two most prevalent genotypes in cattle and INMV 3 represented a minority of strains (26, 44–47). The present results may simply reflect regional specificities/distribution of these types. However, consistent with previous studies, INMV types 7, 8, 13, and 17 were found only in a minority of herds.
The combination of loci MIRU-VNTR 1, 4, 259, 1067, and 3527 has never been used in previous studies. Therefore, a new nomenclature termed “MV type” is proposed. The discriminatory power of MV typing was much lower than INMV or MLSSR typing. Only three distinct MV types were observed for all strains among which the MV 2 type was found to be specific to the bison-type group. Among these 5 loci, only MIRU-VNTR 1067 was found to be polymorphic, in agreement with other studies (30, 48). Considering the results of the present and previous studies, MIRU 1 and MIRU 4 may be excluded from future studies since these loci appear monomorphic for cattle strains from different areas of the world (24, 31). Although the MV typing discriminatory power was somewhat disappointing, it nevertheless subdivided the predominant INMV 2, MLSSR S1, and MLSSR S2 types in distinct clusters, underscoring its advantage in epidemiological investigations. Since INMV 2 has also been reported as the most prevalent M. avium subsp. paratuberculosis type by others (26, 44–47), MV typing using locus 1067 has the potential to further segregate M. avium subsp. paratuberculosis strains and should be considered in future studies.
In agreement with other studies, MLSSR typing was found to be the most discriminatory typing method (17, 44, 48). However, different groups have used distinct sets of SSR loci. In addition, different cutoff values were used for mononucleotide repeats: >14 by some authors (49–51) or the more conservative ≥11 as set in this work and other studies (24, 44). It is unfortunate that due to the technical limitations of sequencing long mononucleotide repeats, there is a loss of resolution power, which can potentially misclassify M. avium subsp. paratuberculosis strains. Nevertheless, using the conservative ≥11 cutoff value, a total of 21 MLSSR types were observed in this population (S1 to S21), of which S1 and S2 were the predominant types and two were novel: S14 and S16. The bison-type strains were segregated into two different MLSSR types, S14 and S20. Although SSR markers are technically more demanding, more difficult to interpret than VNTRs, and less accessible to most laboratories, MLSSR typing subdivided the predominant INMV and MV types into specific MLSSR types, making SSR markers pivotal in M. avium subsp. paratuberculosis strain typing.
Using all three typing methods in combination allowed the clustering of M. avium subsp. paratuberculosis strains into 33 distinct genotypes, including MapGnt 5 and MapGnt 6 as the most common. These results are in accordance with previous studies reporting that most strains were found to segregate into specific genotypes presumably due to increased virulence, enhanced transmissibility, or better cultivability (20, 26, 31, 49, 52). Combining the three typing methods increased the overall discriminatory power, although adding MV typing added little advantage based on Simpson's index of diversity. However, it should be noted that Simpson's index is most sensitive to variations in the abundant genotypes and less so to rare ones, which could help explain the results (53). Given its relatively low genetic heterogeneity, our results suggest that using the three methods in combination will be key to study the population structure, the evolution, and the local or global epidemiology of M. avium subsp. paratuberculosis.
Using environmental or individual fecal samples did not affect the genetic diversity of strains, suggesting that both types of samples could be used interchangeably in epidemiological investigations. Although the number of samples per herd was small, the present results also suggest that typing 5 strains per herds can be used as a proxy to investigate M. avium subsp. paratuberculosis diversity in a herd at a given point in time. However, it should be noted that a maximum of 30 cows per herd were sampled and rare genotypes could have been missed.
Most herds harbored genetically identical or closely related strains, suggestive of within-herd dispersion and microevolution. In agreement with this finding was the isolation of strains with identical genotypes from both the farm environment and animals on most premises, which suggests that M. avium subsp. paratuberculosis shedders are likely spreading the disease to herd mates via this passive route, as previously suggested by others (50). The predominance of a single or closely related genotypes within herds could be due to a selective advantage of specific strains caused by increased virulence, persistence, or transmissibility, which has been reported previously for mycobacteria, including M. avium subsp. paratuberculosis (54–57). Microevolution in patients infected with Mycobacterium tuberculosis and along transmission chains has been documented (58–60). Latent infections, such as those caused by mycobacteria, and adaptation to sequential host-to-host transmission are believed to provide the bacterial population sufficient time for microevolution (58–60). Long incubation periods and transmission chains are also a landmark of M. avium subsp. paratuberculosis infections and might explain the present observations. Interestingly, herd 24 harbored strains belonging to 4 closely related genotypes, suggesting either long-term infection providing sufficient time for microevolution or selective pressure in that herd. However, we cannot rule out the possibility of interherd dispersion of common and genetically closely related strains (e.g., genotypes 5 and 6). On the other hand, a small number of herds harbored genetically distant strains (e.g., herds 40 and 53), which suggests the introduction of multiple M. avium subsp. paratuberculosis strains.
Regarding the distribution of genotypes between herds, a spatial cluster was observed for MapGnt 16. This cluster was quite large, although most of the herds sharing this genotype within the cluster were located in a smaller area of a linear shape; this could be an artifact of the method, as the spatial scan test searched only for circular clusters. Many of the herds included in this cluster were also temporally clustered, suggesting local dissemination of this genotype within a 1-year period. Interestingly, the two most common genotypes (MapGnt 5 and MapGnt 6) were evenly distributed in space and time. This might be due to random dispersal of the bacteria following large-scale processes such as cattle movements between herds. It has been reported that herds with an elevated turnover of cows show multiple genotypes (25, 61), underscoring the importance of live animal trade in the transmission of M. avium subsp. paratuberculosis between herds.
Clustering analysis, based on a combination of genetic markers, allowed the formation of subgroups (nodes) generally made up of several herds sharing a common genotype (Fig. 4). It will be interesting, in future epidemiological studies, to investigate if this clustering is a result of increased virulence, animal movements between herds, shared sources of infection (e.g., wildlife or other-farmed-species reservoirs), indirect contacts between herds through mechanical vectors, and/or other factors. Of particular interest in that regard is the possible wild origin of bison-type strains and of genotype 1, 3, and 4 strains, appearing as an outgroup distantly related to the consensus sequences. Domestic livestock and wild animals infected with M. avium subsp. paratuberculosis can share similar strains (16, 62–64), supporting the transmission of M. avium subsp. paratuberculosis strains between wildlife and livestock in places where habitats overlap. Likewise, the presence of specific epidemiological links between herds 40 and J should be investigated, as they both harbored strains belonging to genotype 4. Other groups of herds of particular interest are the ones sharing less common genotypes, such as genotypes 17, 18, and 24. At the population level, such molecular epidemiology studies could allow the identification of the most likely transmission pathways and help design control and prevention measures.
Conclusion.
The results from this study show that in addition to a majority of cattle-type strains, bison-type strains were isolated for the first time in domestic cattle on the American continent. Further typing of M. avium subsp. paratuberculosis strains, using a combination of conventional and less conventional markers, detected a total of 33 distinct genetic profiles in the population. Using unconventional markers, such as MIRU-VNTR 1067, proved useful in segregating the predominant genotypes into subtypes, underscoring their usefulness in M. avium subsp. paratuberculosis epidemiological investigations. The overall typing results suggest that using a combination of VNTR and SSR markers increases the overall discriminatory power and should be used in future studies. The genetic structure of the population was generally homogeneous and suggested the predominance of within-herd dispersion of strains, most likely via environmental contamination, although evidence of strain dissemination between herds was also uncovered. The finding of a cluster of strains, including the bison type, more distantly related to the consensus group suggests the existence of an unknown reservoir. Finally, although a spatiotemporal cluster was observed for a minor genotype, the two most prevalent genotypes were evenly distributed in space and time.
The typing tools that are now available should be used for a better understanding of the epidemiology and control of this important pathogen.
ACKNOWLEDGMENTS
This work was supported by the Programme de soutien à l'innovation en agroalimentaire from the Ministère de l'agriculture, des pêcheries et de l'alimentation du Québec.
We thank Sébastien Buczinski, Geneviève Côté, Elizabeth Doré, and Saray Rangel for providing valuable samples. Special thanks also to Evelyne Burelle for primary isolation of M. avium subsp. paratuberculosis strains and providing data on the origin of the samples.
Footnotes
Published ahead of print 14 May 2014
REFERENCES
- 1.Johne HA, Frothingham J. 1895. Ein eigenthuemlicher Fall von Tuberculose beim Rind. Dtsch. Z. Tiermed. Pathol. 21:438–454 [Google Scholar]
- 2.Benedictus A, Mitchell RM, Linde-Widmann M, Sweeney R, Fyock T, Schukken YH, Whitlock RH. 2008. Transmission parameters of Mycobacterium avium subspecies paratuberculosis infections in a dairy herd going through a control program. Prev. Vet. Med. 83:215–227. 10.1016/j.prevetmed.2007.07.008 [DOI] [PubMed] [Google Scholar]
- 3.Dore E, Pare J, Cote G, Buczinski S, Labrecque O, Roy JP, Fecteau G. 2012. Risk factors associated with transmission of Mycobacterium avium subsp. paratuberculosis to calves within dairy herd: a systematic review. J. Vet. Intern. Med. 26:32–45. 10.1111/j.1939-1676.2011.00854.x [DOI] [PubMed] [Google Scholar]
- 4.McKenna SL, Keefe GP, Barkema HW, McClure J, Vanleeuwen JA, Hanna P, Sockett DC. 2004. Cow-level prevalence of paratuberculosis in culled dairy cows in Atlantic Canada and Maine. J. Dairy Sci. 87:3770–3777. 10.3168/jds.S0022-0302(04)73515-8 [DOI] [PubMed] [Google Scholar]
- 5.VanLeeuwen JA, Keefe GP, Tremblay R, Power C, Wichtel JJ. 2001. Seroprevalence of infection with Mycobacterium avium subspecies paratuberculosis, bovine leukemia virus, and bovine viral diarrhea virus in maritime Canada dairy cattle. Can. Vet. J. 42:193–198 [PMC free article] [PubMed] [Google Scholar]
- 6.Hasanova L, Pavlik I. 2006. Economic impact of paratuberculosis in dairy cattle herds: a review. Vet. Med. 51:193–211 [Google Scholar]
- 7.Ott SL, Wells SJ, Wagner BA. 1999. Herd-level economic losses associated with Johne's disease on US dairy operations. Prev. Vet. Med. 40:179–192. 10.1016/S0167-5877(99)00037-9 [DOI] [PubMed] [Google Scholar]
- 8.Arsenault J, Girard C, Dubreuil P, Daignault D, Galarneau JR, Boisclair J, Simard C, Belanger D. 2003. Prevalence of and carcass condemnation from maedi-visna, paratuberculosis and caseous lymphadenitis in culled sheep from Quebec, Canada. Prev. Vet. Med. 59:67–81. 10.1016/S0167-5877(03)00060-6 [DOI] [PubMed] [Google Scholar]
- 9.Debien E, Helie P, Buczinski S, Leboeuf A, Belanger D, Drolet R. 2013. Proportional mortality: a study of 152 goats submitted for necropsy from 13 goat herds in Quebec, with a special focus on caseous lymphadenitis. Can. Vet. J. 54:581–587 [PMC free article] [PubMed] [Google Scholar]
- 10.Forde T, Kutz S, De Buck J, Warren A, Ruckstuhl K, Pybus M, Orsel K. 2012. Occurrence, diagnosis, and strain typing of Mycobacterium avium subspecies paratuberculosis infection in Rocky Mountain bighorn sheep (Ovis canadensis canadensis) in southwestern Alberta. J. Wildl. Dis. 48:1–11. 10.7589/0090-3558-48.1.1 [DOI] [PubMed] [Google Scholar]
- 11.Forde T, Orsel K, De Buck J, Cote SD, Cuyler C, Davison T, Elkin B, Kelly A, Kienzler M, Popko R, Taillon J, Veitch A, Kutz S. 2012. Detection of Mycobacterium avium subspecies paratuberculosis in several herds of Arctic Caribou (Rangifer tarandus ssp.). J. Wildl. Dis. 48:918–924. 10.7589/2011-09-261 [DOI] [PubMed] [Google Scholar]
- 12.Sibley JA, Woodbury MR, Appleyard GD, Elkin B. 2007. Mycobacterium avium subspecies paratuberculosis in Bison (Bison bison) from Northern Canada. J. Wildl. Dis. 43:775–779. 10.7589/0090-3558-43.4.775 [DOI] [PubMed] [Google Scholar]
- 13.Raizman EA, Wells SJ, Jordan PA, DelGiudice GD, Bey RR. 2005. Mycobacterium avium subsp. paratuberculosis from free-ranging deer and rabbits surrounding Minnesota dairy herds. Can. J. Vet. Res. 69:32–38 [PMC free article] [PubMed] [Google Scholar]
- 14.Chiodini RJ, Rossiter CA. 1996. Paratuberculosis: a potential zoonosis? Vet. Clin. North Am. Food Anim. Pract. 12:457–467 [DOI] [PubMed] [Google Scholar]
- 15.Feller M, Huwiler K, Stephan R, Altpeter E, Shang A, Furrer H, Pfyffer GE, Jemmi T, Baumgartner A, Egger M. 2007. Mycobacterium avium subspecies paratuberculosis and Crohn's disease: a systematic review and meta-analysis. Lancet Infect. Dis. 7:607–613. 10.1016/S1473-3099(07)70211-6 [DOI] [PubMed] [Google Scholar]
- 16.Motiwala AS, Amonsin A, Strother M, Manning EJ, Kapur V, Sreevatsan S. 2004. Molecular epidemiology of Mycobacterium avium subsp. paratuberculosis isolates recovered from wild animal species. J. Clin. Microbiol. 42:1703–1712. 10.1128/JCM.42.4.1703-1712.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thibault VC, Grayon M, Boschiroli ML, Willery E, Allix-Beguec C, Stevenson K, Biet F, Supply P. 2008. Combined multilocus short-sequence-repeat and mycobacterial interspersed repetitive unit-variable-number tandem-repeat typing of Mycobacterium avium subsp. paratuberculosis isolates. J. Clin. Microbiol. 46:4091–4094. 10.1128/JCM.01349-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sevilla I, Singh SV, Garrido JM, Aduriz G, Rodriguez S, Geijo MV, Whittington RJ, Saunders V, Whitlock RH, Juste RA. 2005. Molecular typing of Mycobacterium avium subspecies paratuberculosis strains from different hosts and regions. Rev. Sci. Tech. 24:1061–1066 [PubMed] [Google Scholar]
- 19.Stevenson K, Hughes VM, de Juan L, Inglis NF, Wright F, Sharp JM. 2002. Molecular characterization of pigmented and nonpigmented isolates of Mycobacterium avium subsp. paratuberculosis. J. Clin. Microbiol. 40:1798–1804. 10.1128/JCM.40.5.1798-1804.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mobius P, Luyven G, Hotzel H, Kohler H. 2008. High genetic diversity among Mycobacterium avium subsp. paratuberculosis strains from German cattle herds shown by combination of IS900 restriction fragment length polymorphism analysis and mycobacterial interspersed repetitive unit-variable-number tandem-repeat typing. J. Clin. Microbiol. 46:972–981. 10.1128/JCM.01801-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cowan LS, Mosher L, Diem L, Massey JP, Crawford JT. 2002. Variable-number tandem repeat typing of Mycobacterium tuberculosis isolates with low copy numbers of IS6110 by using mycobacterial interspersed repetitive units. J. Clin. Microbiol. 40:1592–1602. 10.1128/JCM.40.5.1592-1602.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kremer K, Arnold C, Cataldi A, Gutierrez MC, Haas WH, Panaiotov S, Skuce RA, Supply P, van der Zanden AG, van Soolingen D. 2005. Discriminatory power and reproducibility of novel DNA typing methods for Mycobacterium tuberculosis complex strains. J. Clin. Microbiol. 43:5628–5638. 10.1128/JCM.43.11.5628-5638.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castellanos E, Romero B, Rodriguez S, de Juan L, Bezos J, Mateos A, Dominguez L, Aranaz A. 2010. Molecular characterization of Mycobacterium avium subspecies paratuberculosis types II and III isolates by a combination of MIRU-VNTR loci. Vet. Microbiol. 144:118–126. 10.1016/j.vetmic.2009.12.028 [DOI] [PubMed] [Google Scholar]
- 24.Fernandez-Silva JA, Abdulmawjood A, Akineden O, Drager K, Klawonn W, Bulte M. 2012. Molecular epidemiology of Mycobacterium avium subsp. paratuberculosis at a regional scale in Germany. Res. Vet. Sci. 93:776–782. 10.1016/j.rvsc.2011.12.005 [DOI] [PubMed] [Google Scholar]
- 25.Ricchi M, Barbieri G, Taddei R, Belletti GL, Carra E, Cammi G, Garbarino CA, Arrigoni N. 2011. Effectiveness of combination of mini-and microsatellite loci to sub-type Mycobacterium avium subsp. paratuberculosis Italian type C isolates. BMC Vet. Res. 7:54. 10.1186/1746-6148-7-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thibault VC, Grayon M, Boschiroli ML, Hubbans C, Overduin P, Stevenson K, Gutierrez MC, Supply P, Biet F. 2007. New variable-number tandem-repeat markers for typing Mycobacterium avium subsp. paratuberculosis and M. avium strains: comparison with IS900 and IS1245 restriction fragment length polymorphism typing. J. Clin. Microbiol. 45:2404–2410. 10.1128/JCM.00476-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whittington RJ, Marsh IB, Saunders V, Grant IR, Juste R, Sevilla IA, Manning EJ, Whitlock RH. 2011. Culture phenotypes of genomically and geographically diverse Mycobacterium avium subsp. paratuberculosis isolates from different hosts. J. Clin. Microbiol. 49:1822–1830. 10.1128/JCM.00210-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh N, Singh SV, Gupta VK, Sharma VD, Sharma RK, Katoch VM. 1996. Isolation and identification of Mycobacterium paratuberculosis from naturally infected goatherds in India. Indian J. Vet. Pathol. 20:104–108 [Google Scholar]
- 29.Singh SV, Sohal JS, Singh PK, Singh AV. 2009. Genotype profiles of Mycobacterium avium subspecies paratuberculosis isolates recovered from animals, commercial milk, and human beings in North India. Int. J. Infect. Dis. 13:e221–e227. 10.1016/j.ijid.2008.11.022 [DOI] [PubMed] [Google Scholar]
- 30.Overduin P, Schouls L, Roholl P, van der Zanden A, Mahmmod N, Herrewegh A, van Soolingen D. 2004. Use of multilocus variable-number tandem-repeat analysis for typing Mycobacterium avium subsp. paratuberculosis. J. Clin. Microbiol. 42:5022–5028. 10.1128/JCM.42.11.5022-5028.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bull TJ, Sidi-Boumedine K, McMinn EJ, Stevenson K, Pickup R, Hermon-Taylor J. 2003. Mycobacterial interspersed repetitive units (MIRU) differentiate Mycobacterium avium subspecies paratuberculosis from other species of the Mycobacterium avium complex. Mol. Cell. Probes 17:157–164. 10.1016/S0890-8508(03)00047-1 [DOI] [PubMed] [Google Scholar]
- 32.Amonsin A, Li LL, Zhang Q, Bannantine JP, Motiwala AS, Sreevatsan S, Kapur V. 2004. Multilocus short sequence repeat sequencing approach for differentiating among Mycobacterium avium subsp. paratuberculosis strains. J. Clin. Microbiol. 42:1694–1702. 10.1128/JCM.42.4.1694-1702.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graur D, Li W-H. 1999. Fundamentals of molecular evolution, 2nd ed. Sinauer Associates, Sunderland, MA [Google Scholar]
- 34.Grundmann H, Hori S, Tanner G. 2001. Determining confidence intervals when measuring genetic diversity and the discriminatory abilities of typing methods for microorganisms. J. Clin. Microbiol. 39:4190–4192. 10.1128/JCM.39.11.4190-4192.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hammer Ø, Harper DAT, Ryan PD. 2001. PAST: paleontological statistics software package for education and data analysis. Palaeontologia Electronica 4:9 [Google Scholar]
- 36.Krebs CJ. 1989. Ecological methodology. Harper & Row, New York, NY [Google Scholar]
- 37.Kulldorff M. 1997. A spatial scan statistic. Commun. Statistics Theory Methods 26:1481–1496. 10.1080/03610929708831995 [DOI] [Google Scholar]
- 38.Whittington RJ, Marsh IB, Whitlock RH. 2001. Typing of IS 1311 polymorphisms confirms that bison (Bison bison) with paratuberculosis in Montana are infected with a strain of Mycobacterium avium subsp. paratuberculosis distinct from that occurring in cattle and other domesticated livestock. Mol. Cell. Probes 15:139–145. 10.1006/mcpr.2001.0346 [DOI] [PubMed] [Google Scholar]
- 39.Kim JM, Ku BK, Lee HN, Jang YB, Her M, Kim JW, Kim JY, Kim JJ, Hyun BH, Jung SC. 2012. Mycobacterium avium subsp. paratuberculosis in wild boars from Korea, p 41 In 11th International Colloquium on Paratuberculosis. International Association for Paratuberculosis, Sydney, Australia [Google Scholar]
- 40.Singh SV, Singh AV, Singh PK, Singh B, Ranjendran AS, Swain N. 2011. Recovery of Indian bison type genotype of Mycobacterium avium subsp. paratuberculosis from Wild Bison (Bos gourus) in India. Vet. Res. 4:61–65 [Google Scholar]
- 41.Sohal JS, Sheoran N, Narayanasamy K, Brahmachari V, Singh S, Subodh S. 2009. Genomic analysis of local isolate of Mycobacterium avium subspecies paratuberculosis. Vet. Microbiol. 134:375–382. 10.1016/j.vetmic.2008.08.027 [DOI] [PubMed] [Google Scholar]
- 42.Okuni JB, Dovas CI, Loukopoulos P, Bouzalas IG, Kateete DP, Joloba ML, Ojok L. 2012. Isolation of Mycobacterium avium subspecies paratuberculosis from Ugandan cattle and strain differentiation using optimised DNA typing techniques. BMC Vet. Res. 8:99. 10.1186/1746-6148-8-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sohal JS, Singh SV, Singh AV, Singh PK. 2009. Interspecies sharing of ‘Indian Bison Type' Mycobacterium avium subspecies paratuberculosis as revealed by ‘short sequence repeat' typing. Indian J. Anim. Sci. 79:272–274 [Google Scholar]
- 44.Douarre PE, Cashman W, Buckley J, Coffey A, O'Mahony J. 2011. Molecular characterization of Mycobacterium avium subsp. paratuberculosis using multi-locus short sequence repeat (MLSSR) and mycobacterial interspersed repetitive units-variable number tandem repeat (MIRU-VNTR) typing methods. Vet. Microbiol. 149:482–487. 10.1016/j.vetmic.2010.12.001 [DOI] [PubMed] [Google Scholar]
- 45.Fernandez-Silva JA, Abdulmawjood A, Akineden O, Bulte M. 2012. Genotypes of Mycobacterium avium subsp. paratuberculosis from South American countries determined by two methods based on genomic repetitive sequences. Trop. Anim. Health Prod. 44:1123–1126. 10.1007/s11250-011-0060-6 [DOI] [PubMed] [Google Scholar]
- 46.Fernandez-Silva JA, Abdulmawjood A, Bulte M. 2011. Diagnosis and molecular characterization of Mycobacterium avium subsp. paratuberculosis from dairy cows in Colombia. Vet. Med. Int. 2011:352561. 10.4061/2011/352561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stevenson K, Alvarez J, Bakker D, Biet F, de Juan L, Denham S, Dimareli Z, Dohmann K, Gerlach GF, Heron I, Kopecna M, May L, Pavlik I, Sharp JM, Thibault VC, Willemsen P, Zadoks RN, Greig A. 2009. Occurrence of Mycobacterium avium subspecies paratuberculosis across host species and European countries with evidence for transmission between wildlife and domestic ruminants. BMC Microbiol. 9:212. 10.1186/1471-2180-9-212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ricchi M, Barbieri G, Cammi G, Garbarino CA, Arrigoni N. 2011. High-resolution melting for analysis of short sequence repeats in Mycobacterium avium subsp. paratuberculosis. FEMS Microbiol. Lett. 323:151–154. 10.1111/j.1574-6968.2011.02371.x [DOI] [PubMed] [Google Scholar]
- 49.Harris NB, Payeur JB, Kapur V, Sreevatsan S. 2006. Short-sequence-repeat analysis of Mycobacterium avium subsp. paratuberculosis and Mycobacterium avium subsp. avium isolates collected from animals throughout the United States reveals both stability of loci and extensive diversity. J. Clin. Microbiol. 44:2970–2973. 10.1128/JCM.00584-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pradhan AK, Kramer AJ, Mitchell RM, Whitlock RH, Smith JM, Hovingh E, Van Kessel JS, Karns JS, Schukken YH. 2009. Multilocus short sequence repeat analysis of Mycobacterium avium subsp. paratuberculosis isolates from dairy herds in northeastern United States of a longitudinal study indicates low shedders are truly infected, p 30–33 In 10th International Colloquium on Paratuberculosis. International Association for Paratuberculosis, Minneapolis, MN [Google Scholar]
- 51.Pradhan AK, Mitchell RM, Kramer AJ, Zurakowski MJ, Fyock TL, Whitlock RH, Smith JM, Hovingh E, Van Kessel JA, Karns JS, Schukken YH. 2011. Molecular epidemiology of Mycobacterium avium subsp. paratuberculosis in a longitudinal study of three dairy herds. J. Clin. Microbiol. 49:893–901. 10.1128/JCM.01107-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cernicchiaro N, Wells SJ, Janagama H, Sreevatsan S. 2008. Influence of type of culture medium on characterization of Mycobacterium avium subsp. paratuberculosis subtypes. J. Clin. Microbiol. 46:145–149. 10.1128/JCM.01769-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grunwald NJ, Goodwin SB, Milgroom MG, Fry WE. 2003. Analysis of genotypic diversity data for populations of microorganisms. Phytopathology 93:738–746. 10.1094/PHYTO.2003.93.6.738 [DOI] [PubMed] [Google Scholar]
- 54.Gollnick NS, Mitchell RM, Baumgart M, Janagama HK, Sreevatsan S, Schukken YH. 2007. Survival of Mycobacterium avium subsp. paratuberculosis in bovine monocyte-derived macrophages is not affected by host infection status but depends on the infecting bacterial genotype. Vet. Immunol. Immunopathol. 120:93–105. 10.1016/j.vetimm.2007.07.017 [DOI] [PubMed] [Google Scholar]
- 55.Janagama HK, Jeong K, Kapur V, Coussens P, Sreevatsan S. 2006. Cytokine responses of bovine macrophages to diverse clinical Mycobacterium avium subspecies paratuberculosis strains. BMC Microbiol. 6:10. 10.1186/1471-2180-6-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Motiwala AS, Li L, Kapur V, Sreevatsan S. 2006. Current understanding of the genetic diversity of Mycobacterium avium subsp. paratuberculosis. Microbes Infect. 8:1406–1418. 10.1016/j.micinf.2005.12.003 [DOI] [PubMed] [Google Scholar]
- 57.O'Brien R, Mackintosh CG, Bakker D, Kopecna M, Pavlik I, Griffin JF. 2006. Immunological and molecular characterization of susceptibility in relationship to bacterial strain differences in Mycobacterium avium subsp. paratuberculosis infection in the red deer (Cervus elaphus). Infect. Immun. 74:3530–3537. 10.1128/IAI.01688-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Al-Hajoj SA, Akkerman O, Parwati I, al-Gamdi S, Rahim Z, van Soolingen D, van Ingen J, Supply P, van der Zanden AG. 2010. Microevolution of Mycobacterium tuberculosis in a tuberculosis patient. J. Clin. Microbiol. 48:3813–3816. 10.1128/JCM.00556-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perez-Lago L, Herranz M, Lirola MM, Bouza E, Garcia de Viedma D. 2011. Characterization of microevolution events in Mycobacterium tuberculosis strains involved in recent transmission clusters. J. Clin. Microbiol. 49:3771–3776. 10.1128/JCM.01285-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schurch AC, Kremer K, Kiers A, Daviena O, Boeree MJ, Siezen RJ, Smith NH, van Soolingen D. 2010. The tempo and mode of molecular evolution of Mycobacterium tuberculosis at patient-to-patient scale. Infect. Genet. Evol. 10:108–114. 10.1016/j.meegid.2009.10.002 [DOI] [PubMed] [Google Scholar]
- 61.van Hulzen KJ, Nielen M, Koets AP, de Jong G, van Arendonk JA, Heuven HC. 2011. Effect of herd prevalence on heritability estimates of antibody response to Mycobacterium avium subspecies paratuberculosis. J. Dairy Sci. 94:992–997. 10.3168/jds.2010-3472 [DOI] [PubMed] [Google Scholar]
- 62.Fritsch I, Luyven G, Kohler H, Lutz W, Mobius P. 2012. Suspicion of Mycobacterium avium subsp. paratuberculosis transmission between cattle and wild-living red deer (Cervus elaphus) by multitarget genotyping. Appl. Environ. Microbiol. 78:1132–1139. 10.1128/AEM.06812-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerritsmann H, Stalder GL, Spergser J, Hoelzl F, Deutz A, Kuebber-Heiss A, Walzer C, Smith S. 2014. Multiple strain infections and high genotypic diversity among Mycobacterium avium subsp. paratuberculosis field isolates from diseased wild and domestic ruminant species in the eastern Alpine region of Austria. Infect. Genet. Evol. 21:244–251. 10.1016/j.meegid.2013.11.009 [DOI] [PubMed] [Google Scholar]
- 64.Greig A, Stevenson K, Henderson D, Perez V, Hughes V, Pavlik I, Hines ME, II, McKendrick I, Sharp JM. 1999. Epidemiological study of paratuberculosis in wild rabbits in Scotland. J. Clin. Microbiol. 37:1746–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]