

Abstract
Bovine digital dermatitis (DD) is a leading cause of lameness in dairy cattle throughout the world. Despite 35 years of research, the definitive etiologic agent associated with the disease process is still unknown. Previous studies have demonstrated that multiple bacterial species are associated with lesions, with spirochetes being the most reliably identified organism. This study details the deep sequencing-based metagenomic evaluation of 48 staged DD biopsy specimens collected during a 3-year longitudinal study of disease progression. Over 175 million sequences were evaluated by utilizing both shotgun and 16S metagenomic techniques. Based on the shotgun sequencing results, there was no evidence of a fungal or DNA viral etiology. The bacterial microbiota of biopsy specimens progresses through a systematic series of changes that correlate with the novel morphological lesion scoring system developed as part of this project. This scoring system was validated, as the microbiota of each stage was statistically significantly different from those of other stages (P < 0.001). The microbiota of control biopsy specimens were the most diverse and became less diverse as lesions developed. Although Treponema spp. predominated in the advanced lesions, they were in relatively low abundance in the newly described early lesions that are associated with the initiation of the disease process. The consortium of Treponema spp. identified at the onset of disease changes considerably as the lesions progress through the morphological stages identified. The results of this study support the hypothesis that DD is a polybacterial disease process and provide unique insights into the temporal changes in bacterial populations throughout lesion development.
INTRODUCTION
According to the most recent National Animal Health Monitoring System survey of U.S. dairy farms (1), lameness is the second most common health problem identified in dairy cattle. Digital dermatitis (DD) was found to be the primary cause of lameness within the study herds, accounting for 61.8% of the lameness in bred heifers and 49.1% of the lameness in cows. Bovine DD was first described in 1974, but despite over 35 years of research, the etiological agent(s) associated with this disease has yet to be definitively identified. The first morphological description of DD as an ulcerative disease of the bovine coronary band occurred at the 8th International Meeting on Diseases of Cattle in Milan, Italy (2). The first etiologic descriptions of the disease were published in 1994 and were soon followed by a report describing the isolation and identification of an anaerobic spirochete, believed to be a Treponema sp., in the lesions (3, 4). Since that time, a number of additional papers have been published demonstrating the association of the lesions with additional bacteria, including Bacteroides spp. (now called Porphyromonas spp.) (5), Campylobacter spp. (6, 7), and Borrelia spp. (8), as well as viral etiologies (9, 10). While there is a consistent presence of several Treponema phylotypes in DD lesions (9, 11–15), attempts to induce disease by skin inoculation with pure cultures of these microorganisms have largely failed to result in significant disease in a majority of the animals inoculated (7, 16). Furthermore, there is evidence to suggest that the clinical use of vaccines against spirochetes provides limited protection against the disease process (17). The consistent clinical response to topical or systemic antibiotics suggests a bacterial agent as the true etiology of the disease (18–22). The association of DD lesions with a variety of bacterial agents, the response of the lesions to antibiotics, and the failure to induce disease or protect against it using monovalent vaccines strongly suggest that DD is a polymicrobial disease process (9, 23–25). This conclusion is further supported by two recent studies that utilized the culture-independent method of sequencing clones of 16S rRNA genes by Sanger technology to classify a subset of bacteria derived from biopsy specimens of both lesions and healthy feet (15, 26). While these studies demonstrate that there are mixed microbial populations present in DD lesions that differ from that of normal skin, the lack of information regarding stage and chronicity of the biopsy specimens, combined with the relatively low number of 16S rRNA gene sequences, limits the confidence in the overall conclusions.
The fact that DD develops through a systematic series of morphological stages was first described by Dopfer et al. in 1997 (7). The authors describe four stages of lesion development, with stage M1 (“M” in honor of Mortellaro's initial description of the lesions) being the formation of a small ulcerative area, stage M2 being a mature papillomatous or ulcerative lesion, stage M3 being a healing lesion, and stage M4 being a chronic recurrent lesion. The histopathologic changes associated with the development of DD have been described (7, 27–29). Briefly, DD lesions have been described histologically to include acute, suppurative inflammation of the epidermis with superficial necrosis and hyperkeratosis (10), along with perivascular aggregations of lymphocytes and plasma cells (30). A consistent microscopic observation of spirochetes within lesions has been demonstrated by multiple researchers through the use of hematoxylin and eosin staining, Warthin-Starry silver staining (31), immunohistochemistry (13, 28), electron microscopy (13, 32), and fluorescence in situ hybridization (FISH) (11, 12, 26, 29, 33–37).
The objective of this project was to use deep DNA sequencing to analyze changes in the microbiota of DD lesions as they progressed through the temporal development of disease. The hypothesis was that the development of DD is polybacterial, and consistent and predictable changes to the bacterial microbiota of the bovine foot could be identified as lesions systematically progress through multiple distinct morphological stages. Following the onset of clinical disease, an evaluation of the lesion microbiota following conventional topical treatment with tetracycline was desired to determine the effect of treatment on the microbial community. For this component, it was hypothesized that specific bacterial species associated with DD lesions would be displaced by 9 days after treatment.
MATERIALS AND METHODS
Lesion staging and biopsy sample collection.
DD biopsy specimens were collected from adult Holstein dairy cattle housed at the Iowa State University Dairy Farm. This farm milks approximately 400 cows and has a 305-day rolling herd average of approximately 25,000 lb. The average parity of the herd is 2.6 lactations. The herd had a history of DD-associated lameness, and the prevalence of DD lesions in the herd was estimated to be 50% at the initiation of the study. In order to study the development and progression of DD lesions, an initial subset of 30 cows were identified as a convenience sample to participate in the longitudinal study of DD lesion development. Cows were selected based on availability (i.e., not participating in another trial at the time) and lesion status (the goal was to have cows with no history of DD as well as cows with early and active lesions). Over the next year, an additional 23 cows were enrolled in the epidemiologic study, giving a total of 53 cows from which biopsy specimens were collected. The biopsy specimens that were sequenced and utilized in this study represent 26 of these 53 cows. Cattle in the trial were diverted away from the farm's footbath (5% copper sulfate) through the use of radiofrequency ID tags and sort gates. No additional preventative measures were used to control DD in these cows, allowing observation of lesion development in the absence of intervention. Cattle in this study were examined every 3 to 4 weeks throughout the study, and biopsy specimens were taken from rear feet each time an individual foot changed lesion stage. Additionally, digital images of all feet were taken at each observation to allow retrospective evaluation of lesion morphology and blinded staging of lesions. All observations were made by a single observer (A.C.K.) to ensure consistency. Biopsy specimens utilized in this study were derived from these cattle over the course of the first 2 years of the longitudinal study. Forty-eight biopsy specimens, representing 26 cows, were selected for metagenomic analysis based on their morphological appearance (i.e., DD lesions progress through a transition between stages; however, biopsy specimens were selected due to their clear representation of the DD stage). All animal procedures and biopsy methods were approved by the Institutional Animal Care and Use Committee of Iowa State University.
Lesions were identified as part of a 3-year observational longitudinal study and classified using a novel scoring system (Fig. 1) developed as part of this study. The Iowa DD scoring system was developed after initial efforts to utilize the previously described “M” scoring system proved to be problematic due to the recognition of two distinct previously unrecognized morphological appearances of early-stage lesions. The study's 3-year duration made it possible to monitor lesions over an extended period of time and to demonstrate that both new morphological forms ultimately led to the development of classic DD lesions. The morphological differences of these subtypes suggested that different bacterial species might be involved, and it was thus important for the study to be able to differentiate these early lesion subtypes. Instead of modifying the existing “M” scoring system, it was decided to develop an independent scoring system. The Iowa DD scoring system differentiates lesions progressing from normal skin (stage 0) to initial onset (stage 1), developing lesions (stage 2), classic DD lesions in the acute form (stage 3), and the chronic form (stage 4) of disease. Furthermore, stages 1 and 2 are subdivided into two morphological subtypes. The “A” type lesions initiate in the interdigital space and have a more ulcerated appearance than the “B” type lesions, which develop more diffusely across the heel and have a thickened, crusted appearance. Both subtypes progress to the same morphological stage 3 and 4 lesions. In some cases in this study, the subtypes of stages 1 and 2 are combined for data analysis in order to simplify the presentation of the temporal development of disease. However, the subtypes were evaluated independently for assessment of biomarkers of disease for each stage. A separate category (termed stage 5 in this study) is used to represent a distinct subset of biopsy specimens that were taken exactly 9 days after topical treatment of stage 3 or stage 4 lesions. The data collected over the course of the longitudinal study suggest that DD lesions develop and progress very slowly. In several cases, we had cows with the same lesion stage for the full 3 years of the study. On average, when new lesions developed, cattle took about 135 days to progress from normal skin to an advanced lesion, with the longest time being almost 2 years. A more complete description of the Iowa DD scoring system and the epidemiology of disease development will be reported in a separate article.
FIG 1.

Illustration of the lesion stages identified throughout the 3-year study. Images are of lesions biopsied and utilized in this study. Stage 0 represents normal bovine skin, the A1/A2 and B1/B2 lesions represent morphological variations of developing DD lesions, and stages 3 and 4 represent the clinical forms of disease responsible for lameness. A subset of biopsy specimens from lesions that were treated and biopsied 9 days posttreatment are presented as stage 5 in this study.
Biopsy sample collection was performed under local anesthesia provided by a regional limb perfusion with 20 ml of 1% lidocaine. DD lesions were gently rinsed with water to remove gross organic debris; no antibacterial products or surgical scrubs were used prior to biopsies. Two separate 3-mm biopsy specimens were collected utilizing a biopsy punch to collect epidermis from a representative area of the lesion. Biopsy specimens intended for metagenomic analysis were placed into 1.5-ml microcentrifuge tubes containing 0.5-mm stainless steel beads and transferred to the laboratory within 6 h for DNA extraction. Biopsy specimens intended for histopathologic analysis were immediately placed in 10% neutral buffered formalin. Biopsy specimens were processed for DNA extraction by adding buffer ATL and proteinase K (Qiagen DNeasy blood and tissue kit), homogenized using a Next Advance Bullet blender, and incubated overnight at 56°C. Following overnight incubation, biopsy specimens were processed as directed by the manufacturer's recommendations (Qiagen DNeasy blood and tissue kit), and sample DNA was eluted and stored in AE buffer. Total DNA yield was quantified with the Qubit fluorometer (Life Technologies, Grand Island, NY), and DNA was stored at −80°C prior to downstream processing. Biopsy specimens utilized for histologic examination were stained with hematoxylin and eosin (H&E) using the Gemini Autostainer with the H&E-based staining protocol. Warthin-Starry staining was conducted using the Iowa State Veterinary Diagnostic laboratory standard operating procedure.
Shotgun sequencing.
Three samples were chosen from each of the seven morphological stages for analysis using shotgun metagenomic sequencing. Along with these 21 samples, three additional samples were chosen from stage 4 lesions that had been treated with topical tetracycline 9 days prior to biopsying (identified as stage 5 for simplicity). Table 1 provides details regarding the biopsy specimens included in the project. An aliquot of all 24 samples was normalized to 2.5 ng/μl for use in the Nextera DNA sample prep kit (Epicentre Biotechnologies, Madison, WI). Tagmentation of the samples was done using 50 ng of template, as directed by the manufacturer. Following tagmentation, each sample was purified with the Qiagen MinElute PCR purification kit and eluted with 25 μl of Nextera resuspension buffer. PCR amplification was done per the manufacturer's instruction using a unique combination of indexing primers for each of the 24 samples to allow multiplexing of samples. Following amplification, each DNA library had short DNA fragments (<150 bp) removed using AMPure XP bead purification. DNA concentration and library fragment sizes were analyzed using a Qubit fluorometer and Agilent high-sensitivity chip. Sample library concentrations were normalized, pooled, and diluted to 2 nM for sequencing on the Illumina HiSeq platform. Libraries were run on a single flow cell lane of 100-bp paired-end sequences at the Iowa State University DNA Facility.
TABLE 1.
Metadata for each individual biopsy sample collected for this study
| Cowa | Footb | Subclass | Stage | 16S | Shotgun | No. of days observed |
||
|---|---|---|---|---|---|---|---|---|
| Before biopsy | After biopsy | Total | ||||||
| 1 | RR | 0 | X | X | 353 | 19 | 372 | |
| 15 | LR | 0 | X | X | 123 | 200 | 323 | |
| 19 | LR | 0 | X | X | 202 | 121 | 323 | |
| 21 | LR | 0 | X | 195 | 441 | 636 | ||
| 24 | RR | 0 | X | 63 | 204 | 267 | ||
| 26 | LR | 0 | X | 0 | 225 | 225 | ||
| 4 | LR | A | 1 | X | X | 406 | 200 | 606 |
| 5 | RR | A | 1 | X | X | 491 | 99 | 590 |
| 7 | RR | A | 1 | X | X | 319 | 528 | 847 |
| 2 | LR | B | 1 | X | 470 | 190 | 660 | |
| 14 | RR | B | 1 | X | 260 | 560 | 820 | |
| 16 | LR | B | 1 | X | X | 74 | 580 | 654 |
| 17 | RR | B | 1 | X | X | 151 | 219 | 370 |
| 20 | RR | B | 1 | X | 103 | 533 | 636 | |
| 21 | LR | B | 1 | X | X | 77 | 559 | 636 |
| 23 | LR | B | 1 | X | 83 | 565 | 648 | |
| 25 | LR | B | 1 | X | 0 | 225 | 225 | |
| 3 | LR | A | 2 | X | X | 174 | 85 | 259 |
| 9 | RR | A | 2 | X | X | 385 | 524 | 909 |
| 16 | LR | A | 2 | X | X | 222 | 432 | 654 |
| 22 | LR | A | 2 | X | 90 | 341 | 431 | |
| 2 | LR | B | 2 | X | 491 | 169 | 660 | |
| 8 | LR | B | 2 | X | X | 223 | 249 | 472 |
| 14 | RR | B | 2 | X | 280 | 540 | 820 | |
| 20 | RR | B | 2 | X | 195 | 441 | 636 | |
| 21 | LR | B | 2 | X | X | 223 | 413 | 636 |
| 23 | RR | B | 2 | X | X | 83 | 565 | 648 |
| 23 | LR | B | 2 | X | 202 | 446 | 648 | |
| 2 | LR | 3 | X | 597 | 63 | 660 | ||
| 8 | LR | 3 | X | X | 338 | 134 | 472 | |
| 13 | RR | 3 | X | X | 88 | 570 | 658 | |
| 14 | RR | 3 | X | 506 | 314 | 820 | ||
| 20 | RR | 3 | X | 315 | 321 | 636 | ||
| 22 | LR | 3 | X | X | 222 | 209 | 431 | |
| 23 | LR | 3 | X | 322 | 326 | 648 | ||
| 24 | RR | 3 | X | 156 | 111 | 267 | ||
| 25 | LR | 3 | X | 114 | 111 | 225 | ||
| 26 | LR | 3 | X | 114 | 111 | 225 | ||
| 11 | LR | 4 | X | X | 385 | 536 | 921 | |
| 12 | LR | 4 | X | X | 393 | 203 | 596 | |
| 14 | RR | 4 | X | 596 | 224 | 820 | ||
| 18 | LR | 4 | X | X | 89 | 566 | 655 | |
| 20 | RR | 4 | X | 418 | 218 | 636 | ||
| 21 | LR | 4 | X | 315 | 321 | 636 | ||
| 22 | LR | 4 | X | 328 | 103 | 431 | ||
| 6 | RR | 5 | X | X | 357 | 389 | 746 | |
| 10 | LR | 5 | X | X | 399 | 162 | 561 | |
| 18 | LR | 5 | X | X | 119 | 536 | 655 | |
| Avg | 252.3 | 316.7 | 569.0 | |||||
Certain cows had several sequential biopsies taken, and each number corresponds to an individual cow.
LR, left rear; RR, right rear.
16S sequencing.
Forty-eight samples were chosen for metagenomic analysis using amplification of the V3-V4 hypervariable region of the bacterial 16S rRNA gene (Table 1). This included the 24 samples sequenced in the shotgun sequencing experiment plus an additional 24 samples representative of each of the scoring system stages. All samples were processed using a universal 16S forward primer (515F) and 48 unique Golay barcoded reverse primers (806R) as described by Caporaso et al. (38). Following 35 cycles of PCR amplification, the PCR product was confirmed by visualization of an approximately 300-bp band on agarose gel (0.8%). A negative control for every sample and primer combination was also processed in an identical manner, and the absence of amplification was verified on agarose gels. Sample library DNA concentrations were quantified using a Qubit fluorometer, and samples were pooled with equal amounts of DNA. The pooled libraries were cleaned up with the MO-BIO UltraClean PCR clean-up kit, and the samples were then diluted to 2 nM. Due to the potentially low diversity of our libraries, Illumina PhiX Control v3 was added at a proportion of 20% prior to loading on the MiSeq platform. A single flow cell lane of 300-bp paired-end sequences was run on the Illumina MiSeq at the Iowa State University DNA Facility.
Metagenomic data analysis. (i) Shotgun data.
Raw data files were demultiplexed and converted to FASTQ using Casava v.1.8.2 (Illumina, Inc., San Diego, CA). FASTQ files were concatenated and uploaded to the MG-RAST (39) server for analysis. The server was set to query for the organism information for all biopsy bins. A “best hit” analysis was performed using the M5NR database (40) as the source database. The MG-RAST graphical user interface was used to provide rarefaction curves and relative species abundance data. The data were then exported in BIOM format for further analysis in QIIME (38, 41). Bray-Curtis beta diversity plots (42) and Procrustes analysis (43) were performed using published QIIME scripts.
(ii) 16S metagenomic data.
Forward and reverse reads from the paired-end sequencing were first merged using the fastq-join script. QIIME 1.7 was then used for additional data analysis. Demultiplexing and quality filtering were then performed using the split_libraries_fastq.py script. The pick_reference_otus_through_otu_table.py script was used for operational taxonomic unit (OTU) calling, and taxonomic assignment based on the Greengenes database (44) was performed. Jackknifed (45) (10,000 iterations) Bray-Curtis beta diversities for the samples were plotted by stage using the jackknifed_beta_diversity.py script and compared using a nonparametric analysis of similarity (ANOSIM) with 1,000 permutations. In order to test for biomarker taxa associated with each stage of lesion development, the Galaxy analysis tool (46, 47) was utilized to analyze LDA effect size (LEfSe).
RESULTS
Shotgun sequencing.
Samples from shotgun sequencing varied in the number of 100-bp reads from 837,084 to 22,799,300, with an average of 6,518,062 reads per sample and a total of 156,433,474 sequences. Of these reads, a total of 48,530,678 passed initial quality control presets of the MG-RAST software, with an average duplicate-read inferred sequencing error estimation (DRISEE) (48) of 6.32%. With the M5NR database utilized as the annotation source, 661,098 reads matched a reference sequence with an E value cutoff of 1e−5, 60% minimum identity, and a minimum 15-bp alignment. The percentage of eukaryotic reads was highest in controls (88.0%), decreased as lesions progressed to stages 1 and 2 (40.9%, 6.9%), and was lowest in classic stage 3 and 4 lesions (1.7%, 2.3%). Conversely, the percentage of bacterial reads increased as lesions progressed from 2.1% in controls to 55.1% and 88.6% in stage 1 and 2 lesions and 94.8% and 97.2% in stage 3 and 4 lesions. Analysis showed that almost no sequences (fewer than 10 total sequences) were associated with fungal DNA and less than 0.5% of all sequences were associated with viral elements, consisting primarily of phage DNA.
To determine if quantitative differences existed between the normal cow skin control biopsy specimens and different stages of lesions, an ANOSIM analysis was used to determine whether the Bray-Curtis distances within the same category of the samples is significantly different from those among different categories of the samples. The results of these analyses demonstrated that statistically significant differences do exist between the stages (P < 0.001) at both the genus and species levels. This allows pairwise comparisons of the different stages to be performed where significant differences exist between all comparisons (P < 0.025), except the comparison of the control and posttreatment biopsy specimens.
A plot of taxonomic hits from each stage of disease development is presented in Fig. 2A, showing all bacterial families that had a relative abundance of at least 5% of the bacterial flora for any given stage. The morphological stages A1 and B1 as well as A2 and B2 have been combined as stage 1 and stage 2, respectively, to better demonstrate the temporal development of the disease process. Eleven families were identified that met these criteria. Families that did not reach the 5% threshold have been combined into “other” to give a representation of the total bacterial population dynamics of each stage. The family with the largest increase in abundance as lesions developed is Spirochaetaceae, systematically increasing from 0.0% in control feet to 94.3% in chronic (stage 4) lesions. In contrast, several families, including Staphylococcaceae, Streptococcaceae, Bacteroidaceae, Corynebacteriaceae, and Pasteurellaceae, were overrepresented in controls and replaced with other bacterial families as lesions progressed. Families that had a lower abundance in controls and increased in early-stage development included Spirochaetaceae, Mycoplasmataceae, Moraxellaceae, and Porphyromonadaceae. As lesions progressed to the classic DD morphological appearance, the diversity of the bacterial microbiota greatly decreased. Spirochaetaceae became the predominant family in the stage 3 and 4 lesions (69.7% and 94.3%). The only other family found to be a substantial (≥10.0%) constituent of stage 3 lesions was Mycoplasmataceae. Following treatment, there was a considerable shift in the bacterial population, with Spirochaetaceae and Mycoplasmataceae being considerably reduced, whereas Corynebacteriaceae, Moraxellaceae, and Porphyromonadaceae noticeably increased.
FIG 2.

Relative abundance, by stage, of bacterial families that represent at least 5% of the bacterial reads acquired. For simplicity, the colors of individual families are the same in both graphs. (A) Relative abundance as derived from 24 biopsy specimens sequenced using shotgun metagenomics; (B) relative abundance derived from 48 biopsy specimens sequenced using 16S based metagenomics.
16S sequencing.
A total of 20,042,461 paired-end sequencing reads were returned from the MiSeq. By using a fastq-join script, 17,003,839 overlapping forward and reverse sequences were aligned and joined. QIIME (38, 41) was then used to demultiplex and quality filter using the default quality filter settings. The total number of reads that passed quality filtering was 15,792,486, with a maximum number of reads per sample of 644,571 and a minimum of 40,584. The Greengenes (44) 16S rRNA gene database was used to designate OTUs in QIIME. The total number of unique OTUs identified from the data set was 6,001. The mean number of total OTUs per sample was 198,222, with a maximum of 340,971 and a minimum of 38,563 OTUs.
The alpha diversity (42) of each sample was calculated using rarefactions ranging from 10 to 100,000, with 10 iterations of each calculation. The alpha rarefaction plot (Fig. 3) demonstrates the average estimated number of species identified based on the number of sequences sampled for each of the six lesion stages. The endpoint of this plot is 100,000 sequences, based on the fact that 45 of 48 samples had more than 100,000 reads. As most of the data sets reached an asymptote on the chart, the depth of sequencing was demonstrated to be adequate, and relatively few unique species would be identified with additional sequencing. This plot also demonstrates that the diversity of samples greatly decreases as the lesions progress from normal skin (stage 0) to early lesions (stages 1 and 2) to the end-stage lesions (stages 3 and 4). Biopsy specimens with the most diversity were those obtained 9 days after treatment (stage 5).
FIG 3.
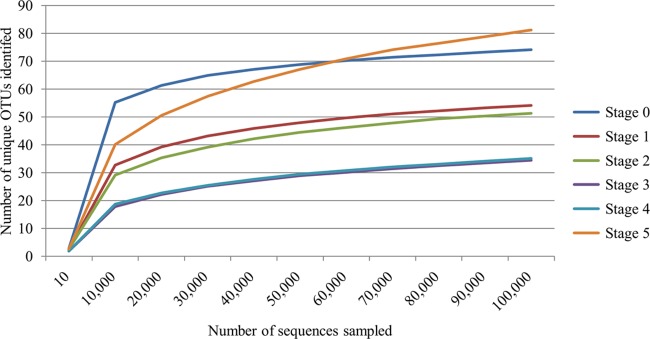
Alpha rarefaction plot of the number of species observed when the OTU table derived from 16S-based metagenomics was rarified in 10,000 increments from 10 to 100,000 reads. For each rarefaction, 10 (10) iterations were performed, and the number plotted represents the average number of OTUs identified.
Figure 2B is a plot of taxonomic hits from each stage of disease development in a plot similar to that in Fig. 2A, although Fig. 2B represents only 16S data and is independent from the shotgun data presented in Fig. 2A. Bacterial families that represent at least 5% of the bacterial flora for any given stage are shown. Eleven families were identified as composing at least 5% of the flora at each stage, of which six were the same as those identified in the shotgun data. Similar to what was shown by the shotgun data, the family Spirochaetaceae increased dramatically from only 1.3% in control feet and to 69.7% in stage 3 lesions. The increase in Spirochaetaceae was not as linear and systematic as the increase in the shotgun data, with the peak in abundance occurring in stage 3 lesions. In contrast to the shotgun data, there was a continued rise in abundance of Spirochaetaceae into stage 4 lesions. The only other bacterial family to be in sizable abundance in stage 3 and 4 lesions was again Mycoplasmataceae. Families that were overrepresented in control feet included Moraxellaceae, Corynebacteriaceae, Lachnospiraceae, and Ruminococcaceae. As lesions developed to stage 1 and stage 2, an increase in the families Spirochaetaceae, Mycoplasmataceae, Porphyromonadaceae, Campylobacteraceae, Aerococcaceae, and Tissierellaceae was noted. Following treatment, a rapid decline in Spirochaetaceae and Mycoplasmataceae occurred, concurrent with a rise in Moraxellaceae, Porphyromonadaceae, Bacteroidaceae, Corynebacteriaceae, Aerococcaceae, and Tissierellaceae, leading to a population that closely resembles that of control feet.
As the 16S data set provided 254 bp of overlapping sequences of the highly variable V3-V4 region of the bacterial 16S region, a deeper analysis of the genus and species was done on the family Spirochaetaceae to identify potential key species. All of the species identified in the family Spirochaetaceae belonged to the genus Treponema, with 45 unique species being identified from the Greengenes database. Figure 4 shows Treponema spp. that make up 5% or more of the total Treponema population at any given stage. To identify the Treponema spp. that were associated with each individual OTU call, the Greengenes reference 16S sequence was subjected to a BLAST search in the Ribosomal Database Project (RDP) (49). Each Treponema sp. listed is the bacterial isolate that had the highest RDP score that was also greater than 0.95. Any BLAST search that did not return an isolate with an RDP score greater than 0.95 was listed with a unique Treponema sp. number. Twelve unique Treponema spp. were identified as contributing to at least 5% of the Treponema population for any given lesion stage. The remaining 32 Treponema spp. have been combined to represent the other Treponema category in Fig. 4. The results of this analysis show that constituents of the Treponema population undergo dramatic shifts as the lesions develop. These shifts were consistently observed within individual samples of a given stage (data not shown). While the Treponema spp. in the control feet were primarily Treponema sp. 33 along with a mixture of other Treponema spp., as lesions developed to stage 1 and stage 2, there was a shift in the population, with five species making up the majority of the bacterial community. These include three Treponema spp. previously identified by Klitgaard et al. (26) and designated PT1, PT2, and PT3, all of which belong to the Treponema refringens-like phylogenetic cluster. The other two abundant species identified in these lesions are Treponema phagedenis and Treponema sp. 44. Of these five species, Treponema sp. PT2 and Treponema sp. 44 are the primary constituents of stage 1 lesions, encompassing 77.4% of the Treponema population, whereas Treponema sp. PT1, Treponema sp. PT3, and Treponema phagedenis make up 82.6% of the stage 2 Treponema population. The morphological shift from developing lesions to classic DD lesions is accompanied by a substantial increase in four Treponema spp. that were present in very low numbers in developing lesions. These included Treponema sp. PT8, Treponema denticola, Treponema pedis, and Treponema medium. These four species went from comprising only 0.1% and 2.6% of the population in stage 1 and stage 2 lesions, respectively, to comprising 68.1% and 69.3% of the Treponema population in stage 3 and stage 4 lesions, respectively. Accompanying this increase in the population of these four species was a rapid decline in four of the five highly abundant Treponema spp. identified in stage 1 and stage 2 lesions. Only Treponema phagedenis remained at a substantial level (24.9% and 23.4%) during this shift, as the other four species comprised less than 1% of the Treponema population in stage 3 and 4 lesions. Following treatment (stage 5), the diversity of Treponema spp. increased, with all 12 contributing to at least 1% of the population, whereas all other lesion stages had only five to seven species contributing at that level. Across all lesion stages, only Treponema phagedenis contributed to more than 0.1% of the population at every stage and ranged from 3.8% to 48.8% of the population throughout lesion development.
FIG 4.
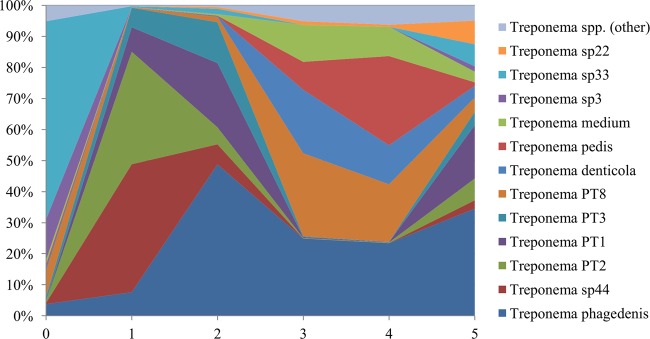
The percentage (of total Treponema hits) of each Treponema sp.-associated OTU plotted by stage of lesion. For each of the most abundant OTUs the “best-hit” RDP database match for the OTU sequence is provided in order to allow comparison to other studies. These best-hit matches are derived from 254-bp sequences aligned to the RDP database in the V3-V4 region, which is known to be highly diverse in Treponema spp.
A principal component analysis (PCoA) plot (50) was constructed using QIIME to analyze the beta diversity (42) between lesion stages. A 3-dimensional jackknifed (45) PCoA plot (Fig. 5) was assembled using Bray-Curtis distances using 100 repetitions, each sampling 10,000 sequences, from each sample to analyze the variation from one replicate to the next. The chart contains confidence ellipsoids around each sample to visualize the variation between sample replicates. The same highly abundant 11 taxa that were plotted in Fig. 2B were also plotted on the PCoA plot, where the coordinates of each taxon are plotted as the weighted average of the coordinates of all samples. The size of the ellipsoid around each taxon is proportional to the mean relative abundance of the taxon across all samples. This plot groups the individual biopsy specimens relatively well based on the Iowa DD scoring system, with the control feet grouping high on the PC1 and PC2 axes, early-stage lesions grouping along the entire PC1 axis and low on the PC2 axis, and end-stage lesions grouping very low on the PC1 axis and high on the PC2 axis. Following treatment, the biopsy specimens revert toward the high end of the PC1 axis grouping in the same area as control feet and early lesions. The main taxa responsible for the shift from the high to the low end of the PC2 axis on the PCoA plot were Mycoplasmataceae, Campylobacteraceae, Porphyromonadaceae, and Tissierellaceae, whereas the single biggest driver of the shift from the high to the low end of the PC1 axis was Spirochaetaceae. The taxa most closely associated with normal skin or very early lesion development include Ruminococcaceae, Aerococcaceae, Corynebacteriaceae, and Moraxellaceae. In order to test for statistical differences between the groups, a nonparametric ANOSIM analysis with 1,000 iterations was performed. This resulted in a P value of <0.001, indicating statistical differences between groups, and an R2 of 0.544, suggesting that roughly 54% of the differences between samples could be ascribed to their lesion stage.
FIG 5.

Three-dimensional jackknifed (45) replicate PCoA plot of Bray-Curtis distances. Jackknifing analysis utilized 100 iterations of 10,000 sequences each. The chart contains confidence ellipsoids around each sample to estimate the variation between stage replicates. The same highly abundant 11 taxa that are plotted in Fig. 1B are also plotted on the PCoA plot, where the coordinates of each taxon are plotted as the weighted average of the coordinates of all samples. The size of the ellipsoid around each taxon is proportional to the mean relative abundance of the taxon across all samples.
In order to determine the statistically and biologically significant organisms involved in the development of DD, the 16S data were analyzed using the web-based genome analysis tool Galaxy (46, 47), accessible from the Huttenhower lab (http://huttenhower.sph.harvard.edu/galaxy/). This analysis first uses a nonparametric factorial Kruskal-Wallis sum-rank test (51) to detect significant differential abundances between samples at the P < 0.05 level. Taxa that met this criterion underwent a linear discriminant analysis (LDA) to generate an LDA score to evaluate the effect size of each significant biomarker. This LEfSe (LDA effect size) analysis provides an output of all statistical and biologically significant taxa with an associated LDA score that is indicative of its relevance. For this data set, 412 taxa met the Kruskal-Wallis criterion of a P value of <0.05, and LDA scores ranged from approximately 2.58 to 5.51. A chart of all of the significant taxa and their LDA scores is provided in the supplemental material. Figure 6 shows a cladogram of the 47 taxa that had an LDA score greater than 4.5. The Greengenes database does not provide many of the species-level designations; therefore, all significant OTUs identified at the species level have been given a unique species number to represent potentially different species. The 4.5 level was used as an arbitrary cutoff point to represent approximately the top 10% most biologically significant taxa. When lower cutoff points were used, the number of significant taxa in stage 0 biopsy specimens greatly increased, from 4.25% to 67.4%, due to the larger diversity (albeit lower individual taxa significance) of the normal skin microbiota (alpha diversity shown in Fig. 3). The morphological subclassifications A and B were included in this analysis in an attempt to identify unique taxa associated with each lesion stage. All of the highly abundant families shown in Fig. 2B are also represented in the cladogram as being statistically and biologically significant. The only highly abundant family that did not have an LDA score greater than 4.5 was the family Lachnospiraceae, which had an LDA score of 4.42.
FIG 6.

Cladogram of the 47 taxa that had a Kruskal-Wallis P value of <0.05 and an LDA score greater than 4.5 following analysis by LEfSe. The center of the cladogram represents the bacterial phylum, and each sequential ring represents the next taxonomic level, down to the species level at the outermost ring. All nonsignificant taxa and taxa with LDA scores of <4.5 are represented in yellow, whereas significant taxonomic hits with LDA scores of >4.5 are colored to match the stage at which they were significantly overrepresented. The actual LDA score for each taxon shown on the cladogram is shown on the bar charts of the legend. For each significant OTU at the species level, a unique species number is shown along with the corresponding genus.
Based on the LEfSe analysis, there are distinct differences between the bacterial populations of A and B lesions. The only significant taxa identified in A1 lesions are Treponema sp. PT2 and Treponema sp. 44, while the A2 lesions were associated with Treponema phagedenis as well as Campylobacter ureolyticus. In contrast, there were no Treponema species significantly identified as biomarkers in B lesions. The significant biomarkers for B lesions were all included in the phyla Firmicutes and Actinobacteria. The families Corynebacteriaceae and Tissierellaceae were significant for B1 lesions, and the family Aerococcaceae was significant for B2 lesions. The biomarkers associated with stage 3 and 4 lesions were primarily from the genus Treponema. Stage 3 lesions were associated with Treponema denticola, Treponema medium, and Treponema sp. PT8, and stage 4 lesions were associated with Treponema pedis. The only other genus associated with classic DD lesions was Mycoplasma in stage 3 lesions. Following treatment (stage 5), several biomarkers from the classes Bacteroidia and Gammaproteobacteria were significantly associated with this stage.
To compare the data sets between the shotgun and 16S data, a Procrustes analysis was performed using the Bray-Curtis distances and 10,000 iterations of a Monte Carlo simulation between the 24 samples included in both data sets. The PCoA plots from both data sets have been combined in Fig. 7 to visualize the individual sample differences. Each biopsy run on both platforms is connected with a line between the 16S and shotgun data point. The points are also colored to correspond to the lesion stage represented. Monte Carlo simulations (52) resulted in a P value of <0.001, which indicates that regardless of the method used, both pipelines gave very similar results as to the microbial community involved in the development of DD.
FIG 7.

Procrustes analysis performed using the Bray-Curtis distances and 10,000 iterations of a Monte Carlo simulation between the 24 samples included in both the shotgun and 16S metagenomic data sets. Monte Carlo simulations resulted in a P value of <0.001.
Histopathology.
One hundred ninety-three biopsy specimens were collected over the time frame of this project and evaluated histologically by a pathologist who was blinded to the gross lesion stage. Biopsy specimens were stained with H&E and examined for pathological changes as well as stained with Warthin-Starry silver stain for evidence of spirochetes. A majority of the biopsy specimens (174/193) were categorized into one of three pathological descriptions (grades 1, 2, and 3). The remaining 19 biopsy specimens were described as having a unique pathological description and are grouped in the “other” category. The grade 1 category encompassed all biopsy specimens identified as normal bovine skin. Grade 2 lesions were described as hyperkeratotic and acanthotic with surface hemorrhage and erythrocytic crusts. This histologic description was found to be more prevalent in the early stages of DD development, with 48% of the early lesions being categorized this way. Grade 3 lesions were described as segmental, localized, necrotizing to necrosuppurative epidermitis with individual cell necrosis, ballooning degeneration of epithelial cells, necrotizing vasculitis, and intralesional bacteria consisting of delicate spirochetes, bacilli, and coccobacilli. This histologic description was of increasing prevalence in developing lesions (Fig. 8) and the most prevalent in end-stage lesions (70%). The biopsy specimens taken 9 days after treatment primarily fell into the grade 3 category as well, despite the metagenomic data indicating a large shift in the microbiota at this time. A summary of the Warthin-Starry staining for spirochetes is summarized in Fig. 9 whereas a biopsy specimen was classified as positive if there was any evidence of spirochetes within or on the surface of the skin regardless of the quantity of spirochetes present. Despite the chart plotting presence versus abundance (as in the metagenomic plots), the trend toward an increase in spirochetes as lesions develop is consistent with the microbiota shift in the metagenomic analysis.
FIG 8.
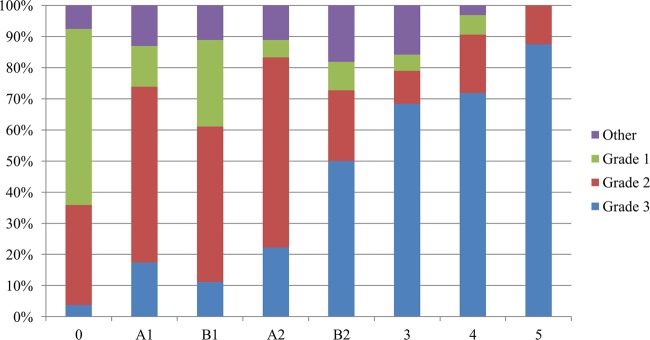
Percentage of each stage of lesion classified by histologic score following blinded assessment by a pathologist.
FIG 9.

Percentage of spirochete-positive biopsy specimens identified via histopathology for each stage of lesion development. Identification was based on silver staining of the biopsy specimen and evaluation by a pathologist as having spirochetes present or absent.
DISCUSSION
From the results presented here, we conclude that the bacterial communities associated with DD biopsy specimens change significantly as the lesions develop and that these bacterial communities are closely tied to the morphological stage of lesion development. Each morphological stage has been statistically validated as containing a unique microbiota, different from those of the prior or subsequent lesions stages. This conclusion is based on an extensive evaluation of more than 175 million sequences, evaluated with two different sequencing pipelines, encompassing 48 biopsy specimens from seven well-defined lesion stages as well as normal skin collected over a 3-year observational period.
Several key findings include a dramatic increase in Treponema sequences as lesions progress, a shift in the Treponema spp. between lesion stages, and a nearly complete absence of viral or fungal DNA. There also appear to be specific phylogenic lineages that serve as biomarkers for each of the lesion stages. Overall, these data provide strong evidence to support the view that DD is a polybacterial disease with significant changes in Treponema species and relative abundance as lesions develop through various morphological stages.
This evaluation of the temporal changes to the bovine foot metagenome throughout the early development of DD using culture-independent methods is the most in-depth one published to date. Several researchers (15, 53) have used 16S-based cloning to evaluate the DD microbiome by classic Sanger sequencing, creating at most 1,525 clones from nine bovine feet. Klitgaard et al. (54) utilized high-throughput 16S-based 454 sequencing to create 212,827 sequences from 40 cows; however, instead of using universal 16S primers, they utilized a custom Treponema-specific primer to explore the diversity of this bacterial genus in each biopsy specimen (54). Consequently, the relative abundance of the Treponema spp. was not provided in context with the overall bacterial community, although histopathologic fluorescent in situ hybridization analysis (FISH) did demonstrate that the majority of the bacteria colonizing the deep tissues of the lesions were Treponema spp. Additionally, it is not possible to ascertain if all Treponema spp. were indeed detected using the custom primer. In contrast, this study utilized biopsy specimens from 48 different feet with seven well-defined stages of development as well as normal control feet. Furthermore, the biopsy specimens were derived from cows with known lesion history (monitoring prior to and subsequent to sampling) as well as utilization of two separate metagenomic methods for analyzing the entire microbiome. The total numbers of sequences evaluated from these 48 biopsy specimens (156,433,474 and 20,042,461) also yielded much more depth than any previous study.
The results of the two separate sequencing techniques generated microbial communities with no significant differences when evaluated using a Procrustes analysis, further verifying the validity of the conclusions. The use of shotgun metagenomic methods allowed verification of previous findings (9) that bovine papillomavirus does not play a role in disease, nor is it likely that any other DNA-based viruses play a role, based on the very minimal amount of viral sequences identified. The shotgun data also allowed us to rule out the potential involvement of any fungal pathogens based on the very minimal amount of fungal DNA present.
Through the use of sequencing and histopathology, several key observations can be made from this project. In both 16S and shotgun sequencing, the amount of Treponema DNA isolated from each stage of development increased as the lesion score increased. This matched the histology results, where the percentage of biopsy specimens that had spirochetes identified with Warthin-Starry staining increased as the lesion scores increased. This population of Treponema spp. also appears to have different constituents at each of the various stages of development, which may help explain the variability noted in the Treponema spp. visualized from previous fluorescent in situ hybridization studies (33, 34, 54). The significance of these changes in Treponema species and relative abundance in relation to the possible role of Treponema spp. in the etiology of DD remains unclear at this time and warrants further investigation. It is clear that advanced lesions have a high percentage of Treponema spp. in relationship to other bacterial species, but it is still unclear if this is strictly an association or a true causation.
The Iowa DD staging system developed throughout the course of the 3 years of this study was validated by the statistically significant microbiota changes that occurred within each of the stages. While there is some overlap with the M scoring system (7), that system did not describe all of the morphological changes that were observed to take place throughout the course of lesion development. The Iowa DD scoring system was especially better at describing the morphological variability of the early-stage lesions. These stages were of particular interest due to the evidence that various bacterial complexes can lead to the same end-stage disease, such as the complexes involved in human gingivitis (55–58). It was also felt that the numerical scoring system better corresponded to the temporal development of disease and provided an easier method for evaluating feet as they progressed from one stage to the next. Although this study used “stage 5” to represent lesions that had been treated and were regressing, this was done for convenience of graphical representation, and it should be emphasized that after treatment, morphologically these lesions regress to earlier stages of development (stages 1 and 2) or back to normal skin. An article on the epidemiology of DD development that describes the time frame in which lesions progress and regress through these stages is in preparation. The results of our ANOSIM calculations in both the shotgun and 16S data verified that the microbial communities of each individual stage were statistically different from all other stages. Therefore, it is believed that is important to continue to utilize this more detailed scoring system when the progression of DD from normal skin to the diseased state is being evaluated.
The results of the previous DD 16S sequencing projects by Yano et al. (15) and Santos et al. (53) yielded similar results in their evaluation of normal healthy skin in comparison to DD lesions. Both studies found Proteobacteria to be abundant in healthy feet, and Yano et al. found Actinobacteria to be abundant in healthy feet. They also both demonstrated a large increase in Treponema spp. in DD lesions as well as some increase in the Tenericutes. These findings match the data sets reported here relatively well. Santos et al. found Tenericutes at a significant level only in the deep biopsy specimens, while our data show a peak of Tenericutes in the acute (stage 3) lesions, suggesting that they may play a role in the transition from early lesions to end-stage lesions. Both of the previous papers revealed a large number of Firmicutes in both healthy and diseased feet. In contrast, our data show Firmicutes at a high level in healthy tissue and early-stage lesions and a much lower level in end-stage lesions, suggesting a nonpathogenic role for this bacterial family. While both of these culture-independent studies had similar conclusions, the depth at which our study explored lesion development using high-throughput sequencing gives a closer look at the microbiota changes throughout the time course of disease development.
The Treponema spp. identified in stage 3 and 4 lesions are very similar to those identified by 454 pyrosequencing (54) from classic DD lesions. Klitgaard et al. identified four major Treponema clusters from DD lesions, which included T. denticola/T. pedis-like, T. phagedenis-like, T. refringens-like, and T. medium/T. vincentii-like clusters. The biopsy specimens used in that study were from classically described DD lesions, which would include the stage 3 and 4 lesions from the Iowa DD scoring system presented herein. The most abundant Treponema OTU sequence closest matches from stage 3 and 4 lesions included T. medium, T. pedis, T. denticola, T. phagedenis, and Treponema sp. PT8. Interestingly, of these five species, only T. phagedenis made up more than 2% of the Treponema population in early-stage lesions. In our analysis, the majority of the Treponema population in these early-stage lesions were classified as PT1, PT2, PT3, and an unclassified Treponema sp. most closely related to T. refringens (RDP score = 0.750). In another paper by Klitgaard et al. (26), an abnormal DD presentation (biopsy specimen 40) was described as “showing circumscribed, focal, moist plaque with increased thickness of epidermis.” The lesion photographed was found to be 100% colonized by Treponema sp. PT3 and would be classified as a B1 lesion in the Iowa DD scoring system. The finding that this early lesion did not contain any of the Treponema species identified in classic lesions is consistent with the results of the current study. This further documents the importance of understanding the progression of the disease and indicates that many of the Treponema spp. identified from the chronic end stage of disease may not be important at the onset of disease.
The identification of microbiota changes in the early stages of lesion development gives new insight into the microbial consortia required to initiate the onset of disease. The idea that various species of Treponema are important is not a novel concept (59–61), but the role that each of these species plays throughout the course of development has been expanded. There is a group of Treponema spp. that made up the majority of the Treponema population at the onset of disease development, whereas a completely different group of Treponema spp. was prevalent at the end stage of the disease process. Although the Greengenes database identified only one Treponema OTU at the species level (Treponema phagedenis), 47 unique OTUs were identified in the data set. Of these 47 OTUs, 11 of them comprise the majority of the Treponema population of these lesions. When these sequences were compared to the ribosomal database, many of them were identified as potential causative agents of DD. Klitgaard et al. (26) identified 10 unique Treponema isolates using 16S amplification and cloning of Treponema spp. from DD lesions. Three of the Treponema spp. that made up a majority of the population of early lesions are closely related to PT1, PT2, and PT3. Although this study may not be able to identify the various Treponema OTUs to the species level, there is a definite reproducible trend in the groups of Treponema spp. identified at the various stages of lesion development. Further work focused on identifying these specific Treponema OTUs at the species level is warranted to determine if certain Treponema spp. are important in initiation of disease, while other, later-colonizing Treponema spp. may simply colonize a niche setup by the early colonizing bacteria. In light of the fact that all biopsy specimens sampled as part of this study originate from a single dairy farm, it is possible that farm-to-farm variation in Treponema spp. or the predominant Treponema species might exist. Inclusion of additional farms in future sampling efforts is warranted in order to address the degree and role of this potential variability.
Despite the overabundance of Treponema spp. in end-stage lesions, there appears to be a number of other bacterial families that play a role in this polymicrobial disease process. The B-type morphological lesions appear to have an association of Corynebacteriaceae and Aerococcaceae with the superficial acanthosis and crusting that distinguishes this stage of development. The A-type lesions, which develop within the interdigital fold, appear to be colonized by several unique Treponema spp. as well as bacteria of the genus Campylobacter. This finding may be due to the unique, potentially microaerophilic area of the lesions located in the interdigital fold. Campylobacter spp. are known to grow best in vitro at an oxygen content of 5% (62, 63), and Treponema phagedenis has been found to grow best in the authors' laboratory at approximately 2% oxygen (A. Krull and P. Plummer, unpublished data). While these bacteria may not be the cause of DD, they certainly could play a role in deteriorating skin integrity and setting up an ideal environment for more pathogenic bacteria to thrive. While stage 3 and 4 lesions are obviously dominated by Treponema spp., it appears that Mycoplasma spp. may be of potential importance in the transition from an early lesion to an active lesion as well.
Although a number of papers have suggested that Bacteroides spp. and Porphyromonas spp. play a role in the polymicrobial nature of the disease, these data suggest that they may be opportunistic bacteria colonizing bovine feet in the absence of other bacteria, as they are seen becoming statistically significant 9 days after treatment. Another genus that has been proposed as potentially contributing to the disease process is Dichelobacter (29, 33, 34). Despite not being statistically significant in any particular stage of development, Dichelobacter did contribute to 1.73% of the bacteria in stage 1 and 2 lesions but less than 0.15% in all other lesion stages. This lack of statistical significance in any particular stage is most likely due to its presence in all four of the early lesion stages, and further investigation into Dichelobacter's role in the initiation of disease is warranted. It is also important to realize that the lower relative abundance of this organism does not preclude a critical role for it in the development of DD (64–68).
Regardless of the particular species identified in both the 16S and shotgun data, there is clear evidence that the bacterial diversity of the sample decreases as the lesions progress. The largest diversity of species was identified in control feet and posttreatment lesions, followed by early-stage lesions, and finally the lowest number of species was identified in end-stage lesions. Although on a different scale, the alpha rarefaction plot published by Santos et al. (53) showed a similar outcome with a much more diverse population in the healthy control feet than in DD lesions. Due to the fact that only 700 clones were sequenced, the alpha rarefaction plots from the paper by Santos et al. do not come close to the asymptote. In our study, the alpha rarefaction curves are very close to plateauing, indicating that additional sequencing would not reveal many more species.
While two deep-sequencing approaches were used, the shotgun method had the advantage of giving insight into the relative abundance of bacteria, viruses, and fungi in comparison to the eukaryotic host DNA. This method led to the conclusion that there is a relatively small percentage of bacteria associated with control feet, compared to upwards of 97% of the sequenced DNA associated with stage 3 and 4 lesions being bacterial DNA. It also verified that there was very little viral or fungal DNA present at any stage of lesion development. The limitation of this method was that with the relatively short reads of DNA from the entire genome of a wide range of bacterial species, there may have been a bias to identify more reads as bacteria whose entire genome has been sequenced. Therefore, it was felt that to attempt to mitigate this bias, these data were better utilized for analysis at the family level and not for analyzing OTU calls at the genus and species levels. Despite the fact that there were a high number of reads that did not match any known sequence, high-throughput sequencing did provide a significant amount of usable data. The use of 16S rRNA gene-based sequencing on the MiSeq platform enabled the use of overlapping 300-bp sequences to more confidently identify bacteria to the genus and species levels, since the V3-V4 region is well established for bacterial taxonomic classification (38, 69–71).
The results of the current study do not provide insights into the source of the bacteria identified in the DD lesions. Evans et al. utilized quantitative PCR to test environmental and animal samples for three of the Treponema spp. identified in DD lesions of this study (72). In that study, they were unable to identify the three DD-associated Treponema spp. in environmental or fecal samples; however, several samples from the bovine rectal mucosal junction and gingiva were positive. More recently, Klitgaard et al. demonstrated the presence of DD-associated treponemes in 43 of 64 samples sequenced by high-throughput next-generation sequencing and a custom treponeme-specific primer (73). Although the DD treponemes were present in very low abundance in environmental samples, the present study demonstrates their presence in the dairy slurry and environment, where they may act as a source of infection.
The blinded histopathologic classification of lesions provided in Fig. 8 demonstrates that some normal skin biopsy specimens (stage 0) demonstrate evidence of histopathologic changes. This finding is not surprising when one considers the increased sensitivity of histopathology to identify cellular change compared to gross observation. Several issues need to be considered in evaluating this data. First, Fig. 8 provides the histopathologic data from 193 biopsy specimens (i.e., not just the 48 included in the study) that were scored morphologically and then subjected to blinded histopathologic evaluation. Of the six stage 0 samples included in the metagenomic study, 5 had no diagnostic lesion (grade 1) and one had a grade 2 histopathologic change. Second, the presence of this discrepancy does not negate the utility of either staging system (i.e., the morphological staging or the histopathologic staging) but simply reflects different means of evaluating these lesions. Why several of these biopsy specimens exhibited these changes is uncertain; however, it is possible that maceration from constant exposure to a moist environment or other trauma could be associated with these cellular changes. Third, in each case of stage 0 biopsy specimens utilized for sequencing, the foot of the animals was observed for an average of 140 days prior to the biopsy sample collection and an average of 103 days after biopsy with no evidence of DD lesion development. This verifies that the samples are indeed representative of skin with no DD lesions. Finally, it is important to realize that the histopathologic classification system utilized in this study is not intended to be an implication of severity. Instead, each of the histopathologic classifications describes a type of change. Within each classification, there could be a gradient of severity that was not implied by the classification.
From these results, we conclude that the DD disease process is polybacterial, with no significant viral or fungal component. In addition, DD manifests itself through consistent and predictable changes in the bacterial microbiota of the bovine foot, as lesions systematically progress through multiple distinct stages. The identification of the early-stage lesions and the phylogenic biomarkers associated with each lesion stage will be instrumental in identifying the essential microbial consortia required to initiate the onset of disease. Further work toward identifying the role each of these bacteria play in the progression of disease is needed in order to create novel prevention strategies targeting specific pathogens.
Supplementary Material
ACKNOWLEDGMENTS
We thank Cory Haglund, Michael Slattery, Tim Kruse, Lance Dahlquist, Mackenzie Dickson, and the ISU Dairy Farm staff for their assistance in sample collection and animal management. Thanks also go to the technical support staff of the Iowa State Veterinary Diagnostic Laboratory histopathology laboratory.
Funds for this research project were provided by Boehringer Ingelheim Vetmedica Inc. and Iowa State University startup funds.
Footnotes
Published ahead of print 27 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02077-14.
REFERENCES
- 1.USDA. 2009. Dairy 2007, part IV: reference of dairy cattle health and management practices in the United States. USDA:APHIS:VS, CEAH, Fort Collins, CO. [Google Scholar]
- 2.Cheli R, Mortellaro CM. 1974. La dermatite digitale del bovino, p 208–213 In 8th International Meeting on Diseases of Cattle, Piacenza, Italy. [Google Scholar]
- 3.Read DH, Walker RL. 1994. Papillomatous digital dermatitis: pathologic findings, p 156 Proceedings of the 8th International Symposium on Disorders of the Ruminant Digit, Banff, Canada. [Google Scholar]
- 4.Walker RL, Read DH, Loretz KJ, Nordhausen RW. 1995. Spirochetes isolated from dairy cattle with papillomatous digital dermatitis and interdigital dermatitis. Vet. Microbiol. 47:343–355. 10.1016/0378-1135(95)00114-X. [DOI] [PubMed] [Google Scholar]
- 5.Grund S, Nattermann H, Horsch F. 1995. Electron microscopic detection of spirochetes in dermatitis digitalis of cattle. Zentralbl. Veterinarmed. B 42:533–542. [PubMed] [Google Scholar]
- 6.Cornelisse JL, Van Asten AJAM, Peterse DJ, Toussaint Raven E. 1982. Campylobacter faecalis as a participator of the bacterial flora of dermatitis digitalis in cows, p 1–3 4th International Symposium on Disorders of the Ruminant Digit, Maison Alfort, France. [Google Scholar]
- 7.Dopfer D, ter Huurne AAHM, Cornelisse JL, van Asten AJAM, Koopmans A, Meijer FA, Schukken YH, Szakall I, Klee W, Bosma RB. 1997. Histological and bacteriological evaluation of digital dermatitis in cattle, with special reference to spirochaetes and Campylobacter faecalis. Vet. Rec. 140:620–623. 10.1136/vr.140.24.620. [DOI] [PubMed] [Google Scholar]
- 8.Blowey RW, Carter SD, White AG, Barnes A. 1994. Borrelia burgdorferi infections in UK cattle: a possible association with digital dermatitis. Vet. Rec. 135:577–578. [PubMed] [Google Scholar]
- 9.Brandt S, Apprich V, Hackl V, Tober R, Danzer M, Kainzbauer C, Gabriel C, Stanek C, Kofler J. 2011. Prevalence of bovine papillomavirus and Treponema DNA in bovine digital dermatitis lesions. Vet. Microbiol. 148:161–167. 10.1016/j.vetmic.2010.08.031. [DOI] [PubMed] [Google Scholar]
- 10.Rebhun WC, Payne RM, King JM, Wolfe M, Begg SN. 1980. Interdigital papillomatosis in dairy cattle. J. Am. Vet. Med. Assoc. 177:437–440. [PubMed] [Google Scholar]
- 11.Choi BK, Nattermann H, Grund S, Haider W, Gobel UB. 1997. Spirochetes from digital dermatitis lesions in cattle are closely related to treponemes associated with human periodontitis. Int. J. Syst. Bacteriol. 47:175–181. 10.1099/00207713-47-1-175. [DOI] [PubMed] [Google Scholar]
- 12.Moter A, Leist G, Rudolph R, Schrank K, Choi BK, Wagner M, Gobel UB. 1998. Fluorescence in situ hybridization shows spatial distribution of as yet uncultured treponemes in biopsies from digital dermatitis lesions. Microbiology 144:2459–2467. 10.1099/00221287-144-9-2459. [DOI] [PubMed] [Google Scholar]
- 13.Evans NJ, Brown JM, Demirkan I, Singh P, Getty B, Timofte D, Vink WD, Murray RD, Blowey RW, Birtles RJ, Hart CA, Carter SD. 2009. Association of unique, isolated treponemes with bovine digital dermatitis lesions. J. Clin. Microbiol. 47:689–696. 10.1128/JCM.01914-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans NJ, Timofte D, Carter SD, Brown JM, Scholey R, Read DH, Blowey RW. 2010. Association of treponemes with bovine ulcerative mammary dermatitis. Vet. Rec. 166:532–533. 10.1136/vr.b4822. [DOI] [PubMed] [Google Scholar]
- 15.Yano T, Moe KK, Yamazaki K, Ooka T, Hayashi T, Misawa N. 2010. Identification of candidate pathogens of papillomatous digital dermatitis in dairy cattle from quantitative 16S rRNA clonal analysis. Vet. Microbiol. 143:352–362. 10.1016/j.vetmic.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 16.Gomez A, Cook NB, Bernardoni ND, Rieman J, Dusick AF, Hartshorn R, Socha MT, Read DH, Dopfer D. 2012. An experimental infection model to induce digital dermatitis infection in cattle. J. Dairy Sci. 95:1821–1830. 10.3168/jds.2011-4754. [DOI] [PubMed] [Google Scholar]
- 17.Berry SL, Ertze RA, Read DH, Hird DW. 2004. Field evaluation of prophylactic and therapeutic effects of a vaccine against (papillomatous) digital dermatitis of dairy cattle in two California dairies, p 130–137 13th International Symposium on Ruminant Lameness. [Google Scholar]
- 18.Berry SL, Read DH, Walker RL, Famula TR. 2010. Clinical, histologic, and bacteriologic findings in dairy cows with digital dermatitis (footwarts) one month after topical treatment with lincomycin hydrochloride or oxytetracycline hydrochloride. J. Am. Vet. Med. Assoc. 237:555–560. 10.2460/javma.237.5.555. [DOI] [PubMed] [Google Scholar]
- 19.Nishikawa A, Taguchi K. 2008. Healing of digital dermatitis after a single treatment with topical oxytetracycline in 89 dairy cows. Vet. Rec. 163:574–576. 10.1136/vr.163.19.574. [DOI] [PubMed] [Google Scholar]
- 20.Silva LA, Silva CA, Borges JR, Fioravanti MC, Borges GT, Atayde IB. 2005. A clinical trial to assess the use of sodium hypochlorite and oxytetracycline on the healing of digital dermatitis lesions in cattle. Can. Vet. J. 46:345–348 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1082879/. [PMC free article] [PubMed] [Google Scholar]
- 21.Ando T, Fujiwara H, Kohiruimaki M, Hayashi T, Ohtsuka H, Watanabe D, Oikawa M, Koiwa M. 2009. Peripheral blood leukocyte subpopulation of dairy cows with digital dermatitis and effect of hoof trimming with antibiotic treatment. J. Vet. Med. Sci. 71:391–395. 10.1292/jvms.71.391. [DOI] [PubMed] [Google Scholar]
- 22.Laven RA. 2006. Efficacy of systemic cefquinome and erythromycin against digital dermatitis in cattle. Vet. Rec. 159:19–20. 10.1136/vr.159.1.19. [DOI] [PubMed] [Google Scholar]
- 23.Laven RA, Logue DN. 2006. Treatment strategies for digital dermatitis for the UK. Vet. J. 171:79–88. 10.1016/j.tvjl.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 24.Logue DN, Offer JE, Laven RA, Ellis WA. 2005. Digital dermatitis—the aetiological soup. Vet. J. 170:12–13. 10.1016/j.tvjl.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 25.Trott DJ, Moeller MR, Zuerner RL, Goff JP, Waters WR, Alt DP, Walker RL, Wannemuehler MJ. 2003. Characterization of Treponema phagedenis-like spirochetes isolated from papillomatous digital dermatitis lesions in dairy cattle. J. Clin. Microbiol. 41:2522–2529. 10.1128/JCM.41.6.2522-2529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klitgaard K, Boye M, Capion N, Jensen TK. 2008. Evidence of multiple Treponema phylotypes involved in bovine digital dermatitis as shown by 16S rRNA gene analysis and fluorescence in situ hybridization. J. Clin. Microbiol. 46:3012–3020. 10.1128/JCM.00670-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Amstel SR, van Vuuren S, Tutt CL. 1995. Digital dermatitis: report of an outbreak. J. S. Afr. Vet. Assoc. 66:177–181. [PubMed] [Google Scholar]
- 28.Demirkan I, Carter SD, Murray RD, Blowey RW, Woodward MJ. 1998. The frequent detection of a treponeme in bovine digital dermatitis by immunocytochemistry and polymerase chain reaction. Vet. Microbiol. 60:285–292. 10.1016/S0378-1135(98)00146-1. [DOI] [PubMed] [Google Scholar]
- 29.Capion N, Boye M, Ekstrom CT, Jensen TK. 2012. Infection dynamics of digital dermatitis in first-lactation Holstein cows in an infected herd. J. Dairy Sci. 95:6457–6464. 10.3168/jds.2012-5335. [DOI] [PubMed] [Google Scholar]
- 30.Blowey RW, Sharp MW. 1988. Digital dermatitis in dairy cattle. Vet. Rec. 122:505–508. 10.1136/vr.122.21.505. [DOI] [PubMed] [Google Scholar]
- 31.Blowey RW, Done SH, Cooley W. 1994. Observations on the pathogenesis of digital dermatitis in cattle. Vet. Rec. 135:115–117. 10.1136/vr.135.5.115. [DOI] [PubMed] [Google Scholar]
- 32.Demirkan I, Williams HF, Dhawi A, Carter SD, Winstanley C, Bruce KD, Hart CA. 2006. Characterization of a spirochaete isolated from a case of bovine digital dermatitis. J. Appl. Microbiol. 101:948–955. 10.1111/j.1365-2672.2006.02976.x. [DOI] [PubMed] [Google Scholar]
- 33.Knappe-Poindecker M, Gilhuus M, Jensen TK, Klitgaard K, Larssen RB, Fjeldaas T. 2013. Interdigital dermatitis, heel horn erosion, and digital dermatitis in 14 Norwegian dairy herds. J. Dairy Sci. 96:7617–7629. 10.3168/jds.2013-6717. [DOI] [PubMed] [Google Scholar]
- 34.Rasmussen M, Capion N, Klitgaard K, Rogdo T, Fjeldaas T, Boye M, Jensen TK. 2012. Bovine digital dermatitis: possible pathogenic consortium consisting of Dichelobacter nodosus and multiple Treponema species. Vet. Microbiol. 160:151–161. 10.1016/j.vetmic.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 35.Schrank K, Choi BK, Grund S, Moter A, Heuner K, Nattermann H, Gobel UB. 1999. Treponema brennaborense sp. nov., a novel spirochaete isolated from a dairy cow suffering from digital dermatitis. Int. J. Syst. Bacteriol. 49:43–50. 10.1099/00207713-49-1-43. [DOI] [PubMed] [Google Scholar]
- 36.Nordhoff M, Moter A, Schrank K, Wieler LH. 2008. High prevalence of treponemes in bovine digital dermatitis—a molecular epidemiology. Vet. Microbiol. 131:293–300. 10.1016/j.vetmic.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 37.Schlafer S, Nordhoff M, Wyss C, Strub S, Hubner J, Gescher DM, Petrich A, Gobel UB, Moter A. 2008. Involvement of Guggenheimella bovis in digital dermatitis lesions of dairy cows. Vet. Microbiol. 128:118–125. 10.1016/j.vetmic.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 38.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2010. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U. S. A. 108(Suppl 1):S4516–S4522. 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards RA. 2008. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilke A, Harrison T, Wilkening J, Field D, Glass EM, Kyrpides N, Mavrommatis K, Meyer F. 2012. The M5nr: a novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinformatics 13:141. 10.1186/1471-2105-13-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuczynski J, Stombaugh J, Walters WA, Gonzalez A, Caporaso JG, Knight R. 2011. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Microbiol. Chapter 1:Unit 1E.5. 10.1002/9780471729259.mc01e05s27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lozupone CA, Knight R. 2008. Species divergence and the measurement of microbial diversity. FEMS Microbiol. Rev. 32:557–578. 10.1111/j.1574-6976.2008.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gower JC. 1975. Generalized Procrustes analysis. Psychometrika 40:33–51. 10.1007/BF02291478. [DOI] [Google Scholar]
- 44.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72:5069–5072. 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clarke KR, Warwick RM. 1998. A taxonomic distinctness index and its statistical properties. J. Appl. Ecol. 35:523–531. 10.1046/j.1365-2664.1998.3540523.x. [DOI] [Google Scholar]
- 46.Goecks J, Nekrutenko A, Taylor J. 2010. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11:R86. 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. 2010. Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. Chapter 19:Unit 19.10.1–21. 10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keegan KP, Trimble WL, Wilkening J, Wilke A, Harrison T, D'Souza M, Meyer F. 2012. A platform-independent method for detecting errors in metagenomic sequencing data: DRISEE. PLoS Comput. Biol. 8:e1002541. 10.1371/journal.pcbi.1002541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maidak BL, Larsen N, McCaughey MJ, Overbeek R, Olsen GJ, Fogel K, Blandy J, Woese CR. 1994. The Ribosomal Database Project. Nucleic Acids Res. 22:3485–3487. 10.1093/nar/22.17.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gonzalez A, Knight R. 2012. Advancing analytical algorithms and pipelines for billions of microbial sequences. Curr. Opin. Biotechnol. 23:64–71. 10.1016/j.copbio.2011.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McDonald JH. 2008. Handbook of biological statistics. Sparky House Publishing, Baltimore, MD. [Google Scholar]
- 52.Singleton DR, Furlong MA, Rathbun SL, Whitman WB. 2001. Quantitative comparisons of 16S rRNA gene sequence libraries from environmental samples. Appl. Environ. Microbiol. 67:4374–4376. 10.1128/AEM.67.9.4374-4376.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santos TM, Pereira RV, Caixeta LS, Guard CL, Bicalho RC. 2012. Microbial diversity in bovine papillomatous digital dermatitis in Holstein dairy cows from upstate New York. FEMS Microbiol. Ecol. 79:518–529. 10.1111/j.1574-6941.2011.01234.x. [DOI] [PubMed] [Google Scholar]
- 54.Klitgaard K, Foix Breto A, Boye M, Jensen TK. 2013. Targeting the treponemal microbiome of digital dermatitis infections by high-resolution phylogenetic analyses and comparison with fluorescent in situ hybridization. J. Clin. Microbiol. 51:2212–2219. 10.1128/JCM.00320-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ellen RP, Galimanas VB. 2005. Spirochetes at the forefront of periodontal infections. Periodontol. 2000 38:13–32. 10.1111/j.1600-0757.2005.00108.x. [DOI] [PubMed] [Google Scholar]
- 56.Simonson LG, McMahon KT, Childers DW, Morton HE. 1992. Bacterial synergy of Treponema denticola and Porphyromonas gingivalis in a multinational population. Oral Microbiol. Immunol. 7:111–112. 10.1111/j.1399-302X.1992.tb00519.x. [DOI] [PubMed] [Google Scholar]
- 57.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr 1998. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25:134–144. 10.1111/j.1600-051X.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 58.Takeuchi Y, Umeda M, Sakamoto M, Benno Y, Huang Y, Ishikawa I. 2001. Treponema socranskii, Treponema denticola, and Porphyromonas gingivalis are associated with severity of periodontal tissue destruction. J. Periodontol. 72:1354–1363. 10.1902/jop.2001.72.10.1354. [DOI] [PubMed] [Google Scholar]
- 59.Evans NJ, Brown JM, Demirkan I, Murray RD, Vink WD, Blowey RW, Hart CA, Carter SD. 2008. Three unique groups of spirochetes isolated from digital dermatitis lesions in UK cattle. Vet. Microbiol. 130:141–150. 10.1016/j.vetmic.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 60.Yano T, Yamagami R, Misumi K, Kubota C, Moe KK, Hayashi T, Yoshitani K, Ohtake O, Misawa N. 2009. Genetic heterogeneity among strains of Treponema phagedenis-like spirochetes isolated from dairy cattle with papillomatous digital dermatitis in Japan. J. Clin. Microbiol. 47:727–733. 10.1128/JCM.01574-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dopfer D, Anklam K, Mikheil D, Ladell P. 2012. Growth curves and morphology of three Treponema subtypes isolated from digital dermatitis in cattle. Vet. J. 193:685–693. 10.1016/j.tvjl.2012.06.054. [DOI] [PubMed] [Google Scholar]
- 62.Davis L, DiRita V. 2008. Growth and laboratory maintenance of Campylobacter jejuni. Curr. Protoc. Microbiol. Chapter 8:Unit 8A.1.1–8A.1.7. 10.1002/9780471729259.mc08a01s10. [DOI] [PubMed] [Google Scholar]
- 63.Humphreys H, O'Morain C. 1988. Culture of the organisms and histochemical identification. Scand. J. Gastroenterol. Suppl. 142:16–20. [DOI] [PubMed] [Google Scholar]
- 64.Celli J, Zahrt TC. 2013. Mechanisms of Francisella tularensis intracellular pathogenesis. Cold Spring Harb. Perspect. Med 3:a010314. 10.1101/cshperspect.a010314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Schaik EJ, Samuel JE. 2012. Phylogenetic diversity, virulence and comparative genomics. Adv. Exp. Med. Biol. 984:13–38. 10.1007/978-94-007-4315-1_2. [DOI] [PubMed] [Google Scholar]
- 66.Foley JE, Nieto NC. 2010. Tularemia. Vet. Microbiol. 140:332–338. 10.1016/j.vetmic.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 67.Sperandio V, Mellies JL, Nguyen W, Shin S, Kaper JB. 1999. Quorum sensing controls expression of the type III secretion gene transcription and protein secretion in enterohemorrhagic and enteropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 96:15196–15201. 10.1073/pnas.96.26.15196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin J, Smith MP, Chapin KC, Baik HS, Bennett GN, Foster JW. 1996. Mechanisms of acid resistance in enterohemorrhagic Escherichia coli. Appl. Environ. Microbiol. 62:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim M, Morrison M, Yu Z. 2011. Evaluation of different partial 16S rRNA gene sequence regions for phylogenetic analysis of microbiomes. J. Microbiol. Methods 84:81–87. 10.1016/j.mimet.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 70.Liu Z, DeSantis TZ, Andersen GL, Knight R. 2008. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res. 36:e120. 10.1093/nar/gkn491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Youssef N, Sheik CS, Krumholz LR, Najar FZ, Roe BA, Elshahed MS. 2009. Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Appl. Environ. Microbiol. 75:5227–5236. 10.1128/AEM.00592-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Evans NJ, Timofte D, Isherwood DR, Brown JM, Williams JM, Sherlock K, Lehane MJ, Murray RD, Birtles RJ, Hart CA, Carter SD. 2012. Host and environmental reservoirs of infection for bovine digital dermatitis treponemes. Vet. Microbiol. 156:102–109. 10.1016/j.vetmic.2011.09.029. [DOI] [PubMed] [Google Scholar]
- 73.Klitgaard K, Nielsen MW, Ingerslev H-C, Boye M, Jensen TK. 9 May 2014. Discovery of bovine digital dermatitis-associated Treponema in the dairy herd environment using a targeted deep-sequencing approach. Appl. Environ. Microbiol. 10.1128/AEM.00873-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.