

Abstract
Many Gram-negative bacteria utilize a type III secretion system (T3SS) to translocate virulence proteins into host cells to cause diseases. In responding to infection, macrophages detect some of the translocated proteins to activate caspase-1-mediated cell death, called pyroptosis, and secretion of proinflammatory cytokines to control the infection. Edwardsiella tarda is a Gram-negative enteric pathogen that causes hemorrhagic septicemia in fish and both gastrointestinal and extraintestinal infections in humans. In this study, we report that the T3SS of E. tarda facilitates its survival and replication in murine bone marrow-derived macrophages, and E. tarda infection triggers pyroptosis of infected macrophages from mice and fish and increased secretion of the cytokine interleukin 1β in a T3SS-dependent manner. Deletion of the flagellin gene fliC of E. tarda results in decreased cytotoxicity for infected macrophages and does not attenuate its virulence in a fish model of infection, whereas upregulated expression of FliC in the fliC mutant strain reduces its virulence. We propose that the host controls E. tarda infection partially by detecting FliC translocated by the T3SS, whereas the bacteria downregulate the expression of FliC to evade innate immunity.
INTRODUCTION
Edwardsiella tarda is a Gram-negative food- and waterborne pathogen that is recognized worldwide as a causative agent of hemorrhagic septicemia in fish. Humans with underlying immune disorders can be infected or colonized with E. tarda through consumption of undercooked seafood or by contact with infected marine life (1, 2). As in many other Gram-negative bacteria, the type III secretion system (T3SS) of E. tarda is vital for its pathogenesis (3, 4). Deletion of its T3SS ATPase gene esaN increased the 50% lethal dose (LD50) by approximately 1 log in the blue gourami infection model (5). The T3SS facilitates the survival and replication of different E. tarda isolates in different eukaryotic cells, such as Hep-2 epithelial cells (6), the epithelioma papillosum of carp cells (3), J774A.1 macrophages (4), and fish primary macrophages (3, 7).
In response to intracellular bacterial infection, at least two major types of cell death might occur: pyroptosis and apoptosis (8, 9). Through detection of bacterial components such as flagellin and the conserved inner rod protein of T3SS, infected macrophages are induced to assemble inflammasome complexes in which caspase-1 is activated. Active caspase-1 triggers pyroptosis and proteolytic maturation and secretion of the proinflammatory cytokines interleukin 1β (IL-1β) and IL-18 (10). Unlike pyroptosis, apoptosis is triggered typically through activation of caspase-3 and -7 rather than caspase-1 (11). Bacteria released from either pyroptotic or apoptotic cells can be phagocytosed and killed. This provides a mechanism to inhibit replication and dissemination of intracellular bacteria. However, some bacterial pathogens are able to modulate and/or avoid constantly triggering host cell death to establish bacterial infection. For instance, Salmonella enterica serovar Typhimurium translocates the effector AvrA through its pathogenicity island 1 (SPI-1)-encoded T3SS into host cells to dampen the proapoptotic innate immune response (12). To avoid activating caspase-1, intracellular S. Typhimurium downregulates expression of flagellin and SPI-1 genes (13, 14). E. tarda is a flagellated bacterium. The T3SS of E. tarda induces an upregulation of anti-apoptotic NF-κB target genes to protect J774A.1 murine macrophages from staurosporine-induced apoptosis, and this ability is required for intracellular replication in J774A.1 macrophages (4). However, nothing is known about pyroptosis and E. tarda infection.
In this study, we explored the possibility of using murine bone marrow-derived macrophages (BMDMs) as a model to study infection of E. tarda PPD130/91, a strain which has been used to investigate virulence factors, gene regulation of E. tarda, and interactions between E. tarda and hosts (15). We found that E. tarda infection induces pyroptosis of murine and fish macrophages in a T3SS-dependent manner.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains and plasmids used in this study are described in Table 1. E. tarda PPD130/91 (16) and its derived strains were grown in tryptic soy broth (TSB; BD Biosciences), or Dulbecco's modified Eagle medium (DMEM) to induce T3SS at 30°C (for the in vitro study) and 25°C (for the in vivo study), while E. coli strains were cultured in Luria-Bertani broth (LB; BD Biosciences) at 37°C. Cultivation of bacteria in DMEM was carried out in a 5% (vol/vol) CO2 atmosphere. When required, appropriate antibiotics were supplemented at the following concentrations: carbenicillin or ampicillin (50 μg/ml or 100 μg/ml), colistin (12.5 μg/ml), gentamicin (100 μg/ml), tetracycline (15 μg/ml), and chloramphenicol (34 μg/ml).
TABLE 1.
Strains and plasmids used in this studya
| Strain or plasmid | Description and/or genotype | Reference or source |
|---|---|---|
| E. tarda strains | ||
| PPD130/91 | Wild type; Kms Colr Amps; LD50 = 105.0 | 16 |
| ΔesaN strain | PPD130/91, in-frame deletion of esaN | 5 |
| ΔesaN/esaN strain | ΔesaN strain with pACYC-esaN | 5 |
| wt/GFP | PPD130/91 transformed with pFPV25.1 | This study |
| GFP-tagged ΔesaN strain | ΔesaN strain transformed with pFPV25.1 | This study |
| GFP-tagged ΔesaN/esaN strain | ΔesaN/esaN strain transformed with pFPV25.1 | This study |
| ΔfliC strain | PPD130/91, in-frame deletion of fliC | This study |
| ΔfliC/pACYC-fliC strain | ΔfliC strain with pACYC-fliC | This study |
| wt/pACYC184 | PPD130/91 with pACYC184 | This study |
| wt/pWSK29 | PPD130/91 with pWSK29 | This study |
| ΔfliC/pWSK29 strain | ΔfliC strain with pWSK29 | This study |
| ΔfliC/pWSK-fliC strain | ΔfliC strain with pWSK-fliC | This study |
| ΔflhB strain | PPD130/91, in-frame deletion of flhB | This study |
| ΔflhB ΔesaN strain | PPD130/91, in-frame deletion of flhB and esaN | This study |
| wt/pACYC-fliC2HA | PPD130/91 with pACYC-fliC2HA | This study |
| ΔflhB/pACYC-fliC2HA strain | ΔflhB strain with pACYC-fliC2HA | This study |
| ΔesaN/pACYC-fliC2HA strain | ΔesaN strain with pACYC-fliC2HA | This study |
| ΔflhB ΔesaN/pACYC-fliC2HA strain | ΔflhB ΔesaN strain with pACYC-fliC2HA | This study |
| E. coli strains | ||
| DH5α | α complementation | Stratagene |
| MC1061 λpir | (λpir) thi thr-1 leu6 proA2 his-4 argE2 lacY1 galK2 ara14 xyl5 supE44 λpir | 21 |
| S17-1 λpir | RK2 tra regulon, λpir, host for pir-dependent plasmid | 22 |
| Plasmids | ||
| pFPV25.1 | Derivative of pBR322 with gfpmut3A under the control of the constitutive promoter | 24 |
| pMD-18T | Ampr | TaKaRa |
| pACYC184 | Tetr Cmr | Amersham |
| pACYC-fliC | pACYC184 with wide-type fliC | This study |
| pACYC-fliC2HA | pACYC184 with C-terminal 2HA-tagged wild-type fliC | This study |
| pRE112 | pGP704 suicide plasmid, pir dependent; Cmr oriT oriV sacB | 19 |
| pRE112-fliCΔ1–416aa | pRE112 with fliC-flanking fragment, 1 to 416 amino acids deleted | This study |
| pRE112-flhBΔ4–430aa | pRE112 with flhB-flanking fragment, 4 to 430 amino acids deleted | This study |
| pWSK29 | Ampr | 23 |
| pWSK-fliC | pWSK29 with orf29/30 promoter and wild-type fliC | This study |
Km, kanamycin; Col, colistin; Amp, ampicillin; Cm, chloramphenicol; Tet, tetracycline; r, resistance; s, sensitivity.
Cells and culture conditions.
BMDMs were isolated from the femurs of 6- to 8-week-old C57BL/6 mice (Charles River) and grown in complete medium as described by Helaine et al. (17): RPMI (Gibco) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 10 mM HEPES, 50 μM β-mercaptoethanol, 20% (vol/vol) L929 cell-conditioned medium (National Institute for Medical Research), and 100 U/ml penicillin-streptomycin if necessary. J774A.1 macrophages were cultured in DMEM (Invitrogen) with 10% FBS and 10 mM l-glutamine.
Healthy mandarin fish (Siniperca chuatsi) were obtained from a commercial fish farm and maintained in well-aerated dechlorinated water at 25 ± 2°C. Leukocytes were isolated from the head kidneys of mandarin fish and purified following the procedure of Secombes (18) with slight modifications. Briefly, head kidneys were removed aseptically, pressed through a 100-μm nylon mesh (BD), and resuspended in DMEM supplemented with 2% FBS and 25 U/ml heparin. The cell suspensions were then layered onto a 51/34% discontinuous Percoll (Pharmacia) density gradients and centrifuged at 400 × g for 30 min. The band of leukocytes lying at the interface was collected, and the cells were washed three times with DMEM supplemented with 2% FBS and 10 U/ml heparin. Leukocytes were diluted in DMEM supplemented with 0.1% FBS and were seeded at 1.5 × 106 cells per well of a 96-well plate for 2 to 3 h to allow to adhere. Old medium and unattached cells were aspirated and DMEM with 1% FBS applied before infection.
Construction of mutants and plasmids.
Nonpolar deletion mutants of fliC and flhB were generated by sacB-based allelic exchange (19) as described previously (20). For example, two PCR fragments were generated from PPD130/91 genomic DNA for the construction of the ΔfliC strain. The primer pairs fliC-for plus fliC-int-rev and fliC-int-for plus fliC-rev were used. The resulting products were a 689-bp fragment containing the upstream region of fliC and a 682-bp fragment containing the downstream region of fliC. A 17-bp overlapping sequence introduced into the flanking DNA fragments made it possible to fuse them together by a second PCR with the primers fliC-for and fliC-rev. The resulting 1,354-bp product, deleting the whole coding sequence of fliC, was digested and ligated into the KpnI site of the suicide vector pRE112 (19) to create pRE112-fliCΔ1–416aa in E. coli MC1061 λpir (21). pRE112-fliCΔ1–416aa was then transferred into S17-1 λpir (22) to conjugate with E. tarda PPD130/91. Deletion mutants were screened on 10% sucrose–tryptic soy agar (TSA) plates. Mutants were verified by PCR (primer pair fliC-com-for and fliC-check-rev). The double-deletion ΔflhB ΔesaN mutant was screened based on ΔesaN (5). No mutants showed growth defects when cultured in TSB or DMEM. All primers used for the construction of mutants are listed in Table 2.
TABLE 2.
Oligonucleotides used in this study
| Designation | Sequence (5′–3′) |
|---|---|
| fliC-for | AAGCATGCTCACCGACGGTAATGGTCA |
| fliC-int-rev | AGTGTGCTTTCCTTCGAATGTTG |
| fliC-int-for | TCGAAGGAAAGCACACTTCGCGCATTACCGCGTGC |
| fliC-rev | AAGCATGCACCTTCACGCCGTTGAAGTC |
| pRE112-For | CAACAGTACTGCGATGAGT |
| pRE112-Rev | GGTGTAAGTGAACTGCATGA |
| fliC-check-rev | GCGGCAACCTTAGTCGTGTC |
| fliC-com-for | CGGGATCCCGTGCCAAGGGGAGCGCCGATA |
| fliC-com-rev | ACATGCATGCATGTTTAACGCAGCAGAGACAGGAC |
| fliC-2HA-rev | CGGAATTCTTACTAGAGGCTAGCATAATCAGGAACATCATACGGATAACGCAGCAGAGACAGGACG |
| Porf2930-for | CCAAGCTTAATGCCCTTTGGAGTATGGA |
| Porf2930-int-rev | GACACAAGGCACCGGTCGTTCGCCGGAACATGGTGCGA |
| Porf2930-fliC-int-for | CGACCGGTGCCTTGTGTCATGGCACAAGTAATTAATACCAAC |
| flhB-for | GGTACCCACGAACACCAGCGTCGGCTTCTCG |
| flhB-int-rev | AGCCGCCACGCTTAAGGCTATTTTAAGTTTCTC |
| flhB-int-for | CCTTAAGCGTGGCGGCTGACTCCGAAGAAGGAACACCG |
| flhB-rev | GGTACCGGCACCAGCCCTAACAGTCC |
| flhB-check-for | ATGTTCTCCAGCCAGGTGTTG |
| flhB-check-rev | GCGTAGTGGGTCGGGTTAGT |
To construct the complementing plasmid pACYC-fliC, the fliC gene and its upstream sequence were amplified with the primers fliC-com-for and fliC-com-rev and ligated into the BamHI and SphI sites of pACYC184 (Amersham). To study the secretion of fliC, we constructed another complementing plasmid, pACYC-fliC-2HA, with fliC-com-rev-2HA as the reverse primer. For creating pWSK-fliC, overlapping PCR was used to fuse the promoter of effector gene orf29/30 with fliC by using the primers Porf2930-for, Porf2930-int-rev, Porf2930-fliC-int-for, and fliC-com-rev. The resulting PCR product was ligated into the HindIII and EcoRI sites of pWSK29 (23). All the plasmids constructed were verified by DNA sequencing.
Replication assay by CFU.
BMDMs from C57BL/6 mice were seeded at a density of 5 × 105 cells per well in 24-well tissue culture plates 24 h before use. Overnight wild-type cultures, ΔesaN mutant cultures, and ΔesaN/esaN cultures expressing green fluorescent protein (GFP) from pFPV25.1 (24) in DMEM at 30°C were opsonized in DMEM with 10% normal mouse serum for 20 min at room temperature (RT). Bacteria were added to the monolayers at a multiplicity of infection (MOI) of ∼2.5, centrifuged at 170 × g for 5 min at RT, and incubated in a 5% CO2 incubator at 30°C for 30 min. The macrophages were washed once with prewarmed phosphate-buffered saline (PBS) and incubated in complete RPMI medium with 100 μg/ml gentamicin for 1 h. The macrophages were then incubated in complete RPMI medium supplemented with 16 μg/ml gentamicin for the rest of the experiment. For enumeration of intracellular bacteria at 1 h, 2 h, and 6.5 h after uptake, macrophages were washed four times with PBS and lysed with 0.1% Triton X-100 for 10 min, and a dilution series was spread onto TSB agar plates. Three replicates for each infection condition were analyzed, and the results were averaged. Replication of the wild-type strain expressing GFP was also assayed at 37°C following the procedure described for 30°C.
Replication assay by counting GFP-labeled bacteria.
BMDMs were seeded onto coverslips and infected as described for the CFU assay. At 0 h, 2 h, and 6.5 h after uptake, the cell monolayer was washed and fixed in 4% paraformaldehyde in PBS for 15 min at RT. Monolayers were then washed three times with PBS, permeabilized and blocked with 10% horse serum (Sigma) in 0.1% saponin–PBS (Sigma), and stained with rhodamine phalloidin (Invitrogen) at a 1:400 dilution for 1 h. After 3 washes, coverslips were mounted on glass microscope slides using ProLong Gold anti-fade reagent (Invitrogen), and bacterial replication was analyzed by enumerating the GFP-labeled bacteria against the cytoskeleton background under a fluorescence microscope equipped with a 100× objective lens (BX50; Olympus).
Cytotoxicity assay by propidium iodide uptake.
BMDMs were seeded at a density of 1 × 106 cells per glass bottom cell culture dish (20-mm diameter) 24 h before use. Infection was performed as described for replication assays. At 2 h after uptake, cells were stained live with 2 μg/ml propidium iodide (PI) in Opti-MEM (Invitrogen) for 20 min at RT in the dark. Cytotoxic cells were identified and scored for PI uptake into the nucleus. PI uptake was counted from more than 10 fields of view for each infection. Images were taken using a confocal laser scanning microscope (LSM510; Zeiss). The data presented are the averages from three independent experiments.
Cytotoxicity assays by lactate dehydrogenase (LDH) release.
BMDMs from C57BL/6 mice were seeded onto 24-well plates at a density of 2 × 105 cells per well and incubated overnight at 30°C. Infection was performed as described for the replication assay. At time zero h and 1 h and 2 h after uptake, supernatants from triplicate wells per infection condition were collected and analyzed for lactate dehydrogenase release using the cytotoxicity detection kit plus (LDH) (Roche), following the manufacturer‘s instructions. Spontaneous LDH release was measured from supernatants of uninfected cells, and total LDH release was measured from lysed uninfected cells. The optical density at 600 nm (OD600) was subtracted from the OD490 for each infection condition, and the percentage of LDH per infection condition was calculated as follows: percent LDH release = [(infected cell LDH release − spontaneous LDH release)/(total LDH release − spontaneous LDH release)] × 100.
For fish leukocytes, cells were infected with E. tarda at an MOI of 5.
To test the effect of a caspase-1 inhibitor on infection-induced cytotoxicity, cells were pretreated with 317 μM Ac-YVAD-AOM (acetyl–Tyr-Val-Ala-Asp–2,6-dimethylbenzoyloxymethyl ketone; Calbiochem) in 1% FBS-DMEM. The LDH release was measured at 2 h after uptake.
Detection of activated caspase-1.
BMDMs were infected as described for the replication assay. After 30 min of infection, the cell monolayers were washed twice and incubated in 200 μl of serum-free DMEM supplemented with gentamicin (50 μg/ml). After 2 h, the cell supernatants were collected, and the cells were lysed with 200 μl 4× lysis buffer. The supernatants and cell lysates were subjected to NuPAGE 12% bis-Tris gel (Novex) electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membranes (pore size, 0.2 μm). The membrane was probed with mouse anti-caspase-1 (p20) monoclonal antibody (Adipogen) at a 1:2,000 dilution and rabbit anti-β-actin polyclonal antibody (Proteintech) at a 1:5,000 dilution.
Cytokine measurements.
Levels of IL-1β and tumor necrosis factor alpha (TNF-α) secreted into tissue culture media during infection assays were measured with the Quantikine mouse IL-1β and TNF-α immunoassay kit following the manufacturer's instructions (R&D Systems). Supernatants from three replicate wells per infection condition were collected at 6.5 h after uptake, centrifuged to remove cellular debris, transferred to new tubes, and stored at −80°C until analyzed. A seven-point standard curve of 2-fold dilutions from 1,000 pg/ml to 15.625 pg/ml of recombinant mouse IL-1β and mouse TNF-α was used. A volume of 100 μl of the standards and samples were added to a 96-well microtiter plate. Standard curves were used to estimate levels of cytokines in each sample. The replicate values for each infection condition were averaged.
Secretion of FliC.
To examine the secretion of FliC, overnight cultures of E. tarda PPD130/91 (wild type), the ΔflhB and ΔesaN mutants, and the ΔflhB ΔesaN strain transformed with pACYC-fliC2HA were diluted 1:200 in TSB with 34 μg/ml chloramphenicol. Bacteria were then grown without shaking for 12 h at 25°C. Each bacterial culture was centrifuged. The supernatant was filtered (0.22-μm pore size; Millipore) and concentrated with an Amicon Ultra-15 centrifugal filtration device with a 10-kDa-molecular-mass-cutoff filter (Millipore). The bacterial pellet was resuspended in PBS and labeled as the total bacterial protein (P). Proteins were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. Membranes were probed with rabbit anti-hemagglutinin (HA) antibody (Proteintech) diluted at 1:2,000 and goat anti-rabbit IgG–horseradish peroxidase (HRP) at 1:3,000 (Millipore), with mouse anti-DnaK monoclonal antibody (Abcam) at 1:2,000 and goat anti-mouse IgG-HRP at 1:5,000 (Millipore), or with rabbit anti-EvpC antibody (25) diluted at 1:5,000 and goat anti-rabbit IgG-HRP.
Competitive index (CI) in blue gourami fish.
Mixed competitive infections in naive blue gourami (9.20 ± 1.55 g) were performed to determine the contribution of FliC to pathogenesis. Bacteria inoculated from a fresh plate were grown overnight at 25°C in TSB and subcultured at 1:40 for 4 h. The bacteria were then washed three times in PBS, and the OD540 was adjusted to 0.5. Equal amounts of bacteria were mixed together and injected intramuscularly (i.m.) at 5 × 104 CFU per fish. At 72 h postinoculation, livers were harvested and homogenized, and a series dilution was spread onto TSA plates supplemented with colistin. The colonies were then patched onto TSA colistin-chloramphenicol plates or TSA colistin-ampicillin plates to determine the ratio of different strains to the wild type. The CI is defined as the ratio of the test and wild-type strains within the output divided by their ratio within the input (26).
Statistical analysis.
Statistical tests were applied to data from at least three independent experiments. Probability (P) values were calculated by two-way analysis of variance and least-significant-difference (LSD) or Student's t test, as stated in the legend to Fig. 6, and were considered significant if they were less than 0.05.
FIG 6.
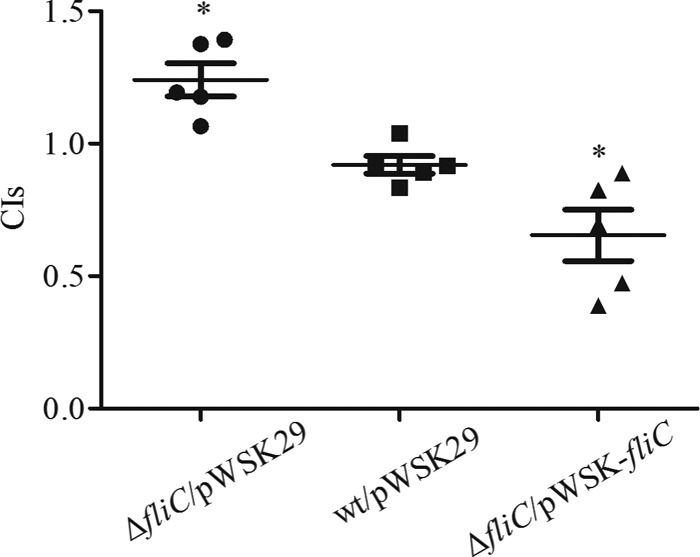
Competitive index analysis. Five naive blue gourami fish were used for each group. Fish were injected intramuscularly with mixtures of equal numbers of cells of the indicated strains and the wild-type strain and sacrificed 72 h after injection. CIs from livers for individual fish are presented, and the mean ± SD is also shown. Student's t test was used to calculate the P value with the hypothetical mean of 1.0 (*, P < 0.05).
RESULTS
Replication and survival of E. tarda PPD130/91 in BMDMs depends on temperature and its T3SS.
Replication of E. tarda PPD130/91 in fish epithelial cells and primary macrophages depends on its T3SS (3), whose expression is regulated by the ambient cultivating temperature in vitro (5, 27, 28). To recapitulate E. tarda replication in BMDMs and investigate the effect of temperature on this process, the E. tarda wild-type strain PPD130/91 was grown at 30°C to activate the T3SS and used to infect BMDMs at 37°C or 30°C. At different time points (1 h, 2 h, and 6.5 h) after uptake, intracellular bacteria were plated for CFU enumeration (Fig. 1A). At 37°C, the number of intracellular bacteria was drastically reduced at 6.5 h after uptake compared with the number at 1 h or 2 h after uptake. In contrast, bacterial numbers slightly increased over the time course at 30°C. This result suggests that temperature affects the intracellular survival/replication of E. tarda PPD130/91 in BMDMs, possibly through the regulation of its T3SS.
FIG 1.
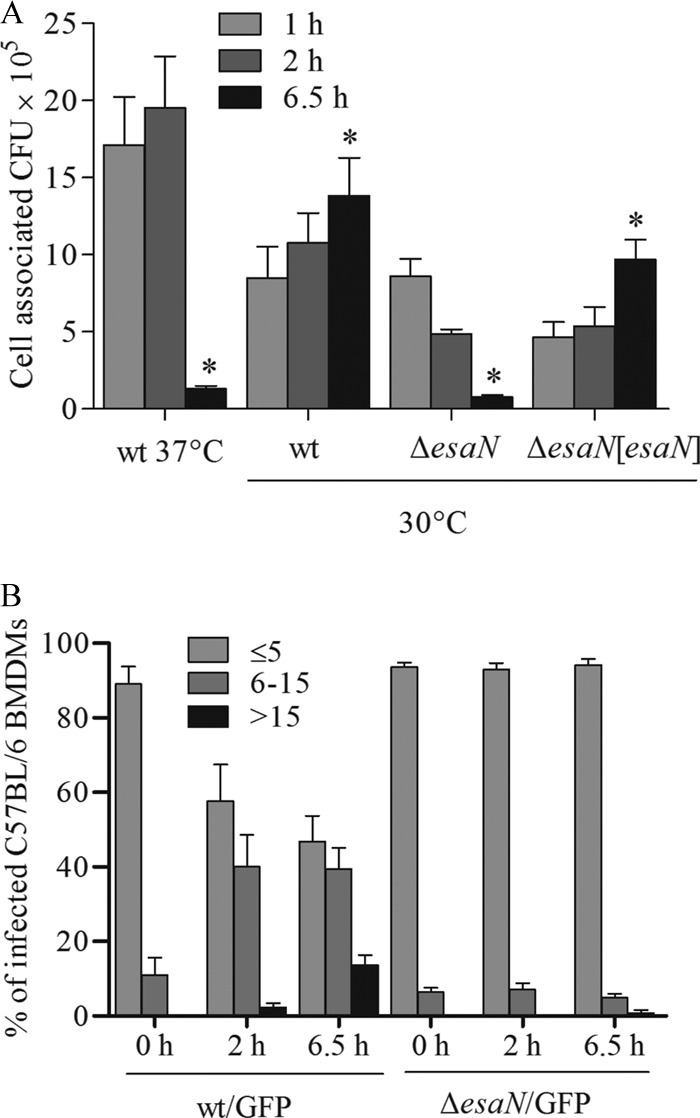
Survival and replication of E. tarda PPD130/91 in C57BL/6 BMDMs. (A) E. tarda PPD130/91 fails to replicate in BMDMs at 37°C, but T3SS-dependent replication is observed at 30°C. BMDMs were infected with opsonized bacteria at an MOI of ∼2.5. Intracellular bacteria were determined by CFU counting at the indicated times after infection. Experiments were performed in quadruplicate wells for each infection, and the data are from one representative infection. *, P < 0.05 (relative to the 1-h value). (B) Microscopic analysis of E. tarda PPD130/91 replication in BMDMs at 30°C. BMDMs were infected with bacteria expressing GFP and fixed at different time points. Cells were labeled with rhodamine phalloidin and subjected to confocal microscopy. At least 300 infected cells per infection were counted for the number of intracellular bacteria. Data are means ± standard errors of the means (SEM) from three independent experiments.
To test the effect of T3SS on bacterial growth in BMDMs at 30°C, an isogenic esaN mutant was used for replication assays (Fig. 1A). Both the mutant and wild-type bacteria were internalized at similar level (1 h after infection; P = 0.869). However, the proportion of the esaN mutant dropped to approximately 56.7% at 2 h and 16.1% at 6.5 h after internalization of bacteria. The growth defect of the esaN mutant was rescued by expressing EsaN from a plasmid (Fig. 1A). As an independent test for T3SS-dependent intracellular growth, we used fluorescence microscopy to count bacteria. To do this, BMDMs were infected at 30°C with bacterial strains expressing GFP from pFPV25.1, fixed at different time points after uptake, and labeled with rhodamine phalloidin for analysis. According to the number of bacteria in each cell, the infected cells were categorized into three groups: (i) no more than 5 bacteria; (ii) more than 5 but no more than 15; (iii) more than 15 bacteria. The percentages in each group are shown in Fig. 1B. This shows that wild-type bacteria replicated over the course of the experiment, while the ΔesaN mutant did not. Taken together, our results indicate that T3SS is required for growth of E. tarda PPD130/91 in BMDMs at 30°C as it is in fish primary macrophages (3).
E. tarda-induced pyroptosis in murine macrophages depends on the T3SS but not temperature.
In the replication assays at both 37°C and 30°C, we noticed that wild-type-infected BMDMs frequently became rounded and displayed membrane blebbing, a sign of apoptosis or pyroptosis (29, 30, 31). Therefore, propidium iodide (PI) staining was done to distinguish the type of cell death caused by E. tarda infection. BMDMs were infected at 30°C with different strains expressing GFP for 2 h, PI was then added, and cells were incubated for 20 min. Images were acquired with a confocal microscope (Fig. 2A), and PI-positive cells were scored. In infected cells, both wild-type and complemented ΔesaN/esaN strains exhibited a significantly higher ratio of PI-positive cells (57.89 ± 4.03% and 53.75 ± 10.58%, respectively) than the ΔesaN mutant (16.14 ± 6.0%) (Fig. 2B). This indicates that E. tarda induces pyroptosis of BMDMs in a T3SS-dependent manner.
FIG 2.

T3SS-dependent cytotoxicity in BMDMs and J774A.1 cells. (A) Confocal micrographs of PI uptake by BMDMs. BMDMs were infected with GFP-expressing strains for 2 h and stained with PI in Opti-MEM without fixation. Green, bacteria; red, PI-positive BMDMs. Bar, 50 μm. (B) Quantification of PI-positive infected cells. More than 300 infected cells for each infection were counted. (C) T3SS-dependent cytotoxicity of E. tarda PPD130/91 in BMDMs at 30°C determined by LDH assay. BMDMs were infected with different bacterial strains, and the culture supernatants were collected and processed for the LDH release assay. (D) T3SS dependent cytotoxicity of E. tarda PPD130/91 in J774A.1 at 30°C. (E) T3SS-dependent cytotoxicity of E. tarda PPD130/91 in BMDMs at 37°C. Samples were collected 1 h after uptake for analysis. Means ± SEM from three experiments are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
One feature of pyroptosis is rapid lysis of the affected cells and release of the cytosolic contents into the extracellular space. Hence, release of cytosolic LDH by infected BMDMs was also examined. Consistent with the above observations, release of LDH by BMDMs infected with the ΔesaN mutant at 30°C was much lower than that by cells infected with the wild type and the complemented strains at 2 h after uptake (Fig. 2C). This was not due to the different number of intracellular bacteria, as a similar result was obtained at 1 h after uptake (Fig. 2C), when wild-type and mutant strains had similar amounts of intracellular bacteria (Fig. 1A). The T3SS-dependent cytotoxicity occurred not only in BMDMs but also in J774A.1 macrophages (Fig. 2D). Furthermore, when the infection of BMDMs was processed at 37°C, a similar phenotype was also observed (Fig. 2E). Collectively, these results indicate that E. tarda-induced pyroptosis in murine macrophages depends on its T3SS but not the infection temperature.
Pyroptosis-triggered release of cytosolic LDH can be blocked by the caspase-1 inhibitor Ac-YVAD-AOM.
To test if a caspase-1 inhibitor could inhibit LDH release by E. tarda-infected BMDMs, cells were pretreated with Ac-YVAD-AOM and then infected with wild-type E. tarda for 2 h. The supernatants were then analyzed for release of cytosolic LDH. As shown in Fig. 3A, Ac-YVAD-AOM inhibited the LDH release by E. tarda-infected BMDMs. To verify that caspase-1 of BMDMs is activated upon E. tarda infection, culture supernatants were collected from uninfected or infected BMDMs for immunoblotting. A cleaved product of caspase-1 (p20) was detected in the supernatants of BMDMs infected with the wild type (wt) or the complemented esaN strain but not in the supernatants of uninfected BMDMs or those infected with the ΔesaN strain (Fig. 3B). These results demonstrate that the T3SS of E. tarda is required to activate caspase-1 to induce pyroptosis of infected cells.
FIG 3.
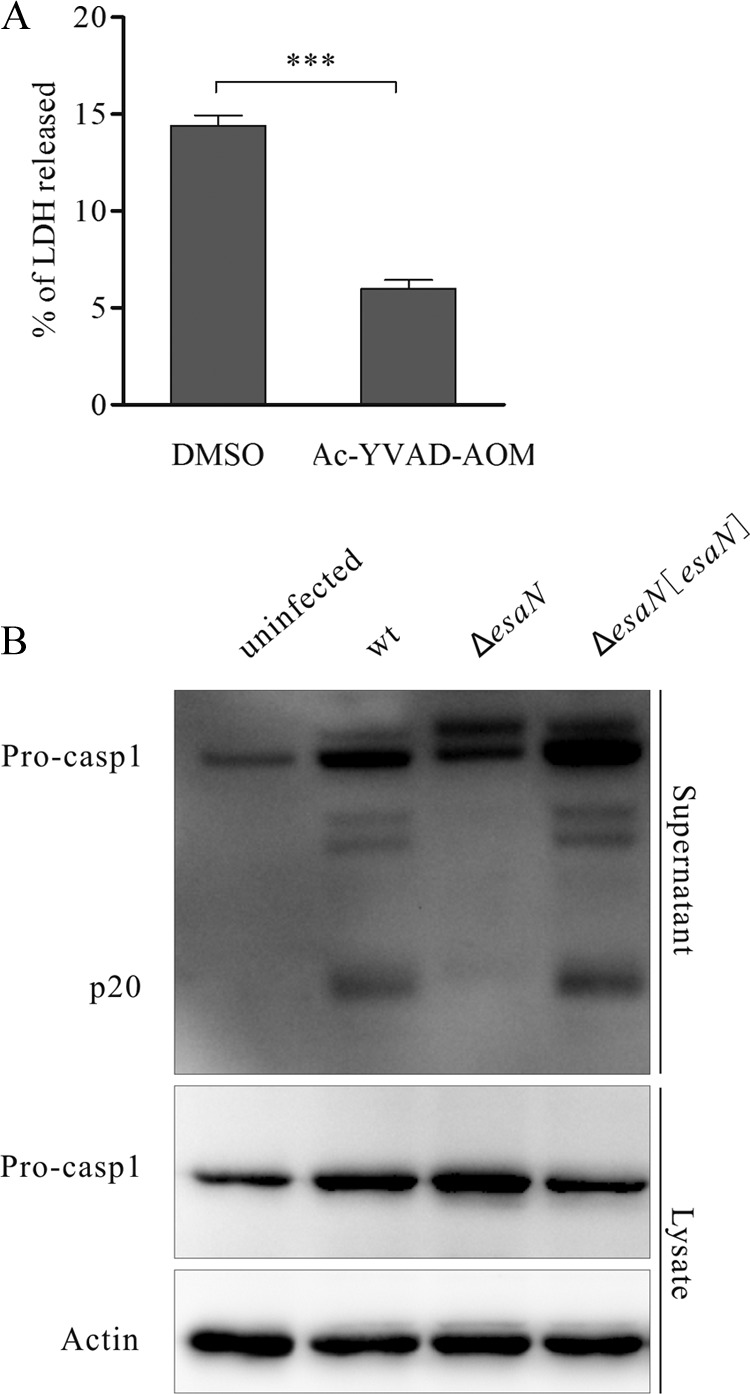
Caspase-1 of BMDMs is activated during E. tarda infection. (A) Effect of the caspase-1 inhibitor Ac-YVAD-AOM on the release of LDH. BMDMs were pretreated with 317 μM Ac-YVAD-AOM or dimethyl sulfoxide (DMSO) for 90 min and then infected with the wild type for 2 h. Culture supernatants were collected to determine LDH release. ***, P < 0.001. (B) E. tarda-induced activation of caspase-1. BMDMs were either mock infected or infected with indicated strains for 2 h. Cell culture supernatants were collected, and cell monolayers were lysed with 4× SDS sample buffer. Membrane was probed with anti-caspase-1 (p20) mouse monoclonal antibody. β-Actin was used to show the same loading of the cell lysates.
Secretion of IL-1β by infected BMDMs depends on the T3SS of E. tarda.
Next, we tested secretion of IL-1β by infected BMDMs, as pyroptosis is induced by activation of caspase-1, which also results in secretion of the proinflammatory cytokines IL-1β and IL-18. For this, BMDMs were infected with different strains for 6.5 h, and supernatants were collected for enzyme-linked immunosorbent assay (ELISA). There was much more IL-1β secreted by BMDMs infected with both the wild type and the complemented strains than in uninfected BMDMs or cells infected with the ΔesaN mutant (Fig. 4A). As a control, BMDMs were infected with the wild-type, ΔesaN mutant, and complemented mutant strains, and the levels of secreted TNF-α were measured. There was no difference between the ΔesaN mutant- and wild-type-infected BMDMs in the levels of this cytokine (Fig. 4B). These data suggest that specific secretion of IL-1β by BMDMs infected with E. tarda depends on its T3SS, consistent with the dependence of pyroptosis on the T3SS described above (Fig. 2 and 3).
FIG 4.

The E. tarda T3SS is required for secretion of IL-1β but not TNF-α by BMDMs. BMDMs were infected with different strains for 6.5 h, and supernatants were collected for ELISA. Supernatant from a noninfected cell culture was used as a control. Values are means ± standard deviations (SD) for triplicate cultures. **, P < 0.01; ***, P < 0.001. (A) Secretion of IL-1β. (B) Secretion of TNF-α.
Requirement of T3SS and flagellin FliC for pyroptosis of fish macrophages.
Having demonstrated that the T3SS of E. tarda is required to induce pyroptosis of murine macrophages, we asked if this was the case for fish macrophages and if flagellin protein FliC was required for pyroptosis, as has been shown for pyroptosis induced by other bacteria, such as Salmonella, Pseudomonas aeruginosa, and Legionella spp. (32, 33, 34, 35, 36). To this end, a nonpolar deletion mutant of fliC was created by allelic exchange and tested for its infection efficiency in fish primary macrophages. After 1 h infection, intracellular bacteria were enumerated by plating. There was no difference between the fliC mutant and the wild type (see Fig. S1 in the supplemental material), indicating that the infection efficiencies of the fliC mutant and the wild type are similar. Next, we checked the release of LDH by fish primary macrophages infected with different E. tarda strains. As shown in Fig. 5A, both ΔesaN and ΔfliC mutants induced significantly less release of LDH than did the wild-type strain or the fliC-complemented strain, in which fliC is constitutively expressed from pACYC-fliC. Interestingly, fish primary macrophages infected with the ΔfliC mutant released larger amounts of LDH than cells infected with the ΔesaN mutants did at 2 h after infection. Consistent with the role of caspase-1 in the release of cytosolic LDH by BMDMs, the caspase-1 inhibitor Ac-YVAD-AOM prevented the T3SS-induced release of LDH by wt-infected fish primary macrophages (Fig. 5B). These results suggest that FliC and other proteins translocated by the T3SS of E. tarda are required for inducing pyroptosis in infected fish macrophages.
FIG 5.
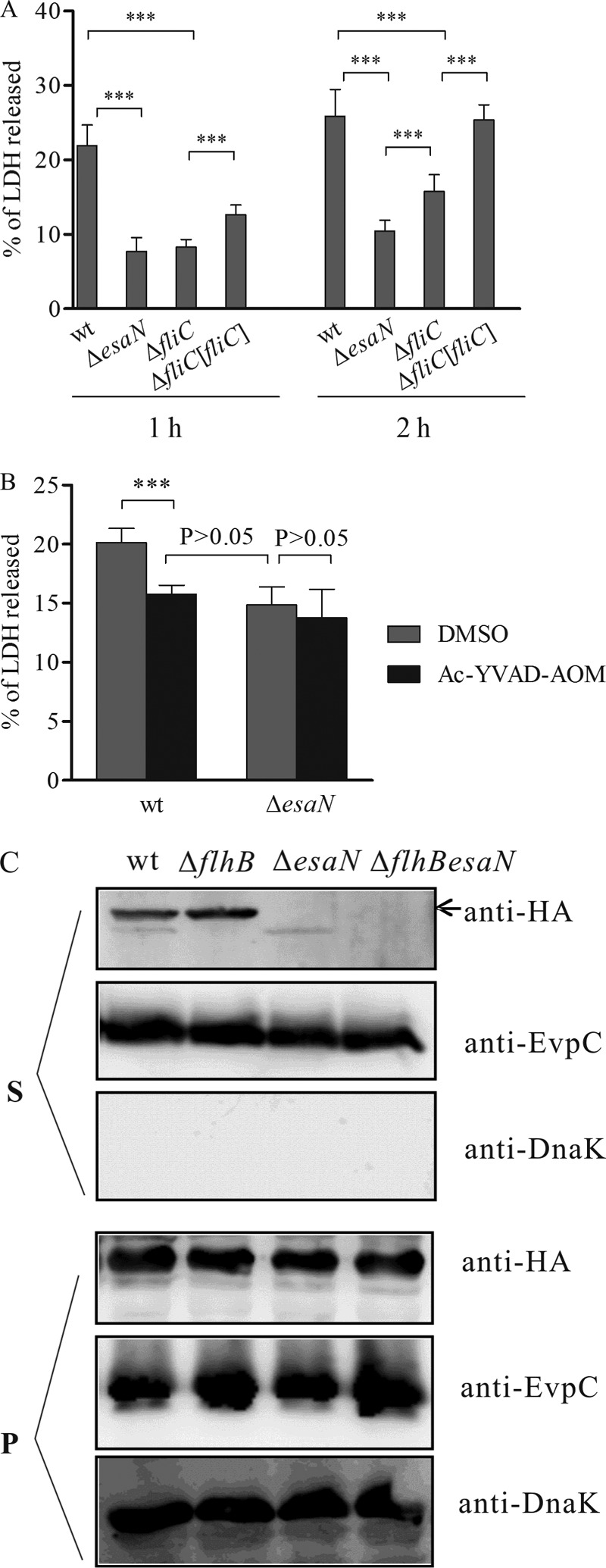
E. tarda T3SS and FliC are required for cytotoxicity for fish leukocytes. (A) Fish leukocytes isolated from head kidney were left noninfected or were infected at an MOI of 5 with different strains. After 1 h and 2 h, the levels of LDH released were analyzed. Values are means ± SD for 8 replicate cultures. ***, P < 0.001. (B) The release of LDH by fish leukocytes is inhibited by Ac-YVAD-AOM. Primary leukocytes were pretreated with 317 μM Ac-YVAD-AOM or DMSO for 90 min and then infected with the wild type or the esaN mutant for 2 h. Culture supernatants were collected to determine LDH release. ***, P < 0.001. (C) FliC2HA is secreted into the culture supernatant in a T3SS-dependent manner. Bacterial strains carrying pACYC-fliC2HA were grown in TSB for preparing protein samples from bacterial pellets (P) or culture supernatants (S). Membranes were probed with anti-HA for FliC2HA, anti-DnaK (a bacterial cytosolic marker), and anti-EvpC (a T6SS protein, which served as an internal loading control for different strains). DnaK was not detected in any culture supernatants, showing that detection of FliC2HA is not due to contamination from bacterial cells.
To test if FliC can be secreted by the T3SS of E. tarda, we constructed plasmid pACYC-fliC2HA and transformed the plasmid into the wild type or its isogenic mutant strains. Expression and secretion of FliC2HA were tested in vitro by immunoblotting using an antibody against the HA epitope. As shown in Fig. 5C, secreted FliC2HA was detected in the culture supernatants of both the wt and a flagellar-secretion-defective (ΔflhB) mutant but not in the ΔesaN mutant or the double ΔflhB ΔesaN mutant. Unlike FliC2HA, a type VI secretion system substrate, EvpC, was secreted by all strains tested, and the intrabacterial protein DnaK was not secreted by any of the strains. These data suggest that a C-terminal 2HA tag of FliC blocks its secretion by the flagellar secretion system but that it can be secreted in a T3SS-dependent manner.
Role of FliC in E. tarda virulence.
To test the effect of deleting FliC on E. tarda virulence, mixed infection in the naive blue gourami model was performed to determine the competitive index (CI) between the wt and the ΔfliC mutant. For this, pWSK29 was transformed into the ΔfliC mutant to distinguish the mutant from the wild-type strain. When equal numbers of wild-type and ΔfliC pWSK29 bacteria were mixed and used to infect naive blue gourami, the ΔfliC pWSK29 strain was slightly more competitive than the wild type (Fig. 6) (CI = 1.24 ± 0.14; P = 0.018). As a control, introducing pWSK29 into the wild-type strain did not affect its virulence. This shows that deletion of fliC does not attenuate its virulence in vivo but rather leads to slightly increased virulence.
Next, we investigated whether increased production of FliC could attenuate E. tarda virulence. As introducing pACYC184-based plasmid into E. tarda resulted in its attenuation in the naive blue gourami model (data not shown), we constructed plasmid pWSK-fliC, in which expression of FliC is under the control of the promoter controlling expression of the T3SS effector Orf29/30, and transformed the plasmid into the ΔfliC mutant. The ΔfliC pWSK-fliC strain was less competitive than the wild-type strain (Fig. 6) (CI = 0.65 ± 0.22). This result suggests that FliC-mediated pyroptosis is a host defense mechanism in the naive blue gourami fish model.
DISCUSSION
In this work, we demonstrated that E. tarda replicates in BMDMs and triggers macrophage pyroptosis and increased secretion of IL-1β in a T3SS-dependent manner. We also found that secretion of FliC could occur through the T3SS and that host cells respond to this by undergoing pyroptosis. This might be an important means by which E. tarda infection is controlled in the E. tarda/naive blue gourami infection model.
T3SS is a multiproteinaceous machinery that mediates the injection of effector proteins from the bacterial cell into the host cell. Through the action of different effectors, the T3SS of different bacterial pathogens enables them to invade nonphagocytic cells or inhibit phagocytosis by phagocytes, to downregulate innate immunity or modulate intracellular trafficking, and to establish a survival/replication niche. The T3SS of E. tarda has been shown to be essential for invading fish epithelial cells (3) and for replication in fish primary macrophages and J774A.1 macrophages. In this work, we demonstrated that the T3SS of E. tarda PPD130/91 is required for bacterial survival and replication in more restrictive BMDMs at 30°C (Fig. 1). The dependence of E. tarda PPD130/91 survival/replication in BMDMs on temperature is consistent with the role of temperature in the regulation of its T3SS (5, 27, 28). At 37°C, the expression and secretion of T3SS were suppressed via the downregulated expression of the two-component system regulatory protein EsrB by the PhoP-PhoQ system (28).
S. Typhimurium T3SSs not only translocate effectors into host cells to benefit bacterial infection but also occasionally translocate flagellin into host cells (33, 37, 38). Secretion of FliC of S. Typhimurium depends on its chaperone, FliS (39), which binds to the C-terminal region of FliC (40, 41). Spontaneous mutations in the C-terminal region of FliC result in its secretion defect in S. Typhimurium (42). Here, we showed that FliC2HA is unable to be secreted by the E. tarda flagellar system (Fig. 5C), indicating that the 2HA tag has an effect on FliC that prevents it from interacting with its chaperone and hence impairs FliC2HA secretion by the flagellar system. However, FliC2HA is secreted by the T3SS of E. tarda in vitro (Fig. 5C), suggesting that FliC can be translocated by the T3SS of E. tarda in vivo.
Translocated flagellin interacts with NAIP5 to activate NLRC4 inflammasomes (43, 44). Caspase-1 is then activated by NLRC4 inflammasomes, induces pyroptosis, and processes IL-1β and IL-18 to their active forms. One feature of pyroptosis is that the membrane-impermeative dye PI is able to enter through the pores in the cell membrane and stain pyroptotic cells (31). We showed here that wild-type-infected BMDMs have significantly higher numbers of PI-positive cells than those infected with the T3SS-null-mutant ΔesaN strain (Fig. 2B), and the level of secreted IL-1β by infected cells is significantly different between wild-type and the mutant strains (Fig. 4). Furthermore, the wild type is more cytotoxic than the ΔesaN mutant in the different macrophages tested (Fig. 2 and 5), and FliC plays a major role in inducing cytotoxicity (Fig. 5). The ΔfliC mutant is more cytotoxic to fish primary macrophages than the ΔesaN mutant (Fig. 5), suggesting that in addition to FliC, other proteins translocated by T3SS are able to induce cytotoxicity. The conserved inner rod protein EsaI and needle protein EsaG of T3SS might be candidates for such a role, as their homologs in other bacteria can also trigger pyroptosis by interacting with different NLRC4 inflammasome receptors (38, 43, 44). We showed here that, consistent with the role of caspase-1 in pyroptosis, the caspase-1 inhibitor Ac-YVAD-AOM blocks the LDH release induced by E. tarda infection (Fig. 3A and 5B), and a cleaved product of caspase-1 is detected in wt-infected but not esaN mutant-infected BMDMs (Fig. 3B). Caspase-1 homologs have been detected in sea bream and zebrafish (45, 46). Intriguingly, the caspase-1 inhibitor impaired S. Typhimurium-induced cell death of sea bream macrophages but did not prevent processing of IL-1β (47), whereas the processing of IL-1β by Francisella noatunensis-infected zebrafish primary leukocytes was considerably abrogated (48). It will be interesting to determine if E. tarda infection and E. tarda-induced pyroptosis induce processing of IL-1β in our model. Based on the data obtained here, we propose that E. tarda-induced pyroptosis depends on its T3SS and flagellin FliC.
Pyroptosis and secretion of proinflammatory cytokines IL-1β and IL-18 induced by pathogens play a vital role in controlling bacterial infection. To avoid inducing this innate immunity, intracellular S. Typhimurium downregulates the expression of flagellin FliC (14, 49). E. tarda possibly uses the same mechanism to avoid activating caspase 1, as the expression of flagellar genes in infected macrophages was greatly decreased (50). When FliC was constantly expressed from the promoter of the effector gene orf29/30, it attenuated E. tarda in the blue gourami fish model, as indicated by mixed infection (Fig. 6). Therefore, detection of FliC by host cells is likely to provide a mechanism to control E. tarda infection. This is consistent with the finding that constant expression of FliC by the promoter of sseJ, an effector gene of SPI-2 T3SS, attenuates S. Typhimurium in vivo (33). It was reported recently that FliC was essential for growth of E. tarda H1 in culture medium and normal secretion of putative translocator proteins EseC and EseB (51). These observations probably explain why the E. tarda H1 ΔfliC mutant was attenuated in the zebrafish model, as measured by the LD50. However, our E. tarda PPD130/91 ΔfliC mutant had a growth curve and amounts of secreted EseC and EseB similar to those of the wild-type E. tarda strain PPD130/91 (see Fig. S2 in the supplemental material) and was not attenuated in the blue gourami fish model (Fig. 6). Indeed, deletion of fliC tends to increase E. tarda virulence, as the CIs for livers were slightly higher than 1 (P = 0.018). This further supports the notion that E. tarda FliC-induced innate immunity controls E. tarda infection.
In conclusion, we have shown that E. tarda is able to survive and replicate in murine BMDMs in a T3SS- and temperature-dependent manner. This provides a useful model for future studies on the host factors that restrict intracellular E. tarda growth. FliC-triggered cytotoxicity of infected macrophages has an important role in controlling E. tarda infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Holden lab for helpful discussions and technical support and Li-Juan Zhao for experimental assistance.
This work was financially supported by National Natural Science Foundation of China no. 30972278 and no. 31172442 awarded to H.-X.X. and the National Basic Research Program of China, 973 program, no. 2009CB118703, to P.N. D.W.H. and X.-J.Y. acknowledge support from the Biotechnology and Biological Sciences Research Council of the United Kingdom.
Footnotes
Published ahead of print 2 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01065-13.
REFERENCES
- 1.Mowbray EE, Buck G, Humbaugh KE, Marshall GS. 2003. Maternal colonization and neonatal sepsis caused by Edwardsiella tarda. Pediatrics 111:e296–e298. 10.1542/peds.111.3.e296. [DOI] [PubMed] [Google Scholar]
- 2.Yousuf RM, How SH, Amran M, Hla KT, Shah A, Francis A. 2006. Edwardsiella tarda septicemia with underlying multiple liver abscesses. Malays. J. Pathol. 28:49–53. [PubMed] [Google Scholar]
- 3.Tan YP, Zheng J, Tung SL, Rosenshine I, Leung KY. 2005. Role of type III secretion in Edwardsiella tarda virulence. Microbiology 151:2301–2313. 10.1099/mic.0.28005-0. [DOI] [PubMed] [Google Scholar]
- 4.Okuda J, Arikawa Y, Takeuchi Y, Mahmoud MM, Suzaki E, Kataoka K, Suzuki T, Okinaka Y, Nakai T. 2006. Intracellular replication of Edwardsiella tarda in murine macrophage is dependent on the type III secretion system and induces an up-regulation of anti-apoptotic NF-kappaB target genes protecting the macrophage from staurosporine-induced apoptosis. Microb. Pathog. 41:226–240. 10.1016/j.micpath.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Xie HX, Yu HB, Zheng J, Nie P, Foster LJ, Mok YK, Leung KY. 2010. EseG, an effector of the type III secretion system of Edwardsiella tarda triggers microtubule destabilization. Infect. Immun. 78:5011–5021. 10.1128/IAI.00152-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okuda J, Kiriyama M, Yamanoi E, Nakai T. 2008. The type III secretion system-dependent repression of NF-kappaB activation to the intracellular growth of Edwardsiella tarda in human epithelial cells. FEMS Microbiol. Lett. 283:9–14. 10.1111/j.1574-6968.2008.01147.x. [DOI] [PubMed] [Google Scholar]
- 7.Srinivasa Rao PS, Lim TM, Leung KY. 2001. Opsonized virulent Edwardsiella tarda strains are able to adhere to and survive and replicate within fish phagocytes but fail to stimulate reactive oxygen intermediates. Infect. Immun. 69:5689–5697. 10.1128/IAI.69.9.5689-5697.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fink SL, Cookson BT. 2007. Pyroptosis and host cell death responses during Salmonella infection. Cell. Microbiol. 9:2562–2570. 10.1111/j.1462-5822.2007.01036.x. [DOI] [PubMed] [Google Scholar]
- 9.Ashida H, Mimuro H, Ogawa M, Kobayashi T, Sanada T, Kim M, Sasakawa C. 2011. Cell death and infection: a double-edged sword for host and pathogen survival. J. Cell Biol. 195:931–942. 10.1083/jcb.201108081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis BK, Wen H, Ting JP. 2011. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29:707–735. 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garrido C, Kroemer G. 2004. Life's smile, death's grin: vital functions of apoptosis-executing proteins. Curr. Opin. Cell Biol. 16:639–646. 10.1016/j.ceb.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Jones RM, Wu H, Wentworth C, Luo L, Collier-Hyams L, Neish AS. 2008. Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade. Cell Host Microbe 3:233–244. 10.1016/j.chom.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 13.Eriksson S, Lucchini S, Thompson A, Rhen M, Hinton JC. 2003. Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol. Microbiol. 47:103–118. 10.1046/j.1365-2958.2003.03313.x. [DOI] [PubMed] [Google Scholar]
- 14.Cummings LA, Wilkerson WD, Bergsbaken T, Cookson BT. 2006. In vivo, fliC expression by Salmonella enterica serovar Typhimurium is heterogeneous, regulated by ClpX, and anatomically restricted. Mol. Microbiol. 61:795–809. 10.1111/j.1365-2958.2006.05271.x. [DOI] [PubMed] [Google Scholar]
- 15.Leung KY, Siame BA, Tenkink BJ, Noort RJ, Mok YK. 2012. Edwardsiella tarda—virulence mechanisms of an emerging gastroenteritis pathogen. Microbes Infect. 14:26–34. 10.1016/j.micinf.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Ling SHM, Wang XH, Xie L, Lim TM, Leung KY. 2000. Use of green fluorescent protein (GFP) to track the invasive pathways of Edwardsiella tarda in in vivo and in vitro fish models. Microbiology 146:7–19. [DOI] [PubMed] [Google Scholar]
- 17.Helaine S, Thompson JA, Watson KG, Liu M, Boyle C, Holden DW. 2010. Dynamics of intracellular bacterial replication at the single cell level. Proc. Natl. Acad. Sci. U. S. A. 107:3746–3751. 10.1073/pnas.1000041107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Secombes CJ. 1990. Isolation of salmonid macrophages and analysis of their killing activity, p 137–154 In Stolen JS, Fletcher TC, Anderson DP, Roberson BS, Van Muiswinkel WB. (ed), Techniques in fish immunology. SOS Publications, Fair Haven, NJ. [Google Scholar]
- 19.Edwards RA, Keller LH, Schifferli DM. 1998. Improved allelic exchange vectors and their use to analyze 987P fimbria gene expression. Gene 207:149–157. 10.1016/S0378-1119(97)00619-7. [DOI] [PubMed] [Google Scholar]
- 20.Zheng J, Tung SL, Leung KY. 2005. Regulation of a type III and a putative secretion system in Edwardsiella tarda by EsrC is under the control of a two-component system, EsrA-EsrB. Infect. Immun. 73:4127–4413. 10.1128/IAI.73.7.4127-4137.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rubirés X, Saigi F, Piqué N, Climent N, Merino S, Albertí S, Tomás JM, Regué M. 1997. A gene (wbbL) from Serratia marcescens N28b (O4) complements the rfb-50 mutation of Escherichia coli K-12 derivatives. J. Bacteriol. 179:7581–7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simon R, Priefer U, Pühler A. 1983. A broad-host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Biotechnology 1:784–791. 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 23.Wang RF, Kushner SR. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195–199. 10.1016/0378-1119(91)90366-J. [DOI] [PubMed] [Google Scholar]
- 24.Valdivia RH, Falkow S. 1997. Fluorescence-based isolation of bacterial genes expressed within host cells. Science 277:2007–2011. 10.1126/science.277.5334.2007. [DOI] [PubMed] [Google Scholar]
- 25.Zheng J, Leung KY. 2007. Dissection of a type VI secretion system in Edwardsiella tarda. Mol. Microbiol. 66:1192–1206. 10.1111/j.1365-2958.2007.05993.x. [DOI] [PubMed] [Google Scholar]
- 26.Beuzón CR, Unsworth KE, Holden DW. 2001. In vivo genetic analysis indicates that PhoP-PhoQ and the Salmonella pathogenicity island 2 type III secretion system contribute independently to Salmonella enterica serovar Typhimurium virulence. Infect. Immun. 69:7254–7261. 10.1128/IAI.69.12.7254-7261.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rao PSS, Yamada Y, Tan YP, Leung KY. 2004. Use of proteomics to identify novel virulence determinants that are required for Edwardsiella tarda pathogenesis. Mol. Microbiol. 53:573–586. 10.1111/j.1365-2958.2004.04123.x. [DOI] [PubMed] [Google Scholar]
- 28.Chakraborty S, Li M, Chatterjee C, Sivaraman J, Leung KY, Mok YK. 2010. Temperature and Mg2+ sensing by a novel PhoP-PhoQ two-component system for regulation of virulence in Edwardsiella tarda. J. Biol. Chem. 285:38876–38888. 10.1074/jbc.M110.179150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cookson BT, Brennan MA. 2001. Pro-inflammatory programmed cell death. Trends Microbiol. 9:113–114. [DOI] [PubMed] [Google Scholar]
- 30.Lamkanfi M, Dixit VM. 2010. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 8:44–54. 10.1016/j.chom.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 31.Miao EA, Rajan JV, Aderem A. 2011. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 243:206–214. 10.1111/j.1600-065X.2011.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galle M, Schotte P, Haegman M, Wullaert A, Yang HJ, Jin S, Beyaert R. 2008. The Pseudomonas aeruginosa type III secretion system plays a dual role in the regulation of caspase-1 mediated IL-1beta maturation. J. Cell. Mol. Med. 12:1767–1776. 10.1111/j.1582-4934.2007.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. 2010. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11:1136–1142. 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amer A, Franchi L, Kanneganti TD, Body-Malapel M, Ozören N, Brady G, Meshinchi S, Jagirdar R, Gewirtz A, Akira S, Núñez G. 2006. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281:35217–35223. 10.1074/jbc.M604933200. [DOI] [PubMed] [Google Scholar]
- 35.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozören N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, Grant EP, Núñez G. 2006. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1 beta in Salmonella-infected macrophages. Nat. Immunol. 7:576–582. 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 36.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. 2006. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1 beta via Ipaf. Nat. Immunol. 7:569–575. 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 37.Sun YH, Rolán HG, Tsolis RM. 2007. Injection of flagellin into the host cell cytosol by Salmonella enterica serotype Typhimurium. J. Biol. Chem. 282:33897–33901. 10.1074/jbc.C700181200. [DOI] [PubMed] [Google Scholar]
- 38.Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A. 2010. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U. S. A. 107:3076–3080. 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Auvray F, Thomas J, Fraser GM, Hughes C. 2001. Flagellin polymerisation control by a cytosolic export chaperone. J. Mol. Biol. 308:221–229. 10.1006/jmbi.2001.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ozin AJ, Claret L, Auvray F, Hughes C. 2003. The FliS chaperone selectively binds to the disordered flagellin C-terminal D0 domain central to polymerization. FEMS Microbiol. Lett. 219:219–224. 10.1016/S0378-1097(02)01208-9. [DOI] [PubMed] [Google Scholar]
- 41.Muskotál A, Király R, Sebestyén A, Gugolya Z, Végh BM, Vonderviszt F. 2006. Interaction of FliS flagellar chaperone with flagellin. FEBS Lett. 580:3916–3920. 10.1016/j.febslet.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 42.Homma M, Fujita H, Yamaguchi S, Iino T. 1987. Regions of Salmonella typhimurium flagellin essential for its polymerization and excretion. J. Bacteriol. 169:291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kofoed EM, Vance RE. 2011. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477:592–595. 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600. 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 45.López-Castejón G, Sepulcre MP, Mulero I, Pelegrín P, Megeguer J, Mulero V. 2008. Molecular and functional characterization of gilthead seabream Sparus aurata caspase-1: the first identification of an inflammatory caspase in fish. Mol. Immunol. 45:49–57. 10.1016/j.molimm.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 46.Masumoto J, Zhou W, Chen FF, Su F, Kuwada JY, Hidaka E, Katsuyama T, Sagara J, Taniguchi S, Ngo-Hazelett P, Postlethwait JH, Núñez G, Inohara N. 2003. Caspy, a zebrafish caspase, activated by ASC oligomerization is required for pharyngeal arch development. J. Biol. Chem. 278:4268–4276. 10.1074/jbc.M203944200. [DOI] [PubMed] [Google Scholar]
- 47.Angosto D, López-Castejón G, López-Muñoz A, Sepulcre MP, Arizcun M, Meseguer J, Meseguer J, Mulero V. 2012. Evolution of inflammasome functions in vertebrates: inflammasome and caspase-1 trigger fish macrophage cell death but are dispensable for the processing of IL-1β. Innate Immun. 18:815–824. 10.1177/1753425912441956. [DOI] [PubMed] [Google Scholar]
- 48.Vojtech LN, Scharping N, Woodson JC, Hansen JD. 2012. Roles of inflammatory caspases during processing of zebrafish interleukin-1β in Francisella noatunensis infection. Infect. Immun. 80:2878–2885. 10.1128/IAI.00543-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stewart MK, Cummings LA, Johnson ML, Berezow AB, Cookson BT. 2011. Regulation of phenotypic heterogeneity permits Salmonella evasion of the host caspase-1 inflammatory response. Proc. Natl. Acad. Sci. U. S. A. 108:20742–20747. 10.1073/pnas.1108963108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Wang Q, Yang M, Xiao J, Liu Q, Wu H, Zhang Y. 2011. QseBC controls flagellar motility, fimbrial hemagglutination and intracellular virulence in fish pathogen Edwardsiella tarda. Fish Shellfish Immunol. 30:944–953. 10.1016/j.fsi.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 51.He Y, Xu T, Fossheim LE, Zhang XH. 2012. FliC, a flagellin protein, is essential for the growth and virulence of fish pathogen Edwardsiella tarda. PLoS One 7:e45070. 10.1371/journal.pone.0045070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.