

Abstract
Chronic obstructive pulmonary disease (COPD) is characterized by long periods of stable symptoms, but exacerbations occur, which result in a permanent worsening of symptoms. Previous studies have shown a link between bacterial colonization of the lower airways of COPD sufferers and an increase in exacerbation frequency. One of the most frequent bacterial colonizers is Streptococcus pneumoniae. To mimic this aspect of COPD, a murine model of low-level pneumococcal colonization in the lung has been developed, in which S. pneumoniae persisted in the lungs for at least 28 days. From day 14 postinfection, bacterial numbers remained constant until at least 28 days postinfection, and animals showed no outward signs of disease. The bacterial presence correlated with a low-level inflammatory response that was localized to small foci across the left and inferior lobes of the lung. The cellular response was predominantly monocytic, and focal fibroplasia was observed at the airway transitional zones. Physiological changes in the lungs were investigated with a Forced Maneuvers system. This new model provides a means of study of a long-term pulmonary infection with a human pathogen in a rodent system. This is an excellent tool for the development of future models that mimic complex respiratory diseases such as COPD and asthma.
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is a major public health problem and is predicted to be the third leading cause of death worldwide by 2020 (1). It is characterized by an airflow obstruction that is not fully reversible (2), but it is a largely heterogeneous condition (3), with patients displaying varied symptoms, including bronchitis and emphysema.
Current in vivo models sometimes offer poor translation to the human condition, highlighted by the observation that some compounds that have shown promise in preclinical animal models failed to demonstrate efficacy in human trials (4). This may be the result of a failure to model all aspects of COPD due to the complexity of the disease. One phenotype that has not been successfully modeled is the asymptomatic pulmonary colonization that occurs in COPD. Patients are frequently colonized by bacteria within their lungs (5) and can be prone to exacerbations induced by either bacteria or viruses that lead to a rapid deterioration in symptoms (6). A study by Patel et al. (7) has shown that the presence of bacteria in the lower airways was associated with an increased frequency of exacerbations. As well as this association with an increase in exacerbation frequency, colonizing bacteria are also considered a comorbid condition that contributes to the pathogenesis and clinical course of COPD, independently of exacerbations (8). The three most common bacterial species to colonize the lungs in stable-state COPD are nontypeable Haemophilus influenzae, Streptococcus pneumoniae, and Moraxella catarrhalis (9). Current in vivo models of infection with these pathogens are acute and do not mimic the low-level-persistent bacterial presence seen in COPD sufferers in the stable state (10, 11, 12). For example, in published murine models of S. pneumoniae infection, animals either quickly succumb to infection or the infection is cleared within a few days (13, 14, 15). Models of nasopharyngeal colonization with S. pneumoniae have been described (13), but these do not mimic the colonization of the lower airways seen in COPD.
This paper describes a model of long-term pulmonary colonization with S. pneumoniae. Pneumococci persisted in the lungs of mice for more than 1 month postinfection, with few or no outward signs of disease. The inflammatory response and pulmonary lung function have also been evaluated.
MATERIALS AND METHODS
Source of mice.
Female HsdOla:MF1 (MF1), Balb/cOlaHsd (BALB/c), and inbred CBA/CaOlaHsd (CBA/Ca) mice were obtained from Harlan Olac (Bicester, United Kingdom). Mice were used when they were at least 9 weeks old. Before use, mice were kept for at least 1 week, under standard conditions, in the University of Leicester's Division of Biomedical Services, with access to water and food ad libitum. All studies were performed in accordance with a United Kingdom Home Office license (60/4327) and were approved by the University of Leicester Ethics Committee. All mice were scored for signs of disease using the method described by Morton and Griffiths (16). Any mouse that became severely lethargic was culled, in accordance with the Home Office License.
Bacteria.
S. pneumoniae strains D39 (NCTC 7466, serotype 2, ST128), BHN191 (serotype 6B, ST138), and LgSt215 (serotype 19F, ST179) were used. BHN191 is an invasive strain obtained from Birgitta Henriques-Normark (Karolinska Institute, Sweden), and LgSt215 is a carriage strain obtained from Herminia de Lencastre (Instituto de Tecnologia Química e Biológica [ITQB], Universidade Nova de Lisbon, Oeiras, Portugal). A pneumolysin-negative mutant of LgSt215 was constructed by mariner mutagenesis of the ply gene in vitro followed by transfer of the mutated DNA into the pneumococcus using competence-stimulating peptide, as described previously (17). The successful mutation was confirmed by PCR and sequence analysis of transformants. Before use, pneumococci were confirmed by optochin sensitivity, Gram stain, catalase reaction, and α-hemolysis on blood agar plates. Bacteria were stored at −80°C, and, when required, aliquots were thawed, centrifuged, and resuspended in phosphate-buffered saline (PBS) (Oxoid, Basingstoke, United Kingdom) as previously described (15).
Infection of mice.
Mice were lightly anesthetized with a mixture of oxygen and 2.5% (vol/vol) isoflurane (Abbott Laboratories, Maidenhead, United Kingdom), and 1 × 106 to 5 × 106 CFU of S. pneumoniae suspended in 20 to 50 μl PBS was instilled in droplets across both nares. Animals were assessed for visible signs of disease (18) and were culled at predetermined time points or if they became severely lethargic.
Characterization of the infection and associated inflammation.
Mice were culled with an intraperitoneal injection of 250 μl Pentoject (20% [vol/vol] sodium pentobarbital; Pharmasol Ltd., Andover, United Kingdom). Death was confirmed by nonresponsiveness to noxious stimuli (hind paw pinch) and exsanguination. The trachea was cannulated and bronchoalveolar lavage fluid (BALF) was collected with three 300-μl washes of PBS. The volume of recovered BALF was determined by gravimetric analysis. A viable count was performed to enumerate bacterial levels, and then BALF was centrifuged at 1,850 × g for 10 min, and supernatants were placed at −80°C for cytokine analysis to be performed at a later date. The pellet was resuspended in 200 μl PBS, and 50 μl of suspension was centrifuged at 160 × g for 3 min in a Cytospin Slide centrifuge (Shandon Southern Products Ltd., United Kingdom). Slides were allowed to dry overnight and then stained using a REASTAIN Quick-Diff kit (Reagena, Finland) according to the manufacturer's instructions. Once dry, a coverslip was mounted onto the slides with DPX mountant (Fisher Chemical, Loughborough, United Kingdom).
Postmortem, blood was collected from the vena cava using a 1-ml insulin syringe (U-100 insulin [Terumo]). Blood was allowed to clot at room temperature and then centrifuged at 7,000 × g for 10 min, and serum was collected and stored at −80°C until needed.
Cytokine levels of BALF supernatant and sera were assayed with Duoset mouse enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Abingdon, United Kingdom) by following the manufacturer's instructions.
For the gravimetrical assessment of pulmonary edema, postmortem lungs were harvested into preweighed vials. The wet weight of lungs was determined, and then lungs were placed into an oven at 60°C for 4 h to dry. The dry weight of lungs was then determined. Levels of pulmonary edema were expressed as the ratio of wet and dry lung weight.
Histological analysis of samples.
For histopathological analysis, the trachea was cannulated and then the lungs were distended with approximately 400 μl 10% (vol/vol) neutral buffered formalin (Sigma, United Kingdom). Lungs were kept in 10% (vol/vol) neutral buffered formalin for a minimum of 24 h. Tissues were embedded overnight with a Leica tissue processor (LEICA TP 1050 fully enclosed vacuum tissue processor). Tissues were dehydrated through graded alcohols (industrial methylated spirits and ethanol) and into paraffin wax and then embedded using a Leica Histoembedder. Blocks were stored at room temperature. Sections (4 μm) were taken with a Leica Jung RM2155. Cut sections were floated on water (37°C) and transferred to charged slides. The slides were dried overnight at 45°C.
Hematoxylin and eosin (H&E) staining.
Slides were dewaxed in xylene and taken through graded alcohols. The slides were stained with Gill's hematoxylin (Pioneer Research Chemicals Ltd., United Kingdom) and then were washed in running water for around 10 min to “blue” the hematoxylin. The slides were submerged in eosin Y (high purity; Acros Organics, New Jersey, USA) for approximately 2 min, washed briefly in running water, and cleared through alcohols to xylene. Slides were mounted with Hystomount (Hughes and Hughes, Somerset, United Kingdom) and covered with a coverslip.
Immunohistochemistry.
Sections were stained with rabbit anti-CD3 antibody (Ab690; Abcam, Cambridge, United Kingdom). After rehydration through graded alcohols, slides were heated for 1 min in high pH (1 mM EDTA [Sigma, United Kingdom], pH 9.0) antigen retrieval solution. Slides were quenched in 3% (vol/vol) hydrogen peroxide (Sigma, United Kingdom) in methanol (Fisher, Loughborough, United Kingdom) for 5 min and blocked for 20 min with 20% (vol/vol) goat serum (Dako, Ely, United Kingdom). Slides were incubated for 60 min with primary antibody (2 μg/ml) or rabbit IgG (Dako, Ely, United Kingdom) as an isotype control. VectaStain Elite ABC (Vector Laboratories, Peterborough, United Kingdom) and diaminobenzidine (DAB) (Vector Laboratories) were used to visualize the bound antibodies, and slides were counterstained with Gill's hematoxylin (Pioneer Research Chemicals Ltd.). Slides were washed briefly in water and dehydrated through the alcohols to xylene, mounted with DPX (Fisher, Loughborough, United Kingdom), and covered with a coverslip.
Indirect ELISA.
Indirect ELISA was performed to analyze the titer of IgG antibody against strain LgSt215 (method adapted from reference 19). MaxiSorp 96-well plates (Nunc, United Kingdom) were coated with 100 μl PBS containing 1 × 106 CFU/well pneumococcal strain LgSt215 and were incubated overnight at 4°C. Plates were washed 3 times with PBS and 0.05% (vol/vol) Tween 20 (Sigma). Serum samples were added at a starting dilution of 1:50; it was then serially diluted 2-fold. Plates were subsequently incubated for 2 h at 37°C, washed three times with 0.05% (vol/vol) Tween 20 in PBS, and then coated with biotinylated goat anti-mouse IgG at a 1:5,000 dilution and incubated for 1 h at room temperature. Wells were incubated with streptavidin-horseradish peroxidase (HRP; R&D Systems, Abingdon, United Kingdom) for 20 min, before being washed 3 times with 0.05% (vol/vol) Tween 20 in PBS. TMB substrate solution (BD Opt EIA) was then added to each well, and after 5 min 50 μl of 0.5 M H2SO4 was added to stop the reaction. The absorbance at 450 nm was determined.
Lung function.
Lung function was assessed using an eSpira Forced Maneuvers system (EMMS, Borden, United Kingdom). Mice were anesthetized with an intraperitoneal injection of anesthetic solution containing ketamine (100 mg/kg of body weight; Fort Dodge Animal Health, Southampton, United Kingdom) and medetomidine (0.25 mg/kg; Dechra Veterinary Products Ltd., Shrewsbury, United Kingdom), and the tracheas were cannulated. Mice were allowed to breathe spontaneously and were monitored in a whole-body plethysmograph with a pneumotachograph connected to a transducer. Transpulmonary pressure was assessed via an esophageal catheter. Mice were pretreated with a volume history maneuver in which the lungs were inflated to 20 cm H2O for 1,000 ms to improve airway patency prior to the start of the maneuvers. Baseline lung function was measured for 1 min; baseline airway resistance was calculated using the eDaq software (EMMS, Borden, United Kingdom). Three semiautomatic maneuvers were performed in triplicate per mouse: forced expiratory volume (FEV), functional residual capacity (FRC), and quasistatic pressure volume curves. The FEV maneuver recorded FEV at 25 ms (FEV25), 40 ms (FEV40), 50 ms (FEV50), and 60 ms (FEV60) and forced peak expiratory flow (PEF). Tidal volume (TV) was calculated by the software from quasistatic pressure volume curves.
Statistical analyses.
GraphPad Prism software version 6 was used to analyze all data. The nonparametric Kruskal-Wallis test, with Dunn's posttest, was used to compare differences between antibody and cytokine levels and cell counts. Results were considered significant when P values were <0.05. Error bars in all figures show the standard errors of the means, unless otherwise stated.
RESULTS
Development of a model of long-term pulmonary colonization with S. pneumoniae.
To develop this model, three factors were investigated: dose volume, serotype of S. pneumoniae, and strain of mouse. Initially, outbred MF1 mice and the pneumococcal strain D39 were chosen, because this was the combination used in the well-studied acute model of pneumococcal infection (14). In the acute model, 1 × 106 CFU suspended in 50 μl of PBS is administered intranasally; this dose regime is lethal within 72 h postinfection (13). Here, MF1 mice were intranasally challenged with 1 × 106 viable pneumococci (strain D39) suspended in 20, 30, 40, or 50 μl of PBS, and percentage survival was assessed.
Figure 1 shows that there was a direct correlation between dose volume and lethality. As expected, 1 × 106 CFU in 50 μl had a high lethality rate, but by lowering the dose volume, while keeping the bacterial CFU constant, the number of animals that survived increased. At 7 days postinfection, there were no detectable viable pneumococci in the lungs and blood, but viable pneumococci were recovered from the nasopharynx with all dose volumes tested, whereas the desired model of long-term colonization required viable pneumococci to be recovered from the lower airways at least 7 days postinfection. Nevertheless, the experiment showed that the dose volume can be a key variable in the establishment of the model. Because pneumococcal serotype can be a determinant of carriage (20), two other serotypes were tested at the different dose volumes: 6B (BHN191) and 19F (LgSt215). After infection with strain BHN191, all four dose volumes tested resulted in >50% lethality within 7 days of infection, and only one surviving animal had recoverable numbers of pneumococci (>1 CFU/mg tissue) in the lungs at 7 days postinfection. In contrast, with strain LgSt215, all mice survived to days postinfection at each dose volume, without signs of disease, and when culled, 40% of mice that received LgSt215 in 30 μl had recoverable numbers of S. pneumoniae (50 to 150 CFU/mg lung tissue) in the lower airways. In comparison, after infection with the dose volumes of 20 and 40 μl, only 20% of animals had recoverable numbers of pneumococci in the lungs. For all dose volumes, no bacteria were recovered from the blood. Following these observations, 30 μl was chosen as the dose volume for further development of the model.
FIG 1.
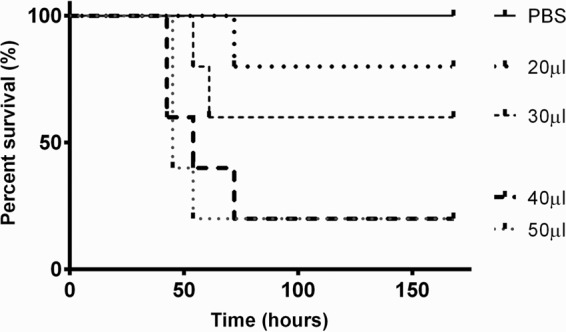
Percentage survival of outbred MF1 mice intranasally infected with 1 × 106 CFU S. pneumoniae strain D39 suspended in 20, 30, 40, or 50 μl PBS (n = 5). Experiments were ended at 168 h postinfection.
The next stage in model development was to increase the proportion of mice with viable pneumococci in the lower airways at 7 days. Doses of LgSt215 between 1 × 104 and 5 × 106 CFU in 30 μl PBS were given intranasally to MF1 mice. However, at best, pneumococci were recovered from the lungs of only 40% of the animals at 7 days postinfection. As an alternative strategy, CBA/Ca mice were tested, because this strain was known to be susceptible to acute pneumococcal pneumonia after intranasal infection (21). When CBA/Ca mice were given 4 × 106 CFU LgSt215 intranasally in 30 μl, there were recoverable numbers of pneumococci in the lungs of 90% of mice at day 7 postinfection. As this suggested that this was the correct combination of dose volume, bacterial strain, and mouse strain, the experiment was repeated and bacterial numbers in the BALF and blood were enumerated at predetermined time points up to 28 days postinfection. Pneumococci were recovered from the lower airways in 100% of mice at 24 h and 7 days postinfection. From day 14 onward, viable pneumococci were recovered from the lungs of over 80% of mice (Fig. 2A).
FIG 2.
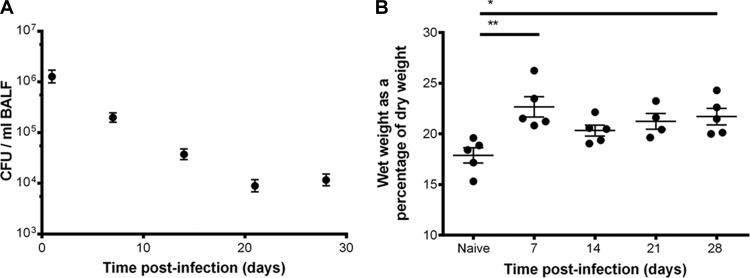
(A) The number of viable pneumococci in the bronchoalveolar lavage fluid of CBA/Ca mice intranasally infected with strain LgSt215; data are from 3 experiments (n = 21 to 25). (B) Pulmonary edema over time postinfection (n = 5). Kruskal-Wallis nonparametric test, with Dunn's posttest, was used to compare differences between time postinfection: *, P values of <0.05 and >0.01; **, P values of <0.01 and >0.001.
Figure 2B shows that the persistence of viable S. pneumoniae in the lower airways of mice was associated with an increase in the levels of pulmonary edema. At 28 days postinfection, the ratio of dry to wet weight of lungs was significantly different from that of naive mice (P < 0.05). From 14 to 21 days postinfection, there was no significant change in edema (P > 0.05).
Characterization of the cellular responses during persistent infection of the lungs with the serotype 19F pneumococcal strain LgSt215.
There was a transient increase in neutrophil numbers, which peaked at 24 h postinfection (P < 0.005), but numbers declined to control levels by day 14 (Fig. 3A). In contrast, macrophage numbers increased progressively and were significantly higher (P < 0.05) at 21 and 28 days postinfection (Fig. 3B).
FIG 3.

Inflammatory cells and cytokines in BALF collected from CBA/Ca mice after intranasal infection with S. pneumoniae strain LgSt215 (n = 10). (A) Number of macrophages; (B) number of neutrophils. Cells were counted in 10 fields of view at ×400 magnification. C, D, E, and F show the levels of the cytokines KC, IL-12 p40, IL-6, and TNF-α, respectively. Mann-Whitney nonparametric t tests were used to compare differences between pairs of time points: *, P values of <0.05 and >0.01; **, P values of <0.01 and >0.001; ***, P values of <0.001 and >0.0001; ****, P values of <0.0001.
A range of different cytokines associated with inflammation also were analyzed, and cytokines of note were KC, interleukin 6 (IL-6), tumor necrosis factor alpha (TNF-α), and the IL-12 p40 subunit. The levels of the neutrophil chemoattractant KC and IL-6 observed in the BALF can be related to the numbers of neutrophils counted. As can be seen, the levels of KC (Fig. 3C) and IL-6 (Fig. 3E) and the number of neutrophils (Fig. 3A) peaked at 24 h and 7 days postinfection and then declined to control levels from 14 days postinfection onward. The level of the natural killer cell and CD4 T cell differentiation inducer, IL-12 p40, was significantly raised (P < 0.001) at 24 h postinfection and remained significantly elevated until 14 days postinfection. The levels of TNF-α in the BALF were also significantly raised (P < 0.01) at 24 h and 7 and 14 days postinfection, but not at 21 or 28 days postinfection.
The histopathology of the lungs was examined using standard H&E staining. Naive mice did not show any notable pathology (Fig. 4A). In contrast, at 24 h postinfection, all lobes showed a diffuse, severe neutrophilic infiltration in the alveolar bed, including the presence of neutrophils and inflammatory debris in the alveolar airspaces. Neutrophils were also present in perivascular and peribronchiolar cuffs (Fig. 4B). Seven days after infection, all lobes in all animals showed some response to the bacterial infection, and there was interlobular variation in the pathology seen. In particular, alveolar consolidation, either focal (Fig. 4C) or lobular, was apparent in many of the mice in one or more lobes. The consolidated tissue consisted primarily of inflammatory cells (neutrophils, alveolar macrophages, and foam cells), cellular debris, and hypertrophic type II pneumocytes (Fig. 4D). Aside from the areas of consolidation, there was a significant inflammatory infiltrate within the alveolar bed and surrounding blood vessels and airways. Within the airways, the epithelial cells appeared hypertrophic and hyperplastic and the lumen were occasionally filled with cellular debris. After 14 days (Fig. 4E), the pathology was similar to that seen at day 7, but additionally areas of fibroplasia were seen focal to transitional zones (Fig. 4F, star). After 21 days, although all mice showed some response to the infection, the extent of the inflammation was reduced and some lobes did not show any pathological changes, indicating resolution of the inflammation. Where present, the inflammatory infiltrate was more mononuclear, with larger numbers of macrophages and lymphocytes being present. By day 28, most mice showed occasional foci of consolidation, with many lobes showing only a low-grade alveolitis. The foci of fibroplasia were seen to persist at 21 and 28 days postinfection but to a lesser extent than at 14 days postinfection. The presence of lymphocytes was confirmed with immunohistochemistry; Fig. 5B shows the presence of CD3-positive cells in the perivascular and peribronchiolar cuffs at 21 days postinfection. This was also observed at 28 days postinfection (data not shown) but was not seen in mice that had not received pneumococci.
FIG 4.

H&E-stained sections of lungs of CBA/Ca mice after intranasal infection with S. pneumoniae strain LgSt215. Sections are from naive animals (A), 24 h postinfection (B), and 7 days postinfection (arrow indicates focal consolidation) (C). (D) At 7 days postinfection, epithelial hypertrophy and hyperplasia along with perivascular and peribronchiolar inflammation were also evident. (E) At 21 days postinfection, the extent of the inflammation was beginning to reduce but consolidation and inflammatory foci persisted. (F, asterisk) Foci of fibroplasia; these were observed at days 14, 21, and 28 and appeared to be focal to the transitional airways.
FIG 5.

Measurement of antibody response of CBA/Ca mice intranasally infected with strain LgSt215. (A) At predetermined time points postinfection, CBA/Ca mice (n = 10) were bled via the saphenous vein and levels of anti-LgSt215 IgG were assessed by direct ELISA. Data were analyzed with the Kruskal-Wallis nonparametric test followed by Dunn's posttest: ***, P value of <0.001. Sections of lungs harvested at 21 days postinfection were stained with anti-CD3 (B) or isotype control antibody (C).
A distribution study was subsequently done to determine the extent of the lesions and whether there was bias for particular lobes at these later time points. The lungs from mice infected with pneumococcal strain LgSt215 were harvested at days 7 and 28, and lung blocks were step-sectioned (300-μm interval). There were only a few lesions per lobe, but these appeared to project through and involve significant portions of the lobe. Although the two larger lobes (left and right superior lobes) were most often affected, there did not appear to be any overrepresentation of any particular lobe, because all lobes showed persistence of inflammation across the group (data not shown).
Levels of anti-LgSt215 IgG were assessed in the sera collected at predetermined times postinfection. Figure 5A shows that at 7, 14, and 21 days postinfection, the levels were significantly increased (P < 0.0001) compared to those in the serum of naive mice.
Characterization of lung functioning during infection of the lungs with the serotype 19F pneumococcal strain LgSt215.
Lung function was assessed with a Forced Maneuvers system (eSpira; EMMS) that measured forced expiratory volume (FEV), tidal volume, and forced peak expiratory flow.
As shown in Fig. 6A and B, the FEV was measured at set points between 25 and 75 ms. At 7, 14, and 21 days postinfection, the values for FEV25 and FEV50 were significantly reduced (P < 0.05) compared to those of naive mice. At 28 days postinfection, a significant difference compared to naive mice was also seen, but only at FEV25 (P < 0.05). From 14 days postinfection, there was also a significant decrease (P < 0.01) in tidal volume compared to that for naive mice (Fig. 6C); however, by 28 days postinfection, this effect was no longer seen. At 24 h postinfection, there was a significant decrease in tidal volume (P < 0.001), as shown in Fig. 6C, but no differences were observed in FEV. Figure 6D shows that significant decreases (P < 0.05) in peak expiratory flow (PEF), compared to that for naive mice, were observed at 21 and 28 days postinfection.
FIG 6.

Lung functioning of CBA/Ca mice intranasally infected with strain LgSt215. At predetermined time points postinfection, lung function was assessed using a Forced Maneuvers system (EMMS). (A and B) FEV; (C) tidal volume; (D) forced peak expiratory flow (n = 10). Kruskal-Wallis nonparametric test, with Dunn's posttest, was used to compare differences between time postinfection and naïve mice: *, P values of <0.05 and >0.01; **, P values of <0.01 and >0.001; ***, P values of <0.001.
To investigate how the pneumococcus was able to persist, a mutant of LgSt215 that was unable to produce the cytolysin, pneumolysin, was constructed. Figure 7A shows that the pneumolysin-negative mutant persisted in the lower airways of CBA/Ca mice for a minimum of 28 days postinfection. At 24 h postinfection, there was no difference (P > 0.05) in the number of pneumococci in the BALF of CBA/Ca mice, but between 24 h and 7 days postinfection, there was a much steeper decline in the numbers of the pneumolysin-negative mutant, but thereafter pneumolysin was not required for persistence, with numbers in the BALF remaining unchanged. At 24 h postinfection, there were significantly (P < 0.05) less neutrophils in the BALF of mice given the pneumolysin-negative mutant (Fig. 7B), but from 7 days postinfection, there was no difference in the numbers of neutrophils present in the BALF of mice dosed with wild-type or pneumolysin-negative pneumococci (P > 0.05). At 7 and 14 days postinfection, there was no difference (P > 0.05) in the number of pulmonary macrophages (Fig. 7C), but at 21 and 28 days postinfection, there were (P < 0.05) less macrophages in the BALF in response to pneumolysin-negative pneumococci compared to wild type. The level of KC in the BALF of mice infected with the wild-type decreased significantly between 1 and 28 days (P < 0.001), whereas KC levels were unchanged (P > 0.05) in response to the pneumolysin-negative mutant (Fig. 7D). Figure 7E shows that at 24 h postinfection, there was a higher level of IL-12 p40 in the BALF of mice dosed with pneumolysin-negative pneumococci than in the BALF of mice dosed with wild-type pneumococci (P < 0.001), but at 7 and 14 days postinfection, there were higher levels of IL-12 p40 in the BALF of mice dosed with wild-type pneumococci than in the BALF of mice dosed with pneumolysin-negative pneumococci (P < 0.0001).
FIG 7.

Comparison of wild-type and a pneumolysin-negative mutant of strain LgSt215 in CBA/Ca mice. BALF was harvested at predetermined time points during the course of infection, and the number of viable pneumococci as well as the number of inflammatory cells and levels of selected cytokines were enumerated (n = 8), and the error bars show standard errors of the means. (A) Number of viable pneumococci in the BALF; (B) number of neutrophils; (C) number of macrophages counted in 10 fields of view at ×400 magnification. Levels of the cytokines KC (D) and IL-12 p40 (E) were measured in the BALF. Two-way analysis of variance with Sidak's multiple comparisons test was used to compare differences between the wild-type and pneumolysin-negative pneumococci: *, P values of <0.05 and >0.01; **, P values of <0.01 and >0.001; ***, P values of <0.001 and >0.0001; ****, P values of <0.0001.
DISCUSSION
It is known that some microorganisms, including S. pneumoniae, can colonize the airways of COPD patients in a stable state (9). This colonization has been associated with an increase in frequency of symptom exacerbations (7), which are linked with a reduction in quality of life (22, 23) and increased health care costs (1). Even though the asymptomatic colonization contributes to the complexity of COPD, there are no published in vivo models that mimic the low-level persistence of S. pneumoniae in the lower airways (10). Currently, pneumococcal infection models in the lower airways of mice are acute, with animals succumbing to disease within 48 h of infection (13).
To address this deficiency, this study characterized a model of long-term colonization in the lower airways of mice with S. pneumoniae. In advance of the experiments, it was concluded that for the model there should be pneumococci present in the lower airways, with an accompanying inflammatory cell response, for a minimum of 14 days postinfection. For the model to have practical utility, at least 70% of mice in a cohort would fulfill these criteria.
For the establishment of the desired model, the outbred MF1 strain was chosen initially because it is a strain frequently used in acute models of pneumococcal infection (15). The desired infection criteria could be met with this strain, but it was not a practical model, because the criteria were only met in 40% of each cohort. To increase the proportion of the cohort with a persistent pneumococcal presence in the lower airways, the impact of mouse strain was investigated.
CBA/Ca mice were chosen for three reasons. First, CBA/Ca mice are very susceptible to pneumococcal infection (21). Second, low levels of recoverable pneumococci in the lungs of CBA/J mice for two to 4 days postinfection had been reported, although mice succumbed to pneumococcal infection within 10 days of an aerosolized challenge (24, 25). Third, another study showed that 90% of CBA/N mice had pneumococci localized in the lungs, without bacteremia, after infection with three strains of serotype 19F pneumococcus, L82013, EF3030, or DS2217 (26). Together, these reports suggested that the CBA background would be suitable for the model of persistent infection. A prediction that was confirmed by the consistent observations was that viable pneumococci could be recovered from the lower airways of over 80% of CBA/Ca mice infected with the 19F pneumococcal strain LgSt215, and an inflammatory response was observed in the lungs.
It was presumed that the intranasal infection deposited bacteria throughout the lung, and a diffuse neutrophilic pneumonic reaction was seen across all lobes at 24 h. Mice culled at this point had the highest numbers of pneumococci in the lower airways, and this correlated with the significantly raised numbers of neutrophils and levels of KC. This acute pneumonic phase appeared to resolve, and by 7 days postinfection animals showed no outward signs of disease, yet approximately 1 × 105 to 1 × 106 CFU pneumococcus/ml BALF was consistently recovered. Although signs of disease were absent, foci of consolidated airspaces were evident in some lobes, in all animals at all time points up to day 28. The nature of the inflammatory infiltrate was seen to change with time, with increasing numbers of macrophages and, later, lymphocytes being detected. These events are typical for a chronic infection and suggested that at the later time points a more adaptive immune response was occurring.
A consequence of the severe pneumonic reaction was foci of fibroplasia associated with the transitional airways at days 14 and 21 and to a lesser extent at day 28. That these fibroplastic lesions did not develop into fibrosis, and were apparently resolving at day 28, suggested that although pneumococci could be recovered from the lower airways of mice, the nature of the host response to them had changed. Interestingly, increased levels of KC/IL-8 have been associated with increased neovascularization and fibroplasia in a mouse bleomycin model (27). It was shown in the model of chronic pulmonary infection that KC levels do remain elevated at 14 and 21 days and declined at 28 days, correlating with the fibroplasia observed in the tissue. However, sputa collected during exacerbations in COPD patients have shown an increase in neutrophils, as well as cytokines, including IL-8, in both tissue and sputum samples (28, 29). Both increased neutrophilia and IL-8/KC are prominent features of the mouse model of chronic pulmonary infection. The pathology seen in this model appears to be a good reflection of the human situation, although studies detailing the histopathology of exacerbations are few due to the difficulties of obtaining tissue from patients (3). Human studies are limited by the fact that only small bronchial biopsy specimens can be harvested, which do not include the lung parenchyma, in contrast to the mouse, where whole lungs can be taken and studied.
An important aspect of the diagnosis of COPD, and its severity, is to assess the decline of lung function (1). In the mouse model at 24 h postinfection, there was a significant decrease in tidal volume, but not FEV, compared to that for the control groups, suggesting that the presence of the diffuse pneumonic inflammation resulted in obstruction of the larger airways during normal respiration, but this inflammation did not result in a limitation of flow, as no reduction in FEV was observed (30). From 7 to 21 days postinfection, there was a significant decrease in FEV and tidal volume, suggesting that airflow obstruction was still present but that the inflammation could have inflamed the airways, causing an airflow limitation. At 7, 21, and 28 days postinfection, there was also a decrease in the peak expiratory flow (PEF), which is the maximal airflow achieved during the maximally forced expiration initiated at full inspiration. This reduction in PEF also suggests that the inflammation observed was causing an airflow limitation, as well as airflow obstruction.
The requirement for pneumolysin in lower airway persistence was investigated, because it is known that the toxin is essential for pneumococcal virulence (14). However, in contrast, it appears that it is not essential for pneumococcal persistence in the lower airways. One interesting observation was that maintaining levels of KC did not result in stable numbers of neutrophils in the lungs. Thus, unlike the lungs of mice infected with LgSt215 in which neutrophils and KC declined together, in the absence of pneumolysin the neutrophils declined even though KC levels did not.
At 24 h postinfection, there was a higher level of IL-12 p40 in the BALF of mice in response to the pneumolysin-negative pneumococci than that in the BALF of mice with wild-type infection. This early peak could explain the much-reduced number of viable pneumolysin-negative pneumococci in the BALF over the 7 days postinfection, in line with the suggestion that IL-12 p40 is protective against pneumococci because it enhances levels of gamma interferon (31). However, IL-12 p40 levels do not explain the pneumococcal persistence after 7 days, because the wild-type and pneumolysin-negative strains both persisted even though they induced significantly different amounts of IL-12 p40. These data show that our model is ideal to evaluate the microbial factors responsible for long-term colonization.
A new model of inflammation has been described, which mimics the low-level bacterial colonization often observed in COPD patients (23). The model is of pneumococcal persistence in the lower airways of mice, causing low-level inflammation but few clinical signs in the host. As a standalone model of inflammation, it is useful, but the power of this model lies in the ability to combine it with other experimental models of inflammation, to build more complex disease models of asthma and COPD (10). Diseases such as asthma and COPD are driven by a complicated interplay between different, heterogeneous phenotypes, and preclinical models involving a single stimulus of inflammation will no longer suffice. This new model of infection and exacerbation has been developed to provide a useful tool for the development of new treatments for severe respiratory diseases such as COPD, as it robustly produces a more clinically relevant phenotype.
ACKNOWLEDGMENTS
L.H. was funded by a BBSRC CASE Industrial studentship with AstraZeneca, United Kingdom. K.H. was funded by a grant from AstraZeneca.
Footnotes
Published ahead of print 27 May 2014
REFERENCES
- 1.GOLD. 2013. Global strategy for the diagnosis, management and prevention of COPD. Global Initiative for Chronic Obstructive Lung Disease. http://www.goldcopd.org/. [Google Scholar]
- 2.Celli BR, MacNee W, ATS/ERS Task Force 2004. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur. Respir. J. 23:932–946. 10.1183/09031936.04.00014304. [DOI] [PubMed] [Google Scholar]
- 3.Wedzicha JA. 2000. The heterogeneity of chronic obstructive pulmonary disease. Thorax 55:631–632. 10.1136/thorax.55.8.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rennard SI, Fogarty C, Kelsen S, Long W, Ramsdell J, Allison J, Mahler D, Saadeh C, Siler T, Snell P, Korenblat P, Smith W, Kaye M, Mandel M, Andrews C, Prabhu R, Donohue JF, Watt R, Lo KH, Schlenker-Herceg R, Barnathan ES, Murray J, COPD Investigators 2007. The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 175:926–934. 10.1164/rccm.200607-995OC. [DOI] [PubMed] [Google Scholar]
- 5.Domenech A, Ardanuy C, Balsalobre L, Marti S, Calatayud A, De la Campa G, Brueggeman B, Linares J. 2012. Pneumococci can persistently colonise adult patients with chronic respiratory disease. J. Clin. Microbiol. 50:4047. 10.1128/JCM.02056-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papi A, Bellettat CM, Braccioni F, Romagnoli M, Casolari P, Caramori G, Fabbri LM, Johnston SL. 2006. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am. J. Respir. Crit. Care Med. 173:1114–1121. 10.1164/rccm.200506-859OC. [DOI] [PubMed] [Google Scholar]
- 7.Patel IS, Seemungal TAR, Wilks M, Lloyd-Owen SJ, Donaldson GC, Wedzicha JA. 2002. Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax 57:759–764. 10.1136/thorax.57.9.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sethi S. 2010. Infection as a comorbidity of COPD. Eur. Respir. J. 35:1209–1215. 10.1183/09031936.00081409. [DOI] [PubMed] [Google Scholar]
- 9.Hirschmann JV. 2000. Do bacteria cause exacerbations of COPD? Chest 118:193–203. 10.1378/chest.118.1.193. [DOI] [PubMed] [Google Scholar]
- 10.Stevenson CS, Birrell MA. 2011. Moving towards a new generation of animal models for asthma and COPD with improved clinical relevance. Pharm. Ther. 130:93–105. 10.1016/j.pharmthera.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 11.Chin CL, Manzel LJ, Lehman EE, Humlicek AL, Shi L, Starner TD, Denning GM, Murphy TF, Sethi S, Look DC. 2005. Haemophilus influenzae from patients with chronic obstructive pulmonary disease exacerbation induce more inflammation than colonizers. Am. J. Respir. Crit. Care Med. 172:85–91. 10.1164/rccm.200412-1687OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drannik AG, Poulad MA, Robbins CS, Goncharova SI, Kianpour S, Stampfi MR. 2004. Impact of cigarette smoke on clearance and inflammation after Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 170:1164–1171. 10.1164/rccm.200311-1521OC. [DOI] [PubMed] [Google Scholar]
- 13.Kadioglu A, Taylor S, Iannelli F, Pozzi G, Mitchell TJ, Andrew PW. 2002. Upper and lower respiratory tract infection by Streptococcus pneumoniae is affected by pneumolysin deficiency and differences in capsule type. Infect. Immun. 70:2886–2890. 10.1128/IAI.70.6.2886-2890.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Canvin JR, Marvin AP, Sivakumaran M, Paton JC, Boulnois GJ, Andrew PW, Mitchell TJ. 1995. The role of pneumolysin and autolysin in the pathology of pneumonia and septicemia in mice infected with a type 2 pneumococcus. J. Infect. Dis. 172:119–123. 10.1093/infdis/172.1.119. [DOI] [PubMed] [Google Scholar]
- 15.Kadioglu A, Gingles NA, Grattan K, Kerr A, Mitchell TJ, Andrew PW. 2000. Host cellular response to pneumococcal lung infection in mice. Infect. Immun. 68:492–501. 10.1128/IAI.68.2.492-501.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morton DB, Griffiths PHM. 1985. Guidelines on the recognition of pain, distress and discomfort in experimental animals and a hypothesis for assessment. Vet. Rec. 116:431–443. 10.1136/vr.116.16.431. [DOI] [PubMed] [Google Scholar]
- 17.Yesilkaya H, Spissu F, Carvalho SM, Terra VS, Homer KA, Benisty R, Porat N, Neves AR, Andrew PW. 2009. Pyruvate formate lyase is required for pneumococcal fermentative metabolism and virulence. Infect. Immun. 77:5418–5427. 10.1128/IAI.00178-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchell TJ, Paterson GK. 2007. Mouse models of pneumococcal infection, p 25 In Hakenbeck R, Chhatwal S. (ed), Molecular biology of streptococci. Horizon Scientific Press, Norfolk, United Kingdom. [Google Scholar]
- 19.Russell H, Tharpe JA, Wells DE, White EH, Johnson JE. 1990. Monoclonal antibody recognizing a species-specific protein from Streptococcus pneumoniae. J. Clin. Microbiol. 28:10 2191–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hogberg L, Geli P, Ringberg H, Melander E, Lipsitch M, Ekdahl K. 2007. Age- and serogroup-related differences in observed durations of nasopharyngeal carriage of penicillin-resistant pneumococci. J. Clin. Microbiol. 45:948–952. 10.1128/JCM.01913-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gingles NA, Alexander JE, Kadioglu A, Andrew PW, Kerr A, Mitchell TJ, Hopes E, Denny P, Brown S, Jones HB, Little S, Booth GC, McPheat WL. 2001. The role of genetic resistance in invasive pneumococcal infection: identification and study of susceptible and resistant inbred mouse strains. Infect. Immun. 69:426–434. 10.1128/IAI.69.1.426-434.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halpin DMG, Decramer M, Celli B, Kesten S, Liu D, Tashkin DP. 2012. Exacerbation frequency and course of COPD. Int. J. COPD 7:653–661. 10.2147/COPD.S34186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sethi S. 2000. Bacterial infection and the pathogenesis of COPD. Chest 117:286S-291S. 10.1378/chest.117.5_suppl_1.286S. [DOI] [PubMed] [Google Scholar]
- 24.Tateda K, Takashima K, Miyazaki H, Matsumoto T, Hatori T, Yamaguchi K. 1996. Noncompromised penicillin-resistant pneumococcal pneumonia CBA/J mouse model and comparative efficacies of antibiotics in this model. Antimicrob. Agents Chemother. 40:1520–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nuermberger E, Helke K, Bishai WR. 2005. Low-dose aerosol model of pneumococcal pneumonia in the mouse: utility for evaluation of antimicrobial efficacy. Int. J. Antimicrob. Agents 26:497–503. 10.1016/j.ijantimicag.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 26.Briles DE, Hollingshead SK, Paton JC, Ades EW, Novak L, van Ginkel FW, Benjamin WH., Jr 2003. Immunisations with pneumococcal surface protein A and pneumolysin are protective against pneumonia in a murine model of pulmonary infection with Streptococcus pneumoniae. J. Infect. Dis. 188:339–348. 10.1086/376571. [DOI] [PubMed] [Google Scholar]
- 27.Keane MP, Belperio JA, Moore TA, Moore BB, Arenberg DA, Smith RE, Burdick MD, Kunkel SL, Strieter RM. 1999. Neutralisation of the CXC chemokine, macrophage inflammatory protein-2 attenuates bleomycin-induced pulmonary fibrosis. J. Immunol. 162:5511–5518. [PubMed] [Google Scholar]
- 28.Aaron SD, Angel JB, Lunau M, Wright K, Fex C, Le Saux N, Dales RE. 2001. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 163:349–355. 10.1164/ajrccm.163.2.2003122. [DOI] [PubMed] [Google Scholar]
- 29.Qiu Y, Zhu J, Bandi V, Atmar RL, Hattotuwa K, Guntnpalli KK, Jeffery PK. 2003. Biopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 168:968–975. 10.1164/rccm.200208-794OC. [DOI] [PubMed] [Google Scholar]
- 30.Vanoirbeek JAJ, Rinaldi M, Vooght VD, Haenen S, Bobic S, Gayan-Ramirez G, Hoet PHM, Verbeken E, Decramer M, Nemery B, Janssens W. 2010. Noninvasive and invasive pulmonary function in models of obstructive and respiratory diseases. Am. J. Respir. Cell Mol. Biol. 42:96–104. 10.1165/rcmb.2008-0487OC. [DOI] [PubMed] [Google Scholar]
- 31.Sun K, Salmon SL, Lotz SA, Metzger DW. 2007. Interleukin-12 promotes gamma interferon-dependent neutrophil recruitment in the lung and improves protection against respiratory Streptococcus pneumoniae infection. Infect. Immun. 75:1196–1202. 10.1128/IAI.01403-06. [DOI] [PMC free article] [PubMed] [Google Scholar]