

Abstract
Staphylococcus aureus is a leading cause of human bacterial infection, causing a wide spectrum of disease ranging from skin and soft tissue infections to life-threatening pneumonia and sepsis. S. aureus toxins play an essential role in disease pathogenesis, contributing to both immunomodulation and host tissue injury. Prominent among these toxins are the membrane-active pore-forming cytolysin alpha-toxin (Hla) and the amphipathic α-helical phenol-soluble modulin (PSM) peptides. As deletion of either the hla or psm locus leads to a phenotypically similar virulence defect in skin and soft tissue infection, we sought to determine the relative contribution of each locus to disease pathogenesis. Here we show that production of Hla can be modulated by PSM expression. An S. aureus mutant lacking PSM expression exhibits a transcriptional delay in hla mRNA production and therefore fails to secrete normal levels of Hla at early phases of growth. This leads to attenuation of virulence in vitro and in murine skin and lung models of infection, correlating with reduced recovery of Hla from host tissues. Production of Hla and restoration of staphylococcal virulence can be achieved in the psm mutant by plasmid-driven overexpression of hla. Our study suggests the coordinated action of Hla and PSMs in host tissue during early pathogenesis, confirming a major role for Hla in epithelial injury during S. aureus infection. These findings highlight the possibility that therapeutics targeting PSM production may simultaneously prevent Hla-mediated tissue injury.
INTRODUCTION
Staphylococcus aureus is a human pathogen that causes considerable morbidity and mortality in infected individuals, most recently occurring in the wake of the epidemic spread of methicillin-resistant S. aureus (MRSA) strains (1–3). The clinical disease manifestations of staphylococcal infection range from skin and soft tissue infections to highly invasive diseases such as pneumonia, sepsis, bone and joint infections, and endocarditis (2, 4). While minor skin infections are often amenable to oral antimicrobial therapy, severe invasive disease necessitates hospitalization and the delivery of intravenous antimicrobials. The management of all forms of S. aureus disease is now complicated by the prevalence of drug resistance. While β-lactam-resistant strains were once common only in the hospital setting, community-associated MRSA (CA-MRSA) infection is now widespread, responsible for >10,000 deaths per year and a total societal economic burden in excess of $10 billion per year in the United States alone (3, 5–7). This unmet disease burden has focused the field on the development of novel strategies to treat and prevent infection.
Central to the ability of S. aureus to cause multiple clinical disease manifestations is the multitude of virulence factors that the organism elaborates in a regulated manner. Active and passive immunization strategies targeting an array of staphylococcal virulence factors have emerged as the leading strategies to prevent S. aureus disease, supported by preclinical assessments in animal models of disease. In the context of human clinical trials, however, these approaches have not been efficacious, suggesting the need to better understand how distinct virulence factors are integrated in disease pathogenesis and how these virulence factors can be most successfully targeted in the human host (8, 9). Secreted staphylococcal toxins have been a long-standing and attractive focus of such immunization efforts given their known roles in immunomodulation and tissue injury (10). Global regulatory systems such as the accessory gene regulator (Agr) quorum-sensing system in S. aureus tightly regulate secreted toxin expression, coordinating the expression of multiple secreted toxins (11). In light of this fact, strategies targeting Agr function have been examined recently as a novel therapeutic approach (12).
Two prominent core genome-encoded agr-regulated toxins are S. aureus alpha-toxin (alpha-hemolysin [Hla]) and the phenol-soluble modulins (PSMs). Hla is a pore-forming cytotoxin that has been extensively studied for its contribution to the pathogenesis of pneumonia, skin and soft tissue infection, sepsis, endocarditis, corneal infections, central nervous system infection, and mastitis (13–21). Binding of Hla to its receptor ADAM10 (a disintegrin and metalloprotease 10) on target host cells facilitates pore formation, cellular membrane injury, and damage to the tissue barrier function (16, 22–24). At high toxin concentrations on susceptible cells, lytic injury predominates, while subcytolytic concentrations of Hla promote the rapid upregulation of the metalloprotease activity of ADAM10 in a toxin pore-dependent manner (16, 22–24). ADAM10 activation in turn promotes the untimely, pathological cleavage of native ADAM10 substrates such as E-cadherin and VE-cadherin, causing injury to the barrier functions of the pulmonary epithelium, the epidermis, and the vascular endothelium (16, 23, 24). Mice harboring a conditional knockout of ADAM10 in the lung and skin epithelium confirm the requirement for this protein in toxin-mediated disease pathogenesis, as these mice are resistant to lethal pneumonia and severe dermonecrotic skin injury following S. aureus infection (23, 24).
PSMs are also membrane-injuring toxins, structurally characterized as a family of 7 small amphipathic α-helical peptides encoded by 3 distinct loci (psmα, psmβ, and hld) in the staphylococcal genome (25). PSMs are classified in S. aureus based on size: PSMα1 to PSMα4 and delta-toxin are ∼2- to 3-kDa peptides consisting of 20 to 30 amino acids, while PSMβ1 and PSMβ2 are approximately 40 amino acids in length with a mass of ∼4.5 kDa. Secreted from staphylococci via a dedicated secretion system (26), the effects of PSMs on host immune cells are mediated by toxin binding to formyl peptide receptor 2 (FPR2) (27). At micromolar concentrations, PSMs cause the lysis of human neutrophils, with the PSMα peptides displaying the most potent cytolytic capacity (28, 29). At submicromolar concentrations, the peptides are proinflammatory, stimulating neutrophil chemotaxis by signaling through FPR2 (27, 30). A growing body of evidence has implicated PSMα peptides in particular as a major contributor to disease pathogenesis, especially in highly virulent CA-MRSA strains (25, 27–29, 31). Deletion of the psmα locus leads to an attenuation of virulence in multiple animal models of disease, including models of dermonecrotic skin infection and lethal sepsis (21, 28). Interestingly, there is striking phenotypic similarity in the disease outcomes of skin infection observed in mice infected with MRSA strains defective in the expression of either Hla or PSMs: both strains are highly attenuated in the ability to cause dermonecrotic injury to the epidermis (15, 24, 28).
Both in vitro and in vivo, the expressions of Hla and PSMα are strongly correlated (32), consistent with the central role of the Agr system in transcriptional regulation of the hla and psm loci. While deletion of the psmα, psmβ, and hld loci did not cause transcriptional regulatory effects during stationary-phase culture (33), a fourth psm locus present in hospital-associated MRSA (HA-MRSA) isolates directly interacts with agrA mRNA, leading to a reduction in virulence factor production, including hla (34). These findings, together with the in vivo virulence defect described above, led us to hypothesize that a deletion of the psm loci might directly or indirectly alter Hla expression or activity in a growth-dependent manner, impairing the ability of psm mutant strains to cause severe epithelial injury. In the present study, we show that Hla production can be modulated by the presence of the psm loci, with psmα appearing to have a predominant role. An S. aureus mutant lacking PSMα expression fails to secrete high levels of Hla, most notably during early phases of growth and in the context of the host tissue microenvironment, despite having an unaltered bacterial growth rate. The failure to secrete Hla correlates with a defect in toxin-dependent cell killing in vitro and contributes to reduced virulence in murine skin and lung infection models. These data illustrate the complexity of the regulation of virulence factor production by S. aureus and suggest that ongoing investigation of in vivo virulence factor expression at the host-pathogen interface may refine current efforts to mitigate staphylococcal disease.
MATERIALS AND METHODS
Bacterial strains.
Staphylococcus aureus USA300 (LAC) and USA400 (MW2); their S. aureus Δhla, Δpsmα, Δpsmβ, Δhld, and Δpsmα psmβ hld isogenic mutants; and complemented S. aureus USA300 Δpsmα pTxΔ (empty vector) and USA300 Δpsmα pTxΔ-psmα strains were described previously (14, 28). The S. aureus USA300 Δpsmα psmβ hld strain expressing plasmid-encoded Hla (Δpsmα psmβ hld phla) was made by electroporation of S. aureus USA300 Δpsmα psmβ hld cells with high-copy-number plasmid pOS1 carrying the hla gene under the control its endogenous promoter (14). For growth curves, stationary cultures of bacteria were washed once in phosphate-buffered saline (PBS) and diluted 1:250 in tryptic soy broth (TSB). Bacterial growth was monitored every 15 min at 37°C by optical density (at 660 nm) readings on a Tecan InfinitePro200 plate reader.
Cytotoxicity assay.
Lung alveolar epithelial cells (A549) were washed and plated in F12K tissue culture medium (Corning) supplemented with 10% fetal bovine serum at a density of 1.5 × 104 cells per well in a 96-well plate. Bacterial cultures were grown overnight in TSB to stationary phase. The cultures were diluted 1:100 and grown with shaking at 37°C to an optical density of 0.5 at 660 nm. Culture aliquots (5 ml) were sedimented by centrifugation, and the bacteria were washed with incomplete F12K medium and resuspended in 5 ml incomplete F12K medium. Washed A549 cells were cultured in triplicate with 100 μl of the indicated dilution of the bacterial suspension. After 2.5 h of incubation at 37°C, lactate dehydrogenase (LDH) activity was determined according to the manufacturer's recommendations (Roche).
Detection of secreted Hla.
Cultures of S. aureus strains grown overnight were washed once in PBS, diluted 1:250 in TSB, and then grown with shaking at 37°C for the indicated times. When noted, washed bacteria were grown in TSB containing 10% filtered supernatant of stationary-phase S. aureus USA300 Δhla cells. Bacteria were sedimented by centrifugation, and the supernatant was precipitated with trichloroacetic acid (TCA). Following an acetone wash, protein precipitants were resuspended in Laemmli's sample buffer and separated on a 12% SDS-PAGE gel. Hla was detected by Western blotting using a 1:5,000 dilution of rabbit polyclonal antibody raised against Hla, as previously described (14), followed by an AlexFluor 680 goat anti-rabbit secondary antibody (Invitrogen) and imaged with the Li-Cor Odyssey imaging system.
Quantitative reverse transcription-PCR.
Bacterial pellets were harvested after 3 h of growth by using RNA Protect (Qiagen), and RNA was isolated by using a Qiagen RNeasy minikit according to the manufacturer's protocol. cDNA was made from 100 ng of RNA by using the iScript cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR was performed by using SYBR green master mix and the Bio-Rad CFX96 real-time system with a C1000 thermal cycler. The following primers were used to quantify mRNA transcript levels: hla forward primer 5′-CAACAACACTATTGCTAGGTTCCA and reverse primer 5′-TTTTTGTGCATGCCATTTTC and psmα forward primer 5′-TATCAAAAGCTTAATCGAACAATTC and reverse primer 5′-CCCCTTCAAATAAGATGTTCATATC. Expression levels were compared by normalization to the amount of 16S transcript, as determined by using forward primer 5′-GAAAGCCACGGCTAACTACG and reverse primer 5′-CATTTCACCGCTACACATGG.
Mouse skin infection model.
All animal studies were performed in accordance with principles set forth by the Animal Welfare Act and National Institutes of Health guidelines for the care and use of animals in biomedical research and were reviewed and approved by the University of Chicago Institutional Animal Care and Use Committee. All animals were housed under standard conditions and provided food and water ad libitum. Mouse skin and soft tissue infections were performed as previously described (24). In brief, shaved female C57BL/6J mice (Jackson Laboratories, Wilmington, MA) were anesthetized with ketamine and xylazine and inoculated by subcutaneous injection in the right flank with 2 × 107 S. aureus USA300 (LAC), Δhla, Δpsmα psmβ hld, or Δpsmα psmβ hld phla cells in 50 μl PBS. Abscess formation and dermonecrotic skin lesion area were monitored at 24-h intervals for 14 days. The sizes of the abscesses and dermonecrotic skin lesions were calculated by using a standard formula for area (A = π/2 × length × width), as previously described (15, 24). For some mice, skin abscesses were harvested at 30 h postinfection by using an 8-mm biopsy punch to remove the epidermal layer containing the abscess. Abscesses were homogenized by using a rotor stator homogenizer (Pro Scientific), and serial dilutions were spotted in 10-μl drops onto TSA plates for quantification of CFU present in the skin abscess. The remaining skin homogenate was stored at −80°C until it was processed for quantification of Hla levels in tissue.
Mouse model of lung infection.
Seven-week-old female C57BL/6J mice (Jackson Laboratories, Wilmington, MA) were anesthetized with ketamine and xylazine. After appropriate anesthesia was documented, mice were inoculated in the left nare with 30 μl of a bacterial slurry containing 1 × 108 to 3 × 108 S. aureus USA300 (LAC) wild-type (WT), Δhla, Δpsmα psmβ hld, or Δpsmα psmβ hld phla cells, as previously described (14). Mice were monitored approximately every 6 h for the first 36 h and at 24-h intervals thereafter. For animals subjected to disease endpoint analysis, animals were sacrificed for removal of the lung tissue at 24 h postinfection. Lung tissue was homogenized, and serial dilutions were spotted onto TSA plates for quantification of CFU present in the lung. The remaining lung homogenate was stored at −80°C until it was processed for quantification of Hla.
Quantification of Hla.
Frozen, homogenized skin and lung tissue samples were thawed and diluted to 1 mg/ml total protein in PBS plus a protease inhibitor (Roche). The concentration of Hla present in each sample was determined by plating 100 μg of each sample onto a MaxiSorp microtiter plate (Thermo-Fisher Scientific) coated with 1 μg/ml 7B8, a monoclonal antibody to Hla (35), and compared to a standard curve of purified HlaH35L, a recombinant inactive toxin variant that represents the monomeric form of Hla (36). The total amount of Hla (μg) present per skin abscess or lung tissue was calculated by taking the concentration of Hla in the sample determined by an enzyme-linked immunosorbent assay (ELISA) and multiplying it by the dilution factor of the sample diluted at 1 mg/ml.
Statistical analysis.
Statistical analysis was performed by using one-way analysis of variance (ANOVA) with Bonferroni correction to adjust for multiplicity at a significance level of 0.05. Assessment of statistical significance in mortality studies was conducted by using the Mantel-Cox log-rank test.
RESULTS
PSM expression regulates Hla production in vitro.
The amount of Hla secreted by individual S. aureus isolates correlates in a direct fashion with the strain's virulence capacity in lung and skin models of infection (14, 32, 37, 38). Similarly, the level of PSM production by a given strain also predicates the pathogenicity of staphylococcal isolates (30, 32). In order to determine the differential contributions of Hla and PSMs to epithelial cell injury, we examined the ability of S. aureus USA300 wild-type (WT), Hla− (Δhla), and PSM− (Δpsmα psmβ hld) strains to induce A549 alveolar epithelial cytotoxicity in an in vitro coculture assay. Previous studies using this assay system demonstrated an essential role for Hla in cellular injury, as loss of expression of Hla by S. aureus, antagonism of Hla by monoclonal antibodies or small-molecule inhibitors, or small interfering RNA (siRNA)-mediated knockdown of A549 ADAM10 expression markedly attenuated staphylococcal killing (14, 22, 35, 39, 40). The addition of early-exponential-phase WT S. aureus inocula across a range of staphylococcal culture dilutions directly correlated with the amount of A549 cell injury observed following a 3-h coculture (Fig. 1A). In agreement with previous results, the S. aureus Δhla strain failed to induce significant cell death, consistent with a primary role for Hla in epithelial cell injury. The S. aureus Δpsmα psmβ hld strain, devoid of all PSM expression, similarly failed to induce cytotoxicity in this assay (Fig. 1A). To define the specific psm locus (or loci) that modulates cytotoxicity, we analyzed individual Δpsmα, Δpsmβ, and Δhld mutants (28), demonstrating that a loss of expression of PSMα significantly impaired A549 cell lysis, while the loss of either PSMβ or delta-toxin had no impact on virulence (Fig. 1B). Together, these data suggested either a direct contribution of PSMα peptides to epithelial injury, a defect in Hla expression in the S. aureus Δpsmα psmβ hld strain, or a degree of synergy between Hla and PSMα such that a reduction or loss of expression of either factor would lead to a marked impairment of cytotoxicity and downstream effects attributed to Hla intoxication.
FIG 1.
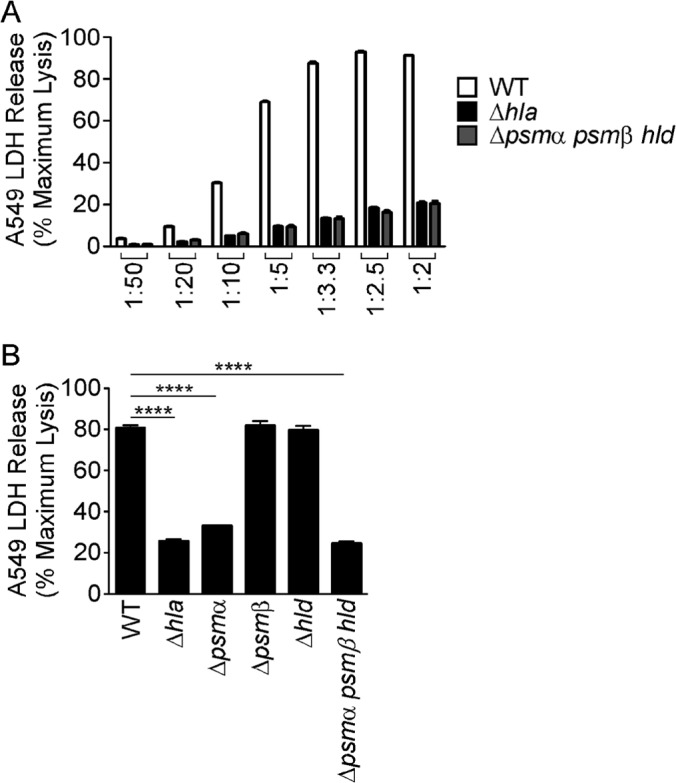
An S. aureus Δpsmα psmβ hld strain has decreased virulence in an in vitro cytotoxicity assay. Shown are data for intoxication of A549 cells with the indicated dilutions (A) or a 1:2.5 dilution (B) of the S. aureus USA300 WT, Δhla, or Δpsm strain for 2 1/2 h. Cell death was measured by LDH release into the supernatant. Data shown are averages ± standard errors of the means of technical replicates and are representative of at least three independent experiments. ****, P < 0.0001.
As previous experiments demonstrated an essential role for Hla in the A549 cell cytotoxicity assay, we chose to examine Hla expression levels in PSM-defective strains. To rule out potential growth defects associated with genetic deletion of the psm loci, we first evaluated bacterial growth of WT and Δpsmα psmβ hld strains in tryptic soy broth (TSB). Following subculturing of stationary-phase bacteria, S. aureus Δpsmα psmβ hld bacteria displayed growth kinetics identical to those of WT bacteria (Fig. 2A). After 6 h of growth, the Δpsmα psmβ hld strain grew to a slightly higher optical density than the WT, perhaps owing to a reduction in energy expenditure by the mutant strain, as PSMs are one of the major components of stationary-phase supernatants (28). Analysis of Hla secretion in WT and mutant strains was performed as a function of time following subculturing in TSB. After 3 h of growth, S. aureus USA300 Δpsmα and Δpsmα psmβ hld strains showed significantly decreased amounts of secreted Hla, while S. aureus USA300 Δpsmβ and Δhld strains mirrored WT S. aureus (Fig. 2B, top). By 6 h of growth, all strains displayed nearly equivalent amounts of secreted Hla (Fig. 2B, bottom), consistent with previously reported observations in which a PSM− mutant was shown to produce similar amounts of Hla as WT S. aureus in stationary-phase cultures (26). Reduced Hla expression of the USA300 strains at 3 h correlated with decreased hla transcript levels in S. aureus Δpsmα and Δpsmα psmβ hld strains compared to WT S. aureus (Fig. 2C). Previous studies successfully complemented the Δpsm defect in neutrophil killing by expressing psm loci under the control of the xylose promoter in which the repressor element had been genetically deleted (28, 29, 41). Our initial attempts to complement the Hla secretion defect were unsuccessful using this system (see Fig. S1A in the supplemental material), which we hypothesized may result from a lag in PSM production when using the pTxΔ vector. After 3 h of growth, psmα1 mRNA levels expressed by the S. aureus Δpsmα pTxΔ-psmα strain were only 18% of the psmα1 mRNA levels expressed by the corresponding WT strain (see Fig. S1B in the supplemental material). This marked reduction in PSM production likely represents an insufficient expression level to fully complement Hla secretion at this early phase of growth. However, at stationary phase, psmα1 mRNA expressed by the Δpsmα pTxΔ-psmα strain accumulated to levels indistinguishable from those of WT bacteria (see Fig. S1B in the supplemental material), providing a rationale for the ability to complement the psmα defect in previous studies using stationary-phase cultures (28, 29, 41). In order to provide additional confirmation that an unanticipated secondary mutation was not responsible for the early psmα-mediated defect in Hla secretion, this phenotype observed for the USA300 S. aureus strain was confirmed by using isogenic Δpsm mutants of CA-MRSA of the USA400 lineage (MW2) (Fig. 2D).
FIG 2.

An S. aureus Δpsmα psmβ hld strain has a defect in Hla transcription and protein production. (A) Bacterial growth of S. aureus WT and Δpsmα psmβ hld strains. OD660, optical density at 660 nm. (B) Immunoblot of precipitated S. aureus USA300 supernatants harvested after growth for 3 or 6 h at 37°C. Immunoblotting with a polyclonal antibody against Hla is shown, and results are representative of three independent experiments. (C) Hla transcript levels measured by quantitative reverse transcription-PCR after 3 h of growth. The data represent the averages of three independent biological replicates. (D) Immunoblot of precipitated S. aureus USA400 MW2 supernatants harvested after growth for 3 or 6 h at 37°C. Quantification of immunoblots was performed with a Li-Cor Odyssey imager. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
PSMs regulate Hla production in vivo.
The observed reduction in Hla expression by psm mutant strains in vitro suggested that the attenuated dermonecrosis phenotype observed upon skin infection with the S. aureus Δpsmα psmβ hld strain was potentially attributable to the loss of Hla activity. In murine and rabbit model systems, disruption of Hla expression or receptor availability results in a moderate reduction in overall skin abscess size but a near-complete attenuation of epidermal injury, corresponding to the maintenance of E-cadherin-based intercellular adherens junctions (15, 21, 24, 42). To determine whether psm loci have a regulatory effect on Hla production in vivo, we performed a comparative analysis of Hla− and PSM− strains in the skin and soft tissue model of infection. Subcutaneous inoculation with S. aureus USA300 Δhla and Δpsmα psmβ hld strains revealed a significant defect in abscess area (Fig. 3A) and dermonecrotic skin lesion area (Fig. 3B) compared to WT S. aureus, consistent with previously reported observations (15, 21, 28, 42). S. aureus recovery (CFU) from skin abscesses was similar among all three strains at the 30-h time point (Fig. 3C). Quantification of Hla in excised skin tissue, however, revealed a significant reduction in the amount of toxin present in the abscesses of mice infected with the S. aureus Δpsmα psmβ hld strain (Fig. 3D), consistent with the in vitro data indicative of a role for the psmα locus in Hla production.
FIG 3.
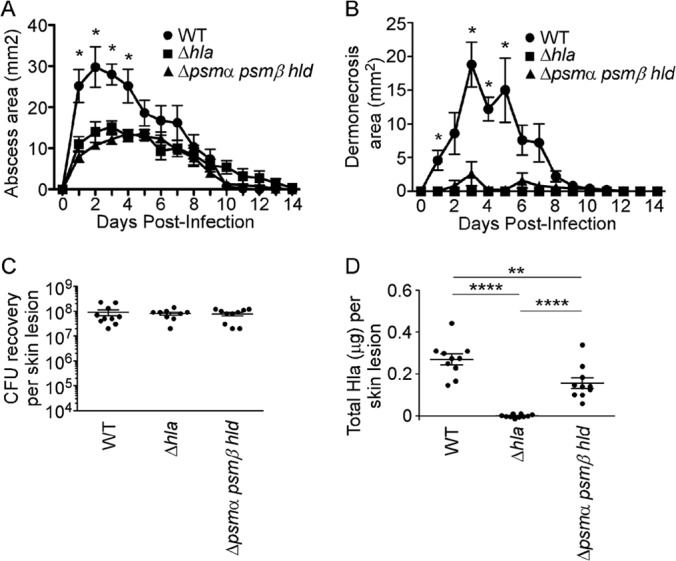
An S. aureus Δpsmα psmβ hld strain has an in vivo defect in Hla production during S. aureus skin infection. C57BL/6J mice received subcutaneous inoculation of 2.5 × 107 CFU of the S. aureus USA300 WT, Δhla, or Δpsmα psmβ hld strain. (A and B) Skin measurements of abscess area (A) and dermonecrosis area (B) of infected mice. (C) CFU recovery from skin abscesses harvested at 30 h postinfection. (D) Quantification of total Hla levels present in skin abscesses by an ELISA. All data are representative of three independent experiments. *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
Underscoring the central role of Hla in epithelial barrier injury, the necessity of this toxin for alveolar epithelial injury in S. aureus pneumonia mirrors that observed in the context of skin infection (13, 43). While the phenotype of a PSM− strain has not been examined in an S. aureus pneumonia model to our knowledge, we hypothesized that this mutant may exhibit a virulence defect owing to a reduction in Hla expression. When mice received an intranasal inoculation of S. aureus to examine the pathogenesis of lung infection, both the Δhla and Δpsmα psmβ hld mutant strains resulted in a lesser degree of tissue injury. Lungs harvested from mice infected with WT S. aureus displayed a deep red color upon gross pathological inspection (Fig. 4A), consistent with severe tissue injury and intrapulmonary hemorrhage. In contrast, lungs harvested from mice infected with either mutant strain were lighter in color (Fig. 4A), evident of less overt tissue injury. Lung CFU recovery was blunted in mice infected with the Δhla and Δpsmα psmβ hld mutant strains (Fig. 4B). While the role of Hla in bacterial tissue recovery in the lung differs from that observed for the skin at 30 h postinfection (Fig. 3C), these findings are consistent with the known contribution of Hla to S. aureus recovery in pneumonia pathogenesis (14). Similar to, albeit more pronounced than, our observation of skin lesions, Hla was undetectable in lung tissue homogenates from Δhla and Δpsmα psmβ hld strain-infected mice (Fig. 4C).
FIG 4.
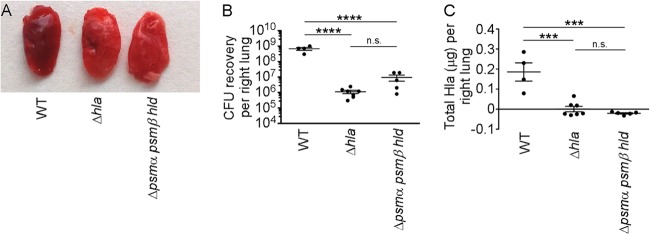
An S. aureus Δpsmα psmβ hld strain exhibits an in vivo defect in Hla production during S. aureus pneumonia. C57BL/6J mice received intranasal inoculation with 3.3 × 108 CFU of the S. aureus USA300 WT, Δhla, or Δpsmα psmβ hld strain. (A) Gross pathology of the left lung at 24 h postinfection. (B) CFU recovery from lung homogenates at 24 h postinfection. (C) Quantification of total Hla levels present in lung homogenates as determined by an ELISA. All data are representative of three independent experiments. ***, P < 0.001; ****, P < 0.0001; n.s., not significant.
Overexpression of Hla restores Hla secretion and virulence of the S. aureus Δpsmα psmβ hld strain.
We considered that psmα could regulate hla expression by augmenting the expression or function of a positive regulator of Hla transcription, such as the Agr system, or dampen a repressor function that attenuates transcription of the toxin. In the former case, supplementation of Δpsm bacterial growth medium with the stationary-phase supernatant from a PSM-expressing strain would provide exogenous autoinducing peptide (AIP), circumventing the necessity for the psm locus and restoring Hla production by the mutant strain. However, this was not the case, as the psmα mutant maintained a defect in Hla production compared to the WT strain when grown in the presence of the stationary-phase supernatant of a PSM-expressing hla-deficient strain (see Fig. S2 in the supplemental material). To address the latter possibility that PSMs dampen a repressor of toxin transcription, we utilized a high-copy-number plasmid containing the hla gene under the control of its native promoter. While this approach might increase basal expression levels of hla simply due to high copy numbers, if the psm loci play a role in dampening a repressor of hla, then multiple copies of the native hla promoter may effectively overcome this repressor function and restore production of Hla by the Δpsm mutant. We have previously demonstrated that strains expressing Hla from S. aureus multicopy plasmid pOS1 display an ∼2-fold level of overexpression of the toxin relative to the endogenous expression in the WT parent strain (14). While the pOS1 empty vector (Δpsmα psmβ hld-pOS1) failed to restore Hla secretion to the Δpsmα psmβ hld strain, overexpression of the hla gene under the control of its own promoter (Δpsmα psmβ hld-phla) restored Hla secretion (Fig. 5A, bottom), leading to an increased amount of A549 cell death in the in vitro cytotoxicity assay (Fig. 5A, top). Furthermore, subcutaneous infection with the S. aureus USA300 Δpsmα psmβ hld-phla strain caused dermonecrotic tissue injury similar to that caused by WT S. aureus (Fig. 5B). Consistent with the in vitro secretion data, quantification of Hla after excision of the abscesses showed that overexpression of Hla restored the ability of the S. aureus Δpsmα psmβ hld strain to secrete Hla in the tissue environment, with significantly more Hla in the abscess than the WT (Fig. 5C). Upon intranasal inoculation in the S. aureus pneumonia model, mice succumbed to WT infection in 48 h (Fig. 5D, circles), while all mice infected with the Δhla strain survived infection (squares). The S. aureus Δpsmα psmβ hld strain displayed a significant defect in virulence (Fig. 5D, triangles), with only 50% of mice succumbing to infection. Overexpression of Hla in the PSM− strain not only restored virulence but also contributed to increased disease severity, as all mice infected with the Δpsmα psmβ hld-phla strain succumbed to infection within 12 h (Fig. 5D, diamonds).
FIG 5.
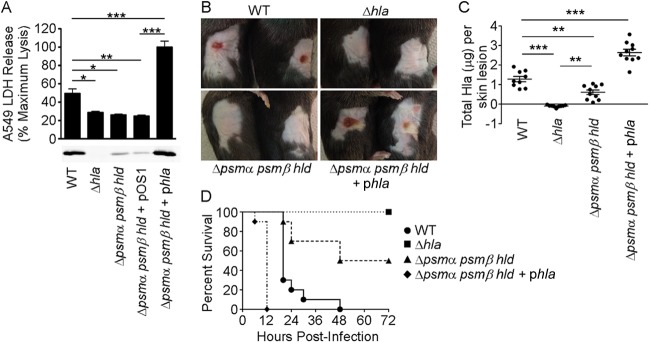
Plasmid-driven overexpression of hla restores virulence of an S. aureus Δpsmα psmβ hld mutant. (A, top) Intoxication of A549 cells with the S. aureus USA300 WT strain; the Δhla or Δpsmα psmβ hld mutant; or the Δpsmα psmβ hld strain complemented with the empty pOS1 vector or pOS1 containing the hla locus with its native promoter. Cell death was measured by LDH release into the supernatant. Data are averages ± standard errors of the means of technical replicates and are representative of at least three independent experiments. (Bottom) Anti-Hla immunoblot of bacterial supernatants harvested after 3 h of growth. The immunoblot is representative of two independent experiments. (B) Gross pathology of C57BL/6J mice infected subcutaneously with 2 × 107 CFU of the S. aureus USA300 WT, Δhla, Δpsmα psmβ hld, or Δpsmα psmβ hld phla strain in 50 μl of PBS. (C) Quantification of total Hla levels present in skin abscesses as determined by an ELISA. Data are representative of two independent experiments. (D) Survival curve of C57BL/6J mice infected intranasally with 3.3 × 108 CFU of the S. aureus USA300 WT, Δhla, Δpsmα psmβ hld, or Δpsmα psmβ hld phla strain. Data are representative of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
The complex regulatory control of virulence factor expression by S. aureus has long been an intense focus of study in the field. S. aureus is remarkable among bacterial pathogens for the wide range of clinical disease manifestations that it can cause in its human host. The pathogen's multitude of virulence factors facilitates a high degree of adaptability to distinct tissue environments, simultaneously providing the ability to circumvent host immunodefenses. The observation that regulatory control circuits such as the Agr, Sar, and Sae systems provide coordinated temporal regulation of distinct virulence factors suggests that the function of these factors is integrated in the host tissue microenvironment to ensure the survival of the pathogen and modulate virulence during infection. In the context of skin and soft tissue infections, both Hla and PSMs have independently been linked to disease pathogenesis, with genetic mutations in either locus leading to significant virulence defects (15, 21, 28). In this study, we show that the psmα locus regulates Hla production during early growth in culture and during an in vivo infection, potentially ensuring a mechanism by which S. aureus can modulate host innate immune function through PSMs and simultaneously engender tissue injury through Hla.
The defect in Hla expression at early phases of growth was notable, as previous studies showed no difference in hla transcript or protein levels when tested in vitro at later points in the growth cycle (26, 28, 33). In S. aureus, the production of Hla is governed by several global regulatory systems (44). The Agr quorum-sensing system controls virulence factor production through the secretion of an autoinducer peptide (AIP) that mediates the activation of the response regulator AgrA (45–50). AgrA upregulates the production of the RNAIII transcript, a regulatory RNA that both provides transcriptional and translational control to augment Hla production (51–53) and directly binds the psmα and psmβ promoters (33), providing strict control of PSM expression. Hla expression is also modulated by the Sar and Sae regulatory systems, with SarA and its homolog SarT acting as a repressor of Hla production in both an Agr-dependent and an Agr-independent manner (44, 54, 55). Together with several other transcriptional regulators, including SigB (44, 55) and the ArtR regulatory RNA, these systems orchestrate S. aureus virulence factor production.
Microarray-based transcriptional profiling of psmα, psmβ, and hld isogenic mutants during stationary-phase growth did not indicate a role for these loci in genetic regulation compared to WT S. aureus (33). However, a fourth psm locus has recently been shown to directly feed into the global regulatory networks of S. aureus (34). psm-mec is a phenol-soluble modulin-encoding gene located in the staphylococcal cassette chromosome mec (SCCmec) mobile genetic element of HA-MRSA but not community-acquired MRSA (CA-MRSA) strains. The transcriptional product of the psm-mec locus binds directly to agrA mRNA, impairing agrA translation and reducing the abundance of the AgrA protein, culminating in a decrease in secreted virulence factors, including PSMα and Hla (34). The transcriptional differences that we observed to underlie a reduction in Hla production in the PSM mutant are subtle and observed only early in the growth cycle in vitro. As this situation is akin to nascent infection, we propose that direct modulation of Hla production at this early time point by PSMα may ensure that the pathogen is simultaneously secreting the immunomodulatory PSM peptides with the cell-injurious Hla. As the in vivo defects in both Hla production and virulence observed following infection with the Δpsmα psmβ hld mutant mirror those of the Δhla mutant, it appears that there is no recovery of Hla expression during infection. This is in contrast to our observations in vitro, where Hla expression was restored to WT levels in the Δpsmα psmβ hld mutant in late-stage cultures. While secreted toxins such as Hla and PSMs are classically thought to be expressed only in later stages of the growth cycle based on in vitro studies, transient pharmacologic antagonism of agr activation with an inhibitory AIP during the first 3 h following S. aureus inoculation in a skin abscess model results in a potent defect in virulence (56, 57). While the specific proteins regulated by agr that underlie this phenotype were not defined, injection of a sterile supernatant from agr+ staphylococci recapitulates sterile abscess formation, leading to the conclusion that secreted agr-regulated exoproteins mediate the early effect of agr regulation (56). This so-called “single-dose paradox” strongly illustrates the critical role of bacterial regulatory control in the successful establishment of infection and may in part explain the significant defect in virulence observed following infection with the Δpsmα psmβ hld strain.
Ongoing investigations will be necessary to determine the precise molecular mechanism by which psmα regulates hla transcription, which is anticipated to shed light on how this early regulatory function is superseded in later stages of growth in vitro but not in vivo. Of interest, engineered agr and sarA mutant strains exhibit a more prominent defect in Hla production at 8 to 16 h following culture initiation, with a greater degree of restoration to WT levels being evident by 24 h (44). The kinetics of toxin expression in that study do not mirror those examined in our work but highlight the temporal complexity of secreted virulence factor regulation. It is likely that the phenotypic and microbiologic outcomes of infection reflect the balance of regulatory control of virulence factor expression by the infecting S. aureus strain, integrated with environmental conditions and the nature of the host immune response in the specific tissue microenvironment. It is clear from our studies that PSM-mediated regulatory control of Hla production is important for virulence in at least two distinct tissue settings, the skin and the lung, tissues that are among the leading sites of S. aureus infection in humans (3, 4) and in which a primary role for Hla in host injury has been demonstrated.
While PSMs may contribute to direct extracellular cytotoxicity of nonimmune cells in specific infection contexts, their ability to stimulate a proinflammatory state, augment neutrophil chemotaxis, and cause neutrophil intracellular lysis might play an essential role in establishing the host-pathogen balance during the course of infection (26, 28, 29, 58). Similarly, Hla elicits chemokine and cytokine release from host immune cells, epithelial cells, and endothelial cells (59–65), including the proinflammatory cytokine interleukin-1β (IL-1β), an indicator of inflammasome activation and a major cytokine involved in the recruitment of neutrophils to the site of infection (66). Primary neutrophils display resistance to the lytic effects of Hla unless this toxin is present at high concentrations that would not likely be achieved early in the course of infection (65, 67). Once recruited to the site of infection, host neutrophils would therefore either be subject to the lytic action of extracellular PSM peptides or sustain lysis from the intracellular compartment as engulfed bacteria produce PSMs (29). As PSM peptides are inactivated by serum lipoproteins (68), both a concentrating effect and maximal activity of these toxins may be optimally achieved in tissues. Innate immune recognition of invading pathogens and the subsequent inflammatory response are the host's first line of defense (69, 70). Thus, the initial interaction between S. aureus and the host can significantly alter the outcome of infection (56). In light of these observations, the virulence defect of the S. aureus Δpsmα psmβ hld strain is likely 2-fold: (i) the lack of PSM production, dampening the ability of the pathogen to cause neutrophil lysis, and (ii) diminished Hla secretion, blunting pathogen-induced tissue damage. Together, PSM-deficient staphylococci are missing two key virulence factors needed to provide the first “punch” to simultaneously evade the innate immune system and cause epithelial injury.
Hla and PSMs represent strategic targets for therapeutic intervention during S. aureus disease. Monoclonal antibody therapy as well as small-molecule inhibitors of both the toxin and its cellular receptor ADAM10 have shown preclinical efficacy in staphylococcal skin and soft tissue infections and staphylococcal pneumonia (23, 24, 35, 39, 40, 43). Several recent reports have highlighted the possibility of targeting PSMs and their host cell receptor formyl peptide receptor 2 to mitigate infection (26, 27, 31). Based on our data, PSM blockade may dampen virulence mediated by both PSMs and Hla.
Supplementary Material
ACKNOWLEDGMENTS
We thank Russell E. N. Becker for assistance with animal experimentation.
This work was supported by NIH award AI097434-01 to J.B.W., the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID), NIH, to M.O., and a Burroughs Wellcome Foundation Investigators in the Pathogenesis of Infectious Disease fellowship to J.B.W. We acknowledge membership in and support from the Region V Great Lakes RCE (NIH award 2-U54-AI-057153). B.J.B. was partially supported by NIH grant T32 GM007183.
Footnotes
Published ahead of print 27 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00089-14.
REFERENCES
- 1.Noskin GA, Rubin RJ, Schentag JJ, Kluytmans J, Hedblom EC, Jacobson C, Smulders M, Gemmen E, Bharmal M. 2007. National trends in Staphylococcus aureus infection rates: impact on economic burden and mortality over a 6-year period (1998-2003). Clin. Infect. Dis. 45:1132–1140. 10.1086/522186. [DOI] [PubMed] [Google Scholar]
- 2.Lowy FD. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520–532. 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 3.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 4.David MZ, Daum RS. 2010. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 23:616–687. 10.1128/CMR.00081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. 2010. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375:1557–1568. 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee BY, Singh A, David MZ, Bartsch SM, Slayton RB, Huang SS, Zimmer SM, Potter MA, Macal CM, Lauderdale DS, Miller LG, Daum RS. 2013. The economic burden of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). Clin. Microbiol. Infect. 19:528–536. 10.1111/j.1469-0691.2012.03914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.CDC. 2013. Antibiotic resistance threats in the United States. CDC, Atlanta, GA: http://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. [Google Scholar]
- 8.Proctor RA. 2012. Is there a future for a Staphylococcus aureus vaccine? Vaccine 30:2921–2927. 10.1016/j.vaccine.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Bagnoli F, Bertholet S, Grandi G. 2012. Inferring reasons for the failure of Staphylococcus aureus vaccines in clinical trials. Front. Cell. Infect. Microbiol. 2:16. 10.3389/fcimb.2012.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheung GY, Otto M. 2012. The potential use of toxin antibodies as a strategy for controlling acute Staphylococcus aureus infections. Expert Opin. Ther. Targets 16:601–612. 10.1517/14728222.2012.682573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung GY, Wang R, Khan BA, Sturdevant DE, Otto M. 2011. Role of the accessory gene regulator agr in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. Infect. Immun. 79:1927–1935. 10.1128/IAI.00046-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gray B, Hall P, Gresham H. 2013. Targeting agr- and agr-like quorum sensing systems for development of common therapeutics to treat multiple gram-positive bacterial infections. Sensors 13:5130–5166. 10.3390/s130405130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bubeck Wardenburg J, Bae T, Otto M, Deleo FR, Schneewind O. 2007. Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat. Med. 13:1405–1406. 10.1038/nm1207-1405. [DOI] [PubMed] [Google Scholar]
- 14.Bubeck Wardenburg J, Schneewind O. 2008. Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 205:287–294. 10.1084/jem.20072208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kennedy AD, Bubeck Wardenburg J, Gardner DJ, Long D, Whitney AR, Braughton KR, Schneewind O, DeLeo FR. 2010. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 202:1050–1058. 10.1086/656043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Powers ME, Kim HK, Wang Y, Bubeck Wardenburg J. 2012. ADAM10 mediates vascular injury induced by Staphylococcus aureus alpha-hemolysin. J. Infect. Dis. 206:352–356. 10.1093/infdis/jis192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Callegan MC, Engel LS, Hill JM, O'Callaghan RJ. 1994. Corneal virulence of Staphylococcus aureus: roles of alpha-toxin and protein A in pathogenesis. Infect. Immun. 62:2478–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jonsson P, Lindberg M, Haraldsson I, Wadstrom T. 1985. Virulence of Staphylococcus aureus in a mouse mastitis model: studies of alpha hemolysin, coagulase, and protein A as possible virulence determinants with protoplast fusion and gene cloning. Infect. Immun. 49:765–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berube BJ, Bubeck Wardenburg J. 2013. Staphylococcus aureus alpha-toxin: nearly a century of intrigue. Toxins (Basel) 5:1140–1166. 10.3390/toxins5061140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kielian T, Cheung A, Hickey WF. 2001. Diminished virulence of an alpha-toxin mutant of Staphylococcus aureus in experimental brain abscesses. Infect. Immun. 69:6902–6911. 10.1128/IAI.69.11.6902-6911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi SD, Malachowa N, Whitney AR, Braughton KR, Gardner DJ, Long D, Bubeck Wardenburg J, Schneewind O, Otto M, DeLeo FR. 2011. Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J. Infect. Dis. 204:937–941. 10.1093/infdis/jir441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilke GA, Bubeck Wardenburg J. 2010. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. U. S. A. 107:13473–13478. 10.1073/pnas.1001815107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, Bubeck Wardenburg J. 2011. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 17:1310–1314. 10.1038/nm.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inoshima N, Wang Y, Wardenburg JB. 2012. Genetic requirement for ADAM10 in severe Staphylococcus aureus skin infection. J. Invest. Dermatol. 132:1513–1516. 10.1038/jid.2011.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peschel A, Otto M. 2013. Phenol-soluble modulins and staphylococcal infection. Nat. Rev. Microbiol. 11:667–673. 10.1038/nrmicro3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chatterjee SS, Joo HS, Duong AC, Dieringer TD, Tan VY, Song Y, Fischer ER, Cheung GY, Li M, Otto M. 2013. Essential Staphylococcus aureus toxin export system. Nat. Med. 19:364–367. 10.1038/nm.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kretschmer D, Gleske AK, Rautenberg M, Wang R, Koberle M, Bohn E, Schoneberg T, Rabiet MJ, Boulay F, Klebanoff SJ, van Kessel KA, van Strijp JA, Otto M, Peschel A. 2010. Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe 7:463–473. 10.1016/j.chom.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, DeLeo FR, Otto M. 2007. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat. Med. 13:1510–1514. 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 29.Surewaard BG, de Haas CJ, Vervoort F, Rigby KM, DeLeo FR, Otto M, van Strijp JA, Nijland R. 2013. Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell. Microbiol. 15:1427–1437. 10.1111/cmi.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rautenberg M, Joo HS, Otto M, Peschel A. 2011. Neutrophil responses to staphylococcal pathogens and commensals via the formyl peptide receptor 2 relates to phenol-soluble modulin release and virulence. FASEB J. 25:1254–1263. 10.1096/fj.10-175208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otto M. 2010. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu. Rev. Microbiol. 64:143–162. 10.1146/annurev.micro.112408.134309. [DOI] [PubMed] [Google Scholar]
- 32.Li M, Cheung GY, Hu J, Wang D, Joo HS, DeLeo FR, Otto M. 2010. Comparative analysis of virulence and toxin expression of global community-associated methicillin-resistant Staphylococcus aureus strains. J. Infect. Dis. 202:1866–1876. 10.1086/657419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Queck SY, Jameson-Lee M, Villaruz AE, Bach TH, Khan BA, Sturdevant DE, Ricklefs SM, Li M, Otto M. 2008. RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus aureus. Mol. Cell 32:150–158. 10.1016/j.molcel.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaito C, Saito Y, Ikuo M, Omae Y, Mao H, Nagano G, Fujiyuki T, Numata S, Han X, Obata K, Hasegawa S, Yamaguchi H, Inokuchi K, Ito T, Hiramatsu K, Sekimizu K. 2013. Mobile genetic element SCCmec-encoded psm-mec RNA suppresses translation of agrA and attenuates MRSA virulence. PLoS Pathog. 9:e1003269. 10.1371/journal.ppat.1003269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ragle BE, Bubeck Wardenburg J. 2009. Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect. Immun. 77:2712–2718. 10.1128/IAI.00115-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Menzies BE, Kernodle DS. 1994. Site-directed mutagenesis of the alpha-toxin gene of Staphylococcus aureus: role of histidines in toxin activity in vitro and in a murine model. Infect. Immun. 62:1843–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeLeo FR, Kennedy AD, Chen L, Bubeck Wardenburg J, Kobayashi SD, Mathema B, Braughton KR, Whitney AR, Villaruz AE, Martens CA, Porcella SF, McGavin MJ, Otto M, Musser JM, Kreiswirth BN. 2011. Molecular differentiation of historic phage-type 80/81 and contemporary epidemic Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 108:18091–18096. 10.1073/pnas.1111084108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montgomery CP, Boyle-Vavra S, Adem PV, Lee JC, Husain AN, Clasen J, Daum RS. 2008. Comparison of virulence in community-associated methicillin-resistant Staphylococcus aureus pulsotypes USA300 and USA400 in a rat model of pneumonia. J. Infect. Dis. 198:561–570. 10.1086/590157. [DOI] [PubMed] [Google Scholar]
- 39.Ragle BE, Karginov VA, Bubeck Wardenburg J. 2010. Prevention and treatment of Staphylococcus aureus pneumonia with a beta-cyclodextrin derivative. Antimicrob. Agents Chemother. 54:298–304. 10.1128/AAC.00973-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tkaczyk C, Hua L, Varkey R, Shi Y, Dettinger L, Woods R, Barnes A, MacGill RS, Wilson S, Chowdhury P, Stover CK, Sellman BR. 2012. Identification of anti-alpha toxin monoclonal antibodies that reduce the severity of Staphylococcus aureus dermonecrosis and exhibit a correlation between affinity and potency. Clin. Vaccine Immunol. 19:377–385. 10.1128/CVI.05589-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grosz M, Kolter J, Paprotka K, Winkler AC, Schafer D, Chatterjee SS, Geiger T, Wolz C, Ohlsen K, Otto M, Rudel T, Sinha B, Fraunholz M. 2014. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin alpha. Cell. Microbiol. 16:451–465. 10.1111/cmi.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tkaczyk C, Hamilton MM, Datta V, Yang XP, Hilliard JJ, Stephens GL, Sadowska A, Hua L, O'Day T, Suzich J, Stover CK, Sellman BR. 2013. Staphylococcus aureus alpha toxin suppresses effective innate and adaptive immune responses in a murine dermonecrosis model. PLoS One 8:e75103. 10.1371/journal.pone.0075103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bubeck Wardenburg J, Patel RJ, Schneewind O. 2007. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 75:1040–1044. 10.1128/IAI.01313-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiong YQ, Willard J, Yeaman MR, Cheung AL, Bayer AS. 2006. Regulation of Staphylococcus aureus alpha-toxin gene (hla) expression by agr, sarA, and sae in vitro and in experimental infective endocarditis. J. Infect. Dis. 194:1267–1275. 10.1086/508210. [DOI] [PubMed] [Google Scholar]
- 45.Recsei P, Kreiswirth B, O'Reilly M, Schlievert P, Gruss A, Novick RP. 1986. Regulation of exoprotein gene expression in Staphylococcus aureus by agar. Mol. Gen. Genet. 202:58–61. 10.1007/BF00330517. [DOI] [PubMed] [Google Scholar]
- 46.Peng HL, Novick RP, Kreiswirth B, Kornblum J, Schlievert P. 1988. Cloning, characterization, and sequencing of an accessory gene regulator (agr) in Staphylococcus aureus. J. Bacteriol. 170:4365–4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reyes D, Andrey DO, Monod A, Kelley WL, Zhang G, Cheung AL. 2011. Coordinated regulation by AgrA, SarA, and SarR to control agr expression in Staphylococcus aureus. J. Bacteriol. 193:6020–6031. 10.1128/JB.05436-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Novick RP, Projan SJ, Kornblum J, Ross HF, Ji G, Kreiswirth B, Vandenesch F, Moghazeh S. 1995. The agr P2 operon: an autocatalytic sensory transduction system in Staphylococcus aureus. Mol. Gen. Genet. 248:446–458. [DOI] [PubMed] [Google Scholar]
- 49.Lina G, Jarraud S, Ji G, Greenland T, Pedraza A, Etienne J, Novick RP, Vandenesch F. 1998. Transmembrane topology and histidine protein kinase activity of AgrC, the agr signal receptor in Staphylococcus aureus. Mol. Microbiol. 28:655–662. 10.1046/j.1365-2958.1998.00830.x. [DOI] [PubMed] [Google Scholar]
- 50.Lyon GJ, Wright JS, Muir TW, Novick RP. 2002. Key determinants of receptor activation in the agr autoinducing peptides of Staphylococcus aureus. Biochemistry 41:10095–10104. 10.1021/bi026049u. [DOI] [PubMed] [Google Scholar]
- 51.Novick RP, Ross HF, Projan SJ, Kornblum J, Kreiswirth B, Moghazeh S. 1993. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 12:3967–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koenig RL, Ray JL, Maleki SJ, Smeltzer MS, Hurlburt BK. 2004. Staphylococcus aureus AgrA binding to the RNAIII-agr regulatory region. J. Bacteriol. 186:7549–7555. 10.1128/JB.186.22.7549-7555.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morfeldt E, Taylor D, von Gabain A, Arvidson S. 1995. Activation of alpha-toxin translation in Staphylococcus aureus by the trans-encoded antisense RNA, RNAIII. EMBO J. 14:4569–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vandenesch F, Kornblum J, Novick RP. 1991. A temporal signal, independent of agr, is required for hla but not spa transcription in Staphylococcus aureus. J. Bacteriol. 173:6313–6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheung AL, Chien YT, Bayer AS. 1999. Hyperproduction of alpha-hemolysin in a sigB mutant is associated with elevated SarA expression in Staphylococcus aureus. Infect. Immun. 67:1331–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wright JS, III, Jin R, Novick RP. 2005. Transient interference with staphylococcal quorum sensing blocks abscess formation. Proc. Natl. Acad. Sci. U. S. A. 102:1691–1696. 10.1073/pnas.0407661102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mayville P, Ji G, Beavis R, Yang H, Goger M, Novick RP, Muir TW. 1999. Structure-activity analysis of synthetic autoinducing thiolactone peptides from Staphylococcus aureus responsible for virulence. Proc. Natl. Acad. Sci. U. S. A. 96:1218–1223. 10.1073/pnas.96.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Geiger T, Francois P, Liebeke M, Fraunholz M, Goerke C, Krismer B, Schrenzel J, Lalk M, Wolz C. 2012. The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog. 8:e1003016. 10.1371/journal.ppat.1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bhakdi S, Muhly M, Korom S, Hugo F. 1989. Release of interleukin-1 beta associated with potent cytocidal action of staphylococcal alpha-toxin on human monocytes. Infect. Immun. 57:3512–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suttorp N, Seeger W, Dewein E, Bhakdi S, Roka L. 1985. Staphylococcal alpha-toxin-induced PGI2 production in endothelial cells: role of calcium. Am. J. Physiol. 248:C127–C134. [DOI] [PubMed] [Google Scholar]
- 61.Rose F, Dahlem G, Guthmann B, Grimminger F, Maus U, Hanze J, Duemmer N, Grandel U, Seeger W, Ghofrani HA. 2002. Mediator generation and signaling events in alveolar epithelial cells attacked by S. aureus alpha-toxin. Am. J. Physiol. Lung Cell. Mol. Physiol. 282:L207–L214. [DOI] [PubMed] [Google Scholar]
- 62.Suttorp N, Fuhrmann M, Tannert-Otto S, Grimminger F, Bhadki S. 1993. Pore-forming bacterial toxins potently induce release of nitric oxide in porcine endothelial cells. J. Exp. Med. 178:337–341. 10.1084/jem.178.1.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suttorp N, Buerke M, Tannert-Otto S. 1992. Stimulation of PAF-synthesis in pulmonary artery endothelial cells by Staphylococcus aureus alpha-toxin. Thromb. Res. 67:243–252. 10.1016/0049-3848(92)90143-X. [DOI] [PubMed] [Google Scholar]
- 64.Onogawa T. 2002. Staphylococcal alpha-toxin synergistically enhances inflammation caused by bacterial components. FEMS Immunol. Med. Microbiol. 33:15–21. 10.1111/j.1574-695X.2002.tb00566.x. [DOI] [PubMed] [Google Scholar]
- 65.Becker RE, Berube BJ, Sampedro GR, Dedent AC, Bubeck Wardenburg J. 10 May 2014. Tissue-specific patterning of host innate immune responses by Staphylococcus aureus alpha-toxin. J. Innate Immun. 10.1159/000360006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cho JS, Guo Y, Ramos RI, Hebroni F, Plaisier SB, Xuan C, Granick JL, Matsushima H, Takashima A, Iwakura Y, Cheung AL, Cheng G, Lee DJ, Simon SI, Miller LS. 2012. Neutrophil-derived IL-1beta is sufficient for abscess formation in immunity against Staphylococcus aureus in mice. PLoS Pathog. 8:e1003047. 10.1371/journal.ppat.1003047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nygaard TK, Pallister KB, DuMont AL, DeWald M, Watkins RL, Pallister EQ, Malone C, Griffith S, Horswill AR, Torres VJ, Voyich JM. 2012. Alpha-toxin induces programmed cell death of human T cells, B cells, and monocytes during USA300 infection. PLoS One 7:e36532. 10.1371/journal.pone.0036532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Surewaard BG, Nijland R, Spaan AN, Kruijtzer JA, de Haas CJ, van Strijp JA. 2012. Inactivation of staphylococcal phenol soluble modulins by serum lipoprotein particles. PLoS Pathog. 8:e1002606. 10.1371/journal.ppat.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mogensen TH. 2009. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 22:240–273. 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Medzhitov R, Janeway C., Jr 2000. Innate immunity. N. Engl. J. Med. 343:338–344. 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.