

Abstract
The liver efficiently restores function after damage induced during malarial infection once the parasites are cleared from the blood. However, the molecular events leading to the restoration of liver function after malaria are still obscure. To study this, we developed a suitable model wherein mice infected with Plasmodium yoelii (45% parasitemia) were treated with the antimalarial α/β-arteether to clear parasites from the blood and, subsequently, restoration of liver function was monitored. Liver function tests clearly indicated that complete recovery of liver function occurred after 25 days of parasite clearance. Analyses of proinflammatory gene expression and neutrophil infiltration further indicated that hepatic inflammation, which was induced immediately after parasite clearance from the blood, was gradually reduced. Moreover, the inflammation in the liver after parasite clearance was found to be correlated positively with oxidative stress and hepatocyte apoptosis. We investigated the role of heme oxygenase 1 (HO-1) in the restoration of liver function after malaria because HO-1 normally renders protection against inflammation, oxidative stress, and apoptosis under various pathological conditions. The expression and activity of HO-1 were found to be increased significantly after parasite clearance. We even found that chemical silencing of HO-1 by use of zinc protoporphyrin enhanced inflammation, oxidative stress, hepatocyte apoptosis, and liver injury. In contrast, stimulation of HO-1 by cobalt protoporphyrin alleviated liver inflammation and reduced oxidative stress, hepatocyte apoptosis, and associated tissue injury. Therefore, we propose that selective induction of HO-1 in the liver would be beneficial for the restoration of liver function after parasite clearance.
INTRODUCTION
Malaria, a parasitic disease caused by protozoa of the genus Plasmodium, is a major public health concern in developing countries. According to the World Health Organization, the disease caused 216 million clinical cases, which included 655,000 deaths, in 2010 (1). Deaths as a result of malaria occur due to a combination of several clinical complications, which include cerebral malaria, severe anemia, renal failure, respiratory distress, acidosis, and several others (1, 2). Hemolysis in malaria is thought to be a major cause of liver damage in both animal models (3–8) and human patients (9–13). Hepatocyte dysfunction (9) and death during malaria occur as a result of accumulation of free heme and high levels of serum tumor necrosis factor alpha (TNF-α) (3, 5). Oxidative stress triggered by free heme stimulates the proinflammatory response and neutrophil infiltration in the liver (3), which further aggravates liver damage. While TNF-α stimulates the proinflammatory response, thereby causing hepatocyte death during malaria (3), high parasite burdens also cause liver damage in mice after malaria infection (14, 15).
The liver responds to the injury by initiating the process of restoration to regain its lost function, and the recovery of liver function after clearance of malaria parasites has already been observed (16). However, the exact process of restoration of liver function after damage by malaria is still unknown. The process may be similar to the healing of a wound. Restoration of function of an organ or the healing of a wound is a very complicated process, which involves various cells, several anti-inflammatory signaling molecules, and cytokines (17). Wound healing, in general, occurs through hemostasis along with inflammatory, proliferative, and remodeling phases (18, 19). All the events in the four phases must occur precisely and must be well orchestrated for proper healing (18). While the inflammatory phase is an essential event during healing, excessive inflammation may delay healing. The inflammatory phase is under the control of several factors, one of which is the heme-degrading enzyme heme oxygenase 1 (HO-1). HO-1 plays a major role as a cytoprotective and anti-inflammatory protein in many diseases (20–34). Induction of HO-1 is necessary for the resolution phase of wound healing, including amelioration of inflammation, proliferation, and protection against apoptosis (35). The downstream effector molecules of HO-1 are the most probable candidates for performing these actions (36). HO-1 acts on free heme to produce biliverdin, CO, and free iron (37). Biliverdin is converted to bilirubin, which functions as a potent antioxidant (37, 38). CO mediates vasodilation by inducing cyclic GMP (cGMP) (39), and ferritin, overinduced due to accumulation of iron, sequesters the pro-oxidant free iron (40). Overexpression of HO-1 inhibits the expression of cell adhesion molecules and reduces the adhesion of leukocytes to the vascular endothelium in the presence of proinflammatory stimuli (41–45). The HO-1–CO–biliverdin pathway has been shown to inhibit the process of rolling, adhesion, and migration of neutrophils during inflammation (46). Wound healing is accelerated in the absence of neutrophils (47).
Since the molecular events leading to the restoration of liver functions after damage from malaria are yet unknown, a more precise understanding of these mechanisms may help to create strategies for protecting the liver from damage subsequent to Plasmodium attack. Here we report that HO-1 is functionally associated with the restoration of altered liver functions after malaria. Chemical silencing of HO-1 enhanced the inflammation and aggravated liver injury. In contrast, induction of HO-1 significantly reduced liver pathology and accelerated the restoration of liver function after parasite clearance from the blood.
MATERIALS AND METHODS
Cobalt protoporphyrin (CoPP), bovine serum albumin (BSA), thiobarbituric acid (TBA), 5,5-dithiobis-nitrobenzoic acid (DTNB), 2,2-diphenyl-1-picrylhydrazyl (DPPH), dimethyl sulfoxide (DMSO), glucose-6-phosphate dehydrogenase, glucose-6-phosphate, hemin, 3,3′-diaminobenzidine (DAB), and a caspase-3 assay kit were obtained from Sigma (St. Louis, MO). Zinc protoporphyrin (ZnPP) and horseradish peroxidase (HRP)-conjugated secondary anti-rabbit antibody were purchased from Calbiochem. Taurine was procured from SRL, India. TRIzol was purchased from Invitrogen. A RevertAid H Minus First Strand cDNA synthesis kit and nuclease-free water were purchased from Fermentas. Power SYBR green was procured from Applied Biosystems. Alcohol was purchased from Merck. HO-1 antibody and anti-neutrophil antibody (NIMP-R14) were procured from Abcam, United Kingdom. Fluorescein isothiocyanate (FITC)-conjugated anti-rat secondary antibody was purchased from Santa Cruz Biotechnology, and ProLong Gold antifade reagent with DAPI (4′,6-diamidino-2-phenylindole) was procured from Life Technologies. The primers were purchased from Integrated DNA Technologies Inc. (San Diego, CA). Assay kits for liver function were purchased from Randox Laboratories Ltd. (Ardmore, Antrim, United Kingdom). All other reagents were of analytical grade.
In vivo growth of Plasmodium yoelii.
The in vivo culture of P. yoelii was maintained in male BALB/c mice (20 to 25 g) as described previously (38). Blood from infected mice with 60% parasitemia was diluted in sterile acid citrate-dextrose, and naive mice were intraperitoneally (i.p.) administered 0.5 ml of diluted infected blood, containing about 1 × 106 parasitized red blood cells (RBCs) (6, 38, 48). Giemsa staining (Qualigens Fine Chemicals, India) of thin smears of blood from the infected mice was performed to monitor the parasitemia. All animals were maintained at 22 ± 2°C in the animal house of the institute. Animal experiments were conducted in accordance with the guidelines of the institutional animal ethics committee and the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA). The protocol was approved by the animal ethics committee of the CSIR-Indian Institute of Chemical Biology, Kolkata, India, registered with the CPCSEA, India (permit 147/1999/CPCSEA). All animals were handled with care, and efforts were made to minimize their suffering.
Animal treatment and parasite clearance.
Male BALB/c mice (20 to 25 g) without infection were considered the control group. Infected mice attained a maximum parasitemia of 65 to 70% in 8 days. Mice having moderate infection (40 to 45% parasitemia) until the 5th day postinfection were treated with α/β-arteether (ART) at 50 mg/kg of body weight (i.p.) for 3 days (day 5 to day 7 postinfection) to kill the parasites. The dose of ART was selected as mentioned earlier (49). Parasitemia was monitored regularly throughout the course of experiments by Giemsa staining to monitor the infection, and animal death was also noted. P. yoelii is a lethal multidrug-resistant (MDR) 17X strain among the malarial parasites, and introduction of only 1 × 106 parasitized RBCs to mice can induce a 50 to 60% parasitemia, resulting in death within 7 to 8 days. Parasitemia decreased steadily after ART treatment, and no parasites were found alive in blood smears upon Giemsa staining. This group of mice, in which parasites were cleared by ART, was called the “infected + ART” group. To ensure that no living parasites remained in the blood after ART treatment, we injected naive mice with blood from infected mice that had received ART for 3 days (infected + ART group) and monitored the parasitemia of these naive mice by Giemsa staining. We found no parasites in their blood even after a week. There is hardly any chance of residual living parasites in the blood after ART treatment; otherwise, the mice would have died within a week. Two parallel infected + ART groups of mice received ZnPP (5 mg/kg) and CoPP (10 mg/kg) intraperitoneally for 4 days after parasite clearance with ART treatment. These groups were called the “infected + ART + ZnPP” and “infected + ART + CoPP” groups, respectively. An infected + ART group which received only saline intraperitoneally for 4 days after parasite clearance with ART treatment was called the “infected + ART + saline” group. This group of mice was used as a control to compare the effects of ZnPP and CoPP. Lastly, a noninfected group of mice that were treated with ART for 3 days, the “noninfected + ART” group, was studied to eliminate the possible effects of ART in hepatopathy, and another noninfected group of mice, without any treatment, referred to as “noninfected,” was studied as a true control. There were six animals in every group.
Liver function tests.
Biochemical analysis of serum was done to follow the extent of liver damage in mice. Serum was separated from the blood for different groups of experimental mice and stored at −20°C. Alanine transaminase (ALT), aspartate transaminase (AST), and alkaline phosphatase (ALP) activities and amounts of albumin and bilirubin were measured in the serum with the help of commercially available kits (Randox Laboratories Ltd., Ardmore, Antrim, United Kingdom). The manufacturer's instructions were precisely followed.
RNA isolation and real-time RT-PCR.
The TRIzol reagent (Invitrogen, Carlsbad, CA) was used to isolate total RNAs from livers of different groups of mice as described by the manufacturer. An oligo(dT)18 primer and a RevertAid First Strand cDNA synthesis kit (Fermentas) were used for the reverse transcription (RT) of total RNA (2 μg). Real-time PCR was performed in triplicate by using a diluted form of the obtained cDNA. Primers (Table 1) were obtained from Integrated DNA Technologies Inc. (San Diego, CA), and Power SYBR green was obtained from Applied Biosystems. The reactions were performed in an ABI 7500 Fast real-time PCR system (Applied Biosystems), and the following cycling conditions were maintained: 1 cycle of 95°C for 10 min and then 50 cycles of 95°C for 15 s and 60°C for 1 min. The 18S rRNA gene was chosen as an internal control. Analysis of data for relative gene expression was based on the 2−ΔΔCT method (50). Data were normalized to the 18S rRNA gene levels and expressed as fold changes compared with the value for “noninfected” controls.
TABLE 1.
Primers for real-time RT-PCR analysisa
| Target gene | Primer sequence (5′–3′) |
Product size (bp) | |
|---|---|---|---|
| Forward | Reverse | ||
| Cxcl1 | TGTTGTGCGAAAAGAAGTGC | ACAAAATGTCCAAGGGAAGC | 184 |
| Cxcl2 | TCCAGAGCTTGAGTGTGACG | CTTTGGTTCTTCCGTTGAGG | 204 |
| Icam1 | CGATCTTCCAGCTACCATCC | CACTGCTGTTTGTGCTCTCC | 167 |
| Vcam1 | TGGAGGCTGAACACTTTTCC | ATGGAGTCACCGATTTGAGC | 199 |
| Il1β | CAAAATACCTGTGGCCTTGG | CTTGTGCTCTGCTTGTGAGG | 229 |
| Il6 | ACTTCACAAGTCCGGAGAGG | TTCTGCAAGTGCATCATCG | 177 |
| Ifnγ | CGCTACACACTGCATCTTGG | ACCATCCTTTTGCCAGTTCC | 178 |
| 18s rRNA gene | GTTGGTTTTCGGAACTGAGG | TCGTTTATGGTCGGAACTACG | 197 |
Details of the PCR conditions are described in Materials and Methods.
Immunohistochemical analysis of neutrophil infiltration.
Neutrophil infiltration in the liver was followed by immunohistochemical staining of paraffin-embedded liver tissues as mentioned earlier (3). Briefly, paraffin-embedded tissue sections from the indicated time points were deparaffinized in xylene, rehydrated in graded ethanol, and subjected to heat-induced antigen retrieval in citrate buffer (pH 6.0) containing 0.05% Tween 20. Following antigen retrieval, the slides were washed in Tris-buffered saline (TBS) plus Triton X-100 (0.025%), blocked with 2% BSA in TBS for 2 h at room temperature, and incubated with a primary neutrophil marker antibody (NIMP-R14) in blocking solution at 4°C overnight. The next day, the slides were thoroughly washed and incubated with FITC-conjugated secondary anti-rat antibody in blocking solution for 1 h at room temperature. The slides were then thoroughly washed and counterstained with the fluorescent nucleic acid stain DAPI. The slides were viewed under the 20× objective lens of a Leica DM 2500 fluorescence microscope (Leica Microsystems, GmbH, Wetzlar, Germany). Fluorescence micrographs provided are representatives of 4 to 6 fields scanned per slide, collected randomly from experiments repeated thrice.
Assay of MPO activity.
Myeloperoxidase (MPO) chlorinating activity was measured on the basis of taurine chlorination with the MPO-H2O2-Cl− assay system (51). Livers from different groups of mice were perfused and homogenized in phosphate-buffered saline (PBS) (pH 7.4). The homogenate was centrifuged at 10,000 × g for 10 min, and the pellet was dissolved in ice-cold solubilization buffer containing 0.5% hexadecyltrimethylammonium bromide in PBS (pH 7.4). The samples were subsequently sonicated, freeze-thawed thrice, and centrifuged at 12,000 × g for 30 min at 4°C; the supernatants were collected. The reaction mixture contained 80 μl of supernatant, 780 μl PBS (pH 7.4), and 100 μl of 150 mM taurine. The reaction mixture was incubated at 25°C for 5 min in the water bath, and 40 μl of 2.5 mM H2O2 was added and mixed well. The reaction was stopped after 30 min by the addition of 40 μl of catalase and 100 μl 5-thio-2-nitrobenzoic acid (TNB) to each well. Fresh TNB solution was prepared by raising the pH of a 2 mM DTNB solution to pH 12 and then readjusting it to pH 7.4. The absorbance of TNB in the assay mixture was measured at 412 nm after 20 min. In this assay, hypochlorous acid reacted with taurine to form taurine chloramine, which converts yellow TNB to colorless DTNB. MPO activity was expressed in units/g wet liver (52). One unit is the amount of MPO that can produce 1.0 nmol of taurine chloramine, which in turn can oxidize 2.0 nmol of TNB to DTNB under the given assay conditions. The extinction coefficient of TNB is 14,100 M−1 cm−1 at 412 nm.
Measurements of oxidative stress.
Oxidative stress was monitored by measuring lipid peroxidation and carbonylation of proteins as described previously (53, 54). Malondialdehyde production or lipid peroxidation was measured on the basis of thiobarbituric acid-reactive substance (TBARS) levels. Briefly, 1 ml of 5% liver homogenate (in 0.9% saline) from mice was mixed with a 2-ml solution containing trichloroacetic acid-thiobarbituric acid (0.375%–15% [wt/vol]) in 0.25 N HCl and 0.01% butylated hydroxytoluene. The mixture was heated in a boiling water bath for 15 min and subsequently cooled at room temperature. The samples were centrifuged, and the absorbance of the supernatants was taken at 535 nm to measure the TBARS concentration. Tetraethoxypropane was considered the standard for this protocol (53–56). For measurements of carbonylated proteins, a standard colorimetric method was followed as described previously (53, 54, 57). The method is based on the binding of dinitrophenylhydrazine to the carbonyl group, and amounts were quantified by measuring the absorbance at 362 nm.
Western immunoblotting.
Liver homogenates were prepared as described previously (3), and the supernatants obtained after centrifugation of the homogenates were quantified. A sample of 70 μg of protein was loaded into each well of 12% polyacrylamide-SDS gels and subjected to electrophoresis at a constant voltage (100 V). Proteins were then transferred to a nitrocellulose membrane in a wet transfer apparatus, incubated with 5% nonfat dry milk, and washed in TBS containing Tween 20. The membranes were next incubated with anti-HO-1 (Abcam) and anti-β-actin (Biovision) primary antibodies overnight at a dilution of 1:1,000. The membranes were then washed and incubated with secondary antibodies for 2 to 3 h, at a dilution of 1:5,000. Finally, after washing, the proteins on the membranes were detected by staining with a DAB solution and hydrogen peroxide (58).
Assay of HO-1 activity.
Liver homogenates from mice were prepared in a buffer (Tris-HCl [pH 7.4], 5 ml/liter Triton X-100, and protease inhibitor cocktail). A liver homogenate containing 200 μg of protein was mixed with assay buffer at a volumetric ratio of 1:1 to make a final volume of 1 ml. The assay buffer contained 0.8 mM NADPH, 1 mM MgCl2, 2 mM glucose-6-phosphate, 0.2 U of glucose-6-phosphate dehydrogenase, 100 mM potassium phosphate buffer, 20 μM hemin, and 2 mg of mouse liver cytosol (as a source of biliverdin reductase). The mixture was incubated in the dark for 1 h at 37°C and then placed on ice for 5 min. Chloroform was used to extract the bilirubin formed in the mixture, and its concentration was estimated from its extinction coefficient (40 mM/liter/cm) by measuring the A464–530. The activity of HO-1 was expressed as nmol of bilirubin/mg protein/h (58, 59).
Detection of apoptosis by measuring caspase-3 activity.
Apoptosis was determined by measuring the caspase-3 activity in cytosolic fractions of liver tissues by use of a commercially available kit (Sigma, St. Louis, MO) as mentioned previously (53, 54, 56, 60). The instructions provided by the manufacturer were followed. The absorbance at 405 nm was measured in triplicate with the help of a microtiter plate reader to determine the amount of pNA released from a caspase-3-specific substrate (Ac-DEVD-pNA). The average values for triplicates are presented, and the activity of caspase-3 is expressed in pmol/mg protein/min (61).
Data analysis.
All experiments were performed in triplicate, with six animals in each group. Data obtained from all experiments are expressed as means ± standard errors of the means (SEM). Calculations of the levels of significance were performed on the basis of unpaired Student's t test and one-way analysis of variance (ANOVA), as applicable. P values of <0.05 were considered statistically significant.
RESULTS
Liver restores its function after clearance of malaria parasites from blood.
Significant liver damage was induced in mice at 45% parasitemia (5th day postinfection), as evident from liver function tests (Fig. 1B to H), and in the absence of antimalarial therapy, it was found that all the infected mice died within 8 days. However, upon ART treatment, the parasites were eradicated from blood within 3 days (day 5 to day 7 postinfection) (Fig. 1A), and 80% of the infected mice survived. For the infected + ART group, day 8 postinfection was considered day 1 post-parasite clearance for mice that survived after antimalarial treatment (Fig. 1A). Liver function was monitored in mice from the noninfected, infected, noninfected + ART, and infected + ART groups after clearance of parasites from blood (Fig. 1B to H). Since hemolysis alone can cause an increase in the activity of liver enzymes (ALP, ALT, and AST) in serum, the measurement of bilirubin (conjugated and unconjugated) was performed to correctly analyze liver function. ALP (Fig. 1B), ALT (Fig. 1C), AST (Fig. 1D), total bilirubin (Fig. 1E), unconjugated bilirubin (Fig. 1F), conjugated bilirubin (Fig. 1G), and albumin (Fig. 1H) were measured in all the experimental groups and compared thereafter. Results indicated that the levels of the liver enzymes (Fig. 1B to D) and bilirubin levels (Fig. 1E to G) in serum were significantly higher in the infected mice than in the noninfected mice and that the levels increased from day 1 to day 5, after which they decreased steadily until day 25 in the infected + ART group (Fig. 1B to G). The conjugated bilirubin level was increased from 0.22 ± 0.02 mg/dl in noninfected mice to 0.31 ± 0.025 mg/dl in infected mice with ∼45% parasitemia, and the level was further increased, to 0.48 ± 0.051 mg/dl, in infected + ART mice after clearance of parasites from blood. Similarly, the unconjugated bilirubin level was found to be increased from 0.9 ± 0.09 mg/dl in noninfected mice to 1.89 ± 0.2 mg/dl in infected mice with ∼45% parasitemia, and this level was also further increased, to 3.22 ± 0.33 mg/dl, in infected + ART mice after clearance of parasites from blood. The albumin level (Fig. 1H) was much lower in the infected mice than in the noninfected mice, and the level decreased in serum from day 1 to day 5 and then gradually increased. On day 25 after clearance of parasites from blood, the levels of these enzymes, bilirubin, and albumin were close to those in noninfected mice maintained for a similar period.
FIG 1.

Clearance of malaria parasites from the blood of infected mice by use of ART, as well as analyses of liver function before parasite clearance and with time after parasite clearance. (A) Percentages of parasitemia in infected mice before and after ART treatment. Restoration of liver function was studied just before and after parasite clearance (day 1 to day 25 post-parasite clearance). Liver function tests were based on measurements of the activities of alkaline phosphatase (ALP) (B), alanine transaminase (ALT) (C), and aspartate transaminase (AST) (D), along with measurements of total bilirubin (E), unconjugated bilirubin (F), conjugated bilirubin (G), and albumin (H), in the sera of noninfected mice, infected mice, noninfected + ART mice, and infected + ART mice on day 1, day 3, day 5, day 10, day 15, and day 25 after clearance of parasites from the blood. The details of the methodology are described in Materials and Methods. Data are presented as means ± SEM. *, P < 0.05 versus noninfected mice (n = 6).
Inflammatory response in liver during malaria and after the clearance of parasites from blood.
Recovery of an organ after injury involves an inflammatory phase followed by proliferation and remodeling phases (17, 18). The status of inflammatory mediators in the liver after malaria infection is important for understanding the mechanism of restoration of liver function. The levels of mRNA expression of proinflammatory cytokines (interleukin-1β [IL-1β], IL-6, gamma interferon [IFN-γ], CXCL1 [KC], and CXCL2 [MIP2]) and endothelial cell adhesion molecules (ICAM-1 and VCAM-1) in livers from different experimental groups were monitored by real-time RT-PCR analysis (Table 2). The results indicated that these proinflammatory genes were highly expressed initially, but their expression gradually decreased with time (Table 2). Immunohistochemical analysis of the liver tissues was done to follow neutrophil infiltration in the liver following malaria and during healing of the liver after the clearance of parasites from the blood (Fig. 2A). Neutrophil infiltration, as evident from the green fluorescence of the neutrophil marker (NIMP-R14) antibody, was significantly higher in the livers of infected mice than in those of noninfected mice. The level of infiltration was found to be highest in the liver tissues of infected + ART mice on day 5 post-parasite clearance, and the level decreased steadily until day 25 post-parasite clearance. The treatment with ART alone did not have any impact on neutrophil infiltration in the liver. An MPO (a marker of neutrophils) assay was also performed to further confirm the infiltration of neutrophils into the liver during inflammation and the restoration of liver function after malaria infection (Fig. 2B), and MPO activity was found to be correlated positively with the degree of neutrophil infiltration. MPO activity was found to be significantly higher in the infected mice than in the noninfected mice. The level of MPO activity was highest, however, on day 5 after clearance of parasites from the blood, and it decreased with time (Fig. 2B), indicating that neutrophil infiltration in the liver gradually decreased after the initial inflammatory phase.
TABLE 2.
Changes in gene expression as measured by real-time RT-PCRa
| Mouse group | Fold change in gene expression (relative to that in noninfected mice, after normalization to 18S rRNA) |
||||||
|---|---|---|---|---|---|---|---|
| Il6 | CxCl1 | Cxcl2 | Ifnγ | Icam1 | Vcam1 | Il1β | |
| Infected (45% parasitemia) | 1.32 ± 0.14 | 3.46 ± 0.41 | 1.85 ± 0.19 | 8.32 ± 0.9 | 1.21 ± 0.12 | 1.2 ± 0.13 | 1.17 ± 0.19 |
| Noninfected + ART (day 1) | 1.02 ± 0.1 | 1.06 ± 0.1 | 1.14 ± 0.15 | 1.08 ± 0.1 | 1.12 ± 0.12 | 1.03 ± 0.1 | 1.07 ± 0.1 |
| Infected + ART (day 1) | 1.3 ± 0.12 | 4.67 ± 0.52 | 2.58 ± 0.3 | 9.18 ± 1.1 | 1.26 ± 0.13 | 1.23 ± 0.13 | 1.22 ± 0.13 |
| Noninfected + ART (day 3) | 1.05 ± 0.1 | 1.09 ± 0.1 | 1.06 ± 0.1 | 0.97 ± 0.1 | 1.03 ± 0.1 | 0.98 ± 0.1 | 1.03 ± 0.1 |
| Infected + ART (day 3) | 2.31 ± 0.23 | 2.12 ± 0.22 | 4.1 ± 0.45 | 9 ± 0.91 | 2.66 ± 0.3 | 3.98 ± 0.4 | 1.26 ± 0.13 |
| Noninfected + ART (day 5) | 1.03 ± 0.1 | 1.02 ± 0.1 | 1.05 ± 0.1 | 1.06 ± 0.1 | 1.01 ± 0.1 | 1.05 ± 0.1 | 1.04 ± 0.1 |
| Infected + ART (day 5) | 8.46 ± 0.9 | 2.05 ± 0.2 | 6.36 ± 0.7 | 9.31 ± 0.95 | 5.02 ± 0.6 | 5.08 ± 0.6 | 1.31 ± 0.14 |
| Noninfected + ART (day 10) | 1.06 ± 0.1 | 1.01 ± 0.1 | 0.98 ± 0.11 | 1.03 ± 0.1 | 1.06 ± 0.1 | 1.01 ± 0.1 | 0.98 ± 0.1 |
| Infected + ART (day 10) | 2.28 ± 0.24 | 1.52 ± 0.15 | 3.48 ± 0.37 | 1.79 ± 0.18 | 1.27 ± 0.15 | 1.7 ± 0.18 | 1.18 ± 0.12 |
| Noninfected + ART (day 15) | 1.04 ± 0.1 | 0.97 ± 0.09 | 1.03 ± 0.12 | 0.98 ± 0.09 | 1.05 ± 0.1 | 1.07 ± 0.1 | 1.05 ± 0.1 |
| Infected + ART (day 15) | 1.68 ± 0.17 | 1.15 ± 0.12 | 2.37 ± 0.25 | 1.53 ± 0.15 | 1.16 ± 0.14 | 1.54 ± 0.15 | 1.15 ± 0.13 |
| Noninfected + ART (day 25) | 1.03 ± 0.1 | 1.03 ± 0.1 | 1.07 ± 0.1 | 1.05 ± 0.1 | 0.98 ± 0.09 | 1.07 ± 0.1 | 1.06 ± 0.09 |
| Infected + ART (day 25) | 1.16 ± 0.18 | 1.18 ± 0.2 | 1.25 ± 0.12 | 1.18 ± 0.15 | 1.1 ± 0.12 | 1.17 ± 0.19 | 1.08 ± 0.14 |
Results depict the means ± SEM of results from five independent experiments. All reactions were done in triplicate. Details of the PCR conditions are described in Materials and Methods.
FIG 2.

Time-dependent evaluation of neutrophil infiltration in livers of mice before and after clearance of malaria parasites. (A) Immunohistochemistry of livers showing neutrophil infiltration. Neutrophils (as indicated by green fluorescence) were observed in the livers of noninfected mice, infected mice, noninfected + ART mice, and infected + ART mice on day 1, day 3, day 5, day 10, day 15, and day 25 after clearance of parasites from the blood. Blue fluorescence (DAPI) indicates hepatocytes. (B) Measurement of MPO activity to follow neutrophil infiltration in the livers of noninfected mice, infected mice, noninfected + ART mice, and infected + ART mice on day 1, day 3, day 5, day 10, day 15, and day 25 after clearance of parasites from the blood. The details of the methodology are described in Materials and Methods. Data are presented as means ± SEM. **, P < 0.01 versus noninfected mice (n = 6).
Liver inflammation after clearance of malaria parasites is linked to oxidative stress and hepatocyte apoptosis.
Since inflammation is associated with the generation of reactive oxidants, we were interested in measuring oxidative stress in the liver after parasite clearance (3). Lipid peroxidation and protein carbonylation were measured as indicators of oxidative stress (Fig. 3). Biochemical analyses of liver homogenates indicated that lipid peroxidation (Fig. 3A) and protein carbonylation (Fig. 3B) were significantly higher in the livers of infected mice than in those of noninfected mice. The levels further increased initially, until day 5 after parasite clearance from the blood. However, oxidative stress decreased steadily from day 5 to day 25, clearly indicating that restoration of liver function after malaria infection could be correlated negatively with oxidative stress after the initial inflammatory phase (Fig. 3A and B). Oxidative stress and inflammation in the livers of mice with malaria can cause cell death through apoptosis. Biochemical assays were done to check whether apoptosis occurred due to inflammation in the liver during malaria and after clearance of malaria parasites from blood. Apoptosis was detected by measuring the activity of caspase-3. Analysis of caspase-3 activity revealed that the degree of apoptosis was higher in the infected mice than in the noninfected mice, and apoptosis continued to occur even after clearance of malaria parasites but eventually decreased steadily with time (Fig. 3C).
FIG 3.
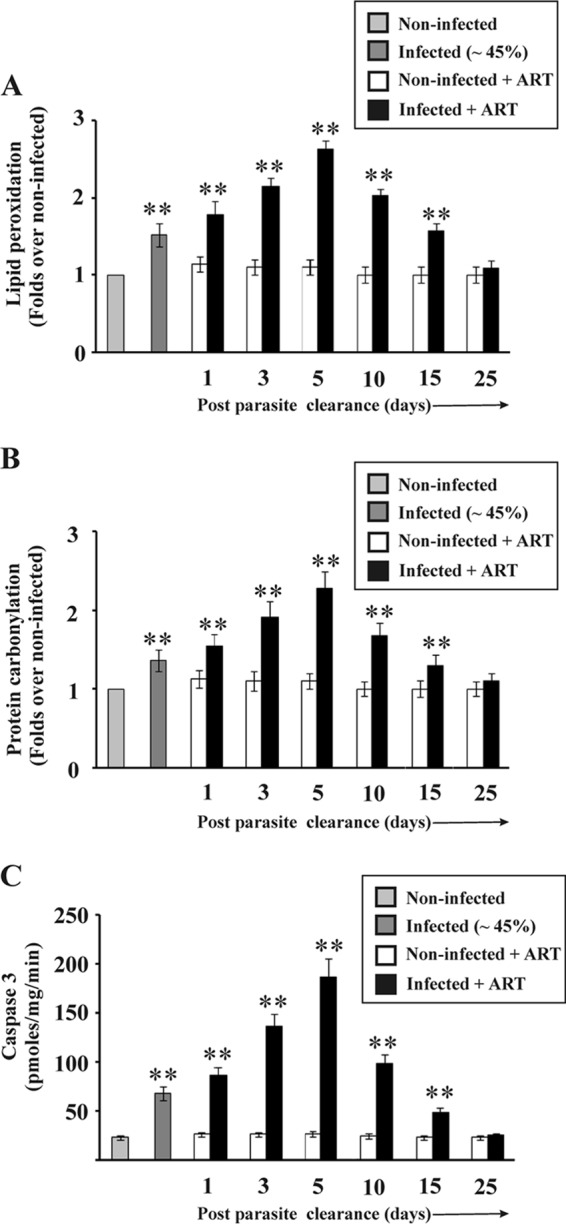
Measurement of oxidative stress and detection of apoptosis in the liver before and after clearance of parasites. Lipid peroxidation products (A) and protein carbonylation (B) (markers of oxidative stress) in the livers of noninfected, infected, noninfected + ART, and infected + ART mice were measured on day 1, day 3, day 5, day 10, day 15, and day 25 after clearance of parasites from the blood. Data represent fold changes relative to the levels in noninfected mice. Statistical analyses were performed on untransformed data. (C) Measurement of caspase-3 activity as a measure of apoptosis in the livers of noninfected, infected, noninfected + ART, and infected + ART mice on day 1, day 3, day 5, day 10, day 15, and day 25 after clearance of parasites from blood. The details of the methodology are described in Materials and Methods. Data are presented as means ± SEM. **, P < 0.01 versus noninfected mice (n = 6).
Association of HO-1 as a cytoprotective factor against inflammation, oxidative stress, and apoptosis in the liver during malaria and after parasite clearance.
Inflammation is mostly associated with tissue injury, and the inflammatory phase after injury is very important for the recovery of damaged tissue (62). However, an excessive or prolonged inflammatory phase can cause a delay in recovery from the injury. HO-1 has a very important role in regulating inflammation. Therefore, it is essential to determine the relationship between HO-1 activity and the inflammatory phase during the process of restoration of liver function after clearance of parasites from blood. HO-1 activity was analyzed and found to be significantly higher in the livers of infected mice than in those of noninfected mice. The level increased until day 5 after parasite clearance (Fig. 4A) and then decreased gradually, indicating the active functioning of HO-1 during the resolution of inflammation and recovery of the damaged liver after parasite clearance. Furthermore, Western immunoblotting revealed that HO-1 expression was high on day 5 after parasite clearance by ART and then gradually decreased (Fig. 4B and C). Since HO-1 could be associated with the inflammatory phase of the process of restoration of liver function after malaria infection, it was worth investigating the role of HO-1 in inflammation. In this regard, after parasite clearance from the blood by ART treatment, HO-1 activity in vivo was monitored on the 5th day, after administering ZnPP or CoPP for 4 consecutive days (Fig. 5A). Inhibition of HO-1 activity by ZnPP was found to be associated with increases in the expression of proinflammatory molecules, such as CXCL1, CXCL2, ICAM-1, and VCAM-1, on day 5 relative to the levels in infected + ART mice (Fig. 5B). MPO activity was thus measured, because the increase in endothelial cell adhesion molecules resulted in increased neutrophil infiltration (Fig. 5C). The increase in neutrophil infiltration due to inhibition of HO-1 activity led to an enhancement of oxidative stress and apoptosis in the liver. Increased protein carbonylation and lipid peroxidation indicated that oxidative stress was significantly enhanced on the 5th day after ZnPP treatment (Fig. 5C). Increased caspase-3 activity also indicated enhanced apoptosis on the 5th day after ZnPP treatment (Fig. 5C). To confirm whether HO-1 offered a cytoprotective, anti-inflammatory effect during the process of restoration of liver function after malaria infection in our model, the mice were treated with CoPP (an HO-1 inducer) for 4 consecutive days after parasite clearance by ART. Stimulation of HO-1 activity by CoPP resulted in decreases in the expression of proinflammatory cytokines and endothelial cell adhesion molecules relative to the levels in the infected + ART mice (Fig. 5B). Neutrophil infiltration in the liver during the inflammatory phase was reduced, as evident from the decrease in MPO activity (Fig. 5C). Furthermore, there was a significant decrease in oxidative stress in the liver, as revealed by reductions in lipid peroxidation and protein carbonylation (Fig. 5C). Caspase-3 activity in the livers of CoPP-treated mice was also reduced significantly (Fig. 5C).
FIG 4.
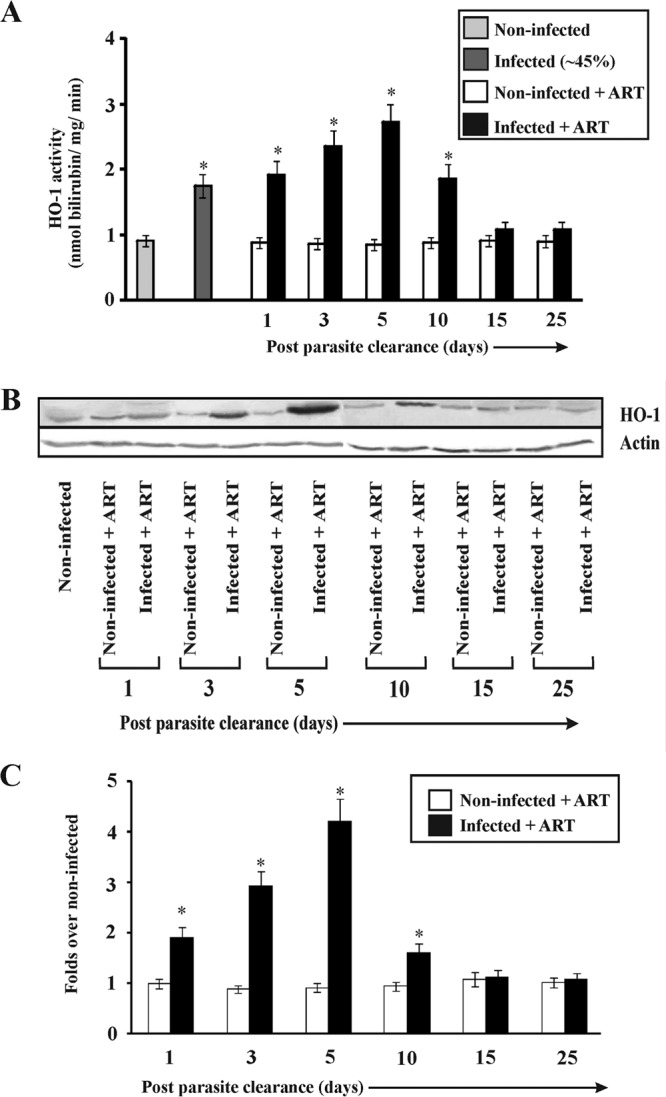
Activity and expression of HO-1 in the liver during malaria and after parasite clearance. (A) Activities of HO-1 in the livers of noninfected, infected, noninfected + ART, and infected + ART mice on day 1, day 3, day 5, day 10, day 15, and day 25 after clearance of parasites from the blood. (B) Western immunoblot of HO-1 in the livers of noninfected, noninfected + ART, and infected + ART mice on day 1, day 3, day 5, day 10, day 15, and day 25 after clearance of parasites from the blood. (C) Densitometric analyses of Western immunoblots to quantitate HO-1 expression. Data are given as fold changes relative to the levels in the noninfected group. The details of the methodology are described in Materials and Methods. Data are presented as means ± SEM. *, P < 0.05 versus noninfected mice (n = 6).
FIG 5.

Effects of inhibition of HO-1 on inflammation, oxidative stress, and apoptosis in the liver before and after parasite clearance. (A) HO-1 activities in livers of noninfected + ART, infected + ART, infected + ART + ZnPP, and infected + ART + CoPP mice on day 5 after clearance of parasites from the blood. Infected mice which were not administered ZnPP or CoPP received only saline. Administrations of ZnPP, CoPP, or saline were carried out from days 1 to 4 after clearance of parasites from the blood. (B) Real-time RT-PCR analyses of expression of the Cxcl1, Cxcl2, Icam1, and Vcam1 genes in livers of infected, noninfected + ART, infected + ART, infected + ART + ZnPP, and infected + ART + CoPP mice on day 5 after clearance of parasites from blood. ZnPP and CoPP were administered to ART-treated infected mice from days 1 to 4 after clearance of malaria parasites. Data are presented as fold changes relative to the levels in noninfected mice (considered 1-fold). Statistical analyses were performed on untransformed data. (C) Measurements of MPO activity, protein carbonylation, lipid peroxidation, and caspase-3 activity in the livers of noninfected + ART, infected + ART, infected + ART + ZnPP, and infected + ART + CoPP mice on day 5 after clearance of parasites from blood. Data are presented as fold changes compared to the levels in noninfected mice. The details of the methodology are described in Materials and Methods. Statistical analyses were performed on untransformed data. Data are presented as means ± SEM. *, P < 0.05 versus noninfected + ART mice (day 5); #, P < 0.05 versus infected + ART mice (day 5); **, P < 0.01 versus noninfected mice; ##, P < 0.01 versus infected + ART mice (day 5) (n = 6).
Involvement of HO-1 in the recovery of liver function after damage due to malarial infection.
Finally, it was essential to check the involvement of HO-1 in the process of restoration of liver function after damage due to Plasmodium attack. Serum ALP (Fig. 6A), ALT (Fig. 6B), and AST (Fig. 6C) activities and levels of total bilirubin (Fig. 6D), conjugated bilirubin (Fig. 6E), and albumin (Fig. 6F) were measured on day 1, day 5, day 10, day 15, and day 25 after ZnPP or CoPP treatment (post-parasite clearance). The data clearly indicated that while there was a significant delay in restoration of liver function in the infected + ART + ZnPP mice, there was a considerable acceleration in restoration of liver function in the infected + ART + CoPP mice after parasite clearance (Fig. 6). The activities of the liver enzymes and the amount of conjugated bilirubin were significantly increased in the sera of infected + ART + ZnPP mice on day 5 compared to the infected + ART mice (Fig. 6). The albumin level decreased significantly in the infected + ART + ZnPP group compared to the infected + ART group. On the other hand, the enhancement in activity of all these enzymes, along with the level of conjugated bilirubin, was significantly reduced during the inflammatory phase after treatment with CoPP (Fig. 6). The fall in albumin level in the infected + ART + CoPP mice was also significantly reduced compared to that in the infected + ART group. These data suggest that HO-1 is considerably associated with alleviation of the inflammatory phase after the clearance of malaria parasites from the blood.
FIG 6.

Liver function tests to follow the role of HO-1 in restoration of liver function after damage induced by malarial attack. Measurements of ALP (A), ALT (B), AST (C), total bilirubin (D), conjugated bilirubin (E), and albumin (F) were performed. All these analyses were performed on sera from noninfected + ART, infected + ART, infected + ART + CoPP, and infected + ART + ZnPP mice. The details of the methodology are described in Materials and Methods. Data are presented as means ± SEM. *, P < 0.05 versus noninfected + ART mice (day 5); #, P < 0.05 versus infected + ART mice (day 5) (n = 6).
DISCUSSION
We have presented evidence that HO-1 is functionally involved in the process of inherent restoration of liver function after damage from malaria. Chemical silencing of HO-1 by ZnPP after parasite clearance enhanced inflammation and oxidative stress in the liver, along with hepatocyte apoptosis, thereby delaying the restoration process. On the other hand, induction of HO-1 by CoPP considerably reduced the above complications. These results therefore suggest probable anti-inflammatory and hepatoprotective roles of HO-1 in spontaneous recovery of liver function after malaria.
The emergence of drug-resistant malaria is a cause of tremendous increase in mortality due to malaria (63–65). Therefore, we infected mice with an MDR strain of P. yoelii to estimate the intensity of damage. MDR P. yoelii is lethal and caused severe liver damage even when the peak parasitemia was not reached. At 45% parasitemia, the liver damage was significant. Although such high-level parasitemia is generally not allowed in human beings (due to treatment against malaria in patients), we allowed the parasitemia to increase to 45% so that significant damage could occur, thereby giving an opportunity to study the restoration of liver function in a better way. With low-level parasitemia, the liver damage would not be very significant, and therefore the process of the restoration of liver function would have been hard to explore. Thus, this murine malaria model was specifically designed to study the mechanism of recovery of liver function after malaria.
ART is one of the most powerful antimalarial drugs of the recent era and helps to counter chloroquine-resistant parasites (66). In our model, ART successfully killed all the parasites when administered for 3 successive days at a high dose (50 mg/kg/day). However, absolute clearance of dead parasites from the circulation requires a few more days. A high dose was selected to ensure clearance of the parasites in a short duration. After clearance of the parasites, liver function was monitored, and the data revealed that liver function was restored within 25 days. Administration of ART did not cause significant alterations in liver function or proinflammatory gene expression. A wound or injury is followed by phases of hemostasis, inflammation, proliferation, and, finally, tissue remodeling (17–19, 67, 68). The inflammatory phase is critical for the restoration of organ function, during which growth factors and cytokine signals synchronize cellular movements required during the repair process (62). However, excessive inflammation has been found to be responsible for the delay in healing in several experimental models of wound repair (62). Activation and infiltration of neutrophils in the damaged tissue are characteristic features of the inflammatory phase. Infiltration of neutrophils is regulated by proinflammatory cytokines and endothelial cell adhesion molecules (62). MPO is one of the important enzymes released by the activated neutrophils that produce reactive oxygen and nitrogen species (69, 70). One of the many reactive products produced by MPO is hypochlorous acid (HOCl), which modifies various biomolecules by chlorination and/or oxidation (71–73). Thus, excessive infiltration of neutrophils during the inflammatory phase results in severe oxidative stress in the injured tissue, which eventually causes apoptosis and subsequently delays the process of restoration of organ function (74–77). Malaria infection causes severe hemolysis. Free heme and hemozoin accumulate in the liver during malaria. Free heme is a pro-oxidant, so massive oxidative stress, hepatocyte apoptosis, and liver dysfunction occur in the liver after malaria infection. Liver damage immediately leads to a proinflammatory response, and the resultant localized neutrophil infiltration further aggravates liver dysfunction (3). The clearance of heme, neutrophils, hemozoin, proinflammatory cytokines, chemokines, and various other pro-oxidants from the liver requires a long time, and these factors are therefore the probable causes of persistent liver dysfunction even after clearance of parasites (3).
In the present study, the process of restoration of liver function after malaria was addressed. The data revealed that the activities of liver enzymes, such as ALT, ALP, and AST, were significantly elevated initially, until day 5 after parasite clearance by ART. These levels, however, gradually decreased with time. The bilirubin levels also followed the same pattern. Studies of malaria patients so far have indicated that severe malaria in human beings can cause conjugated (9, 78, 79), unconjugated, and mixed (10, 80, 81) hyperbilirubinemia. This depends on the kind of pathological changes involved. The extents of hemolysis and hepatopathy decide the type of hyperbilirubinemia in malaria patients (81). In our model of murine malaria, we observed unconjugated hyperbilirubinemia, possibly because of excessive hemolysis. However, we also observed a significant increase in conjugated bilirubin initially, until day 5 post-parasite clearance by ART. The inflammatory phase is a critical period because it determines the time of restoration of function of any injured organ. The data suggested that there were significant increases in gene expression for some of the selected proinflammatory proteins, such as CXCL1, CXCL2, IL-1β, and IL-6, although the times of increase were not the same for all of them. Moreover, the data also indicated that parasite clearance from blood by ART did not resolve inflammation, as evident from the gene expression patterns of the proinflammatory mediators. Rather, the proinflammatory mediators maintained elevated expression until day 5 after parasite clearance, indicating continued inflammation even after parasite clearance from blood. Thus, the data indicate an initial inflammation of the liver followed by a gradual restoration of function. The gene expression of the endothelial cell adhesion molecules was also significantly elevated until day 5, but later it decreased gradually. Since all these molecules aid in neutrophil infiltration in the liver, further studies were performed to check whether neutrophils infiltrate the liver during the inflammatory phase of restoration of liver function. Interestingly, a significant increase in neutrophil infiltration, as evident from immunohistochemical analysis, followed by their persistence was evident in the livers of mice early after parasite clearance. The same pattern reflected in the MPO levels further confirmed the infiltration of neutrophils in the liver. Since neutrophils are exogenous sources of reactive oxidants, their infiltration in the liver is expected to cause severe oxidative stress. Biochemical analyses of liver homogenates clearly indicated an increase in oxidative stress, as evident from increased lipid peroxidation and protein carbonylation. The data also demonstrated that there was an increase in apoptosis as revealed by caspase-3 assay. Although apoptosis may not cause inflammation (82, 83), inflammation causes apoptosis under various pathological conditions (84). In several instances, it has been seen that neutrophil infiltration involved in inflammation causes tissue damage (85). Increased neutrophil infiltration further amplifies inflammation via a positive-feedback loop. Increased neutrophils are associated with MPO activity and subsequent production of highly toxic reactive species, such as HOCl (86). Moreover, production of proinflammatory cytokines, such as TNF-α, at the site of inflammation can also induce apoptosis (87). Under several pathological and certain physiological conditions, reactive oxygen species produced by cellular MPO induce apoptosis in inflammatory and other cells (88). Thus, inflammation and, more specifically, neutrophilic inflammation often induce apoptosis (84).
The HO-1 system has been credited with cytoprotective and anti-inflammatory roles. Its role in preventing excessive and prolonged inflammation after wounding has already been studied (20, 89–91). Although the HO-1 system has been shown to prevent inflammation in the liver under several pathological conditions (36), the actual mechanism behind the anti-inflammatory and cytoprotective actions of HO-1 is still not very clear. HO-1 can mediate its effects either through production of CO or bilirubin (92, 93) or by clearing heme from the circulation, thus reducing heme-induced oxidative damage (94, 95). In this regard, our data from HO-1-specific Western immunoblot analysis revealed that the expression of HO-1 was increased severalfold in liver samples from infected + ART mice on day 5 after clearance of parasites from the blood. This suggested that HO-1 may play an important role either in promoting inflammation or in alleviating inflammation after damage to the liver due to malaria. To understand the effect of HO-1 on inflammation and/or restoration of liver function in this model, the use of an HO-1 inducer as well as an inhibitor was essential. ZnPP and CoPP are a well-known inhibitor and stimulator of HO-1, respectively (59, 96–102). However, both the protoporphyrins may have various effects on the expression of transforming growth factor beta (TGF-β) (103), a cytokine that is intricately associated with the regulation of inflammation (104). In the present study, we specifically focused on the changes in HO-1 activity due to the administration of ZnPP and CoPP and its association with liver dysfunction and restoration of its function after parasite clearance from the blood. The modulation of HO-1 activity by these chemicals has been performed previously, and the dose was selected accordingly. Separate groups of mice were administered ZnPP or CoPP for 4 successive days after parasite clearance by ART treatment, and the animals were sacrificed on day 5. ZnPP treatment caused a significant increase in the expression of proinflammatory genes, such as Icam1, Vcam1, Cxcl1, and Cxcl2, while CoPP treatment showed a decrease in the expression of the aforesaid genes in comparison to that in infected + ART mice treated with saline. Furthermore, MPO activity was found to be relatively higher in ZnPP-treated mouse livers and lower in CoPP-treated mouse livers than in those of the control mice. Lipid peroxidation, protein carbonylation, and caspase-3 activity were also found to be higher in livers of ZnPP-treated mice and significantly lower in those of CoPP-treated mice than in those of infected + ART mice undergoing normal restoration of liver function. Finally, the data revealed that the activities of the liver enzymes ALT, ALP, and AST and the level of bilirubin were significantly higher in ZnPP-treated mice and much lower in CoPP-treated mice than in infected + ART mice, especially during the inflammatory phase. Since the ZnPP or CoPP treatment was discontinued after day 4 post-parasite clearance, the effect of HO-1 inhibition or induction was well understood only during the inflammatory phase, not after that. The liver function was restored in all groups of mice by approximately day 25 after clearance of parasites. All these results collectively indicate that inhibition or induction of HO-1 activity by ZnPP or CoPP immediately before maximum inflammation may enhance or reduce the inflammation, respectively. Therefore, continuous inhibition of HO-1 may possibly result in severe damage to the liver, and the process of restoration of liver function may be delayed. In contrast, continuous stimulation of the HO-1 activity at the site of inflammation by CoPP is expected to alleviate the inflammatory phase, thereby accelerating the process of restoration of liver function. Thus, from the various associations of HO-1 with the gradual evaluation of damage, it can be concluded that HO-1 may be involved, along with several other factors, in alleviating inflammation and restoring liver function after damage by malaria. There are certain reports which show that induction of HO-1 mediates tolerance to the cytotoxic effects of heme during malaria-associated hemolysis, but it may impair the resistance to bacterial infection by limiting bactericidal reactive oxygen species production. Coinfection of mice with P. yoelii and Salmonella enterica serovar Typhimurium causes acute, fatal bacteremia with a high bacterial load. Recently, heme oxygenase was proposed as one of the factors that play significant roles in the pathogenesis of falciparum malaria complications (105). Expression of HO-1 is upregulated in the liver following infection by P. berghei and P. yoelii sporozoites. HO-1 overexpression leads to a proportional increase in parasite loads in the liver. In the absence of HO-1, the levels of inflammatory cytokines involved in the control of liver infection are increased (106). HO-1 expression modulates the host inflammatory response, protecting the infected hepatocytes and promoting the liver stage of infection (106). Although there are various opinions regarding the protective role of HO-1 against liver damage during malaria, from the various observations in our study, we propose that selective induction of HO-1 in the liver just after the clearance of parasites would be beneficial for the restoration of liver function.
ACKNOWLEDGMENTS
We thankfully acknowledge the Council of Scientific and Industrial Research (CSIR), New Delhi, India, for providing financial support to carry out this work through supra-institutional (BEnD; grant BSC 0206) and network (SPLenDID; grant BSC 0104) projects.
Footnotes
Published ahead of print 12 May 2014
REFERENCES
- 1.World Health Organization. 2011. The world malaria report. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Burte F, Brown BJ, Orimadegun AE, Ajetunmobi WA, Battaglia F, Ely BK, Afolabi NK, Athanasakis D, Akinkunmi F, Kowobari O, Omokhodion S, Osinusi K, Akinbami FO, Shokunbi WA, Sodeinde O, Fernandez-Reyes D. 2012. Severe childhood malaria syndromes defined by plasma proteome profiles. PLoS One 7:e49778. 10.1371/journal.pone.0049778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dey S, Bindu S, Goyal M, Pal C, Alam A, Iqbal MS, Kumar R, Sarkar S, Bandyopadhyay U. 2012. Impact of intravascular hemolysis in malaria on liver dysfunction: involvement of hepatic free heme overload, NF-kappaB activation, and neutrophil infiltration. J. Biol. Chem. 287:26630–26646. 10.1074/jbc.M112.341255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Portugal S, Carret C, Recker M, Armitage AE, Goncalves LA, Epiphanio S, Sullivan D, Roy C, Newbold CI, Drakesmith H, Mota MM. 2011. Host-mediated regulation of superinfection in malaria. Nat. Med. 17:732–737. 10.1038/nm.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, Rebelo S, Penido C, Smith NR, Coutinho A, Soares MP. 2009. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc. Natl. Acad. Sci. U. S. A. 106:15837–15842. 10.1073/pnas.0903419106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guha M, Kumar S, Choubey V, Maity P, Bandyopadhyay U. 2006. Apoptosis in liver during malaria: role of oxidative stress and implication of mitochondrial pathway. FASEB J. 20:1224–1226. 10.1096/fj.05-5338fje. [DOI] [PubMed] [Google Scholar]
- 7.Adachi K, Tsutsui H, Kashiwamura S, Seki E, Nakano H, Takeuchi O, Takeda K, Okumura K, Van Kaer L, Okamura H, Akira S, Nakanishi K. 2001. Plasmodium berghei infection in mice induces liver injury by an IL-12- and Toll-like receptor/myeloid differentiation factor 88-dependent mechanism. J. Immunol. 167:5928–5934. 10.4049/jimmunol.167.10.5928. [DOI] [PubMed] [Google Scholar]
- 8.Adachi K, Tsutsui H, Seki E, Nakano H, Takeda K, Okumura K, Van Kaer L, Nakanishi K. 2004. Contribution of CD1d-unrestricted hepatic DX5+ NKT cells to liver injury in Plasmodium berghei-parasitized erythrocyte-injected mice. Int. Immunol. 16:787–798. 10.1093/intimm/dxh080. [DOI] [PubMed] [Google Scholar]
- 9.Kochar DK, Agarwal P, Kochar SK, Jain R, Rawat N, Pokharna RK, Kachhawa S, Srivastava T. 2003. Hepatocyte dysfunction and hepatic encephalopathy in Plasmodium falciparum malaria. QJM 96:505–512. 10.1093/qjmed/hcg091. [DOI] [PubMed] [Google Scholar]
- 10.Bhalla A, Suri V, Singh V. 2006. Malarial hepatopathy. J. Postgrad. Med. 52:315–320. [PubMed] [Google Scholar]
- 11.Rodriguez-Acosta A, Finol HJ, Pulido-Mendez M, Marquez A, Andrade G, Gonzalez N, Aguilar I, Giron ME, Pinto A. 1998. Liver ultrastructural pathology in mice infected with Plasmodium berghei. J. Submicrosc. Cytol. Pathol. 30:299–307. [PubMed] [Google Scholar]
- 12.Premaratna R, Gunatilake AK, de Silva NR, Tilakaratne Y, Fonseka MM, de Silva HJ. 2001. Severe hepatic dysfunction associated with falciparum malaria. Southeast Asian J. Trop. Med. Public Health 32:70–72. [PubMed] [Google Scholar]
- 13.Rupani AB, Amarapurkar AD. 2009. Hepatic changes in fatal malaria: an emerging problem. Ann. Trop. Med. Parasitol. 103:119–127. 10.1179/136485909X385054. [DOI] [PubMed] [Google Scholar]
- 14.Haque A, Best SE, Amante FH, Ammerdorffer A, de Labastida F, Pereira T, Ramm GA, Engwerda CR. 2011. High parasite burdens cause liver damage in mice following Plasmodium berghei ANKA infection independently of CD8(+) T cell-mediated immune pathology. Infect. Immun. 79:1882–1888. 10.1128/IAI.01210-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martins YC, Daniel-Ribeiro CT. 2013. A new hypothesis on the manifestation of cerebral malaria: the secret is in the liver. Med. Hypotheses 81:777–783. 10.1016/j.mehy.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tangpukdee N, Thanachartwet V, Krudsood S, Luplertlop N, Pornpininworakij K, Chalermrut K, Phokham S, Kano S, Looareesuwan S, Wilairatana P. 2006. Minor liver profile dysfunctions in Plasmodium vivax, P. malaria and P. ovale patients and normalization after treatment. Korean J. Parasitol. 44:295–302. 10.3347/kjp.2006.44.4.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirsner RS, Eaglstein WH. 1993. The wound healing process. Dermatol. Clin. 11:629–640. [PubMed] [Google Scholar]
- 18.Guo S, Dipietro LA. 2010. Factors affecting wound healing. J. Dent. Res. 89:219–229. 10.1177/0022034509359125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gosain A, DiPietro LA. 2004. Aging and wound healing. World J. Surg. 28:321–326. 10.1007/s00268-003-7397-6. [DOI] [PubMed] [Google Scholar]
- 20.Naito Y, Takagi T, Yoshikawa T. 2004. Heme oxygenase-1: a new therapeutic target for inflammatory bowel disease. Aliment. Pharmacol. Ther. 20(Suppl 1):S177–S184. [DOI] [PubMed] [Google Scholar]
- 21.Naito Y, Takagi T, Uchiyama K, Yoshikawa T. 2011. Heme oxygenase-1: a novel therapeutic target for gastrointestinal diseases. J. Clin. Biochem. Nutr. 48:126–133. 10.3164/jcbn.10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hull TD, Bolisetty S, Dealmeida AC, Litovsky SH, Prabhu SD, Agarwal A, George JF. 2013. Heme oxygenase-1 expression protects the heart from acute injury caused by inducible Cre recombinase. Lab. Invest. 93:868–879. 10.1038/labinvest.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben-Ari Z, Issan Y, Katz Y, Sultan M, Safran M, Michal LS, Nader GA, Kornowski R, Grief F, Pappo O, Hochhauser E. 2013. Induction of heme oxygenase-1 protects mouse liver from apoptotic ischemia/reperfusion injury. Apoptosis 18:547–555. 10.1007/s10495-013-0814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Onyiah JC, Sheikh SZ, Maharshak N, Steinbach EC, Russo SM, Kobayashi T, Mackey LC, Hansen JJ, Moeser AJ, Rawls JF, Borst LB, Otterbein LE, Plevy SE. 2013. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology 144:789–798. 10.1053/j.gastro.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li B, Lee DS, Choi HG, Kim KS, Jeong GS, An RB, Kim YC. 2012. Involvement of heme oxygenase-1 induction in the cytoprotective and immunomodulatory activities of Viola patrinii in murine hippocampal and microglia cells. Evid. Based Complement Alternat. Med. 2012:128019. 10.1155/2012/128019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Neubauer JA, Sunderram J. 2012. Heme oxygenase-1 and chronic hypoxia. Respir. Physiol. Neurobiol. 184:178–185. 10.1016/j.resp.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 27.Haines DD, Lekli I, Teissier P, Bak I, Tosaki A. 2012. Role of haeme oxygenase-1 in resolution of oxidative stress-related pathologies: focus on cardiovascular, lung, neurological and kidney disorders. Acta Physiol. (Oxf.) 204:487–501. 10.1111/j.1748-1716.2011.02387.x. [DOI] [PubMed] [Google Scholar]
- 28.Durante W. 2011. Protective role of heme oxygenase-1 against inflammation in atherosclerosis. Front. Biosci. 16:2372–2388. 10.2741/3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu ML, Ho YC, Yet SF. 2011. A central role of heme oxygenase-1 in cardiovascular protection. Antioxid. Redox Signal. 15:1835–1846. 10.1089/ars.2010.3726. [DOI] [PubMed] [Google Scholar]
- 30.Durante W. 2010. Targeting heme oxygenase-1 in vascular disease. Curr. Drug Targets 11:1504–1516. 10.2174/1389450111009011504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raval CM, Lee PJ. 2010. Heme oxygenase-1 in lung disease. Curr. Drug Targets 11:1532–1540. 10.2174/1389450111009011532. [DOI] [PubMed] [Google Scholar]
- 32.Jazwa A, Cuadrado A. 2010. Targeting heme oxygenase-1 for neuroprotection and neuroinflammation in neurodegenerative diseases. Curr. Drug Targets 11:1517–1531. 10.2174/1389450111009011517. [DOI] [PubMed] [Google Scholar]
- 33.Ferenbach DA, Kluth DC, Hughes J. 2010. Hemeoxygenase-1 and renal ischaemia-reperfusion injury. Nephron Exp. Nephrol. 115:e33–e37. 10.1159/000313828. [DOI] [PubMed] [Google Scholar]
- 34.Belcher JD, Beckman JD, Balla G, Balla J, Vercellotti G. 2010. Heme degradation and vascular injury. Antioxid. Redox Signal. 12:233–248. 10.1089/ars.2009.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagener FA, van Beurden HE, von den Hoff JW, Adema GJ, Figdor CG. 2003. The heme-heme oxygenase system: a molecular switch in wound healing. Blood 102:521–528. 10.1182/blood-2002-07-2248. [DOI] [PubMed] [Google Scholar]
- 36.Wunder C, Potter RF. 2003. The heme oxygenase system: its role in liver inflammation. Curr. Drug Targets Cardiovasc. Haematol. Disord. 3:199–208. 10.2174/1568006033481410. [DOI] [PubMed] [Google Scholar]
- 37.Kumar S, Bandyopadhyay U. 2005. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 157:175–188. 10.1016/j.toxlet.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 38.Kumar S, Guha M, Choubey V, Maity P, Srivastava K, Puri SK, Bandyopadhyay U. 2008. Bilirubin inhibits Plasmodium falciparum growth through the generation of reactive oxygen species. Free Radic. Biol. Med. 44:602–613. 10.1016/j.freeradbiomed.2007.10.057. [DOI] [PubMed] [Google Scholar]
- 39.Siow RC, Sato H, Mann GE. 1999. Heme oxygenase-carbon monoxide signalling pathway in atherosclerosis: anti-atherogenic actions of bilirubin and carbon monoxide? Cardiovasc. Res. 41:385–394. 10.1016/S0008-6363(98)00278-8. [DOI] [PubMed] [Google Scholar]
- 40.Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F, Eaton JW, Vercellotti GM. 1992. Ferritin: a cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 267:18148–18153. [PubMed] [Google Scholar]
- 41.Wagener FA, Eggert A, Boerman OC, Oyen WJ, Verhofstad A, Abraham NG, Adema G, van Kooyk Y, de Witte T, Figdor CG. 2001. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 98:1802–1811. 10.1182/blood.V98.6.1802. [DOI] [PubMed] [Google Scholar]
- 42.Wagener FA, da Silva JL, Farley T, de Witte T, Kappas A, Abraham NG. 1999. Differential effects of heme oxygenase isoforms on heme mediation of endothelial intracellular adhesion molecule 1 expression. J. Pharmacol. Exp. Ther. 291:416–423. [PubMed] [Google Scholar]
- 43.Hayashi S, Takamiya R, Yamaguchi T, Matsumoto K, Tojo SJ, Tamatani T, Kitajima M, Makino N, Ishimura Y, Suematsu M. 1999. Induction of heme oxygenase-1 suppresses venular leukocyte adhesion elicited by oxidative stress: role of bilirubin generated by the enzyme. Circ. Res. 85:663–671. 10.1161/01.RES.85.8.663. [DOI] [PubMed] [Google Scholar]
- 44.Rucker M, Schafer T, Roesken F, Spitzer WJ, Bauer M, Menger MD. 2001. Reduction of inflammatory response in composite flap transfer by local stress conditioning-induced heat-shock protein 32. Surgery 129:292–301. 10.1067/msy.2001.111079. [DOI] [PubMed] [Google Scholar]
- 45.Vachharajani TJ, Work J, Issekutz AC, Granger DN. 2000. Heme oxygenase modulates selectin expression in different regional vascular beds. Am. J. Physiol. Heart Circ. Physiol. 278:H1613–H1617. [DOI] [PubMed] [Google Scholar]
- 46.Freitas A, Alves-Filho JC, Secco DD, Neto AF, Ferreira SH, Barja-Fidalgo C, Cunha FQ. 2006. Heme oxygenase/carbon monoxide-biliverdin pathway down regulates neutrophil rolling, adhesion and migration in acute inflammation. Br. J. Pharmacol. 149:345–354. 10.1038/sj.bjp.0706882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dovi JV, He LK, DiPietro LA. 2003. Accelerated wound closure in neutrophil-depleted mice. J. Leukoc. Biol. 73:448–455. 10.1189/jlb.0802406. [DOI] [PubMed] [Google Scholar]
- 48.Solomon VR, Puri SK, Srivastava K, Katti SB. 2005. Design and synthesis of new antimalarial agents from 4-aminoquinoline. Bioorg. Med. Chem. 13:2157–2165. 10.1016/j.bmc.2004.12.051. [DOI] [PubMed] [Google Scholar]
- 49.Goyal M, Singh P, Alam A, Das SK, Iqbal MS, Dey S, Bindu S, Pal C, Panda G, Bandyopadhyay U. 2012. Aryl aryl methyl thio arenes prevent multidrug-resistant malaria in mouse by promoting oxidative stress in parasites. Free Radic. Biol. Med. 53:129–142. 10.1016/j.freeradbiomed.2012.04.028. [DOI] [PubMed] [Google Scholar]
- 50.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 51.Weiss SJ, Klein R, Slivka A, Wei M. 1982. Chlorination of taurine by human neutrophils. Evidence for hypochlorous acid generation. J. Clin. Invest. 70:598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El-Beshbishy HA, Autifi MA, Mariee AD. 2010. Green tea (Camellia sinensis) extract protects against azathioprine-induced hepatotoxicity and neutrophil infiltration in rats. Asian J. Trad. Med. 5:1–11. [Google Scholar]
- 53.Maity P, Bindu S, Dey S, Goyal M, Alam A, Pal C, Mitra K, Bandyopadhyay U. 2009. Indomethacin, a non-steroidal anti-inflammatory drug, develops gastropathy by inducing reactive oxygen species-mediated mitochondrial pathology and associated apoptosis in gastric mucosa: a novel role of mitochondrial aconitase oxidation. J. Biol. Chem. 284:3058–3068. 10.1074/jbc.M805329200. [DOI] [PubMed] [Google Scholar]
- 54.Maity P, Bindu S, Dey S, Goyal M, Alam A, Pal C, Reiter R, Bandyopadhyay U. 2009. Melatonin reduces indomethacin-induced gastric mucosal cell apoptosis by preventing mitochondrial oxidative stress and the activation of mitochondrial pathway of apoptosis. J. Pineal Res. 46:314–323. 10.1111/j.1600-079X.2009.00663.x. [DOI] [PubMed] [Google Scholar]
- 55.Pal C, Bindu S, Dey S, Alam A, Goyal M, Iqbal MS, Sarkar S, Kumar R, Halder KK, Debnath MC, Adhikari S, Bandyopadhyay U. 2012. Tryptamine-gallic acid hybrid prevents non-steroidal anti-inflammatory drug-induced gastropathy: correction of mitochondrial dysfunction and inhibition of apoptosis in gastric mucosal cells. J. Biol. Chem. 287:3495–3509. 10.1074/jbc.M111.307199. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Pal C, Bindu S, Dey S, Alam A, Goyal M, Iqbal MS, Maity P, Adhikari SS, Bandyopadhyay U. 2010. Gallic acid prevents nonsteroidal anti-inflammatory drug-induced gastropathy in rat by blocking oxidative stress and apoptosis. Free Radic. Biol. Med. 49:258–267. 10.1016/j.freeradbiomed.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 57.Dey S, Guha M, Alam A, Goyal M, Bindu S, Pal C, Maity P, Mitra K, Bandyopadhyay U. 2009. Malarial infection develops mitochondrial pathology and mitochondrial oxidative stress to promote hepatocyte apoptosis. Free Radic. Biol. Med. 46:271–281. 10.1016/j.freeradbiomed.2008.10.032. [DOI] [PubMed] [Google Scholar]
- 58.Bindu S, Mazumder S, Dey S, Pal C, Goyal M, Alam A, Iqbal MS, Sarkar S, Azhar Siddiqui A, Banerjee C, Bandyopadhyay U. 2013. Nonsteroidal anti-inflammatory drug induces proinflammatory damage in gastric mucosa through NF-kappaB activation and neutrophil infiltration: anti-inflammatory role of heme oxygenase-1 against nonsteroidal anti-inflammatory drug. Free Radic. Biol. Med. 65:456–467. 10.1016/j.freeradbiomed.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 59.Bindu S, Pal C, Dey S, Goyal M, Alam A, Iqbal MS, Dutta S, Sarkar S, Kumar R, Maity P, Bandyopadhyay U. 2011. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J. Biol. Chem. 286:39387–39402. 10.1074/jbc.M111.279893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maity P, Bindu S, Choubey V, Alam A, Mitra K, Goyal M, Dey S, Guha M, Pal C, Bandyopadhyay U. 2008. Lansoprazole protects and heals gastric mucosa from non-steroidal anti-inflammatory drug (NSAID)-induced gastropathy by inhibiting mitochondrial as well as Fas-mediated death pathways with concurrent induction of mucosal cell renewal. J. Biol. Chem. 283:14391–14401. 10.1074/jbc.M800414200. [DOI] [PubMed] [Google Scholar]
- 61.Guha M, Maity P, Choubey V, Mitra K, Reiter RJ, Bandyopadhyay U. 2007. Melatonin inhibits free radical-mediated mitochondrial-dependent hepatocyte apoptosis and liver damage induced during malarial infection. J. Pineal Res. 43:372–381. 10.1111/j.1600-079X.2007.00488.x. [DOI] [PubMed] [Google Scholar]
- 62.Eming SA, Krieg T, Davidson JM. 2007. Inflammation in wound repair: molecular and cellular mechanisms. J. Invest. Dermatol. 127:514–525. 10.1038/sj.jid.5700701. [DOI] [PubMed] [Google Scholar]
- 63.Plowe CV. 2009. The evolution of drug-resistant malaria. Trans. R. Soc. Trop. Med. Hyg. 103(Suppl 1):S11–S14. 10.1016/j.trstmh.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Petersen I, Eastman R, Lanzer M. 2011. Drug-resistant malaria: molecular mechanisms and implications for public health. FEBS Lett. 585:1551–1562. 10.1016/j.febslet.2011.04.042. [DOI] [PubMed] [Google Scholar]
- 65.Price RN, Nosten F. 2001. Drug resistant falciparum malaria: clinical consequences and strategies for prevention. Drug Resist. Updat. 4:187–196. 10.1054/drup.2001.0195. [DOI] [PubMed] [Google Scholar]
- 66.Wells TN, Alonso PL, Gutteridge WE. 2009. New medicines to improve control and contribute to the eradication of malaria. Nat. Rev. Drug Discov. 8:879–891. 10.1038/nrd2972. [DOI] [PubMed] [Google Scholar]
- 67.Reinke JM, Sorg H. 2012. Wound repair and regeneration. Eur. Surg. Res. 49:35–43. 10.1159/000339613. [DOI] [PubMed] [Google Scholar]
- 68.Li J, Chen J, Kirsner R. 2007. Pathophysiology of acute wound healing. Clin. Dermatol. 25:9–18. 10.1016/j.clindermatol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 69.Fu X, Kassim SY, Parks WC, Heinecke JW. 2003. Hypochlorous acid generated by myeloperoxidase modifies adjacent tryptophan and glycine residues in the catalytic domain of matrix metalloproteinase-7 (matrilysin): an oxidative mechanism for restraining proteolytic activity during inflammation. J. Biol. Chem. 278:28403–28409. 10.1074/jbc.M304739200. [DOI] [PubMed] [Google Scholar]
- 70.Klebanoff SJ. 2005. Myeloperoxidase: friend and foe. J. Leukoc. Biol. 77:598–625. 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 71.Spickett CM, Jerlich A, Panasenko OM, Arnhold J, Pitt AR, Stelmaszynska T, Schaur RJ. 2000. The reactions of hypochlorous acid, the reactive oxygen species produced by myeloperoxidase, with lipids. Acta Biochim. Pol. 47:889–899. [PubMed] [Google Scholar]
- 72.Hurst JK, Barrette WC., Jr 1989. Leukocytic oxygen activation and microbicidal oxidative toxins. Crit. Rev. Biochem. Mol. Biol. 24:271–328. 10.3109/10409238909082555. [DOI] [PubMed] [Google Scholar]
- 73.Daugherty A, Dunn JL, Rateri DL, Heinecke JW. 1994. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Invest. 94:437–444. 10.1172/JCI117342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Naito Y, Takagi T, Yoshikawa T. 2007. Neutrophil-dependent oxidative stress in ulcerative colitis. J. Clin. Biochem. Nutr. 41:18–26. 10.3164/jcbn.2007003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kannan K, Jain SK. 2000. Oxidative stress and apoptosis. Pathophysiology 7:153–163. 10.1016/S0928-4680(00)00053-5. [DOI] [PubMed] [Google Scholar]
- 76.Steiling H, Munz B, Werner S, Brauchle M. 1999. Different types of ROS-scavenging enzymes are expressed during cutaneous wound repair. Exp. Cell Res. 247:484–494. 10.1006/excr.1998.4366. [DOI] [PubMed] [Google Scholar]
- 77.Schafer M, Werner S. 2008. Oxidative stress in normal and impaired wound repair. Pharmacol. Res. 58:165–171. 10.1016/j.phrs.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 78.Kochar DK, Kaswan K, Kochar SK, Sirohi P, Pal M, Kochar A, Agrawal RP, Das A. 2006. A comparative study of regression of jaundice in patients of malaria and acute viral hepatitis. J. Vector Borne Dis. 43:123–129. [PubMed] [Google Scholar]
- 79.Shah S, Ali L, Sattar RA, Aziz T, Ansari T, Ara J. 2009. Malarial hepatopathy in falciparum malaria. J. Coll. Physicians Surg. Pak. 19:367–370. [PubMed] [Google Scholar]
- 80.Wilairatana P, Looareesuwan S, Charoenlarp P. 1994. Liver profile changes and complications in jaundiced patients with falciparum malaria. Trop. Med. Parasitol. 45:298–302. [PubMed] [Google Scholar]
- 81.Autino B, Corbett Y, Castelli F, Taramelli D. 2012. Pathogenesis of malaria in tissues and blood. Mediterr. J. Hematol. Infect. Dis. 4:e2012061. 10.4084/MJHID.2012.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Savill J, Fadok V. 2000. Corpse clearance defines the meaning of cell death. Nature 407:784–788. 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 83.Kurosaka K, Takahashi M, Watanabe N, Kobayashi Y. 2003. Silent cleanup of very early apoptotic cells by macrophages. J. Immunol. 171:4672–4679. 10.4049/jimmunol.171.9.4672. [DOI] [PubMed] [Google Scholar]
- 84.Ramaiah SK, Jaeschke H. 2007. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol. Pathol. 35:757–766. 10.1080/01926230701584163. [DOI] [PubMed] [Google Scholar]
- 85.Bian Z, Guo Y, Ha B, Zen K, Liu Y. 2012. Regulation of the inflammatory response: enhancing neutrophil infiltration under chronic inflammatory conditions. J. Immunol. 188:844–853. 10.4049/jimmunol.1101736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chatham WW, Turkiewicz A, Blackburn WD., Jr 1994. Determinants of neutrophil HOCl generation: ligand-dependent responses and the role of surface adhesion. J. Leukoc. Biol. 56:654–660. [DOI] [PubMed] [Google Scholar]
- 87.Schlatter R, Schmich K, Lutz A, Trefzger J, Sawodny O, Ederer M, Merfort I. 2011. Modeling the TNFalpha-induced apoptosis pathway in hepatocytes. PLoS One 6:e18646. 10.1371/journal.pone.0018646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Simon HU, Haj-Yehia A, Levi-Schaffer F. 2000. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 5:415–418. 10.1023/A:1009616228304. [DOI] [PubMed] [Google Scholar]
- 89.Lundvig DM, Immenschuh S, Wagener FA. 2012. Heme oxygenase, inflammation, and fibrosis: the good, the bad, and the ugly? Front. Pharmacol. 3:81. 10.3389/fphar.2012.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guo X, Shin VY, Cho CH. 2001. Modulation of heme oxygenase in tissue injury and its implication in protection against gastrointestinal diseases. Life Sci. 69:3113–3119. 10.1016/S0024-3205(01)01417-5. [DOI] [PubMed] [Google Scholar]
- 91.Otterbein LE, Soares MP, Yamashita K, Bach FH. 2003. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 24:449–455. 10.1016/S1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- 92.Ryter SW, Otterbein LE, Morse D, Choi AM. 2002. Heme oxygenase/carbon monoxide signaling pathways: regulation and functional significance. Mol. Cell. Biochem. 234–235:249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bach FH. 2005. Heme oxygenase-1: a therapeutic amplification funnel. FASEB J. 19:1216–1219. 10.1096/fj.04-3485cmt. [DOI] [PubMed] [Google Scholar]
- 94.Wagener FA, Volk HD, Willis D, Abraham NG, Soares MP, Adema GJ, Figdor CG. 2003. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol. Rev. 55:551–571. 10.1124/pr.55.3.5. [DOI] [PubMed] [Google Scholar]
- 95.Ryter SW, Alam J, Choi AM. 2006. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol. Rev. 86:583–650. 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 96.Lin YT, Chen YH, Yang YH, Jao HC, Abiko Y, Yokoyama K, Hsu C. 2010. Heme oxygenase-1 suppresses the infiltration of neutrophils in rat liver during sepsis through inactivation of p38 MAPK. Shock 34:615–621. 10.1097/SHK.0b013e3181e46ee0. [DOI] [PubMed] [Google Scholar]
- 97.Gu Q, Wu Q, Jin M, Xiao Y, Xu J, Mao C, Zhao F, Zhang Y, Zhang Y. 2012. Heme oxygenase-1 alleviates mouse hepatic failure through suppression of adaptive immune responses. J. Pharmacol. Exp. Ther. 340:2–10. 10.1124/jpet.111.186551. [DOI] [PubMed] [Google Scholar]
- 98.Wei CL, Lee KH, Khoo HE, Hon WM. 2003. Expression of haem oxygenase in cirrhotic rat liver. J. Pathol. 199:324–334. 10.1002/path.1284. [DOI] [PubMed] [Google Scholar]
- 99.Nowis D, Bugajski M, Winiarska M, Bil J, Szokalska A, Salwa P, Issat T, Was H, Jozkowicz A, Dulak J, Stoklosa T, Golab J. 2008. Zinc protoporphyrin IX, a heme oxygenase-1 inhibitor, demonstrates potent antitumor effects but is unable to potentiate antitumor effects of chemotherapeutics in mice. BMC Cancer 8:197. 10.1186/1471-2407-8-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hirai K, Sasahira T, Ohmori H, Fujii K, Kuniyasu H. 2007. Inhibition of heme oxygenase-1 by zinc protoporphyrin IX reduces tumor growth of LL/2 lung cancer in C57BL mice. Int. J. Cancer 120:500–505. 10.1002/ijc.22287. [DOI] [PubMed] [Google Scholar]
- 101.Maines MD. 1981. Zinc protoporphyrin is a selective inhibitor of heme oxygenase activity in the neonatal rat. Biochim. Biophys. Acta 673:339–350. 10.1016/0304-4165(81)90465-7. [DOI] [PubMed] [Google Scholar]
- 102.Ueda K, Ueyama T, Yoshida K, Kimura H, Ito T, Shimizu Y, Oka M, Tsuruo Y, Ichinose M. 2008. Adaptive HNE-Nrf2-HO-1 pathway against oxidative stress is associated with acute gastric mucosal lesions. Am. J. Physiol. Gastrointest. Liver Physiol. 295:G460–G469. 10.1152/ajpgi.00204.2007. [DOI] [PubMed] [Google Scholar]
- 103.Wang QM, Du JL, Duan ZJ, Guo SB, Sun XY, Liu Z. 2013. Inhibiting heme oxygenase-1 attenuates rat liver fibrosis by removing iron accumulation. World J. Gastroenterol. 19:2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yoshimura A, Wakabayashi Y, Mori T. 2010. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J. Biochem. 147:781–792. 10.1093/jb/mvq043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cunnington AJ, de Souza JB, Walther M, Riley EM. 2012. Malaria impairs resistance to Salmonella through heme- and heme oxygenase-dependent dysfunctional granulocyte mobilization. Nat. Med. 18:120–127. 10.1038/nm.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Epiphanio S, Mikolajczak SA, Goncalves LA, Pamplona A, Portugal S, Albuquerque S, Goldberg M, Rebelo S, Anderson DG, Akinc A, Vornlocher HP, Kappe SH, Soares MP, Mota MM. 2008. Heme oxygenase-1 is an anti-inflammatory host factor that promotes murine plasmodium liver infection. Cell Host Microbe 3:331–338. 10.1016/j.chom.2008.04.003. [DOI] [PubMed] [Google Scholar]