

Abstract
Malnutrition is thought to contribute to more than one-third of all childhood deaths via increased susceptibility to infection. Malnutrition is a significant risk factor for the development of visceral leishmaniasis, which results from skin inoculation of the intracellular protozoan Leishmania donovani. We previously established a murine model of childhood malnutrition and found that malnutrition decreased the lymph node barrier function and increased the early dissemination of L. donovani. In the present study, we found reduced numbers of resident dendritic cells (conventional and monocyte derived) but not migratory dermal dendritic cells in the skin-draining lymph nodes of L. donovani-infected malnourished mice. Expression of chemokines and their receptors involved in trafficking of dendritic cells and their progenitors to the lymph nodes was dysregulated. C-C chemokine receptor type 2 (CCR2) and its ligands (CCL2 and CCL7) were reduced in the lymph nodes of infected malnourished mice, as were CCR2-bearing monocytes/macrophages and monocyte-derived dendritic cells. However, CCR7 and its ligands (CCL19 and CCL21) were increased in the lymph node and CCR7 was increased in lymph node macrophages and dendritic cells. CCR2-deficient mice recapitulated the profound reduction in the number of resident (but not migratory dermal) dendritic cells in the lymph node but showed no alteration in the expression of CCL19 and CCL21. Collectively, these results suggest that the malnutrition-related reduction in the lymph node barrier to dissemination of L. donovani is related to insufficient numbers of lymph node-resident but not migratory dermal dendritic cells. This is likely driven by the altered activity of the CCR2 and CCR7 chemoattractant pathways.
INTRODUCTION
Malnutrition contributes to 3.7 million deaths per year among children <5 years of age (1). It is thought to underlie as many as a third of childhood deaths worldwide (2). Protein energy malnutrition and micronutrient deficiencies, which often coexist, increase the susceptibility to and the severity of infectious diseases. Most of the children die from infection, including diarrheal and respiratory infections, measles, tuberculosis, and a number of parasitic diseases (3).
Visceral leishmaniasis (VL), caused by the intracellular protozoan parasites of the Leishmania donovani complex (L. infantum [L. chagasi] and L. donovani), is an important but neglected tropical disease, with approximately 0.5 million cases and 50,000 deaths occurring per year (4). Following inoculation of the parasite by the sand fly vector, these visceralizing Leishmania organisms disseminate from the skin to reside in macrophages in the spleen, liver, and bone marrow (5). Most of those infected with L. donovani and L. infantum develop only subclinical disease or chronic latent infection without any clinical manifestation. However, approximately 10% of infected people develop fever, severe hepatosplenomegaly, pancytopenia, and cachexia, which ultimately progresses to death if left untreated (6–9). Clinical (10–13) and experimental (14–18) studies have documented that malnutrition is a strong predisposing factor for the progression of L. donovani or L. infantum infection to active VL and increased mortality. A child with moderate or severe malnutrition (based on weight for age or height for age) has a 9-fold greater risk of developing VL than a child of normal nutritional status or mild malnutrition (11–13).
We developed a murine model of polynutrient deficiency that mimics a moderate degree of childhood malnutrition (15). A combination of protein, energy, iron, and zinc deficiency was chosen because deficiencies of these nutrients are common and frequently coexist (19–21). In this model, we observed that malnutrition impaired the lymph node (LN) barrier function and led to a marked increase in the dissemination of L. donovani to the spleen and liver. The overall lymph node architecture of the malnourished mice remained intact, but there was a profound reduction in the lymph node mass and cellularity. In particular, the number of fibroblastic reticular cells and myeloid phagocytic cells (macrophages, neutrophils, and dendritic cells [DCs]) was markedly reduced (18). The reduction in the numbers of myeloid cells of the conduit system with altered function suggested that the overall phagocytic capacity of the lymph node was impaired (18). Lymph node composition or function has not been studied in human malnutrition. However, altered DC function was previously demonstrated in cells isolated from the peripheral blood of a malnourished patient (22). In the present study, we investigated the influence of malnutrition on the expression of lymph node inflammatory mediators that govern DC trafficking and retention in the lymph node. We found that polynutrient deficiency dysregulated the main chemotactic mechanisms involved in the migration and accumulation of DCs in the lymph nodes following an inflammatory stimulus.
MATERIALS AND METHODS
Experimental animals.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Research Council (23). The protocol was approved by the Institutional Animal Care and Use Committee of the South Texas Veterans Health Care System and the University of Texas Medical Branch at Galveston, Galveston, TX. Wild-type BALB/c mice were obtained from Charles River Laboratories, Inc. (Wilmington, MA). Female and male C-C chemokine receptor type 2 (CCR2)-deficient (CCR2−/−) BALB/c mice (12 to 16 weeks old) were obtained from a breeding colony at the VA Ann Arbor Healthcare System, Ann Arbor, MI. Wild-type controls for the experiments involving CCR2-deficient mice were age and gender matched. The mice were maintained under specific-pathogen-free conditions at the Veterinary Medical Unit of the Audie L. Murphy Memorial Veterans Affairs Hospital, South Texas Veterans Health Care System, San Antonio, TX.
Diets and feeding of well-nourished and malnourished mice.
Recently weaned female BALB/c mice (3 to 4 weeks old) were matched for age and initial weight and distributed to either the control or polynutrient-deficient (PND) diet. Mice were housed in standard polycarbonate shoebox cages with bedding low in trace element content (Alpha-Dri; Shepard Specialty Papers, Kalamazoo, MI) and with free access to water. The mice were acclimatized to standard laboratory mouse chow (Teklad LM-485; Harlan Teklad, Madison, WI) for 3 days and then fed the experimental diets for 4 weeks. Mice in the well-nourished control group were placed on the control diet (17% protein, 100 ppm iron, 30 ppm zinc), and a group of mice was placed on a polynutrient-deficient diet (3% protein, 10 ppm iron, 1 ppm zinc), as previously described (18). The malnourished mice received 90% of the weight of food consumed per day by the mice in the control group. The body weights of the mice were measured weekly, and food intake was recorded on a twice-weekly basis in order to calculate the amount of chow to provide to the malnourished group on subsequent days.
Experimental infection.
Leishmania donovani (MHOM/SD/001S-2D) promastigotes were cultured in complete M199 medium for 6 days, and the metacyclic forms were purified following lectin agglutination as described previously (24). Metacyclic promastigotes (1 × 106) in 20 μl Dulbecco's modified Eagle medium (DMEM) were inoculated intradermally into the skin over each hind footpad. In some experiments, PKH26 red fluorescent cell linker (Sigma-Aldrich, St. Louis, MO) labeling of L. donovani was performed as described previously (25).
Flow cytometric analysis.
Flow cytometry was performed as described previously (18). The popliteal lymph nodes were harvested in RPMI medium supplemented with 2% heat-inactivated fetal bovine serum (FBS; Gibco, Grand Island, NY), and a single-cell suspension was obtained after digestion at 37°C for 30 min with collagenase D (2 mg/ml; Roche Diagnostics, Indianapolis, IN) in 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 10 mM HEPES, pH 7.4, and pushing of the lymph nodes through 100-μm-pore-size cell strainers (BD, San Jose, CA). The cells were adjusted to a concentration of 0.5 × 106 to 1 × 106 cells per 50 μl in RPMI with 2% FBS, incubated for 15 min with 1 μg Fc Block reagent at room temperature, incubated with the relevant antibodies for 30 min at room temperature in the dark, washed in phosphate-buffered saline (PBS) with 2% FBS and 1 mM sodium azide, fixed with 1-step Fix/Lyse solution (eBioscience, San Diego, CA), and analyzed by flow cytometry (BD, San Jose, CA). For CCR7 staining, cells were first incubated with anti-CCR7 antibody (37°C, 30 min), washed, and then labeled with the other fluorochrome-conjugated antibodies. The antibodies used for cell surface markers were hamster anti-mouse CD11c (clone N418; AbD Serotec, Raleigh, NC), rat anti-mouse CCR2 (clone 475301; R&D Systems, Minneapolis, MN), rat anti-mouse CD11b (clone M1/70; eBioscience), rat anti-mouse CCR7 (clone 4B12; R&D Systems or eBioscience), rat anti-mouse CD169 (clone 3D6.112; Abcam), and rabbit anti-mouse CCR2 (Abcam). For flow cytometry analysis, rat anti-mouse CD19 (clone eBio1D3), rat anti-mouse CD3 (clone 17A2; eBioscience), rat anti-mouse CD49 (clone DX5; eBioscience), and Live/Dead fluorescent reactive dye (Life Technologies, Grandview, NY) were used to exclude B cells, T cells NK cells, and dead cells. Compensation controls were established using the tested antibody mixed with 1 drop of OneComp eBeads (eBioscience). For intracellular monocyte/macrophage marker antibody (MOMA-2) analysis, cell preparations were fixed and permeabilized with fixation/permeabilization buffers (AbD Serotec) and stained with Alexa Fluor 647-conjugated rat anti-mouse monocyte/macrophage (AbD Serotec). For intracellular ER-TR7 antibody analysis, cell preparations were fixed and permeabilized with a mixture of ethanol-acetone (7:3) and stained with rat anti-mouse ER-TR7 (Abcam). The secondary antibodies used in the indirect staining were allophycocyanin-conjugated goat anti-rat IgG and peridinin chlorophyll protein-Cy5.5-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). The relevant isotype antibodies (rat or hamster IgG isotype) were used as controls.
In vivo labeling of dermal DCs.
Skin painting was performed using freshly prepared 5-chloromethylfluorescein (CMFDA) or 5-(6)-{[(4-chloromethyl)benzoyl]amino}tetramethylrhodamine (CMTMR) (CellTracker; Molecular Probes, Eugene, OR), as described previously (26, 27). The use of the different cell trackers was determined by which other fluorophores were used in the flow cytometry. In brief, 2 × 106 PKH26-labeled L donovani metacyclic promastigotes were inoculated into the skin over each footpad, and the skin at the inoculation site was painted immediately after infection with 50 μl of tracker (final concentration, 0.5 mM) dissolved in 1:1 (vol/vol) acetone-dibutyl phthalate. The efficiency of labeling of migratory dermal cells was determined in control and malnourished mice using an ex vivo dermal explant culture as described previously (28). The footpad skin was painted with CMTMR and 3 days after infection was harvested, disinfected, and placed dermal side down in Dispase II solution (Millipore, Billerica, MA) at 4°C overnight. Tissue and released cells were then transferred to complete medium (RPMI, 10% FBS, 1,000 units/ml of penicillin, 1,000 μg/ml of streptomycin), and after 72 h, the cells that had exited the skin were harvested by centrifugation. Samples of 3 to 4 animals were pooled, and the percentage of CD11c+ cells that were CMTMR positive was determined by flow cytometry.
RNA isolation and cDNA synthesis.
The popliteal lymph nodes and spleens were collected in RNA preservation buffer (RNAlater; Ambion, Carlsbad, CA) and frozen at −80°C until use. Total RNA was extracted from 15 to 20 mg of tissue using an RNeasy kit (Qiagen, Valencia, CA). All RNA samples were quantified using a Thermo Scientific NanoDrop spectrophotometer and kept at −80°C until assayed. One hundred nanograms of RNA was treated with DNase (SABiosciences, Valencia, CA) to eliminate contaminating genomic DNA, and following heat inactivation of the DNase, cDNA synthesis was accomplished using an RT2 first-strand kit (SABiosciences). The RNA samples were incubated for 15 min at 42°C (reverse transcription [RT] reaction), followed by 5 min at 95°C (to terminate the reaction). The cDNA was stored at −20°C until subsequent steps were performed.
Real-time PCR.
For real-time PCR assay, 1 μl of the synthesized cDNA was combined with 1 μl of gene-specific 10 μM PCR primer pair stock, and that combination was mixed with RT2 SYBR green–carboxy-X-rhodamine (ROX) quantitative PCR (qPCR) master mix (SABiosciences) in a total reaction volume of 10 μl. The PCR samples were initially incubated for 10 min at 95°C (AmpliTaq Gold preactivation) and then underwent 40 cycles at 95°C for 15 s and 60°C for 1 min in a 7900 HT Fast real-time PCR system (Applied Biosystems, Carlsbad, CA). Mouse β-actin was used in a separate reaction to normalize the differences in the amount of input cDNA in each assay. Relative gene expression was determined by the 2−ΔΔCT threshold cycle (CT) method, and results were expressed as the fold change in expression compared to the mean level of expression for the well-nourished group. Primers specific for mouse gamma interferon (IFN-γ), CCL19, CCL2, CCL8, CCL21, CCL7, CCR2, CCR7, CCL11, CXCL10, Cola3, lymphotoxin beta receptor (LTBR), and β-actin were purchased from SABiosciences.
PCR array analysis.
Five hundred nanograms of total purified RNA, pooled from 4 lymph nodes, was synthesized to cDNA using an RT2 PCR array first-strand kit (SABiosciences). PCR array analysis was performed using 90 μl of the synthesized cDNA mixed with RT2 SYBR green-ROX qPCR master mix (SABiosciences), and 25 μl of the final mix was applied to each well in the 96-well plate. Two different PCR arrays were used: the mouse inflammatory cytokine and receptor RT2 Profiler PCR array and the mouse extracellular matrix and adhesion molecule RT2 Profiler PCR array (SABiosciences). The PCR array was run using the 7900 HT Fast real-time PCR system (Applied Biosystems).
ELISA.
Single-cell suspensions (6 × 106 cells) prepared from the lymph node or spleen tissues were cultured in 600 μl of DMEM supplemented with 10% FBS without exogenous stimulus. After 24 to 36 h, supernatants were harvested and enzyme-linked immunosorbent assay (ELISA) kits were used to determine the levels of CCL2 (eBioscience) and of CCL21 and CCL19 (RayBiotech, Norcross, GA) released into the culture medium. In additional experiments, protein tissue lysates were prepared directly from the spleen and lymph node tissue, as described previously (25), or from the single-cell suspensions. The range of detection by ELISA was 31.25 to 2,000 pg/ml for CCL2, 31.25 to 1,000 pg/ml for CCL21, and 4.1 to 1,000 pg/ml for CCL19. To calculate the concentration of the chemokine per mg protein, the total protein concentrations were measured with the Bradford reagent (Sigma-Aldrich, St. Louis, MO), using the manufacturer's instructions.
Immunohistochemistry.
Lymph nodes and spleens were harvested, immediately embedded in Tissue-Tek optimum-cutting-temperature compound (Sakura FineTek, Torrance, CA), and frozen at −80°C. Cryostat sections of 6 μm were placed on positively charged microscope slides (Superfrost/Plus; Fisher Scientific, Waltham, MA), and the sections were air dried, fixed in ice-cold acetone for 10 min, blocked in 10% donkey or goat serum, and incubated with unlabeled primary antibodies diluted in 2% serum for 1 h at room temperature. The slides were then washed 5 times in PBS with 0.02% bovine serum albumin (BSA) for 5 min each time, incubated with fluorescence-labeled secondary antibodies for 1 h, and washed 5 times in PBS with 0.02% BSA for 5 min each time, and a coverslip was applied with Gold Prolong antifade mounting medium (Molecular Probes, Eugene, OR). Lymph node sections were examined using an Olympus Provis AX 70 fluorescence microscope, and the intensity of the fluorescence was measured as a proportion of the tissue area by Image-Pro Plus software (Media Cybernetics, Inc., Bethesda, MD). The primary antibodies used in immunohistochemistry staining were polyclonal goat anti-mouse CCL21, polyclonal goat anti-mouse CCL19 (R&D Systems), monoclonal rat anti-mouse ER-TR7, rat anti-mouse CD169 (clone 3D6.112; Abcam), biotin rat anti-mouse MOMA-2 (LifeSpan BioSciences, Seattle, WA), Armenian hamster anti-mouse CD11c (clone N418; GeneTex, Irvine, CA), and fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse CD11b (BD Pharmingen, Franklin Lakes, NJ). The secondary antibodies used in the staining process were Cy3-conjugated donkey anti-goat IgG, FITC-conjugated donkey anti-goat IgG, FITC-conjugated donkey anti-rat IgG, FITC-conjugated goat anti-rat IgG, Cy3-conjugated goat anti-rat IgG (Chemicon, Temecula, CA), aminomethylcoumarin acetate (AMCA)-conjugated goat anti-hamster IgG (Jackson ImmunoResearch, West Grove, PA), and FITC-conjugated streptavidin conjugate (BD Pharmingen). The relevant isotype controls were used as negative controls.
Statistical analysis.
Prism software (GraphPad, La Jolla, CA) was used to analyze the data, and either the parametric unpaired t test or the nonparametric Mann-Whitney U test was used for the analysis, depending on the normalcy of the distribution of the data. Statistical significance was considered if P was ≤0.05. The data in the graphs are expressed as the mean and standard error of the mean (error bars).
RESULTS
Malnutrition leads to a dramatic reduction in the total number of lymph node-resident DCs but does not alter the migration of dermal DCs to the lymph node.
In our previous studies, we found increased early visceralization following inoculation of L. donovani in the skin of malnourished mice. The increased numbers of visceral parasites in the malnourished group was not the result of impaired parasite killing but was due to parasites escaping the barrier function of the skin-draining lymph node (15, 18). We also demonstrated a reduced number of macrophages, neutrophils, and DCs in the lymph nodes of malnourished mice (18). This suggested that the increased dissemination of L. donovani might be due to the reduced number of phagocytes and/or the ineffective capture of parasites as they transited to and through the lymph nodes. Confirming our previous observation (18), we found that the total DC population (Fig. 1A) and the conventional DC (CD11c+ CD11b−) (Fig. 1B) and monocyte-derived DC (CD11c+ CD11b+) (Fig. 1C) subpopulations were reduced in the lymph nodes of infected malnourished mice. Similarly, the total number of parasite-loaded DCs was decreased (Fig. 1D), but there was no difference in the proportion of DCs that were parasitized between the two groups (Fig. 1E). To evaluate DC trafficking from the skin to the LN, we infected well-nourished and malnourished mice with L. donovani and, immediately after the parasite inoculation, painted the skin site with a fluorescent marker (CMFDA or CMTMR) so that labeled dermal DCs that had migrated to the draining LN could be distinguished from DCs already resident in the LN (27, 29, 30). Control experiments demonstrated highly efficient and equivalent labeling of migratory skin DCs in the well-nourished and malnourished mice (Fig. 1F). We identified fewer lymph node-resident conventional DCs (CMTMR negative [CMTMR−] CD11c+ CD11b−) and resident monocyte-derived DCs (CMTMR− CD11c+ CD11b+) in the infected malnourished mice but no difference in these subpopulations of migratory dermal (CMTMR-positive [CMTMR+]) DCs (Fig. 1G). These data indicate that the reduction in the total number of DCs in the LN of malnourished mice was due to a reduction in the population of DCs that were present in the lymph node prior to the inflammatory stimulus (probably conventional DCs) or that were acutely recruited by the inflammatory stimulus to the lymph node through the blood (high endothelial venule) and not the skin. The fewer numbers of conventional DCs seemed to make the greatest contribution to the malnutrition-related reduction in resident DCs. The proportion of infected lymph node-resident DCs (parasite positive, CMFDA negative [CMFDA−], CD11c+) or migratory dermal DCs (parasite positive, CMFDA positive [CMFDA+], CD11c+) relative to the total number of lymph node cells (Fig. 1H) and their rate of infection (number of parasitized resident DCs/total number of resident DCs and number of parasitized dermal DCs/total number of dermal DCs) (Fig. 1I) were similar in the two groups. Thus, there was no inherent defect in phagocytosis in the DCs from malnourished mice. Although migratory dermal DCs made up a relatively small proportion of the total number of DCs in the lymph node (Fig. 1G and H), the majority of these cells that arrived from the skin were infected (Fig. 1I). These data indicate that while malnutrition led to a reduction in the overall number of resident DCs, it did not reduce the trafficking of dermal DCs from the skin to the draining lymph node.
FIG 1.

Malnutrition reduces the number of lymph node-resident dendritic cells in L. donovani-infected mice. Well-nourished (WN) and malnourished (polynutrient-deficient [PND]) mice (n = 6 per group) were inoculated in the skin over the footpad with L. donovani, and immediately thereafter the skin at the same site was painted with either CMTMR or CMFDA. Draining popliteal lymph nodes were removed after 3 days, and flow cytometry was used to evaluate infected or uninfected DC populations. The gating protocol included exclusion of NK, T, and B cells, as shown in Fig. 5A. In some experiments, PKH26-labeled parasites were used to enable gating on infected cells, and in those experiments, NK, T, and B cells were not excluded. (A) Percentage of CD11c+ DCs in malnourished and well-nourished infected mice relative to the number of all lymph node cells; (B) percentage of conventional DCs (CD11c+ CD11b−) in malnourished and well-nourished infected mice relative to the number of all lymph node cells; (C) percentage of monocyte-derived DCs (MoDCs; CD11c+ CD11b+) in malnourished and well-nourished infected mice relative to the number of all lymph node cells; (D, E) total number (D) and percentage (E) of parasitized CD11c+ DCs in malnourished and well-nourished infected mice; (F) percentage of dermal migratory CD11c+ DCs that were CMTMR+ in an ex vivo skin explant culture; (G) percentage of conventional (Conv; CMTMR− CD11c+ CD11b−) and monocyte-derived (CMTMR− CD11c+ CD11b+) lymph node-resident dendritic cells (LNDC) and conventional and monocyte-derived (CMTMR+ CD11c+ CD11b+) migratory dermal conventional DCs (DDCs; CMTMR+ CD11c+ CD11b−) relative to the number of all LN cells in malnourished and well-nourished infected mice; (H) percentage of infected lymph node-resident dendritic cells (labeled L. donovani parasites [Ld+], CMFDA−, CD11c+) and infected migratory dermal dendritic cells (labeled L. donovani parasites, CMFDA+, CD11c+) relative to the number of all LN cells in malnourished and well-nourished infected mice; (I) percentage of infected lymph node-resident CD11c+ dendritic cells and migratory dermal dendritic cells relative to the total CD11c+ population. *, P < 0.05; **, P < 0.01; ***, P < 0.01.
Malnutrition does not lead to reduced numbers of myeloid precursors in bone marrow.
Monocytes, macrophages, and DCs in the lymph nodes originate from precursors in the bone marrow (31). DC precursors and conventional DCs typically migrate from the bone marrow to peripheral tissue and then to the draining lymph nodes via the afferent lymphatics, which empty into the subcapsular sinus that surrounds the lymph node cortex (32). We reasoned that the reduced numbers of myeloid cells in the lymph nodes could be the result of reduced numbers of bone marrow precursors, especially since there is apoptosis-induced thymocyte depletion in children with severe malnutrition (33, 34). We found that there was no difference in the total number (Fig. 2A) or proportion (Fig. 2B) of CD11c+ DCs (which would also include immediate precursors) or in the number of CD11b+ Ly6C+ CCR2+ inflammatory monocytes, which could differentiate into resident DCs following bloodstream migration to the lymph nodes.
FIG 2.
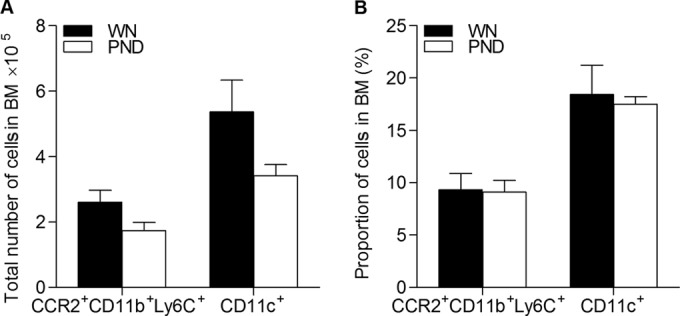
Malnutrition does not alter the total number of inflammatory monocytes or DC precursors in the bone marrow (BM) of L. donovani-infected mice. Flow cytometry was used to determine the total number (A) and percentage (B) of bone marrow cells in well-nourished (WN) and malnourished (polynutrient-deficient [PND]) mice (n = 7 per group) infected with L. donovani for 3 days using markers for DC cells (CD11c) and inflammatory monocytes (CD11b Ly6C CCR2). There were no significant differences between the well-nourished and malnourished groups.
Malnutrition dysregulates the mRNA expression of DC chemoattractants in the lymph node and spleen in response to L. donovani infection.
In light of the reduced numbers of myeloid cells in the lymph nodes but normal numbers in the bone marrow, we reasoned that there might be impaired migration of these cell populations to the lymph nodes in malnutrition. Therefore, we investigated the influence of malnutrition on the lymph node inflammatory mediators that were likely to play a role in the recruitment and retention of DCs. PCR arrays were used to analyze gene expression in pools of skin-draining popliteal lymph nodes. Overall, there was little difference in the transcriptional profile between the control and malnourished mice (see Table S1 in the supplemental material for the full data set). However, we observed the downregulation of several transcripts (CCL2, CCL7, CCL11, CXCL1, CXCL10, CCR2, CCR9, Spp1, Tnfrsf1b, CXCL11, CCL8, LAMA1, MMP7, MMP9, VCAM1, Col3a1, and IFN-γ) and the upregulation of some transcripts (CCL19, IL6ST, IL1R2, Syt1, Lamb2, and Itga3) in the draining nodes of infected malnourished mice compared to their regulation in the control mice. A subset of the differentially expressed genes identified in the PCR array showed significant differential expression when groups of mice (n = 6) were studied by quantitative RT-PCR (qRT-PCR) (Fig. 3). Congruent with the findings obtained with the PCR array, the effect of malnutrition on gene expression was most evident following L. donovani infection. DCs are thought to be the primary cells responsible for transporting L. donovani from the site of cutaneous infection to the draining LN (35). Therefore, it was particularly interesting to find the altered expression of DC chemoattractants and their receptors. The expression of CCR7 and its ligands CCL19 and CCL21 was increased in the lymph nodes of infected malnourished mice. In contrast, the expression of CCR2 and its ligands CCL7 and CCL2 was decreased (Fig. 3A). In uninfected mice, the expression of CCR2 and CCR7 and their ligands was comparable in the lymph nodes of malnourished and well-nourished mice (Fig. 3B). Thus, the local inflammatory stimulus (L. donovani infection) accentuated the chemokine dysregulation in malnourished mice. To exclude the possibility that the lower level of expression of CCL2 and CCL7 was merely a consequence of the lower parasite load in the lymph nodes, we examined the expression of CCR2 and its ligands in the spleen, where the malnourished mice had a higher parasite burden. CCR2 expression in the infected spleen was increased (in contrast to decreased expression in the LN), but the expression of CCL7 was reduced. The expression of CCL2 was no different in the malnourished infected spleen than in the control spleen (Fig. 3C). However, there was significant upregulation of CCR7, CCL21, and CCL19 in the spleens of infected malnourished mice (Fig. 3C). The spleens of the malnourished uninfected mice also showed significantly increased expression of CCL21, whereas the expression of CCR2, CCR7, CCL19, CCL7, and CCL2 mRNAs was no different (Fig. 3D). Collectively, these data indicate that malnutrition modulates the DC chemoattractant signals in the spleen and lymph node. These differences were most evident following L. donovani infection, but the level of expression of these chemokines and their receptors was not determined solely by the parasite load.
FIG 3.

Malnutrition dysregulates the mRNA expression of lymph node and spleen inflammatory mediators in L. donovani-infected mice. Real-time RT-PCR was used to quantify the mRNA expression of multiple inflammatory mediators in the draining popliteal lymph nodes and spleens of well-nourished (WN) and malnourished (polynutrient-deficient [PND]) mice that were uninfected or infected for 3 days (n = 8 per group). (A) Lymph nodes from L. donovani-infected mice; (B) lymph nodes from uninfected mice; (C) spleens of infected mice; (D) spleens of uninfected mice. The data were normalized to the expression of β-actin and are shown as the fold change in expression relative to the mean of uninfected well-nourished mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Malnutrition leads to increased CCL19 and CCL21 production but decreased CCL2 production in the lymph node and spleen of L. donovani-infected mice.
We measured the levels of secreted CCL2, CCL19, and CCL21 in the supernatant of spleen and lymph node cells cultured ex vivo for 24 h without exogenous stimulation. As was noted with the qRT-PCR data, the concentration of secreted CCL2 from the lymph node cells of infected malnourished mice was dramatically lower (Fig. 4A; P = 0.004), whereas the CCL2 concentration in the spleen and serum was comparable (Fig. 4A). Also consistent with the qRT-PCR data, the concentration of secreted CCL21 from both lymph node and spleen cells was significantly higher in the infected malnourished mice that the well-nourished infected mice (Fig. 4B; P = 0.04 and P = 0.006, respectively). The level of CCL19 in the supernatants of the cultured spleen and lymph node cells was below the limit of detection of the ELISA, so we determined the total amount of the CCL19 protein in tissue homogenates. We observed a higher CCL19 protein level in the lymph node tissue lysate of malnourished-infected mice (Fig. 4C and D; P = 0.001 and P = 0.02, respectively). The level of CCL19 in the spleen was comparable between the two infected groups (Fig. 4C and D). Since CCL19 and CCL21 in the lymph nodes are produced mainly by fibroblastic reticular and endothelial cells (36), we characterized the quantity and localization of CCL21 and CCL19 in cryosections by costaining for the chemokines and the fibroblastic reticular cell marker ER-TR7. The findings of increased CCL19 and CCL21 in the lymph node were confirmed by immunohistochemistry (Fig. 4E; P = 0.03 and P = 0.04 for the differences in CCL19 and CCL21, respectively). CCL21 was dramatically increased in the lumen of the high endothelial venule (which is surrounded by endothelial and fibroblastic reticular cells) in the infected malnourished mice, whereas CCL19 showed a comparable distribution (Fig. 4F and G). In the spleens of well-nourished infected mice, CCL21 was distributed in both the white and red pulp, whereas in the spleens of the malnourished mice, CCL21 was noted only in the white pulp (Fig. 4H).
FIG 4.

Malnutrition dysregulates CCL2, CCL19, and CCL21 production in the lymph node and spleen of L. donovani-infected mice. Chemokine production in the draining popliteal lymph nodes and spleens of well-nourished (WN) and malnourished (polynutrient-deficient [PND]) mice infected for 3 days (n = 6 per group) was examined. (A) The level of CCL2 produced by ex vivo culture of lymph node and spleen cells without an exogenous stimulus or in unmanipulated serum was determined by ELISA; (B) the level of CCL21 produced by ex vivo culture of lymph node and spleen cells without an exogenous stimulus was determined by ELISA; (C, D) the levels of CCL19 in the lymph node and spleen tissue lysates were determined by ELISA and calculated relative to the total protein content (C) or the organ weight (D); (E) immunofluorescence analysis was performed for quantification of CCL19 and CCL21 in the lymph node relative to the lymph node area; (F, G) CCL21 and CCL19 localization on consecutive popliteal lymph node cyrosections from infected mice was determined by immunofluorescence microscopy using monoclonal antibody against fibroblastic reticular cells (ER-TR7 antibody positive; green), polyclonal antibody against CCL21 (red) (F), and polyclonal antibody against CCL19 (red) (G); (H) distribution of CCL21 (green) in the spleens of well-nourished and malnourished infected mice. Magnifications, ×10 (G) and ×20 (H). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Malnutrition alters the expression of CCR2 and CCR7 on macrophages and DCs from infected mice.
In prior studies, Leishmania-infected DCs expressed the CCR7 receptor and migrated in response to its ligand CCL21 (37, 38), but DCs from CCR2−/− mice failed to enter the lymph node and accumulated in the subcapsular sinus following cutaneous L. major infection (39). We therefore investigated whether the dysregulated CCR2 and CCR7 mRNA expression in the lymph nodes of the infected malnourished mice was accompanied by altered receptor expression on macrophages and DCs (see the flow cytometry gating protocol in Fig. 5A). We found that infected malnourished mice had significantly reduced numbers of CCR2-bearing nonmyeloid (NK, T, and B) cells (Fig. 5B) and CCR2-bearing CD11b+ CD11c− monocytes/macrophages (Fig. 5C). The frequency of CCR2+ cells within the monocyte/macrophage population in infected malnourished mice was also reduced (Fig. 5D). The population of CCR2-bearing monocyte-derived or inflammatory (CD11c+ CD11b+) DCs was also reduced relative to the number of all lymph node cells (Fig. 5E), but there was no difference in the frequency of CCR2+ cells within the monocyte-derived DC population (Fig. 5F). In contrast, the CCR2-bearing conventional DC (CCR2+ CD11c+ CD11b−) population was not decreased with malnutrition (Fig. 5G), and the frequency of CCR2+ cells within that subpopulation was actually increased (Fig. 5H). When gating on infected cells from mice infected with fluorescence-labeled L. donovani, we found that the infected lymph node CD11c+ DC population had similar levels of CCR2+ expression in the two groups of mice (83.8% ± 3.4% and 89.5% ± 6.8% in the malnourished and well-nourished mice, respectively; P = 0.49). Thus, the decreased lymph node CCR2 mRNA expression (Fig. 3A) was paralleled by a decrease in CCR2-bearing macrophages and DCs and a decreased frequency of surface expression within the macrophage (but not DC) population. The same strategy was used to investigate the expression of CCR7 on macrophages and DCs. Infected malnourished mice showed no change in the percentage of CCR7-bearing CD11b+ CD11c− macrophages relative to the number of all lymph node cells (Fig. 6A); however, the frequency of CCR7+ cells within the macrophage population itself was significantly increased (Fig. 6B). The population of CCR7-bearing monocyte-derived CD11c+ CD11b+ DCs was insignificantly increased (Fig. 6C; P = 0.13), but there was a significant increase in the frequency of CCR7+ cells within the monocyte-derived DC population (Fig. 6D). Similarly, there was a trend toward an increased percentage of CCR7-bearing conventional CD11c+ CD11b− DCs (Fig. 6E; P = 0.057) and a significant increase in the frequency of CCR7+ cells within the conventional DC population (Fig. 6F). The malnourished and well-nourished mice infected with fluorescence-labeled L. donovani showed a similar percentage of CCR7+ expression on infected CD11c+ DCs (82.5% ± 1.7% and 97.4% ± 11.1%, respectively; P = 0.26). Thus, in contrast to the findings of decreased CCR2 expression, CCR7 expression was increased on macrophages and DCs in the infected malnourished mice. This paralleled the increased amount of lymph node CCR7 mRNA (Fig. 3A).
FIG 5.

Malnutrition alters the expression of CCR2 on lymph node macrophages and DCs in L. donovani-infected mice. Flow cytometry was used to determine CCR2 surface expression on nonmyeloid cells, CD11b+ CD11c− monocytes/macrophages, and CD11c+ DCs isolated from the draining popliteal LNs from well-nourished (WN) and malnourished (polynutrient-deficient [PND]) mice (n = 6 per group) infected in the skin with L. donovani for 3 days. (A) Gating strategy. After excluding doublets and gating out B cells, T cells, and NK cells, the myeloid cells were subgated to identify 3 populations: CD11b+ CD11c− cells (monocytes/macrophages [MoMϕ]), CD11c+ CD11b− cells (conventional DCs [cDCs]), and CD11c+ CD11b+ cells (inflammatory monocyte-derived DCs [MoDCs]). Each population was subgated to determine the frequency of CCR2-positive cells in each subgate. SSC, side scatter; FSC, forward scatter. (B) Percentage of CCR2-bearing nonmyeloid cells (pooled NK, B, and T cells) relative to the number of all lymph node cells. (C) Percentage of CCR2-bearing monocytes/macrophages relative to the number of all lymph node cells. (D) Percentage of the monocyte/macrophage (CD11b+ CD11c−) population that expressed CCR2. (E) Percentage of CCR2-bearing monocyte-derived DCs (CCR2+ CD11c+ CD11b+) relative to the number of all lymph node cells. (F) Percentage of the monocyte-derived DC (CD11c+ CD11b+) population that expressed CCR2. (G) Percentage of CCR2-bearing conventional DCs (CCR2+ CD11c+ CD11b−) relative to the number of all lymph node cells. (H) Percentage of the conventional DC (CD11c+ CD11b−) population that expressed CCR2. ***, P < 0.001.
FIG 6.

Malnutrition alters the expression of CCR7 on lymph node macrophages and DCs in L. donovani-infected mice. Flow cytometry was used to determine CCR7 surface expression on nonmyeloid cells, CD11b+ CD11c− monocytes/macrophages, and CD11c+ DCs isolated from the draining popliteal LNs from well-nourished (WN) and malnourished (polynutrient-deficient [PND]) mice (n = 6 per group) infected in the skin with L. donovani for 3 days. The gating strategy was the same as that described in the legend to Fig. 5, except that CCR7 expression was evaluated in place of CCR2. (A) Percentage of CCR7-bearing monocytes/macrophages (CCR7+ CD11b+ CD11c−) relative to the number of all lymph node cells; (B) percentage of the monocyte/macrophage (CD11b+ CD11c−) population that expressed CCR7; (C) percentage of CCR7-bearing monocyte-derived DCs (CCR7+ CD11c+ CD11b+) relative to the number of all lymph node cells; (D) percentage of the monocyte-derived DC (CD11c+ CD11b+) population that expressed CCR7; (E) percentage of CCR7-bearing conventional DCs (CCR7+ CD11c+ CD11b−) relative to the number of all lymph node cells; (F) percentage of the conventional DC (CD11c+ CD11b−) population that expressed CCR7. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
CCR2 deficiency leads to a reduction in the total number of lymph node-resident dendritic cells in L. donovani-infected mice.
In light of the reduced expression of CCR2 and its ligands in the lymph node of malnourished mice, we evaluated the effect of CCR2 deficiency on the cellular composition of the lymph node early following L. donovani infection. Similar to what we found in malnourished mice (Fig. 1) (18), infected CCR2−/− mice had a reduced total number (Fig. 7A; P = 0.04) and lower percentage (Fig. 7B; P = 0.001) of CD11c+ DCs than the wild-type controls. We did not detect any difference in the percentage or total number of fibroblastic reticular cells (ER-TR7 positive [ER-TR7+]), macrophages (MOMA-2 positive [MOMA-2+]), or subcapsular sinus macrophages (CD11b+ CD169+) between infected CCR2−/− and wild-type mice (Fig. 7A and B). To further investigate the source of the reduced DC population in the CCR2-deficient mice, we examined the proportion and total number of resident lymph node DCs (CMFDA− CD11c+) and migratory dermal DCs (CMFDA+ CD11c+) in infected CCR2−/− mice and wild-type controls. No differences in the total number or proportion of dermal DC that had migrated from the skin were noted (Fig. 7C and D). However, the total number and proportion of resident DCs were significantly lower in the infected CCR2−/− mice (Fig. 7C and D; P = 0.02 and 0.0002, respectively). Collectively, these data support the notion that although signaling through CCR2 was not required for the migration of dermal DCs to the draining lymph nodes under inflammatory conditions (L. donovani infection), dysregulation of CCR2 signaling (via the reduction of CCR2 and its ligands) in the infected malnourished mice might account for the remarkable decrease in the amount of resident DCs.
FIG 7.

CCR2 deficiency reduces the total number of lymph node-resident DCs in L. donovani-infected mice. Flow cytometry was used to quantify draining LN cell populations in wild-type (WT) and CCR2-deficient (CCR2−/−) BALB/c mice (n = 6 per group) 3 days after infection in the skin with L. donovani. (A) Total number of macrophages (MOMA-2+), DCs (CD11c+), fibroblastic reticular cells (FRC; ER-TR7+), and subcapsular sinus macrophages (CD169+ CD11b+) per LN. (B) Proportion of specific LN cell populations relative to the total number of LN cells. (C, D) Wild-type and CCR2−/− mice were inoculated with L. donovani, and the skin over the footpad was immediately painted with CMFDA. At 3 days postinfection, the lymph nodes were harvested for flow cytometry. (C) Total number of lymph node-resident dendritic cells (LN DC; CMFDA− CD11c+) and dermal dendritic cells (DDC; CMFDA+ CD11c+) per LN. (D) Proportion of lymph node-resident dendritic cells and dermal dendritic cells relative to the total number of lymph node cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
CCR2 deficiency impairs the overall lymph node phagocytic capacity in response to L. donovani infection.
We used flow cytometry to further quantify the level of infection of lymph node cell populations in wild-type and CCR2−/− mice. We observed a marked reduction in the total number and proportion of infected myeloid cells (DCs, macrophages, and subcapsular sinus macrophages) in CCR2−/− mice compared to wild-type mice (Fig. 8A to C). In contrast, there were no differences in the proportion of fibroblastic reticular cells infected (Fig. 8A to C). There was also a profound reduction in the total number, proportion relative to the total number of lymph node cells, and rate of infection within the resident lymph node DC population (CD11c+ CMFDA−) in the CCR2−/− mice (Fig. 8D to F). However, the total number, proportion, and rate of infection of the infected migratory dermal DCs (CD11c+ CMFDA+) were equivalent in the CCR2−/− and wild-type mice (Fig. 8D to F). Thus, CCR2 deficiency led to a decreased phagocytic capacity of the lymph node myeloid cell population, through both the reduced number of phagocytes (resident DCs) that populate the draining lymph node and the reduced efficiency of the resident DCs in capturing L. donovani (Fig. 8F).
FIG 8.

CCR2 deficiency reduces the level of lymph node cellular infection in L. donovani-infected mice. Wild-type (WT) and CCR2-deficient (CCR2−/−) female BALB/c mice (n = 6 per group) were infected with PKH26-labeled L. donovani in the skin over the footpad, and in some mice the skin at the same site was then immediately painted with CMFDA. Popliteal lymph nodes were harvested after 3 days, and flow cytometry was used to quantify infected DCs (CD11c+), fibroblastic reticular cells (ER-TR7+), subcapsular sinus macrophages (CD169+ CD11b+), and macrophages (MOMA-2+). (A) Proportion of infected individual cell populations relative to the total number of lymph node cells; (B) total number of the infected individual cell populations per lymph node; (C) proportion of infected cells within an individual population relative to the total number of the same cell population; (D) proportion of infected lymph node-resident dendritic cells (LN DC; labeled parasite positive, CMFDA−, CD11c+) and infected dermal dendritic cells (DDC; labeled parasite positive, CMFDA+, CD11c+) relative to the total number of lymph node cells; (E) total number of infected lymph node-resident dendritic cells and dermal dendritic cells; (F) proportion of infected lymph node-resident dendritic cells and dermal dendritic cells relative to the total number of the same cell population. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
CCR2 deficiency does not alter the localization of key cell populations in the LN following L. donovani infection.
We investigated whether the impaired overall phagocytic capacity of the major myeloid cell populations in the lymph node of CCR2−/− mice was associated with altered leukocyte localization in the lymph node after L. donovani infection. To define the location of DCs and macrophages/monocytes, we costained lymph node sections for the DC marker CD11c and the monocyte/macrophage marker MOMA-2 together with the fibroblastic reticular cell marker ER-TR7. CD11c+ DCs and MOMA-2-positive macrophages were distributed similarly in the subcortical regions of both groups of mice (Fig. 9A and B). CD11c+ DCs associated with the conduit network (reticular fibers surrounded by fibroblastic reticular cells) (40) were probably resident DCs, while the DCs located in the paracortex were likely DCs that had migrated from either the dermis or conduit system (Fig. 9A). The subcapsular sinus macrophages (CD169+ CD11b+) were distributed similarly in the subcapsular sinus of CCR2−/− and control infected mice but appeared to be fewer in number in CCR2−/− mice (Fig. 9C and H).
FIG 9.

CCR2 deficiency does not alter the cellular distribution and parasite localization within the lymph node. (A to C) Wild-type (WT) and CCR2-deficient (CCR2−/−) female BALB/c mice (n = 4 per group) were infected with 106 L. donovani promastigotes in the skin over each footpad for 3 days, and the cellular distribution within the lymph node was detected by immunofluorescence microscopy on consecutive popliteal lymph node cryosections. (A) DCs (CD11c+; blue) and fibroblastic reticular cells (ER-TR7+; green). (B) Macrophages (MOMA-2+; green) and fibroblastic reticular cells (ER-TR7+; blue). (C) Subcapsular sinus macrophage (CD169+; blue) and all macrophages (CD11b+; green). (D to H) CCR2−/− and wild-type mice were infected with PKH26-labeled L. donovani for 1 day, and the localization of the parasite in the lymph node was investigated by immunofluorescence microscopy on popliteal lymph node cryosections using monoclonal antibody against fibroblastic reticular cells, DCs, macrophages, and subcapsular sinus macrophages. (D) Parasite (red) distribution in the lymph node subcapsular and subcortical regions; (E) parasite (red) associated with fibroblastic reticular cells (ER-TR7+; blue); (F) parasite (red) colocalized with DCs (CD11c+; blue) and fibroblastic reticular cells (ER-TR7+; green); (G) parasite (red) colocalized with macrophages (MOMA-2+; green); (H) parasite (red) colocalized with subcapsular sinus macrophages (CD169+; blue) and all macrophages (CD11b+; green). Magnifications, ×10 (A, B, and D), ×20 (C, F, and G), and ×40 (E and H).
Next, we investigated whether the lack of CCR2 expression had an influence on the early trafficking of the parasite to the lymph node. We infected mice with fluorescence-labeled L. donovani and examined the parasite distribution in the lymph node compartments and the localization of the parasite relative to fibroblastic reticular cells (ER-TR7+), macrophages (MOMA-2+), DCs (CD11c+), and CD169+ cells at 1 and 3 days postinfection. In prior studies of L. major in CCR2−/− mice, infected cells were localized to the subcapsular sinus of the draining lymph nodes without traversing into the subcortical region (39). In contrast, we found that L. donovani parasites were dispersed throughout the lymph nodes of both CCR2−/− and control mice (Fig. 9D). There was a similar pattern of distribution of parasitized cells in the CCR2−/− and control mice. In both groups, L. donovani was observed in close association with the conduit system, but without a clear colocalization with the fibroblastic reticular cells (Fig. 9E). There was a high degree of colocalization of L. donovani with resident DCs and, to a lesser degree, with macrophages (Fig. 9F and G). Consistent with the flow cytometry data (Fig. 8D and E), the colocalization between DCs and the parasites appeared to be less in the CCR2−/− mice (Fig. 9F). The subcapsular sinus macrophages infected with the labeled parasite were similarly localized in the subcapsular region of both CCR2−/− and control mice (Fig. 9H). Together these data indicate that although CCR2 signaling regulates the abundance of myeloid cell populations in the infected lymph node, its absence does not appear to affect the distribution of these cells.
CCR2 deficiency is associated with reduced CCL19 and CCL21 production in the lymph node and spleen.
The levels of CCL19 and CCL21 in the lymph node and spleen were higher in the infected malnourished mice (which had lower CCR2 and CCL2 levels) than the control mice. To examine whether reduced CCR2 signaling contributed to the remarkably high CCL19 and CCL21 levels, we cultured lymph node cells from both CCR2−/− and control L. donovani-infected mice without an exogenous stimulus and measured the levels of secreted CCL19 and CCL21. Compared with wild-type mice, the levels of CCL19 and CCL21 secreted by the lymph node cells (Fig. 10A; P = 0.01 for both) were lower in L. donovani-infected CCR2−/− mice. No significant differences in the levels of CCL19 and CCL21 in spleen homogenates were noted (Fig. 10B). These data indicate that the relative CCR2 deficiency in the lymph nodes of infected malnourished mice does not account for the high levels of CCL21 and CCL19 in their lymph nodes and spleens.
FIG 10.
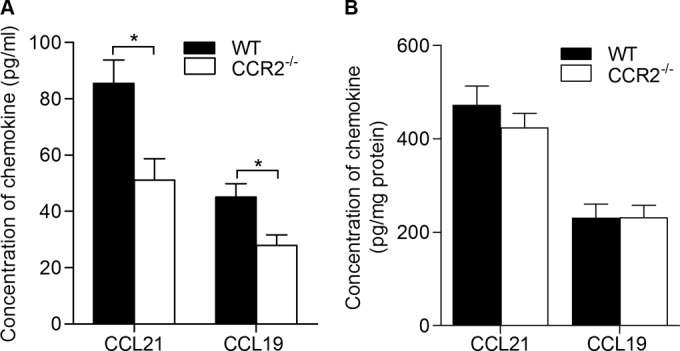
CCR2 deficiency reduces the level of lymph node CCL19 and CCL21 in L. donovani-infected mice. Wild-type (WT) and CCR2-deficient (CCR2−/−) female BALB/c mice (n = 6 per group) were infected with 106 L. donovani promastigotes in the skin over each footpad, and the draining popliteal lymph nodes and spleens were collected at 3 days postinfection. (A) Lymph node cells were cultured in DMEM supplemented with 10% serum for 36 h without an exogenous stimulus, and the levels of CCL19 and CCL21 in the supernatants of lymph node cultured cells from CCR2−/− and wild-type infected mice were measured by ELISA. (B) The levels of CCL19 and CCL21 in the spleen tissue lysates were measured by ELISA and calculated relative to the total protein content. *, P < 0.05.
DISCUSSION
The synergy between malnutrition and infection is thought to underlie as many as a third of childhood deaths worldwide (2). The immune mechanisms responsible for this are not fully understood. The nutritional status of the host is also a key determinant for the progression of L. donovani infection to active visceral leishmaniasis (6, 11, 12, 41–43). In our previous work, we demonstrated that polynutrient (protein, energy, zinc, and iron) deficiency impaired the barrier function of the draining lymph node, resulting in increased early dissemination of L. donovani to the spleen and liver (15, 18). The loss of the lymph node barrier was associated with reduced numbers of phagocytes (e.g., macrophages and DCs) in the subcortical region and subcapsular sinus. Thus, dissemination might be the result of a reduced overall lymph node phagocytic capacity. Here we show that the reduced numbers of DCs in the lymph node of malnourished mice was due to fewer resident DCs in the subcortical region rather than the impaired migration of dermal DCs. The resident DC population that was reduced included both conventional and monocyte-derived DCs, indicating that malnutrition impacted the homeostatic maintenance of cells in the lymph node prior to inflammation and/or diminished blood-to-lymph node cellular recruitment in response to the inflammatory stimulus. Dysregulation of expression of the chemokine receptors and ligands that are responsible for recruitment of DCs and DC precursors to the lymph node offers a plausible explanation for the altered macrophage and DC populations. CCR2 and its ligands CCL2 and CCL7, which are critical to the recruitment of inflammatory monocytes and resident monocyte-derived DCs, were reduced in the lymph node of infected malnourished mice. This reduction in resident but not migratory skin DCs was recapitulated in infected CCR2-deficient mice. Conversely, CCR7 and its ligands CCL19 and CCL22, which are primarily responsible for the migration of DCs from the skin to the draining lymph node, were increased in the infected malnourished lymph node.
The dynamics of DCs in draining lymph nodes are complex. At steady state, DC progenitors reside in the bone marrow (44, 45) and give rise to plasmacytoid DCs and precursor DCs (pre-DCs) (46, 47). Pre-DCs leave the bone marrow and migrate through the bloodstream to generate DCs in nonlymphoid tissues, such as the skin. When activated, skin DCs upregulate CCR7 and migrate to the draining lymph nodes via afferent lymphatics in response to CCL19 and CCL21 (48, 49). DCs can also enter lymph nodes via the blood and high endothelial venules as pre-DCs or as monocytes to differentiate to resident monocyte-derived DCs (32, 46, 50, 51). The migration of monocytes and monocyte-derived DCs to the lymph node is driven by CCR2 and its ligands. During inflammation, there is increased mobilization of pre-DCs from the bone marrow and recruitment of tissue-resident DCs to the lymph nodes (51). Infected dermal DCs are thought to be the primary means of transport of Leishmania parasites from the site of skin infection to the lymph nodes (35). In the current work, we did not observe any alteration in the migratory dermal DCs and parasitized dermal DCs in the lymph nodes of the malnourished mice. Thus, the skin-to-lymph node migration in response to an inflammatory stimulus was intact. However, we found a marked reduction in lymph node-resident DCs (both conventional and monocyte-derived DCs) in the infected malnourished mice.
To investigate the mechanisms behind the reduced numbers of resident DCs, we compared the levels of expression of a variety of inflammatory mediators. We found that malnutrition dysregulated the two principal chemoattractant pathways (CCR7 with its ligands CCL19 and CCL21 and CCR2 with its ligands CCL2 and CCL7) involved in the recruitment of DCs (39, 49, 51, 52). CCR2, in response to CCL2 and CCL7, also regulates the egress of monocytes from the bone marrow to enter the circulation (53–56) and peripheral lymph nodes via high endothelial venules (57–59). The reduced production of CCL2 and CCL7 that we observed in the lymph nodes of infected malnourished mice, coupled with the reduced expression of CCR2, likely contributes to the reduced numbers of lymph node DCs through the impaired migration of either DC precursors or inflammatory monocytes that subsequently differentiate into DCs (32, 51, 60). Our finding of reduced CCR2 expression in monocytes/macrophages and monocyte-derived DCs suggests that malnutrition primarily affects this population rather than conventional DCs that arise directly from DC precursors. Monocyte-derived DCs may enter the lymph node either via the blood and high endothelial venule or from the skin-draining lymphatics. Since the migration of dermal DCs was not reduced, the fewer number of resident DCs in the lymph node is likely the result of reduced migration via the blood and high endothelial venule. This is in keeping with previous findings that migration of inflammatory monocytes and inflammatory monocyte-derived DCs to the lymph node is CCR2 dependent (59, 61) and our finding of reduced resident macrophages and DCs, but not migratory dermal DCs, in L. donovani-infected CCR2−/− mice.
Migration of DCs from the skin to the lymph node is primarily regulated by CCR7 and its ligands and not CCR2 (49, 62). Therefore, the increased expression of CCR7 on lymph node DCs and macrophages, coupled with the higher levels of CCL21 and CCL19 in the lymph nodes and the higher levels of CCL21 in the spleens of malnourished mice, would be expected to promote skin-to-lymph node migration and might act to traffic the CCR7+ parasitized dermal DCs through the lymph node to the visceral organs. Further studies using CCR7-deficient leukocytes or antibodies to neutralize the CCR7-binding chemokines are needed to define their role in the altered lymph node accumulation of DCs (and other myeloid cells) and parasite dissemination.
L. donovani-infected CCR2-deficient mice had significantly reduced amounts of lymph node-resident macrophages and DCs but not migratory dermal DCs. This was strikingly similar to the findings for malnourished mice. Thus, this supports the notion that a relative deficit in CCR2-dependent cell recruitment/retention in the malnourished mice contributes to the altered lymph node cellularity. In both the malnourished and CCR2−/− mice, we found no change in the localization of uninfected or L. donovani-infected myeloid cells within the lymph node (18). This contrasts with the reduced migration of Langerhans cells to the lymph nodes and retention of those cells in the subcapsular sinus in CCR2−/− mice infected with L. major (39). Infected CCR2−/− mice differed from the malnourished mice in several respects. The lymph node myeloid cell populations in the CCR2−/− mice showed a reduced capacity to capture parasites, but this appeared to be normal in the malnourished mice (18). This suggests that complete and global CCR2 deficiency indirectly alters the cell's phagocytic capacity. In contrast to CCR2−/− mice, the reduced expression of CCR2 (and its ligands) in the malnourished mice was localized to the lymph nodes (expression was normal in the spleen). Therefore, localized (skin-lymph node) blockade of the receptor or it ligands would mimic the malnutrition-related effects more closely than they were in the globally CCR2-deficient mouse. Consistent with previous work (52), CCR2−/− mice had lower levels of CCL19 and CCL21 in the lymph nodes and normal levels in their spleens, but malnourished mice had elevated levels of these chemokines in both lymph nodes and spleens. A number of tools, including mice that express a fluorescent transgene in specific myeloid populations or under the control of a chemokine promoter and intravital microscopy, that will help to further define the mechanisms involved in altered cell migration and increased parasite dissemination in this model are now available (63–65).
Lymph flows from peripheral tissue through the afferent lymphatics into the lymph node subcapsular sinus and then to the conduit system and out through the high endothelial venule into the bloodstream (66). The reduced number of resident DCs along the conduits and the consequent reduced phagocytic capacity could explain our previous observation of the increased transit of labeled antigen through the conduit system in malnourished mice (18). This mechanism could also contribute to the malnutrition-related dissemination of parasites (15, 18). We found that about 90% of the parasitized DCs were resident DCs and only 10% were migratory dermal DCs. This finding, which had not been fully appreciated from past studies, suggests that resident DCs play a key role in the capture of parasites and the prevention of parasite dissemination to the visceral organs. Similarly, in a model of cutaneous L. major infection, Iezzi et al. found that monocyte-derived inflammatory DCs and not migratory dermal DCs were responsible for the early capture of Leishmania antigen in the lymph node (27). Our work also corroborated their finding of a small number of skin-derived DCs in the draining lymph node relative to the number of resident DCs early after infection (27). Whether malnutrition leads to a loss of barrier function and increased parasite dissemination through the reduced DC capture of free parasites (analogous to Leishmania antigen [27]) as they transit through the lymph and/or through the reduced capture of parasites released from migrating dermal cells (67) remains to be determined.
In summary, this study demonstrates that malnutrition profoundly impacts the resident lymph node DC population. The reduction in CCR2-bearing inflammatory monocytes and monocyte-derived DCs, coupled with the substantially lower levels of lymph node CCL2, indicates that the deficiency of resident DCs in the infected malnourished mice is mediated in part through the downregulation of the CCR2 chemoattractant pathway. The observed reduction in the number of resident DCs in infected CCR2−/− mice supports this conclusion. The deficiency in resident DCs in the malnourished mice has profound implications for both host defense and vaccine-induced immunity. Fewer resident DCs will lead to the reduced capture of Leishmania and other pathogens as they transit through the lymph node, leading to increased dissemination. Decreased numbers of resident DCs would also reduce the capture of lymph-borne free antigens that traffic to the lymph node and, consequently, impair the initiation of a pathogen-specific adaptive immune response (17, 27). Furthermore, the reduced capture of vaccine antigens in the draining lymph node will limit the induction of vaccine-induced immunity, recognized to be a problem for some vaccines administered to malnourished children (68). Additional studies are warranted to define the mechanisms through which the CCR2 and CCR7 pathways and the migration of monocytes/macrophages and DCs to peripheral lymphoid tissue are disrupted in the malnourished host. Even more critical are investigations to inform rational interventions that can reverse the vicious synergy between malnutrition and infection.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from the U.S. Department of Veterans Affairs and the Department of Internal Medicine at the University of Texas Medical Branch (to P.C.M.).
We thank Sunil Ahuja, Robert Clark, Lynn Soong, Nisha Garg, Robin Stephens, Keith Krolick, Guanming Zhong, Carlos Orihuela, and David Hilmers for insightful discussions and the personnel of the Veterinary Medical Unit at the South Texas Veterans Health Care System for the excellent care of the experimental animals.
Footnotes
Published ahead of print 12 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01778-14.
REFERENCES
- 1.Blossner M, de Onis M. 2005. Malnutrition: quantifying the health impact at the national and local levels. In Prüss-Üstün A, Campbell-Lendrum D, Corvalán C, Woodward A. (ed), WHO environmental burden of disease series, vol 12 World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Black RE, Allen LH, Bhutta ZA, Caulfield LE, de Onis M, Ezzati M, Mathers C, Rivera J. 2008. Maternal and child undernutrition: global and regional exposures and health consequences. Lancet 371:243–260. 10.1016/S0140-6736(07)61690-0. [DOI] [PubMed] [Google Scholar]
- 3.Caulfield LE, de Onis M, Blossner M, Black RE. 2004. Undernutrition as an underlying cause of child deaths associated with diarrhea, pneumonia, malaria, and measles. Am. J. Clin. Nutr. 80:193–198. [DOI] [PubMed] [Google Scholar]
- 4.Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, Peeling RW, Alvar J, Boelaert M. 2007. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 5:873–882. 10.1038/nrmicro1748. [DOI] [PubMed] [Google Scholar]
- 5.Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M. 2012. Leishmaniasis worldwide and global estimates of its incidence. PLoS One 7:e35671. 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearson RD, Cox G, Jeronimo SM, Castracane J, Drew JS, Evans T, de Alencar JE. 1992. Visceral leishmaniasis: a model for infection-induced cachexia. Am. J. Trop. Med. Hyg. 47:8–15. [DOI] [PubMed] [Google Scholar]
- 7.Ostyn B, Gidwani K, Khanal B, Picado A, Chappuis F, Singh SP, Rijal S, Sundar S, Boelaert M. 2011. Incidence of symptomatic and asymptomatic Leishmania donovani infections in high-endemic foci in India and Nepal: a prospective study. PLoS Negl. Trop. Dis. 5:e1284. 10.1371/journal.pntd.0001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hailu A, Gramiccia M, Kager PA. 2009. Visceral leishmaniasis in Aba-Roba, south-western Ethiopia: prevalence and incidence of active and subclinical infections. Ann. Trop. Med. Parasitol. 103:659–670. 10.1179/000349809X12554106963555. [DOI] [PubMed] [Google Scholar]
- 9.Silveira FT, Lainson R, Crescente JA, de Souza AA, Campos MB, Gomes CM, Laurenti MD, Corbett CE. 2010. A prospective study on the dynamics of the clinical and immunological evolution of human Leishmania (L.) infantum chagasi infection in the Brazilian Amazon region. Trans. R. Soc. Trop. Med. Hyg. 104:529–535. 10.1016/j.trstmh.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Collin S, Davidson R, Ritmeijer K, Keus K, Melaku Y, Kipngetich S, Davies C. 2004. Conflict and kala-azar: determinants of adverse outcomes of kala-azar among patients in southern Sudan. Clin. Infect. Dis. 38:612–619. 10.1086/381203. [DOI] [PubMed] [Google Scholar]
- 11.Cerf BJ, Jones TC, Badaro R, Sampaio D, Teixeira R, Johnson WD., Jr 1987. Malnutrition as a risk factor for severe visceral leishmaniasis. J. Infect. Dis. 156:1030–1033. 10.1093/infdis/156.6.1030. [DOI] [PubMed] [Google Scholar]
- 12.Malafaia G. 2009. Protein-energy malnutrition as a risk factor for visceral leishmaniasis: a review. Parasite Immunol. 31:587–596. 10.1111/j.1365-3024.2009.01117.x. [DOI] [PubMed] [Google Scholar]
- 13.Rukunuzzaman M, Rahman M. 2008. Epidemiological study of risk factors related to childhood visceral leishmaniasis. Mymensingh Med. J. 17:46–50. [PubMed] [Google Scholar]
- 14.Actor P. 1960. Protein and vitamin intake and visceral leishmaniasis in the mouse. Exp. Parasitol. 10:1–20. 10.1016/0014-4894(60)90078-3. [DOI] [PubMed] [Google Scholar]
- 15.Anstead GM, Chandrasekar B, Zhao W, Yang J, Perez LE, Melby PC. 2001. Malnutrition alters the innate immune response and increases early visceralization following Leishmania donovani infection. Infect. Immun. 69:4709–4718. 10.1128/IAI.69.8.4709-4718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perez H, De La Rosa M, Malave I. 1984. The effect of protein restriction on the development of protective immunity to Leishmania mexicana. Parasite Immunol. 6:285–294. 10.1111/j.1365-3024.1984.tb00801.x. [DOI] [PubMed] [Google Scholar]
- 17.Serafim TD, Malafaia G, Silva ME, Pedrosa ML, Rezende SA. 2010. Immune response to Leishmania (Leishmania) chagasi infection is reduced in malnourished BALB/c mice. Mem. Inst. Oswaldo Cruz 105:811–817. 10.1590/S0074-02762010000600014. [DOI] [PubMed] [Google Scholar]
- 18.Ibrahim MK, Barnes JL, Anstead GM, Jimenez F, Travi BL, Peniche AG, Osorio EY, Ahuja SS, Melby PC. 2013. The malnutrition-related increase in early visceralization of Leishmania donovani is associated with a reduced number of lymph node phagocytes and altered conduit system flow. PLoS Negl. Trop. Dis. 7:e2329. 10.1371/journal.pntd.0002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cunningham-Rundles S, McNeeley DF, Moon A. 2005. Mechanisms of nutrient modulation of the immune response. J. Allergy Clin. Immunol. 115:1119–1128. 10.1016/j.jaci.2005.04.036. [DOI] [PubMed] [Google Scholar]
- 20.Warren PJ, Hansen JD, Lehmann BH. 1969. The concentration of copper, zinc and manganese in the liver of African children with marasmus and kwashiorkor. Proc. Nutr. Soc. 28:6A–7A. [PubMed] [Google Scholar]
- 21.Sandstead HH. 1994. Understanding zinc: recent observations and interpretations. J. Lab. Clin. Med. 124:322–327. [PubMed] [Google Scholar]
- 22.Hughes S, Kelly P. 2006. Interactions of malnutrition and immune impairment, with specific reference to immunity against parasites. Parasite Immunol. 28:577–588. 10.1111/j.1365-3024.2006.00897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
- 24.Sacks DL, Melby PC. 2001. Animal models for the analysis of immune responses to leishmaniasis. Curr. Protoc. Immunol. Chapter 19:Unit 19.2. 10.1002/0471142735.im1902s28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quinones M, Ahuja SK, Melby PC, Pate L, Reddick RL, Ahuja SS. 2000. Preformed membrane-associated stores of interleukin (IL)-12 are a previously unrecognized source of bioactive IL-12 that is mobilized within minutes of contact with an intracellular parasite. J. Exp. Med. 192:507–516. 10.1084/jem.192.4.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cavanagh LL, Weninger W. 2008. Dendritic cell behaviour in vivo: lessons learned from intravital two-photon microscopy. Immunol. Cell Biol. 86:428–438. 10.1038/icb.2008.25. [DOI] [PubMed] [Google Scholar]
- 27.Iezzi G, Frohlich A, Ernst B, Ampenberger F, Saeland S, Glaichenhaus N, Kopf M. 2006. Lymph node resident rather than skin-derived dendritic cells initiate specific T cell responses after Leishmania major infection. J. Immunol. 177:1250–1256. 10.4049/jimmunol.177.2.1250. [DOI] [PubMed] [Google Scholar]
- 28.Stoitzner P, Romani N, McLellan AD, Tripp CH, Ebner S. 2010. Isolation of skin dendritic cells from mouse and man. Methods Mol. Biol. 595:235–248. 10.1007/978-1-60761-421-0_16. [DOI] [PubMed] [Google Scholar]
- 29.Bajana S, Roach K, Turner S, Paul J, Kovats S. 2012. IRF4 promotes cutaneous dendritic cell migration to lymph nodes during homeostasis and inflammation. J. Immunol. 189:3368–3377. 10.4049/jimmunol.1102613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruedl C, Koebel P, Bachmann M, Hess M, Karjalainen K. 2000. Anatomical origin of dendritic cells determines their life span in peripheral lymph nodes. J. Immunol. 165:4910–4916. 10.4049/jimmunol.165.9.4910. [DOI] [PubMed] [Google Scholar]
- 31.Gordon S, Taylor PR. 2005. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5:953–964. 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 32.Randolph GJ, Ochando J, Partida-Sanchez S. 2008. Migration of dendritic cell subsets and their precursors. Annu. Rev. Immunol. 26:293–316. 10.1146/annurev.immunol.26.021607.090254. [DOI] [PubMed] [Google Scholar]
- 33.Savino W, Dardenne M, Velloso LA, Dayse Silva-Barbosa S. 2007. The thymus is a common target in malnutrition and infection. Br. J. Nutr. 98(Suppl 1):S11–S16. 10.1017/S0007114507832880. [DOI] [PubMed] [Google Scholar]
- 34.Prentice AM. 1999. The thymus: a barometer of malnutrition. Br. J. Nutr. 81:345–347. [PubMed] [Google Scholar]
- 35.Ritter U, Meissner A, Scheidig C, Korner H. 2004. CD8 alpha- and Langerin-negative dendritic cells, but not Langerhans cells, act as principal antigen-presenting cells in leishmaniasis. Eur. J. Immunol. 34:1542–1550. 10.1002/eji.200324586. [DOI] [PubMed] [Google Scholar]
- 36.Luther SA, Tang HL, Hyman PL, Farr AG, Cyster JG. 2000. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc. Natl. Acad. Sci. U. S. A. 97:12694–12699. 10.1073/pnas.97.23.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ato M, Stager S, Engwerda CR, Kaye PM. 2002. Defective CCR7 expression on dendritic cells contributes to the development of visceral leishmaniasis. Nat. Immunol. 3:1185–1191. 10.1038/ni861. [DOI] [PubMed] [Google Scholar]
- 38.Steigerwald M, Moll H. 2005. Leishmania major modulates chemokine and chemokine receptor expression by dendritic cells and affects their migratory capacity. Infect. Immun. 73:2564–2567. 10.1128/IAI.73.4.2564-2567.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sato N, Ahuja SK, Quinones M, Kostecki V, Reddick RL, Melby PC, Kuziel WA, Ahuja SS. 2000. CC chemokine receptor (CCR)2 is required for Langerhans cell migration and localization of T helper cell type 1 (Th1)-inducing dendritic cells. Absence of CCR2 shifts the Leishmania major-resistant phenotype to a susceptible state dominated by Th2 cytokines, B cell outgrowth, and sustained neutrophilic inflammation. J. Exp. Med. 192:205–218. 10.1084/jem.192.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roozendaal R, Mebius RE, Kraal G. 2008. The conduit system of the lymph node. Int. Immunol. 20:1483–1487. 10.1093/intimm/dxn110. [DOI] [PubMed] [Google Scholar]
- 41.Badaro R, Carvalho EM, Rocha H, Queiroz AC, Jones TC. 1986. Leishmania donovani: an opportunistic microbe associated with progressive disease in three immunocompromised patients. Lancet i:647–649. [DOI] [PubMed] [Google Scholar]
- 42.Harrison LH, Naidu TG, Drew JS, de Alencar JE, Pearson RD. 1986. Reciprocal relationships between undernutrition and the parasitic disease visceral leishmaniasis. Rev. Infect. Dis. 8:447–453. 10.1093/clinids/8.3.447. [DOI] [PubMed] [Google Scholar]
- 43.Gomes CM, Giannella-Neto D, Gama ME, Pereira JC, Campos MB, Corbett CE. 2007. Correlation between the components of the insulin-like growth factor I system, nutritional status and visceral leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 101:660–667. 10.1016/j.trstmh.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 44.Fogg DK, Sibon C, Miled C, Jung S, Aucouturier P, Littman DR, Cumano A, Geissmann F. 2006. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 311:83–87. 10.1126/science.1117729. [DOI] [PubMed] [Google Scholar]
- 45.Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, Carotta S, O'Keeffe M, Bahlo M, Papenfuss A, Kwak JY, Wu L, Shortman K. 2007. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat. Immunol. 8:1217–1226. 10.1038/ni1522. [DOI] [PubMed] [Google Scholar]
- 46.Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, Chu FF, Randolph GJ, Rudensky AY, Nussenzweig M. 2009. In vivo analysis of dendritic cell development and homeostasis. Science 324:392–397. 10.1126/science.1170540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Naik SH, Metcalf D, van Nieuwenhuijze A, Wicks I, Wu L, O'Keeffe M, Shortman K. 2006. Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat. Immunol. 7:663–671. 10.1038/ni1340. [DOI] [PubMed] [Google Scholar]
- 48.Alvarez D, Vollmann EH, von Andrian UH. 2008. Mechanisms and consequences of dendritic cell migration. Immunity 29:325–342. 10.1016/j.immuni.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, Blankenstein T, Henning G, Forster R. 2004. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity 21:279–288. 10.1016/j.immuni.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 50.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. 2010. Development of monocytes, macrophages, and dendritic cells. Science 327:656–661. 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmid MA, Takizawa H, Baumjohann DR, Saito Y, Manz MG. 2011. Bone marrow dendritic cell progenitors sense pathogens via Toll-like receptors and subsequently migrate to inflamed lymph nodes. Blood 118:4829–4840. 10.1182/blood-2011-03-344960. [DOI] [PubMed] [Google Scholar]
- 52.Jimenez F, Quinones MP, Martinez HG, Estrada CA, Clark K, Garavito E, Ibarra J, Melby PC, Ahuja SS. 2010. CCR2 plays a critical role in dendritic cell maturation: possible role of CCL2 and NF-kappa B. J. Immunol. 184:5571–5581. 10.4049/jimmunol.0803494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jia T, Serbina NV, Brandl K, Zhong MX, Leiner IM, Charo IF, Pamer EG. 2008. Additive roles for MCP-1 and MCP-3 in CCR2-mediated recruitment of inflammatory monocytes during Listeria monocytogenes infection. J. Immunol. 180:6846–6853. 10.4049/jimmunol.180.10.6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Serbina NV, Pamer EG. 2006. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7:311–317. 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 55.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. 2007. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Invest. 117:902–909. 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Osterholzer JJ, Chen GH, Olszewski MA, Curtis JL, Huffnagle GB, Toews GB. 2009. Accumulation of CD11b+ lung dendritic cells in response to fungal infection results from the CCR2-mediated recruitment and differentiation of Ly-6Chigh monocytes. J. Immunol. 183:8044–8053. 10.4049/jimmunol.0902823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kamei M, Carman CV. 2010. New observations on the trafficking and diapedesis of monocytes. Curr. Opin. Hematol. 17:43–52. 10.1097/MOH.0b013e3283333949. [DOI] [PubMed] [Google Scholar]
- 58.Imhof BA, Aurrand-Lions M. 2004. Adhesion mechanisms regulating the migration of monocytes. Nat. Rev. Immunol. 4:432–444. 10.1038/nri1375. [DOI] [PubMed] [Google Scholar]
- 59.Palframan RT, Jung S, Cheng G, Weninger W, Luo Y, Dorf M, Littman DR, Rollins BJ, Zweerink H, Rot A, von Andrian UH. 2001. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J. Exp. Med. 194:1361–1373. 10.1084/jem.194.9.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Geissmann F, Jung S, Littman DR. 2003. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19:71–82. 10.1016/S1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 61.Nakano H, Lin KL, Yanagita M, Charbonneau C, Cook DN, Kakiuchi T, Gunn MD. 2009. Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat. Immunol. 10:394–402. 10.1038/ni.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Randolph GJ, Angeli V, Swartz MA. 2005. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 5:617–628. 10.1038/nri1670. [DOI] [PubMed] [Google Scholar]
- 63.Megens RT, Kemmerich K, Pyta J, Weber C, Soehnlein O. 2011. Intravital imaging of phagocyte recruitment. Thromb. Haemost. 105:802–810. 10.1160/TH10-11-0735. [DOI] [PubMed] [Google Scholar]
- 64.Mempel TR, Scimone ML, Mora JR, von Andrian UH. 2004. In vivo imaging of leukocyte trafficking in blood vessels and tissues. Curr. Opin. Immunol. 16:406–417. 10.1016/j.coi.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 65.Russo E, Nitschke M, Halin C. 2013. Dendritic cell interactions with lymphatic endothelium. Lymphatic Res. Biol. 11:172–182. 10.1089/lrb.2013.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gretz JE, Norbury CC, Anderson AO, Proudfoot AE, Shaw S. 2000. Lymph-borne chemokines and other low molecular weight molecules reach high endothelial venules via specialized conduits while a functional barrier limits access to the lymphocyte microenvironments in lymph node cortex. J. Exp. Med. 192:1425–1440. 10.1084/jem.192.10.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kastenmuller W, Torabi-Parizi P, Subramanian N, Lammermann T, Germain RN. 2012. A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell 150:1235–1248. 10.1016/j.cell.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Savy M, Edmond K, Fine PE, Hall A, Hennig BJ, Moore SE, Mulholland K, Schaible U, Prentice AM. 2009. Landscape analysis of interactions between nutrition and vaccine responses in children. J. Nutr. 139:2154S–2218S. 10.3945/jn.109.105312. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.