

ABSTRACT
Pancreatic ductal adenocarcinoma (PDA) is the most lethal form of human cancer, with dismal survival rates due to late-stage diagnoses and a lack of efficacious therapies. Building on the observation that avian influenza A viruses (IAVs) have a tropism for the pancreas in vivo, the present study was aimed at testing the efficacy of IAVs as oncolytic agents for killing human PDA cell lines. Receptor characterization confirmed that human PDA cell lines express the alpha-2,3- and the alpha-2,6-linked glycan receptor for avian and human IAVs, respectively. PDA cell lines were sensitive to infection by human and avian IAV isolates, which is consistent with this finding. Growth kinetic experiments showed preferential virus replication in PDA cells over that in a nontransformed pancreatic ductal cell line. Finally, at early time points posttreatment, infection with IAVs caused higher levels of apoptosis in PDA cells than gemcitabine and cisplatin, which are the cornerstone of current therapies for PDA. In the BxPC-3 PDA cell line, apoptosis resulted from the engagement of the intrinsic mitochondrial pathway. Importantly, IAVs did not induce apoptosis in nontransformed pancreatic ductal HPDE6 cells. Using a model based on the growth of a PDA cell line as a xenograft in SCID mice, we also show that a slightly pathogenic avian IAV significantly inhibited tumor growth following intratumoral injection. Taken together, these results are the first to suggest that IAVs may hold promise as future agents of oncolytic virotherapy against pancreatic ductal adenocarcinomas.
IMPORTANCE Despite intensive studies aimed at designing new therapeutic approaches, PDA still retains the most dismal prognosis among human cancers. In the present study, we provide the first evidence indicating that avian IAVs of low pathogenicity display a tropism for human PDA cells, resulting in viral RNA replication and a potent induction of apoptosis in vitro and antitumor effects in vivo. These results suggest that slightly pathogenic IAVs may prove to be effective for oncolytic virotherapy of PDA and provide grounds for further studies to develop specific and targeted viruses, with the aim of testing their efficacy in clinical contexts.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA) is considered the most lethal form of cancer in humans, ranking as the fourth leading cause of cancer-related death in North America and the sixth in Europe (1, 2). The disease generally causes few or no symptoms, and diagnoses are typically made at an advanced stage, when only 1 in 5 patients is eligible for surgical resection (3). The average 5-year survival rates for this disease are less than 5%, and even in cases where surgery is possible, the rates rise to only 15% (2, 4). In addition to surgical intervention, combination chemotherapy and radiation treatments are employed, but the highly aggressive and invasive nature of PDA leads to poor responses, and new innovative therapeutic approaches are in high demand. Disruption of the apoptotic program is a hallmark of numerous cancers, including PDA, and the induction of apoptosis is a primary target of many treatment regimens, including oncolytic virotherapy (5, 6). A number of oncolytic viruses have been tested for efficacy against pancreatic cancer, including adenoviruses, herpesviruses, parvoviruses, reoviruses, and poxviruses. Some of these viruses, when they show particular preferences for a specific mutation along a signaling pathway, show selectivity for cancer cells based on these aberrant pathways (2, 7). While some have demonstrated appreciable efficacy in preclinical models, the great genetic diversity of PDAs encountered in the clinical setting often leads to unsatisfactory results in clinical trials and reaffirms the need to investigate additional viral classes that offer alternative modes of tumor-specific targeting (7).
Influenza A viruses (IAVs) of the family Orthomyxoviridae are naturally circulating viruses of aquatic birds, from which a total of 16 diverse hemagglutinin (HA) and 9 neuraminidase (NA) types have been isolated in various combinations (8, 9). Additional subtypes have recently been discovered in bats but represent highly genetic lineages (10). In their natural hosts, these viruses cause asymptomatic infection of the gastrointestinal and respiratory tracts; however, in land-based poultry, mild respiratory symptoms may be present. In its more severe form, highly pathogenic (HP) avian IAVs cause systemic disease with high mortality and spread to numerous organs of the respiratory, digestive, and nervous systems (8, 11, 12). A difference in core body temperature between humans and avian species is a known factor in limiting interspecies transmission, as avian IAVs that have adapted to replication at 41°C demonstrate decreased polymerase activity at temperatures of 33 to 37°C, typical of the human respiratory tract (13–16). However, the most important restriction for crossing the species barrier lies at the receptor level. Avian IAVs require α-2,3-linked sialic acids (SAs), the dominant form in the avian gastrointestinal and respiratory tracts, while the human respiratory tract contains mostly the α-2,6-linked forms that are recognized by human-tropic IAVs (17, 18). On rare occasions, the HA from an avian IAV has successfully overcome this barrier and adapted to bind α-2,6 linkages, causing the pandemics of 1918, 1957, and 1968. Such viruses then become established in the human population and cause seasonal influenza epidemics (9). The H1N1 2009 pandemic was instead caused by a triple reassortant carrying genes from avian and swine influenza viruses; however, in this case, the swine HA was already specific for the human receptor (19).
Several observational studies have indicated the predilection of both HP IAVs and IAVs with low pathogenicity (LP) for the pancreas in domesticated avian species and migratory waterfowl following experimental or natural infection (20–29). Necrosis of the pancreatic ductal epithelium was observed in ferrets intragastrically infected with HP H5N1 virus (30), and pancreatic postmortem lesions ranging from inflammation to necrosis have also been observed in HP-IAV-infected cats (31, 32). Pathological examinations of human fatalities from the H1N1 2009 pandemic also revealed pancreatic lesions in two of six postmortem examinations (33), and in our recent studies using in vitro and ex vivo models, we demonstrated that human cells originating from the exocrine pancreas were infected and killed by LP IAVs (34). Therefore, although the pancreas is not considered a typical site of replication after standard infection, influenza A virus seems to be capable of infecting and damaging pancreatic cells in severe infections. Furthermore, the fact that IAV is known to induce apoptosis in numerous cell types (35–37) may provide an advantage in overcoming the known resistance to apoptosis of PDA cells.
Building on these findings, in the present study, we tested the ability of influenza virus to infect and kill PDA cell lines in vitro and in vivo, characterizing the expression of virus-specific receptors, viral replication kinetics, and the induction of apoptosis following infection of PDA cell lines.
MATERIALS AND METHODS
Cells.
Madin-Darby canine kidney (MDCK) cells were maintained in Eagle medium after alpha modification (MEM; Sigma) and supplementation with 10% fetal bovine serum (FBS; Euroclone), 1% 200 mM l-glutamine (Sigma), and a 1% antibiotic solution of penicillin-streptomycin-nystatin (10,000 U/ml, 10,000 μg/ml, 10,000 U/ml; Gibco and Sigma). The nontumoral human pancreatic ductal cell line HPDE6, PDA lines BxPC-3 and AsPC-1, and murine PDA PANC02 cells were maintained in RPMI 1640, PANC-1 and MIA PaCa-2 cells were maintained in Dulbecco's MEM (DMEM), and CFPAC-1 cells were maintained in Iscove's modified Dulbecco's medium. All media were supplemented with FBS, l-glutamine, and antibiotics as for the MEM, and all cell lines were maintained in a humidified incubator at 37°C with 5% CO2 and subcultured twice weekly.
Viruses.
A panel of IAVs from multiple host species, including HP and LP isolates, were examined for their ability to infect pancreatic cells. Viral strains used in this study included A/turkey/Italy/2962/2003 (H7N3), A/turkey/Italy/4580/99 (H7N1 HP), A/cockatoo/England/72 (H4N8), A/macaw/England/626/80 (H7N7), A/mallard/Italy/3401/05 (H5N1), A/chicken/Egypt/1701/6 (H5N1 HP), A/Puerto Rico/8/34 (PR/8 H1N1), and A/canine/Florida/43/2004 (H3N8). Virus stocks were grown in 9- to 10-day-old specific-pathogen-free (SPF) embryonated chicken eggs (Charles River), and harvested allantoic fluid was clarified by centrifugation and tested for bacterial contamination prior to use. All viruses were titrated by a standard plaque assay procedure on MDCK cells using a 0.8% agarose overlay with a final concentration of 1× DMEM, 1% antibiotics, 1% l-glutamine, and l-1-tosylamide-2-phenylethyl chloromethyl ketone (TPCK)-trypsin (Sigma) at concentrations ranging from 0 to 2 μg/ml, depending on the virus isolate. Plaques were visualized and counted at 3 days postinfection, and resulting titers in numbers of PFU were used for all multiplicity of infection (MOI) determinations described.
Sialic acid receptor characterization.
The presence of alpha-2,3- and alpha-2,6-linked sialic acid residues was determined by flow cytometry for each PDA cell line included in the study. Following trypsinization, 1 × 106 cells were aliquoted into microcentrifuge tubes and washed twice with 500 μl of phosphate-buffered saline (PBS)–10 mM HEPES (PBS-HEPES). To control for endogenous biotin or avidin binding sites, an avidin/biotin blocking kit (Vector Laboratories, USA) was employed prior to staining. Reagents were prepared as per the manufacturer's instructions, and cells were incubated with 100 μl of each solution for 15 min, with two PBS-HEPES washes after each treatment. Alpha-2,3- and alpha-2,6-sialic acid linkages were detected by incubating cells for 30 min with 100 μl of biotinylated Maackia amurensis lectin II (Vector Laboratories) (5 μg/ml) and then with 100 μl of phycoerythrin (PE)-streptavidin (BD Biosciences) (10 μg/ml) for 30 min at 4°C in the dark or with 100 μl of fluorescein-conjugated lectin (Vector Laboratories) (5 μg/ml). Cells were washed twice with PBS-HEPES between stainings and resuspended in PBS with 1% formalin prior to flow cytometric analyses. To confirm the specificities of lectins, cells were pretreated with 1 U per ml of neuraminidase from Clostridium perfringens (Sigma) for 1 h prior to the avidin/biotin blocking step. Samples were analyzed on a BD FACSCalibur or the BD LSR II (BD Biosciences), and a minimum of 5,000 events were recorded.
Entry of avian HA- and NA-bearing pseudotypes into pancreatic adenocarcinoma cells.
Pseudotypes bearing the HA (GenBank accession number AY651333) and the NA (GenBank accession number AY651445) from the influenza H5N1 virus isolate A/Viet Nam/1194/2004 were generated in 293T cells as previously described (38). PDA cell permissiveness to viral pseudotypes was assessed by adding 250 μl per well of pseudotypes diluted 1:10 in serum-free medium onto confluent monolayers of PANC-1, AsPC-1, BxPC-3, and CFPAC-1 cell lines in 48-well plates. Inoculum was replaced with fresh medium containing 3% FBS following 3 h of incubation, and cells were left at 37°C for 48 to 72 h. Entry of pseudotypes into PDA cell lines was monitored by the expression of the green fluorescent protein (GFP) reporter in the target cells using a Zeiss Axiovert 40 CFL, fluorescence, phase-contrast, trinocular, inverted microscope fitted with an HBO50 mercury short-arc lamp.
Sensitivity of PDA cells to influenza virus infection.
To determine whether pancreatic cell lines were susceptible to infection by IAV, we conducted a pilot experiment where cells seeded on 96-well plates were infected with 10-fold serial dilutions of virus stocks and incubated at 37°C. A minimum of four wells were infected per dilution of virus, and infections were performed in the presence of 0.05 μg/ml of TPCK-trypsin, the maximum concentration tolerated by the pancreatic cells without toxicity. At 72 h postinfection (hpi), the highest dilution of inoculum at which a cytopathic effect (CPE) was noted was recorded, and supernatants from each virus dilution were harvested, pooled, and directly passaged onto 96-well plates of MDCK cells for virus isolation. On the plates, TPCK-trypsin was used at a concentration of 1 μg/ml.
Virus replication kinetics in pancreatic cell lines.
The ability of a panel of IAVs to replicate in select cell lines was monitored over a 72-h time course. BxPC-3, HPDE6, and MDCK cells were seeded on 24-well plates 1 day prior to infection in order to achieve a confluent monolayer. On the day of infection, cells were washed twice and then infected with 200 μl of inoculum per well at an MOI of 0.001 PFU/cell. Following 1 h of incubation, the inoculum was removed and replaced with 1 ml of serum-free medium containing 0.05 μg/ml TPCK-trypsin. Mock-infected control wells were included in all experiments. At 1, 24, 48, and 72 hpi, supernatants from three infected wells were harvested and viral titers were determined via the 50% tissue culture infectious dose (TCID50) assay on MDCK cells, using the formula of Reed and Muench (39).
Replication kinetics at 37°C and 41°C.
To ensure that the lack of virion production by LP IAVs observed in time course experiments was not attributed to the temperature sensitivity of avian viruses in human cells, BxPC-3 and MDCK cells were infected with a selection of viruses in parallel at an MOI of 0.001 as described above, and plates were incubated simultaneously at 37°C or 41°C. Three infected samples were harvested at 24, 48, and 72 hpi, and viral titers were determined by TCID50 assay.
Viral RNA replication in PDA cell lines.
To assess active viral genome replication, AsPC-1, BxPC-3, CFPAC-1, MIA PaCa-2, PANC-1, and HPDE6 cells were infected with the panel of LP virus isolates at an MOI of 0.1. MDCK cells were included as positive controls. Monolayers were washed once with PBS after inoculum removal and overlaid with serum-free medium containing 0.05 μg/ml TPCK-trypsin. At 1, 16, and 24 hpi, supernatants and trypsinized cell pellets from infected wells were collectively harvested and stored at −80°C for RNA extraction. Total RNA from infected cells and supernatants was obtained by automated extraction (Hamilton Robotics, Switzerland) using the MagMAX 96 AI/ND viral RNA isolation kit (Ambion; AM1835) according to the manufacturer's instructions.
One step rRT-PCR.
Real-time reverse transcription-PCR (rRT-PCR) targeting the conserved matrix (M) gene of influenza A virus was performed on isolated RNA using the published primers and probes previously described (40). The amplification reaction was performed using 5 μl of extracted RNA in a final volume of 25 μl using the QuantiTect Multiplex RT-PCR kit (Qiagen, Hilden, Germany). Each reaction mixture contained 300 nM forward and reverse primers (M25F and M124-R, respectively) and 100 nM fluorescently labeled probe (M+64). The PCR was carried out under the following parameters: 50°C for 20 min and 95°C for 15 min, followed by 40 cycles of 94°C for 45 s and 60°C for 45 s.
Cell proliferation assay.
Cells were seeded in 96-well plates at densities of 30,000 cells per well and infected with a panel of influenza A viruses the following day using an MOI of 1. Cell proliferation and subsequently cell viability were determined based on tetrazolium reduction at 24 hpi using the standard 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. Briefly, 10 μl of MTT reagent (Sigma; M2128) freshly prepared in PBS was added directly to culture medium, producing a final concentration of 0.5 mg/ml. Following 4 h of incubation at 37°C, 100 μl of solubilization solution (10% SDS in 0.01 M HCl) was added to each well for overnight incubation at 37°C. Absorbance was read at 570 nm, with correction at 690 nm, and results from infected cells were normalized to results for uninfected controls.
Detection of virus-induced apoptosis by flow cytometry.
Semiconfluent monolayers of cells seeded in 24-well plates were infected at an MOI of 1, and after 1 h of absorption, inoculum was removed and replaced with 1 ml of serum-free medium. Gemcitabine (2 mM) and cisplatin (0.8 μM) (Gem+Cisp), two common chemotherapeutic agents used for PDA treatment (7, 41), were included in combination as a positive control. FBS was added to each well, for a final concentration of 10%, approximately 1 h postcollection to ensure cell membrane integrity for the labeling process. At 16 and 24 hpi, cells were harvested from two infected wells and one control well and incubated with Alexa Fluor 647 annexin V conjugate (Invitrogen) (1 μl per 375,000 cells) and propidium iodide (PI; 0.5 μl per 375,000 cells) in a volume of 300 μl of medium with 10% FBS for 10 min in the dark. Samples were then fixed for 15 min in 3.6% paraformaldehyde, centrifuged, resuspended in 300 μl of PBS-FBS, and analyzed on a BD FACSCalibur. A minimum of 5,000 events were recorded. Virus-induced apoptosis was determined by subtracting the percentage of annexin V-positive control cells from infected cells, and results are reported as specific cell death.
Detection of virus induction of caspase activity by immunocytochemistry.
BxPC-3 and HPDE6 cells seeded on sterile glass chamber slides (BD) and high-binding slides were infected at an MOI of 1. At 16 and 24 hpi, supernatants were removed, and slides were air dried in a biosafety cabinet, fixed in ice-cold acetone for 20 min, and then stored at −20°C until analysis. Uninfected cells and those treated with gemcitabine (2 mM) and cisplatin (0.8 μM) served as negative and positive controls, respectively. Prior to stainings, frozen slides were thawed and washed 3 times for 5 min with deionized water to remove residual acetone, blocked with 3% H2O2 for 8 min at room temperature to remove endogenous peroxidases, and washed 3 times with deionized water and once with PBS-Tween 20. Slides were then blocked for 30 min with 1% bovine serum albumin (BSA), washed with PBS-Tween 20, and permeabilized with 0.1% Triton X-100 for 10 min. Anti-active/cleaved caspase-8 (1:50; Imgenex), anti-active-caspase-9 (1:10; BioVision), and anti-active-caspase-3 (1:30; Cambridge, United Kingdom) primary antibodies were applied for 1 h in a humidified chamber at room temperature. Immunoreactivity was revealed by the avidin/biotin method (LSAB+ system with horseradish peroxidase [HRP]; DakoCytomation, Glostrup, Denmark) using the ready-to-use AEC+ (aminoethylcarbazole) substrate-chromogen (DakoCytomation). Carazzi's hematoxylin was used as a counterstain, and Faramount mounting medium (DakoCytomation) was used to mount coverslips on slides. Ten histological counts of 500 cells each were determined per cell line/treatment/time point using Nis Elements BR software (Nikon) to determine the percentage of caspase-positive cells.
Oncolytic effects of LP IAV in vivo.
Twelve 6-week-old female SCID mice were subcutaneously injected with 5 × 106 BxPC-3 cells in a volume of 100 μl into the right flank. Palpable tumors developed after 8 days, and mice were then randomly divided into two groups (n = 6 per group), one group receiving an intratumoral inoculation of 2.4 × 104 PFU of H7N3 in a volume of 100 μl and the other receiving 100 μl of PBS. The procedure was subsequently repeated 3, 5, and 7 days later for a total of four intratumoral inoculations per treatment group. The overall physical condition and behavior of the mice were monitored daily, and measurements of tumor size were taken on days 8, 15, 19, and 25 following initial injection. Caliper measurements of tumor sizes were taken at regular intervals throughout the experiment, and the length (L) and width (W) were recorded to determine tumor volume (V) using the formula V = L × W2 × (π/6). At 25 days postinfection, mice were sacrificed and tumors were snap-frozen for RNA extraction and IAV-specific rRT-PCR as described above. All experimental protocols employed were previously approved by the Italian Ministry of Health (protocol 130/2011).
Statistical analyses.
GraphPad PRISM version 6 statistical analysis software was used for the analyses of experimental data. Results from in vitro growth curves, annexin V expression, caspase induction, and in vivo experiments were analyzed using one-way analysis of variance (ANOVA) followed by Tukey's honestly significant difference (HSD) post hoc test for multiple comparisons, whereas MTT assay results were analyzed using one-way ANOVA followed by Dunnett's post hoc test. Data from experiments examining effects of different growth temperatures were analyzed using two-way ANOVA plus the Bonferroni posttest. P values of ≤0.05 were considered statistically significant.
RESULTS
Expression of alpha-2,3- and alpha-2,6-linked sialic acid receptors and infection of human PDA cell lines by IAV-GFP pseudotypes.
To determine whether human PDA cell lines expressed receptors specific for avian IAVs, sialic acid-specific lectin staining was performed. Flow cytometry results demonstrated the presence of receptors for both human and avian influenza A viruses on the PDA cell lines BxPC-3, CFPAC-1, MIA PaCa-2, and PANC02 (Fig. 1). The PANC-1 cells contained high levels of the alpha-2,3 receptors (specific for avian IAVs) but expressed a bimodal distribution of alpha-2,6 receptors (specific for human IAVs). Interestingly, AsPC-1 cells showed higher expression of alpha-2,3 receptors than of alpha-2,6 receptors. The presence of both receptor glycoforms on HPDE6 cells was reported in a previous study (32). To confirm the presence of functional alpha-2,3-linked receptors on the human PDA cell lines, we tested the entry of GFP-expressing viral pseudotypes bearing avian signature HA and NA glycoproteins of A/Viet Nam/1194/2004 (H5N1). Results showed that infected monolayers of PANC-1, AsPC-1, BxPC-3, and CFPAC-1 cells all expressed GFP, confirming the ability of virus-like particles bearing an avian HA and NA to enter these cells (Fig. 2). Cell line-dependent differences were observed, with BxPC-3 and CFPAC-1 cells showing higher levels of GFP expression than PANC-1 and AsPC-1 cells.
FIG 1.
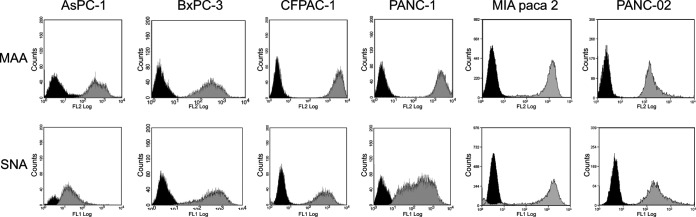
Expression of alpha-2,3- and alpha-2,6-linked SA receptors on PDA cell lines. Cells were incubated with either fluorescein isothiocyanate-labeled Sambucus nigra (SNA) lectin or biotinylated Maackia amurensis (MAA) lectin II lectin and then with phycoerythrin-streptavidin to detect α-2,6- and α-2,3-SAs, respectively. Samples were read on a BD FACSCalibur, with a minimum of 5,000 events recorded. Profiles in black, negative control (no lectin added); profiles in gray, binding of indicated lectin.
FIG 2.

H5N1 pseudotype particle entry into PDA cells. AsPC-1, BxPC-3, CFPAC-1, and PANC-1 cell lines were infected with fluorescent HA- and NA-bearing IAV pseudotypes as described in the text and visualized following 48 h of incubation. Monolayers (left), GFP reporter gene expression (middle), and the merging of these two images (right) are shown for each cell line at a ×10 magnification. Cells were visualized with a Zeiss Axiovert 40 CFL, fluorescence, phase-contrast, inverted microscope equipped with an HBO50 mercury short-arc lamp.
Sensitivity of cell lines to influenza virus infection.
To determine whether the PDA cell lines were susceptible to infection by IAV and could support viral replication, cells were infected with serial dilutions of a panel of HP and LP viruses and monitored for cytopathic effect (CPE) over a period of 72 h. Inoculum for all initial infections was supplemented with 0.05 μg/ml TPCK-trypsin, the maximum concentration tolerated by PDA cells without associated cytotoxicity. CPE was observed in all cell lines following infection with HP or LP isolates, though not all cells displayed CPE with all virus strains (Table 1). Due to the unusual growth characteristics of cells, CPE was difficult to discern in AsPC-1, MIA PaCa-2, and PANC-1 cells. In all cases, however, virus reisolation on MDCK cells confirmed the presence of live virus in the supernatants, often at several logs above the endpoint dilution at which CPE was originally observed. PANC02, a murine PDA cell line, was the least permissive of all cell lines tested and thus was excluded from further studies.
TABLE 1.
Cytopathic effect and virus isolation following infection of various PDA cell lines with HP and LP avian and human influenza virusesa
| Virus isolate | Highest dilution on indicated cell line |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MDCK |
BxPC-3 |
CFPAC-1 |
AsPC-1 |
PANC-1 |
PANC02 |
MIA PaCa-2 |
HPDE6 |
|||||||||
| At which CPE was observed | After reisolation | At which CPE was observed | After reisolation | At which CPE was observed | After reisolation | At which CPE was observed | After reisolation | At which CPE was observed | After reisolation | At which CPE was observed | After reisolation | At which CPE was observed | After reisolation | At which CPE was observed | After reisolation | |
| A/chicken/Egypt/1701/6 (H5N1 HP) | 10−9 | 10−9 | 10−7 | 10−7 | 10−3 | 10−4 | 10−5 | 10−6 | 10−3 | 10−3 | 10−6 | 10−6 | 10−3 | 10−8 | 10−4 | 10−6 |
| A/turkey/Italy/4580/99 (H7N1 HP) | 10−6 | 10−8 | 10−5 | 10−5 | 10−5 | 10−5 | 10−5 | 10−5 | 10−3 | 10−5 | 10−3 | 10−3 | 10−3 | 10−7 | 10−4 | 10−5 |
| A/mallard/Italy/3401/05 (H5N1) | 10−6 | 10−6 | 10−4 | 10−4 | 10−3 | 10−6 | 10−2 | 10−4 | ND | 10−3 | 10−2 | 10−2 | 10−2 | 10−4 | 10−2 | 10−4 |
| A/turkey/Italy/2962/V03 (H7N3) | 10−8 | 10−8 | 10−3 | 10−4 | 10−5 | 10−6 | 10−3 | 10−6 | 10−3 | 10−4 | 10−3 | 10−3 | 10−3 | 10−5 | 10−4 | 10−5 |
| A/Puerto Rico/8/34(H1N1) | 10−6 | 10−6 | 10−2 | 10−5 | ND | 10−3 | 10−3 | 10−5 | ND | 10−2 | 10−2 | 10−2 | ND | 10−4 | ND | 10−5 |
| A/canine/Florida/2004 (H3N8) | 10−4 | 10−4 | ND | 10−2 | ND | 10−2 | ND | 10−2 | ND | 10−1 | 10−1 | 10−1 | ND | 10−3 | ND | 10−3 |
| A/cockatoo/England/72 (H4N8) | 10−6 | 10−6 | 10−2 | 10−5 | 10−2 | 10−4 | 10−4 | 10−5 | 10−2 | 10−4 | — | — | 10−3 | 10−3 | 10−2 | 10−3 |
| A/macaw/England/626/80 (H7N7) | 10−7 | 10−7 | 10−4 | 10−5 | 10−2 | 10−6 | 10−4 | 10−6 | 10−3 | 10−5 | — | — | 10−2 | 10−4 | 10−2 | 10−6 |
MDCK cells were infected in quadruplicate wells with 10-fold serial dilutions of stock virus and isolates from pooled supernatants. Results indicate the highest dilution at which CPE was observed either after initial infection at 72 hpi or following reisolation on MDCK cells. ND, no definitive cytopathic effect; —, experiments were not performed.
Replication kinetics in pancreatic cells.
The replication kinetics of the virus panel over an extended period was examined in three cell lines: BxPC-3, HPDE6, and MDCK. BxPC-3 was chosen as the representative human PDA cell line based on results from the pseudotype and virus sensitivity experiments and compared with the nontumoral HPDE6 cells to test whether IAVs showed an intrinsic tropism for cancer cells. Using an MOI of 0.001 and a TPCK-trypsin concentration of 0.05 μg/ml, BxPC-3 infection led to successful virion production in the cases of PR/8 H1N1, LP H5N1, and HP H5N1 and H7N1 isolates (Fig. 3); however, three other LP viruses (H7N3, H4N8, H7N7) were never reisolated over the 72-h time course. These results closely mirrored the CPEs observed, as infection with HP IAVs led to progressive destruction of the monolayer, whereas LP IAVs did not cause any notable CPE at this MOI (data not shown). Infection of BxPC-3 cells with PR/8 H1N1 also led to productive infection, as confirmed by virus isolation, though no CPE was observed and titers did not show a significant increase from 24 to 72 h. In fact, this trend was observed for almost all trypsin-dependent viruses in all cell lines, where titers did not generally increase after 24 hpi, most likely due to the low TPCK-trypsin concentrations. Further testing of the LP H5N1 isolate, previously described as slightly pathogenic due to the absence of multibasic residues in the HA cleavage site, showed an intermediate phenotype and ability to replicate to low levels even in the absence of exogenous trypsin (data not shown), thus explaining virus isolation results in the presence of small amounts of TPCK-trypsin. Results obtained with the nontransformed HPDE6 cell line were similar to those of BxPC-3 cells; however, for three of the four virus isolates, titers were consistently low (Fig. 3). MDCK cells, which are considered the gold standard for in vitro replication of IAVs, supported the highest levels of replication of all cell lines tested. For all experiments, supernatants were also collected at 1 hpi and titrated. Values were used in growth curves to establish a baseline attributed to residual inoculum and were typically below or just at the limit of detection of the TCID50 assay (≤6.3 × 101).
FIG 3.
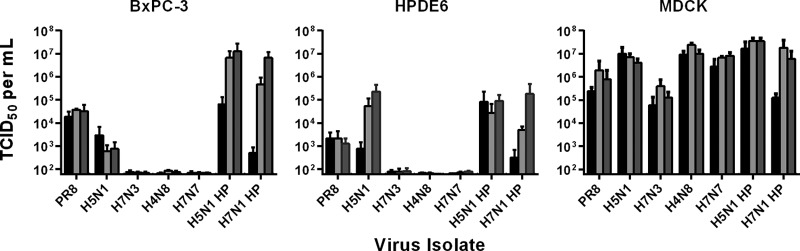
Replication kinetics of influenza A viruses in BxPC-3, HPDE6, and MDCK cells. Cells were infected at an MOI of 0.001 PFU/cell, and virus titers in infected supernatants were determined via the TCID50 assay at 24 (black bars), 48 (light-gray bars), and 72 (dark-gray bars) hours postinfection. Results represent means plus standard deviations of results from three independent experiments of three replicate samples each.
Comparative replication of influenza viruses at 37 and 41°C.
To determine whether variations in virus titers observed between MDCK cells and the human BxPC-3 cells could be attributable to suboptimal temperatures for polymerase function, these cell lines were infected in parallel at physiologically relevant temperatures for the growth of human and avian viruses, i.e., 37°C and 41°C, respectively. A selection of 5 viruses representing HP and LP isolates were studied over a 72-h time course. Replication kinetics of avian viruses followed the general trend; viruses reached higher titers at early time points when they were incubated at 41°C than when they were incubated at 37°C, while PR/8 H1N1 replicated more efficiently at 37°C, consistent with its tropism for mammalian cells (Fig. 4). Interestingly, all avian IAVs tested were able to efficiently replicate in the PDA cell line BxPC-3 even at 37°C, suggesting that their replication in human PDA cells might not be subject to host-dependent temperature sensitivity.
FIG 4.

Comparative replication kinetics of influenza isolates at 37°C and 41°C. BxPC-3 and MDCK cells were infected at an MOI of 0.001 with the viruses indicated in the absence (for HP isolates) or presence of 0.05 μg/ml TPCK-trypsin. At 24, 48, and 72 hpi, supernatants from three independent wells were harvested and titrated via the TCID50 assay on MDCK cells in the presence of 1 μg/ml TPCK-trypsin. Values shown are means and standard deviations from two independent experiments (*, P < 0.05 at 24 hpi).
Viral RNA replication kinetics in PDA cells.
Given our inability to isolate LP IAVs from PDA cells at a low MOI, we wanted to ensure that this was not due to a lack of effective genome replication. All cell lines were infected with the panel of LP isolates as well as PR/8 H1N1 at a higher MOI of 0.1 in order to analyze replication kinetics over a 24-h period. Active replication of viral RNA was noted for all viral isolates in all cell lines based on rRT-PCR results, in which threshold cycles (CTs) at 16 hpi were decreased compared to those of cultures sampled at 1 hpi (Fig. 5). The lack of changes in CT values from 16 to 24 hpi strongly suggests that replication was limited to a single cycle, most likely due to the low levels of TPCK-trypsin. When different viruses were compared, the H7N3 isolate had the highest replication of all cell lines tested, while H4N8 generally presented the lowest levels of replication. Similar trends were also observed for all isolates in MDCK cells, with no changes in CT between 16 and 24 hpi (results not shown).
FIG 5.

Viral RNA replication kinetics in infected PDA cells. AsPC-1, BxPC-3, CFPAC-1, MIA PaCa-2, PANC-1, and nontransformed HPDE6 cells were infected with a panel of LP influenza viruses at an MOI of 0.1. Supernatants and cell pellets were harvested together at 1, 16, and 24 hpi, and extracted RNA was amplified using viral matrix gene-specific rRT-PCR. Data represent means plus standard deviations of results for triplicate samples and indicate changes in CT values from those at 1 hpi.
Assessment of cell proliferation postinfection.
The MTT assay measures tetrazolium reduction by metabolically active and proliferating cells and therefore is used as an indicator of cell viability (42). Given the results of the experiments showing the death of PDA cells infected with high concentrations of virus (Table 1) and the demonstration of active viral RNA replication in all PDA cell lines infected with LP IAVs (Fig. 5), we were interested in examining the intensity of virus-induced cell death in the various PDA cell lines and possible variations between virus isolates. All pancreatic cell lines were infected with PR/8 H1N1 and the complete panel of LP avian IAVs used in previous experiments at an MOI of 1 to ensure that all cells were infected upon analysis. MTT assay results showed a general agreement with observations from initial experiments on cell line sensitivity to the panel of virus isolates, with PANC-1 displaying the highest levels of resistance, whereas BxPC-3 and CFPAC-1 cells showed overall the highest sensitivity to virus-induced cytotoxicity of the PDA cell lines (Fig. 6). Furthermore, the H7N7 and H7N3 isolates, whose RNA replication rates were the highest among the viruses tested, consistently caused the greatest loss of cell viability across the panel of cell lines tested, with highly statistically significant differences from control values (P < 0.01 to P < 0.0001). The H7N3 isolate in particular showed the greatest innate affinity for the PDA cells, causing higher losses of cell proliferation in BxPC-3 and CFPAC-1 cells than in normal ductal HPDE6 cells (P < 0.0001). Of note, absorbance values did not necessarily correlate with visible cell damage, as H7N3 infection of CFPAC-1 and BxPC-3 cells resulted in massive CPEs and complete destruction of the monolayer, yet results indicated that 30% of proliferative activity was retained (Fig. 6).
FIG 6.

PDA cell proliferation following influenza A virus infection. AsPC-1, BxPC-3, CFPAC-1, MIA PaCa-2, PANC-1, and nontransformed HPDE6 cells were infected with a panel of LP influenza viruses at an MOI of 1 and analyzed for cell proliferation at 24 hpi via the MTT assay. Absorbance readings at 570 nm corrected for 690 nm have been normalized to values for mock-infected controls. Results shown represent means plus standard deviations from two independent experiments of four replicates each, with statistically significant differences from mock-infected controls indicated for virus-infected cells based on one-way ANOVA followed by Dunnett's post hoc test (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Induction of apoptosis following influenza virus infection.
To build on results observed in MTT assays and examine the mode of cell death induced, the ability of IAVs to induce apoptosis in PDA cells was assessed following infection at an MOI of 1. Engagement of the apoptotic program was assessed by annexin V binding and flow cytometry at 16 and 24 hpi. As a positive control, cells were subjected to a high concentration of gemcitabine-cisplatin (Gem+Cisp), two commonly used chemotherapeutic agents. Levels of apoptosis were highly varied between cell lines and virus isolates, ranging from only 5% of H7N7-infected PANC-1 cells to over 60% of H7N3-infected BxPC-3 cells by 16 hpi (Fig. 7). BxPC-3 cells were the most sensitive among the PDA cell lines to IAV-induced apoptosis, followed by AsPC-1, CFPAC-1, MIA PaCa-2, and PANC-1 cells; PANC-1 was the most resistant cell line. Interestingly, PR/8 H1N1 induced far less apoptosis than LP avian IAVs, which often were more powerful than Gem+Cisp treatment. As with results from MTT assays, the H7N3 isolate was by and large the most potent inducer of apoptosis in all of the PDA cell lines examined (Fig. 7).
FIG 7.

Comparative induction of apoptosis in PDA cells following infection with influenza A virus. Cells infected at an MOI of 1 with IAV or cultured with gemcitabine (2 mM) plus cisplatin (0.8 μM) were assessed for induction of apoptosis at 16 hpi (black bars) and 24 hpi (gray bars) by Alexa Fluor 647-labeled annexin V binding and flow cytometry. Results are normalized to those for uninfected controls and represent means plus standard deviations from two (Gem+Cisp treatment) or three (virus infection) independent experiments. Statistically significant differences between virus-induced apoptosis and Gem+Cisp-induced apoptosis at 16 hpi are indicated (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). Note that severe cell death induced in H7N3-infected CFPAC-1 cells (×) prevented proper annexin V cell labeling at the time points examined.
Influenza virus-induced caspase activation.
Given that influenza may cause apoptosis via both intrinsic and extrinsic pathways (43–45) and that the disruption of both pathways has been documented in different cancers (6), we tested the mechanism of IAV-induced cell death in PDA. As BxPC-3 represented the most sensitive PDA cell line, while HPDE6 cells were largely insensitive to virus-induced apoptosis, we investigated whether different apoptotic effector mechanisms were at play and were differently engaged by different virus isolates. Results of immunocytochemistry analyses showed a marked induction of caspase-3 in experimentally infected BxPC-3 cells, with a significantly higher level (P ≤ 0.0001) induced by H7N3 infection (50.72% positive) than by PR/8 H1N1 infection (10.14% positive) or the gemcitabine and cisplatin combination (8.92%). As with the annexin V results, infection of HPDE6 cells with influenza viruses resulted in caspase-3 induction (13.52% with H7N3 and 6.12% with PR/8 H1N1) that was much lower than that induced by gemcitabine and cisplatin (17.32%) (P ≤ 0.0001). To differentiate between intrinsic and extrinsic pathways, cells were stained with anti-caspase-8 and -9 antibodies, respectively. BxPC-3 cells infected with H7N3 showed activation of both caspases; however, positivity for caspase-9 was consistently higher than that for caspase-8 (46.1 versus 21.7% at 16 hpi and 72.02 versus 38.1% at 24 hpi), suggesting a stronger involvement of the intrinsic mitochondrial pathway. With the PR/8 H1N1 virus, however, twice as many cells were positive for caspase-8 as for caspase-9 at 16 hpi, suggesting that the extrinsic apoptotic pathway was preferentially engaged by this virus isolate (Fig. 8). Results for HPDE6 cells did not reflect the same patterns, as H7N3 induced a nearly 2-fold-higher activation of caspase-8 than of caspase-9 at 16 hpi, while PR/8 H1N1 showed no difference between activation levels.
FIG 8.

Caspase induction in influenza A virus-infected cells. Semiconfluent monolayers of BxPC-3 and HPDE6 cells grown on glass chamber slides were infected with A/turkey/Italy/2602/2003 (H7N3) or A/Puerto Rico/8/34 (H1N1) at an MOI of 1 and tested by immunocytochemistry for expression of cleaved caspase-3 (A and B), caspase-9 (C and D), and caspase-8 (E and F). Gemcitabine (2 mM) and cisplatin (0.8 μM) were included as positive controls. Results are means plus standard deviations of 10 repeat counts of 500 cells each, with significant differences shown between results for virus treatment and those for gemcitabine plus cisplatin treatment at 16 hpi (****, P < 0.0001).
Oncolytic effects of LP IAV in vivo.
The oncolytic ability of the H7N3 virus isolate was further examined in vivo in a SCID mouse tumor xenograft model. Following four successive virus inoculations into palpable BxPC-3 tumors over 7 days, the oncolytic effect of IAV on tumor reduction was compared to that of PBS on a control group. Overall, H7N3 treatment resulted in a significant reduction in tumor growth versus that of PBS alone (P < 0.001) (Fig. 9), and all tumors collected from H7N3-treated mice sacrificed upon termination of the experiment remained positive for IAV infection by rRT-PCR, with CTs ranging from 22 to 27 (data not shown).
FIG 9.

Oncolytic effect of an LP avian influenza A virus in an orthotopic SCID mouse model of pancreatic ductal adenocarcinoma. BxPC-3 cells (5 × 106) in a volume of 100 μl were subcutaneously implanted in the right flanks of 6-week-old female SCID mice. At 8 days postimplantation, palpable tumors became established, and mice were randomly divided in two groups (n = 6 each) receiving either four intratumoral injections of 2.4 × 104 PFU of LP H7N3 virus (circles) between day 8 and day 15 or four injections of PBS (squares). Caliper measurements of tumor dimensions were taken on the indicated days after BxPC-3 implantation for calculation of tumor volumes. Data shown represent mean tumor volumes plus standard errors of the means.
DISCUSSION
The present work provides the first description of influenza A virus infection of human pancreatic cancer cells, demonstrating its ability to replicate and induce apoptosis in several PDA cell lines. We have shown that LP avian IAVs may be interesting candidates for oncolytic viruses given their enhanced activity in PDA cell lines compared to that in nontumoral pancreatic ductal cells and their demonstrated ability to reduce tumor size following intratumoral injection in a mouse xenograft model.
We also provide the first characterization of the receptor profiles of a panel of human pancreatic adenocarcinoma cell lines. Of interest was the fact that these cells generally contained equal levels of α-2,6- and α α-2,3-sialic acid linkages, making them susceptible to infection by both avian and mammalian viruses. While the upper respiratory tract is the primary site of infection for human influenza viruses as a result of high levels of α-2,6-linked SAs, the expression of α-2,3-linked SAs has been detected in other human tissues, including endothelial cells of the heart, brain, intestines, and liver as well as nonciliated cells in the lung (46). Recently, we also demonstrated their presence on cells isolated from a healthy human pancreas (34). Although all PDA cell lines expressed both types of SA receptors on their surfaces, differences in expression levels were noted. Such heterogeneity in levels of SA expression in different cell lines was not surprising, as altered levels of expression of sialyltransferases and fucosyltransferases have been demonstrated in different types of tumors, including pancreatic, breast, colon, gastric, cervical, and renal cancers (47, 48). Similar results have also been observed in the case of melanoma cell lines, with heterogeneous distribution of surface receptors when different lines were compared (49).
The ability of IAV to replicate in different host systems is thought to be influenced by an optimal temperature at which viral polymerase functions and interacts with the host RNA replication machinery. For this reason, we investigated whether low viral titers achieved with certain isolates in human pancreatic cells resulted from temperature sensitivity in this host system. Growth curves conducted at physiologically relevant temperatures for humans (37°C) and birds (41°C) did in fact indicate that a virus's replicative fitness mimicked that of the host from which it was isolated, with avian viruses often reaching high titers at 41°C at 24 hpi, whereas PR/8 H1N1 replication was hindered at this temperature. However, this trend was not observed for all virus isolates, and further differences were never found to be significant in BxPC-3 cells, indicating that the avian isolates did not suffer from limited polymerase activity in these human PDA cells at temperatures reflective of the in vivo context.
Susceptibility of PDA cell lines to HP avian IAVs was greater than to LP avian IAVs, as demonstrated by replication kinetics experiments. To confirm that this was not a result of the low TPCK-trypsin concentrations used in the experiments, we performed parallel infections of MDCK cells using concentrations of 1 μg/ml and 0.05 μg/ml TPCK-trypsin, and similar titers of virus were obtained under both conditions (results not shown). While these results suggest that low trypsin concentrations were not the limiting factor, multicycle replication of human IAVs, which typically require the addition of trypsin, has been observed in a number of cases (50, 51). It is therefore our opinion that proteolytic activation of the LP AIVs is likely suboptimal under our experimental conditions, and this was confirmed experimentally when we examined viral genome replication, as no increases were found between 16 and 24 hpi, indicating the absence of multicycle replication. While the pancreas is the site of trypsinogen production, this proenzyme is typically activated to trypsin in other organs of the digestive tract. On the other hand, in a diseased state, such as in acute pancreatitis, trypsin activation may occur within the pancreas, suggesting the possibility of supporting multiple rounds of influenza A virus replication in vivo (52).
In a study of the oncolytic properties of a modified human influenza A virus isolate in colorectal cancer cells, viruses were shown to undergo multicycle replication in vitro without the addition of exogenous trypsin, suggesting the production of trypsin or a trypsin-like enzyme by the cells examined (53). The enhanced production of proteolytic enzymes is a hallmark of cancer cells, as they aid in invasion and metastasis (54); altered expression of matrix metalloproteinase and their inhibitors have been particularly associated with progression of pancreatic cancer (55–59), and exploitation of these proteases has been suggested as a mode of targeted cancer treatment (54). In fact, matrix metalloproteinase (MMP) targeting has been used as a strategy for tumor-specific targeting of oncolytic viruses in the case of measles virus (60, 61), retroviruses (62, 63), and Sendai virus (64), and modifications to the HA proteolytic cleavage site to render it MMP specific are being planned not only to increase the specificity of PDA-associated proteases but to allow for multicycle replication in the absence of exogenous trypsin.
Influenza viruses are known to induce apoptosis in a number of cell lines and tissues, while in others they induce death via necrosis (35, 65, 66). Based on MTT assay results showing that LP avian IAVs cause reduction in cell viability, we tested whether these viruses induced apoptotic cell death in PDA cells. Results of annexin V cell staining indicated that LP avian IAVs caused death by apoptosis in the panel of PDA cell lines tested and generally correlated well with levels of cytotoxicity observed in the MTT assay. However, in a few instances, there were notable discrepancies in which annexin V staining showed greater induction of apoptosis than was shown in the MTT assay, and this was especially true for BxPC-3 infections. Differences in the sensitivity levels of these two assays have been observed elsewhere (67), confirming the notion that while metabolic assays may provide a good overall picture of cytotoxicity, the use of additional assays is favored in order to accurately deduce levels of cell death.
Annexin V staining demonstrated that levels of IAV-induced apoptosis in PDA cells varied highly depending on the virus isolate and cell line tested, as with the variations observed in RNA replication rates and sensitivities to infection. Alterations in apoptotic signaling pathways are among the most frequent genetic changes observed in pancreatic cancers (5, 68), contributing to resistance to chemotherapeutic agents (6, 69). BxPC-3 cells were the most sensitive and, unlike the others, contain a wild-type K-RAS (70). K-RAS enzymes are modulators of numerous cellular signal transduction pathways, and K-RAS mutations are known to increase resistance to apoptosis (71). The fact that BxPC-3 cells contain a wild-type K-RAS may thus partially explain the higher levels of apoptosis observed in this cell line; however, the facts that nontransformed HPDE6 cells also contain wild-type K-RAS and display a resistant phenotype suggest that other contributing factors are at play (72).
The interferon status of tumor cells has also been proven highly influential on the oncolytic activities of several viruses, IAV included (53, 73, 74). A recent publication detailing the susceptibility of PDA cell lines to vesicular stomatitis virus found that the lack of type I interferon (IFN) production by AsPC-1, CFPAC-1, MIA PaCa-2, and Panc-1 cells largely correlated with sensitivity to infection; however, both IFN-negative BxPC-3 cells and IFN-producing HPDE6 cells showed resistance (75). In the present study, however, the degree of PDA sensitivity to IAV-induced apoptosis does not correlate with the cells' interferon status, as cells incapable of IFN production were both highly sensitive (BxPC-3) and relatively resistant (Panc-1), results similar to those of a recent report on PDA cell line sensitivity to Newcastle disease virus (76).
PDAs are associated with a constellation of genetic alterations in oncogenes and tumor suppressor genes, the four most frequent being observed in the K-RAS, TP53, SSMAD4/DPC4, and CDKN2A/p16 genes (3, 9, 70). The status of these four genes has been described previously for all six pancreatic cell lines included in this study (3, 70); however, no immediate associations can be made by comparing the individual cell genotypes with their phenotypes with regard to sensitivity to influenza virus-induced apoptosis. It is likely that complex interactions between a multitude of genes controlling cell cycle, signal transduction pathways, apoptosis resistance, and interferon status all determine the ability of influenza virus to induce apoptosis in PDA cells, and an understanding of the specific mechanisms at hand will be crucial for future development of modified oncolytic influenza viruses with potential in a clinical setting.
The lack of apoptotic induction following gemcitabine and cisplatin treatment observed in the present study is not entirely in agreement with the results of other publications using the same PDA cell lines, but this is likely to result from differences in experimental conditions. Several reports documenting gemcitabine sensitivity of PDA cell lines show higher induction of apoptosis following chemotherapeutic treatment than prior to treatment, but these studies involve cells treated for 48 to 72 h and seeded at densities ranging from 5 to 15 times lower than in the present study (77–79). As our study was concerned with the ability of influenza virus to induce apoptosis following infection at a high MOI, we chose to study the response over a period of 24 h, starting with those of confluent monolayers, to monitor CPE following infection. It is likely that the higher cell density with increased cell-cell contact combined with the 24-h observation window resulted in lower observed rates of response to gemcitabine and cisplatin treatment in the present study despite the increased drug concentrations.
Even more interesting than the differences observed between cell lines were the major differences observed between viral isolates in their ability to induce apoptosis in PDA cells. While several of the isolates tested showed a significantly enhanced ability to induce apoptosis compared to that after treatment with gemcitabine and cisplatin, the H7N3 isolate repeatedly outperformed the others in terms of the rapidity and potency of cell death induced. Strain-specific variations in apoptotic induction have previously been documented in primary cultures and established cell lines in the cases of human, avian, and swine influenza viruses (36, 37). The particularity of the H7N3 isolate led us to investigate it further; we examined the specific apoptotic pathway induced in a highly sensitive cell line (BxPC-3) compared to that of a resistant line (HPDE6). Based on the expression of effector and executioner caspases in infected cells, H7N3-induced apoptosis appeared to result from activation of the intrinsic mitochondrial pathway in BxPC-3 cells, but this phenomenon was not observed in the HPDE6 cells. Induction of cell death via the intrinsic pathway is a characteristic of a number of oncolytic viruses, including Newcastle disease virus and vesicular stomatitis virus (80–82). Though caspase-independent mechanisms of apoptosis cannot be ruled out, our results strongly suggest that the ability to induce the intrinsic pathway is a critical factor in the success of the H7N3 isolate at pancreatic cancer cell killing. A constellation of viral proteins, including NS1, PB1-F2, and NA (45, 83), have been demonstrated as regulators of the apoptotic response in infected cells, and the distinct genetic signatures of the H7N3 isolate are currently under study.
Building on results observed in vitro, an experiment using a SCID mouse xenograft model was performed to examine the oncosuppresive activity of the H7N3 virus in an in vivo setting. As reflected by in vitro sensitivity, BxPC-3-based tumors showed significantly reduced growth following H7N3 treatment compared to growth after treatment with PBS alone. The presence of viral RNA in tumors of all H7N3-treated animals sacrificed 1 week following the final injection confirmed that all tumors were successfully infected with IAV, although virus isolations were not performed to determine whether this represented live virus. Given the inability of the LP viruses to undergo multiple rounds of replication in PDA cells in vitro, most likely due to protease-limiting conditions, the H7N3 virus was not expected to undergo multiple rounds of infection in the BxPC-3 tumor cells. However, detection of viral RNA in treated tumors up to 1 week following the final injection suggests the possibility that virus replication did occur within the tumor microenvironment. In any case, the overall positive results and lack of detrimental side effects observed in the in vivo trial are promising signs for future studies with IAVs harboring appropriate PDA-specific modifications.
The fact that LP influenza viruses are able to induce levels of apoptosis in PDA cells that are significantly higher and more targeted to the cancerous cells than those of commonly employed chemotherapeutic agents indicates that these viruses have a higher tropism for the cancerous phenotype that may be further exploited. These observations were further confirmed in an in vivo xenograft model where intratumoral inoculation with a LP H7N3 isolate decreased tumor growth compared to that in the control. Taken together, our results indicate that PDA cells are sensitive to the oncolytic effects of influenza viruses and that further studies are warranted to understand this phenomenon at the molecular level, leading to the generation of specific and targeted viruses with enhanced potential in vivo and, ultimately, to their use in a clinical setting.
ACKNOWLEDGMENTS
Special thanks go to Paola De Benedictis and Elisabetta Viale for their input and technical assistance, Antonella Casu for technical assistance with flow cytometry, and Alessio Palini for his guidance in interpretation of fluorescence-activated cell sorter (FACS) results. A special thank you goes as well to Donna D'Agostino for kindly providing the MTT reagent.
Preliminary studies were funded through the EU FP6 project Training and Technology Transfer of Avian Influenza Diagnostics and Disease Management Skills (FLUTRAIN) (project no. 044212).
Footnotes
Published ahead of print 4 June 2014
REFERENCES
- 1.World Health Organization. 2008. World cancer report. IARC, Lyon, France [Google Scholar]
- 2.Kasuya H, Takeda S, Nomoto S, Nakao A. 2005. The potential of oncolytic virus therapy for pancreatic cancer. Cancer Gene Ther. 12:725–736. 10.1038/sj.cgt.7700830 [DOI] [PubMed] [Google Scholar]
- 3.Qian J, Niu J, Li M, Chiao PJ, Tsao MS. 2005. In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer Res. 65:5045–5053. 10.1158/0008-5472.CAN-04-3208 [DOI] [PubMed] [Google Scholar]
- 4.Feldmann G, Rauenzahn S, Maitra A. 2009. In vitro models of pancreatic cancer for translational oncology research. Expert Opin. Drug Discov. 4:429–443. 10.1517/17460440902821657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Westphal S, Kalhtoff H. 2003. Apoptosis: targets in pancreatic cancer. Mol. Cancer 2:6. 10.1186/1476-4598-2-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fulda S, Debatin KM. 2006. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25:4798–4811. 10.1038/sj.onc.1209608 [DOI] [PubMed] [Google Scholar]
- 7.Wennier S, Li S, McFadden G. 2011. Oncolytic virotherapy for pancreatic cancer. Expert Rev. Mol. Med. 13:e18. 10.1017/S1462399411001876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Capua I, Alexander DJ. 2009. Avian influenza and Newcastle disease, a field and laboratory manual. Springer, Milan, Italy [Google Scholar]
- 9.Cox NJ, Neumann G, Donis RO, Kawaoka Y. 15 March 2010. Orthomyxoviruses: influenza. In Mahy BWJ, ter Meulen V. (ed), Topley and Wilson's microbiology and microbial infections. John Wiley & Sons, New York, NY. 10.1002/9780470688618.taw0238 [DOI] [Google Scholar]
- 10.Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, Yang H, Chen X, Recuenco S, Gomez J, Chen LM, Johnson A, Tao Y, Dreyfus C, Yu W, McBride R, Carney PJ, Gilbert AT, Chang J, Guo Z, Davis CT, Paulson JC, Stevens J, Rupprecht CE, Holmes EC, Wilson IA, Donis RO. 2013. New world bats harbor diverse influenza A viruses. PLoS Pathog. 9(10):e1003657. 10.1371/journal.ppat.1003657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. 1992. Evolution and ecology of influenza A viruses. Microbiol. Rev. 56:152–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taubenberger JK. 1998. Influenza virus hemagglutinin cleavage into HA1, HA2: no laughing matter. Proc. Natl. Acad. Sci. U. S. A. 95:9713–9715. 10.1073/pnas.95.17.9713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Subbarao EK, London W, Murphy BR. 1993. A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J. Virol. 67:1761–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noah DL, Krug RM. 2005. Influenza virus virulence and its molecular determinants. Adv. Virus Res. 65:121–145. 10.1016/S0065-3527(05)65004-X [DOI] [PubMed] [Google Scholar]
- 15.Massin P, van der Werf S, Naffakh N. 2001. Residue 627 of PB2 is a determinant of cold sensitivity in RNA replication of avian influenza viruses. J. Virol. 75:5398–5404. 10.1128/JVI.75.11.5398-5404.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massin P, Kuntz-Simon G, Barbezange C, Deblanc C, Oger A, Marquet-Blouin E, Bougeard S, van der Werf S, Jestin V. 2010. Temperature sensitivity on growth and/or replication of H1N1, H1N2 and H3N2 influenza A viruses isolated from pigs and birds in mammalian cells. Vet. Microbiol. 142:232–241. 10.1016/j.vetmic.2009.10.012 [DOI] [PubMed] [Google Scholar]
- 17.Neumann G, Kawaoka Y. 2006. Host range restriction and pathogenicity in the context of influenza pandemic. Emerg. Infect. Dis. 12:881–886. 10.3201/eid1206.051336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Poucke SG, Nicholls JM, Nauwynck HJ, Van Reeth K. 2010. Replication of avian, human and swine influenza viruses in porcine respiratory explants and association with sialic acid distribution. Virol. J. 7:38. 10.1186/1743-422X-7-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neumann G, Kawaoka Y. 2011. The first influenza pandemic of the new millennium. Influenza Other Respir. Viruses 5:157–166. 10.1111/j.1750-2659.2011.00231.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanimura N, Tsukamoto K, Okamatsu M, Mase M, Imada T, Nakamura K, Kubo M, Yamaguchi S, Irishio W, Hayashi M, Nakai T, Yamauchi A, Nishimura M, Imai K. 2006. Pathology of fatal highly pathogenic H5N1 avian influenza virus infection in large-billed crows (Corvus macrorhynchos) during the 2004 outbreak in Japan. Vet. Pathol. 43:500–509. 10.1354/vp.43-4-500 [DOI] [PubMed] [Google Scholar]
- 21.Teifke JP, Klopfleisch R, Globig A, Starick E, Hoffmann B, Wolf PU, Beer M, Mettenleiter TC, Harder TC. 2007. Pathology of natural infections by H5N1 highly pathogenic avian influenza virus in mute (Cygnus olor) and whooper (Cygnus cygnus) swans. Vet. Pathol. 44:137–143. 10.1354/vp.44-2-137 [DOI] [PubMed] [Google Scholar]
- 22.Abolnik C, Londt BZ, Manvell RJ, Shell W, Banks J, Gerdes GH, Akol G, Brown IH. 2009. Characterisation of a highly pathogenic influenza A virus of subtype H5N2 isolated from ostriches in South Africa in 2004. Influenza Other Respir. Viruses 3:63–68. 10.1111/j.1750-2659.2009.00074.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertran K, Pérez-Ramírez E, Busquets N, Dolz R, Ramis A, Darji A, Abad FX, Valle R, Chaves A, Vergara-Alert J, Barral M, Höfle U, Majó N. 2011. Pathogenesis and transmissibility of highly (H7N1) and low (H7N9) pathogenic avian influenza virus infection in red-legged partridge (Alectoris rufa). Vet. Res. 42:24. 10.1186/1297-9716-42-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zanella A. 2003. Avian influenza attributable to serovar H7N1 in light layers in Italy. Avian Dis. 47:1177–1180. 10.1637/0005-2086-47.s3.1177 [DOI] [PubMed] [Google Scholar]
- 25.Shinya K, Awakura T, Shimada A, Silvano FD, Umemura T, Otsuki K. 1995. Pathogenesis of pancreatic atrophy by avian influenza a virus infection. Avian Pathol. 24:623–632. 10.1080/03079459508419102 [DOI] [PubMed] [Google Scholar]
- 26.Mutinelli F, Capua I, Terregino C, Cattoli G. 2003. Clinical, gross, and microscopic findings in different avian species naturally infected during the H7N1 low- and high-pathogenicity avian influenza epidemics in Italy during 1999 and 2000. Avian Dis. 47:844–848. 10.1637/0005-2086-47.s3.844 [DOI] [PubMed] [Google Scholar]
- 27.Chaves AJ, Busquets N, Campos N, Ramis A, Dolz R, Rivas R, Valle R, Abad FX, Darji A, Majo N. 2011. Pathogenesis of highly pathogenic avian influenza A virus (H7N1) infection in chickens inoculated with three different doses. Avian Pathol. 40:163–172. 10.1080/03079457.2011.551874 [DOI] [PubMed] [Google Scholar]
- 28.Kwon YK, Joh SJ, Kim MC, Kang MS, Lee YJ, Kwon JH, Kim JH. 2010. The susceptibility of magpies to a highly pathogenic avian influenza virus subtype H5N1. Poult. Sci. 89:1156–1161. 10.3382/ps.2009-00549 [DOI] [PubMed] [Google Scholar]
- 29.Kwon YK, Thomas C, Swayne DE. 2010. Variability in pathobiology of South Korean H5N1 high-pathogenicity avian influenza virus infection for 5 species of migratory waterfowl. Vet. Pathol. 47:495–506. 10.1177/0300985809359602 [DOI] [PubMed] [Google Scholar]
- 30.Lipatov AS, Kwon YK, Pantin-Jackwood MJ, Swayne DE. 2009. Pathogenesis of H5N1 influenza virus infections in mice and ferret models differs according to respiratory tract or digestive system exposure. J. Infect. Dis. 199:717–725. 10.1086/596740 [DOI] [PubMed] [Google Scholar]
- 31.Reperant LA, van de Bildt MW, van Amerongen G, Leijten LM, Watson S, Palser A, Kellam P, Eissens AC, Frijlink HW, Osterhaus AD, Kuiken T. 2012. Marked endotheliotropism of highly pathogenic avian influenza virus H5N1 following intestinal inoculation in cats. J. Virol. 86:1158–1165. 10.1128/JVI.06375-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yingst SL, Saad MD, Felt SA. 2006. Qinghai-like H5N1 from domestic cats, northern Iraq. Emerg. Infect. Dis. 12:1295–1297. 10.3201/eid1708.060264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calore EE, Uip DE, Perez NM. 2011. Pathology of the swine-origin influenza A (H1N1) flu. Pathol. Res. Pract. 207:86–90. 10.1016/j.prp.2010.11.003 [DOI] [PubMed] [Google Scholar]
- 34.Capua I, Mercalli A, Pizzuto MS, Romero-Tejeda A, Kasloff S, De Battisti C, Bonfante F, Patrono LV, Vicenzi E, Zappulli V, Lampasona V, Stefani A, Doglioni C, Terregino C, Cattoli G, Piemonti L. 2013. Influenza A viruses grow in human pancreatic cells and cause pancreatitis and diabetes in an animal model. J. Virol. 87:597–610. 10.1128/JVI.00714-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takizawa T, Matsukawa S, Higuchi Y, Nakamura S, Nakanishi Y, Fukuda R. 1993. Induction of programmed cell death (apoptosis) by influenza virus infection in tissue culture cells. J. Gen. Virol. 74:2347–2355. 10.1099/0022-1317-74-11-2347 [DOI] [PubMed] [Google Scholar]
- 36.Mok CK, Lee DC, Cheung CY, Peiris M, Lau AS. 2007. Differential onset of apoptosis in influenza A virus H5N1- and H1N1-infected human blood macrophages. J. Gen. Virol. 88:1275–1280. 10.1099/vir.0.82423-0 [DOI] [PubMed] [Google Scholar]
- 37.Choi YK, Kim TK, Kim CJ, Lee JS, Oh SY, Joo HS, Foster DN, Hong KC, You S, Kim H. 2006. Activation of the intrinsic mitochondrial apoptotic pathway in swine influenza virus-mediated cell death. Exp. Mol. Med. 38:11–17. 10.1038/emm.2006.2 [DOI] [PubMed] [Google Scholar]
- 38.Temperton NJ, Hoschler K, Major D, Nicolson C, Manvell R, Hien VM, Ha do Q, de Jong M, Zambon M, Takeuchi Y, Weiss RA. 2007. A sensitive retroviral pseudotype assay for influenza H5N1-neutralizing antibodies. Influenza Other Respir. Viruses 1:105–112. 10.1111/j.1750-2659.2007.00016.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hygiene 27:493–497 [Google Scholar]
- 40.Spackman E, Senne DA, Myers TJ, Bulaga LL, Garber LP, Perdue ML, Lohman K, Daum LT, Suarez DL. 2002. Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J. Clin. Microbiol. 40:3256–3260. 10.1128/JCM.40.9.3256-3260.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi JH, Oh SY, Kwon HC, Kim JH, Lee JH, Lee S, Lee DM, Kim SH, Rho MH, Kim YH, Rho MS, Kim HJ. 2008. Gemcitabine versus gemcitabine combined with cisplatin treatment locally advanced or metastatic pancreatic cancer: a retrospective analysis. Cancer Res. Treat. 40:22–26. 10.4143/crt.2008.40.1.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riss TL, Moravec RA, Niles AL, Benink HA, Worzella TJ, Minor L. 1 May 2013. Cell viability assays. In Sittampalam GS, Gal-Edd N, Arkin M, Auld D, Austin C, Bejcek B, Glicksman M, Inglese J, Lemmon V, Li Z, McGee J, McManus O, Minor L, Napper A, Riss T, Trask OJ, Jr, Weidner J. (ed), Assay guidance manual. Eli Lilly & Company, Bethesda, MD: http://www.ncbi.nlm.nih.gov/books/NBK144065/ [Google Scholar]
- 43.Zamarin D, García-Sastre A, Xiao X, Wang R, Palese P. 2005. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 1(1):e4. 10.1371/journal.ppat.0010004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lowy RJ. 2003. Influenza virus induction of apoptosis by intrinsic and extrinsic mechanisms. Int. Rev. Immunol. 22:425–449. 10.1080/08830180305216 [DOI] [PubMed] [Google Scholar]
- 45.Xing Z, Harper R, Anunciacion J, Yang Z, Gao W, Qu B, Guan Y, Cardona CJ. 2011. Host immune and apoptotic responses to avian influenza virus H9N2 in human tracheobronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 44:24–33. 10.1165/rcmb.2009-0120OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yao L, Korteweg C, Hsueh W, Gu J. 2008. Avian influenza receptor expression in H5N1-infected and noninfected human tissues. FASEB J. 22:733–740. 10.1096/fj.06-7880com [DOI] [PubMed] [Google Scholar]
- 47.Mas E, Pasqualini E, Caillol N, El Battari A, Crotte C, Lombardo D, Sadoulet MO. 1998. Fucosyltransferase activities in human pancreatic tissue: comparative study between cancer tissues and established tumoral cell lines. Glycobiology 8:605–613. 10.1093/glycob/8.6.605 [DOI] [PubMed] [Google Scholar]
- 48.Pérez-Garay M, Arteta B, Pagès L, de Llorens R, de Bolòs C, Vidal-Vanaclocha F, Peracaula R. 2010. α2,3-Sialyltransferase ST3Gal III modulates pancreatic cancer cell motility and adhesion in vitro and enhances its metastatic potential in vivo. PLoS One 5(9):e12524. 10.1371/journal.pone.0012524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pietra G, Manzini C, Vitale M, Balsamo M, Ognio E, Boitano M, Queirolo P, Moretta L, Mingari MC. 2009. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int. Immunol. 21:793–801. 10.1093/intimm/dxp047 [DOI] [PubMed] [Google Scholar]
- 50.Noma K, Kiyotani K, Kouchi H, Fujii Y, Egi Y, Tanaka K, Yoshida T. 1998. Endogenous protease-dependent replication of human influenza viruses in two MDCK cell lines. Arch. Virol. 143:1893–1909. 10.1007/s007050050428 [DOI] [PubMed] [Google Scholar]
- 51.Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solórzano A, Swayne DE, Cox NJ, Katz JM, Taubenberger JK, Palese P, García-Sastre A. 2005. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 310:77–80. 10.1126/science.1119392 [DOI] [PubMed] [Google Scholar]
- 52.Sherwood MW, Prior IA, Voronina SG, Barrow SL, Woodsmith JD, Gerasimenko OV, Petersen OH, Tepikin AV. 2007. Activation of trypsinogen in large endocytic vacuoles of pancreatic acinar cells. Proc. Natl. Acad. Sci. U. S. A. 104:5674–5679. 10.1073/pnas.0700951104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sturlan S, Stremitzer S, Bauman S, Sachet M, Wolschek M, Ruthsatz T, Egorov A, Bergmann M. 2010. Endogenous expression of proteases in colon cancer cells facilitate influenza A viruses mediated oncolysis. Cancer Biol. Ther. 10:592–599. 10.4161/cbt.10.6.12565 [DOI] [PubMed] [Google Scholar]
- 54.Vartak DG, Gemeinhart RA. 2007. Matrix metalloproteases: underutilized targets for drug delivery. J. Drug Target. 15:1–20. 10.1080/10611860600968967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones LE, Humphreys MJ, Campbell F, Neoptolemos JP, Boyd MT. 2004. Comprehensive analysis of matrix metalloproteinase and tissue inhibitor expression in pancreatic cancer: increased expression of matrix metalloproteinase-7 predicts poor survival. Clin. Cancer Res. 10:2832–2845. 10.1158/1078-0432.CCR-1157-03 [DOI] [PubMed] [Google Scholar]
- 56.Sato N, Maehara N, Su GH, Goggins M. 2003. Effects of 5-aza-2′-deoxycytidine on matrix metalloproteinase expression and pancreatic cancer cell invasiveness. J. Natl. Cancer Inst. 95:327–330. 10.1093/jnci/95.4.327 [DOI] [PubMed] [Google Scholar]
- 57.Yamamoto H, Itoh F, Iku S, Adachi Y, Fukushima H, Sasaki S, Mukaiya M, Hirata K, Imai K. 2001. Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in human pancreatic adenocarcinomas: clinicopathologic and prognostic significance of matrilysin expression. J. Clin. Oncol. 19:1118–1127 [DOI] [PubMed] [Google Scholar]
- 58.Bramhall SR, Neoptolemos JP, Stamp GW, Lemoine NR. 1997. Imbalance of expression of matrix metalloproteinases (MMPs) and tissue inhibitors of the matrix metalloproteinases (TIMPs) in human pancreatic carcinoma. J. Pathol. 182:347–355. 10.1002/(SICI)1096-9896(199707)182:3<347::AID-PATH848>3.0.CO;2-J [DOI] [PubMed] [Google Scholar]
- 59.Keleg S, Büchler P, Ludwig R, Büchler MW, Friess H. 2003. Invasion and metastasis in pancreatic cancer. Mol. Cancer 2:14. 10.1186/1476-4598-2-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Springfeld C, von Messling V, Frenzke M, Ungerechts G, Buchholz CJ, Cattaneo R. 2006. Oncolytic efficacy and enhanced safety of measles virus activated by tumor-secreted matrix metalloproteinases. Cancer Res. 66:7694–7700. 10.1158/0008-5472.CAN-06-0538 [DOI] [PubMed] [Google Scholar]
- 61.Mühlebach MD, Schaser T, Zimmermann M, Armeanu S, Hanschmann KM, Cattaneo R, Bitzer M, Lauer UM, Cichutek K, Buchholz CJ. 2010. Liver cancer protease activity profiles support therapeutic options with matrix metalloproteinase-activatable oncolytic measles virus. Cancer Res. 70:7620–7629. 10.1158/0008-5472.CAN-09-4650 [DOI] [PubMed] [Google Scholar]
- 62.Schneider RM, Medvedovska Y, Hartl I, Voelker B, Chadwick MP, Russell SJ, Cichutek K, Buchholz CJ. 2003. Directed evolution of retroviruses activatable by tumour-associated matrix metalloproteases. Gene Ther. 10:1370–1380. 10.1038/sj.gt.3302007 [DOI] [PubMed] [Google Scholar]
- 63.Szécsi J, Drury R, Josserand V, Grange MP, Boson B, Hartl I, Schneider R, Buchholz CJ, Coll JL, Russell SJ, Cosset FL, Verhoeyen E. 2006. Targeted retroviral vectors displaying a cleavage site-engineered hemagglutinin (HA) through HA-protease interactions. Mol. Ther. 14:735–744. 10.1016/j.ymthe.2006.04.007 [DOI] [PubMed] [Google Scholar]
- 64.Kinoh H, Inoue M, Washizawa K, Yamamoto T, Fujikawa S, Tokusumi Y, Iida A, Nagai Y, Hasegawa M. 2004. Generation of a recombinant Sendai virus that is selectively activated and lyses human tumor cells expressing matrix metalloproteinases. Gene Ther. 11:1137–1145. 10.1038/sj.gt.3302272 [DOI] [PubMed] [Google Scholar]
- 65.van Rikxoort M, Michaelis M, Wolschek M, Muster T, Egorov A, Seipelt J, Doerr HW, Cinatl J., Jr 2012. Oncolytic effects of a novel influenza A virus expressing interleukin-15 from the NS reading frame. PLoS One 7:e36506. 10.1371/journal.pone.0036506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhirnov O, Klenk HD. 2003. Human influenza A viruses are proteolytically activated and do not induce apoptosis in CACO-2 cells. Virology 313:198–212. 10.1016/S0042-6822(03)00264-2 [DOI] [PubMed] [Google Scholar]
- 67.Wu S, Liu B, Zhang Q, Liu J, Zhou W, Wang C, Li M, Bao S, Zhu R. 2013. Dihydromyricetin reduced Bcl-2 expression via p53 in human hepatoma HepG2 cells. PLoS One 8(11):e76886. 10.1371/journal.pone.0076886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. 2008. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321:1801–1806. 10.1126/science.1164368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karikari CA, Roy I, Tryggestad E, Feldmann G, Pinilla C, Welsh K, Reed JC, Armour EP, Wong J, Herman J, Rakheja D, Maitra A. 2007. Targeting the apoptotic machinery in pancreatic cancers using small-molecule antagonists of the X-linked inhibitor of apoptosis protein. Mol. Cancer Ther. 6:957–966. 10.1158/1535-7163.MCT-06-0634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deer EL, González-Hernández J, Coursen JD, Shea JE, Ngatia J, Scaife CL, Firpo MA, Mulvihill SJ. 2010. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 39:425–435. 10.1097/MPA.0b013e3181c15963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moon DO, Kim BY, Jang JH, Kim MO, Jayasooriya RG, Kang CH, Choi YH, Moon SK, Kim WJ, Ahn JS, Kim GY. 2012. K-RAS transformation in prostate epithelial cell overcomes H2O2-induced apoptosis via upregulation of gamma-glutamyltransferase-2. Toxicol. In Vitro 26:429–434. 10.1016/j.tiv.2012.01.013 [DOI] [PubMed] [Google Scholar]
- 72.Ouyang H, Mou Lj, Luk C, Liu N, Karaskova J, Squire J, Tsao MS. 2000. Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am. J. Pathol. 157:1623–1631. 10.1016/S0002-9440(10)64800-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Elankumaran S, Chavan V, Qiao D, Shobana R, Moorkanat G, Biswas M, Samal SK. 2010. Type I interferon-sensitive recombinant Newcastle disease virus for oncolytic virotherapy. J. Virol. 84:3835–3844. 10.1128/JVI.01553-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Efferson CL, Tsuda N, Kawano K, Nistal-Villán E, Sellappan S, Yu D, Murray JL, García-Sastre A, Ioannides CG. 2006. Prostate tumor cells infected with a recombinant influenza virus expressing a truncated NS1 protein activate cytolytic CD8+ cells to recognize noninfected tumor cells. J. Virol. 80:383–394. 10.1128/JVI.80.1.383-394.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Murphy AM, Besmer DM, Moerdyk-Schauwecker M, Moestl N, Ornelles DA, Mukherjee P, Grdzelishvili VZ. 2012. Vesicular stomatitis virus as an oncolytic agent against pancreatic ductal adenocarcinoma. J. Virol. 86:3073–3087. 10.1128/JVI.05640-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Buijs PR, van Eijck CH, Hofland LJ, Fouchier RA, van den Hoogen BG. 2014. Different responses of human pancreatic adenocarcinoma cell lines to oncolytic Newcastle disease virus infection. Cancer Gene Ther. 21:24–30. 10.1038/cgt.2013.78 [DOI] [PubMed] [Google Scholar]
- 77.Cui Y, Brosnan JA, Blackford AL, Sur S, Hruban RH, Kinzler KW, Vogelstein B, Maitra A, Diaz LA, Jr, Iacobuzio-Donahue CA, Eshleman JR. 2012. Genetically defined subsets of human pancreatic cancer show unique in vitro chemosensitivity. Clin. Cancer Res. 18:6519–6530. 10.1158/1078-0432.CCR-12-0827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Réjiba S, Bigand C, Parmentier C, Hajri A. 2009. Gemcitabine-based chemogene therapy for pancreatic cancer using Ad-dCK::UMK GDEPT and TS/RR siRNA strategies. Neoplasia 11:637–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rathos MJ, Joshi K, Khanwalkar H, Manohar SM, Joshi KS. 2012. Molecular evidence for increased antitumor activity of gemcitabine in combination with a cyclin-dependent kinase inhibitor, P276-00, in pancreatic cancers. J. Transl. Med. 10:161. 10.1186/1479-5876-10-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mansour M, Palese P, Zamarin D. 2011. Oncolytic specificity of Newcastle disease virus is mediated by selectivity for apoptosis-resistant cells. J. Virol. 85:6015–6023. 10.1128/JVI.01537-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tumilasci VF, Olière S, Nguyên TL, Shamy A, Bell J, Hiscott J. 2008. Targeting the apoptotic pathway with BCL-2 inhibitors sensitizes primary chronic lymphocytic leukemia cells to vesicular stomatitis virus-induced oncolysis. J. Virol. 82:8487–8499. 10.1128/JVI.00851-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gadaleta P, Perfetti X, Mersich S, Coulombié F. 2005. Early activation of the mitochondrial apoptotic pathway in vesicular stomatitis virus-infected cells. Virus Res. 109:65–69. 10.1016/j.virusres.2004.10.007 [DOI] [PubMed] [Google Scholar]
- 83.Hale BG, Randall RE, Ortín J, Jackson D. 2008. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89:2359–2376. 10.1099/vir.0.2008/004606-0 [DOI] [PubMed] [Google Scholar]