

ABSTRACT
Rotaviruses and orbiviruses are nonturreted Reoviridae members. The rotavirus VP3 protein is a multifunctional capping enzyme and antagonist of the interferon-induced cellular oligoadenylate synthetase-RNase L pathway. Despite mediating important processes, VP3 is the sole protein component of the rotavirus virion whose structure remains unknown. In the current study, we used sequence alignment and homology modeling to identify features common to nonturreted Reoviridae capping enzymes and to predict the domain organization, structure, and active sites of rotavirus VP3. Our results suggest that orbivirus and rotavirus capping enzymes share a domain arrangement similar to that of the bluetongue virus capping enzyme. Sequence alignments revealed conserved motifs and suggested that rotavirus and orbivirus capping enzymes contain a variable N-terminal domain, a central guanine-N7-methyltransferase domain that contains an additional inserted domain, and a C-terminal guanylyltransferase and RNA 5′-triphosphatase domain. Sequence conservation and homology modeling suggested that the insertion in the guanine-N7-methyltransferase domain is a ribose-2′-O-methyltransferase domain for most rotavirus species. Our analyses permitted putative identification of rotavirus VP3 active-site residues, including those that form the ribose-2′-O-methyltransferase catalytic tetrad, interact with S-adenosyl-l-methionine, and contribute to autoguanylation. Previous reports have indicated that group A rotavirus VP3 contains a C-terminal 2H-phosphodiesterase domain that can cleave 2′-5′ oligoadenylates, thereby preventing RNase L activation. Our results suggest that a C-terminal phosphodiesterase domain is present in the capping enzymes from two additional rotavirus species. Together, these findings provide insight into a poorly understood area of rotavirus biology and are a springboard for future biochemical and structural studies of VP3.
IMPORTANCE Rotaviruses are an important cause of severe diarrheal disease. The rotavirus VP3 protein caps viral mRNAs and helps combat cellular innate antiviral defenses, but little is known about its structure or enzymatic mechanisms. In this study, we used sequence- and structure-based alignments with related proteins to predict the structure of VP3 and identify enzymatic domains and active sites therein. This work provides insight into the mechanisms of rotavirus transcription and evasion of host innate immune defenses. An improved understanding of these processes may aid our ability to develop rotavirus vaccines and therapeutics.
INTRODUCTION
RNA viruses have evolved a variety of transcriptional strategies, many of which involve the addition of a cap structure to the 5′ end of viral mRNAs through the activities of viral capping enzymes. A cap-1 structure is typically generated by the concerted activities of an RNA 5′-triphosphatase (RTPase), which removes the γ-phosphate from the 5′ end of the mRNA; a guanylyltransferase (GTase), which conjugates GMP to the mRNA through a 5′-5′ linkage; a guanine-N7-methyltransferase (N7-MTase), which methylates the guanine cap; and a ribose-2′-O-methyltransferase (2′-O-MTase), which methylates the ribose of the initiating nucleotide (1, 2). The addition of a methylated cap structure may help stabilize viral mRNAs, enhance their translation, and prevent their detection by cellular innate immune sensors. A variety of RTPase structures have been reported, including three distinct metal-dependent types and one metal-independent, cysteine phosphatase type (1). The GTases of DNA viruses contain two domains and often conserve active-site residues, including a catalytic lysine within a KxDG(I/L) motif (where x is any residue) (3, 4). However, the GTase domains encoded by two double-stranded RNA (dsRNA) viruses of the family Reoviridae, mammalian orthoreovirus and bluetongue virus, feature folds that differ both from one another and from those of DNA viruses (5–7). MTases share little sequence identity but have a common core fold that consists of seven β-strands flanked by three α-helices on each side of the sheet (1). Both N7- and 2′-O-MTases belong to the class I family of S-adenosyl-l-methionine (SAM)-dependent MTases. While N7-MTases are thought to catalyze methyl transfer by optimally positioning the reacting groups and providing a favorable electrostatic environment (8, 9), 2′-O-MTases catalyze methyl transfer through a catalytic Lys-Asp-Lys-Glu (KDKE) tetrad (10–12). Viral capping enzymes may be found as independent entities with discrete activities or as components of multifunctional, multidomain proteins (1). Due to significant variation in sequence and structure, the architecture and enzymatic mechanisms of many viral capping enzymes remain unknown or poorly understood.
Reoviridae viruses are nonenveloped, icosahedral, multilayered particles that encapsidate segmented, dsRNA genomes and enzymes involved in RNA synthesis and capping. The Sedoreovirinae subfamily comprises the nonturreted Reoviridae and contains 6 genera, including Orbivirus, Seadornavirus, and Rotavirus. Rotaviruses are important gastrointestinal pathogens that cause the deaths of more than 450,000 infants and young children annually worldwide (13). Eight species of rotavirus (rotavirus species A [RVA] to RVH) have been proposed; RVA is responsible for the majority of human disease (14). On the basis of the alignment of individual structural protein-encoding genome segments and concatenated genome sequences, the eight proposed rotavirus species resolve phylogenetically into two distinct clades (15, 16). Clade A comprises RVA, RVC, RVD, and RVF, and clade B comprises RVB, RVG, and RVH. The three protein layers of a mature rotavirus virion encapsidate 11 segments of dsRNA and several copies each of VP1, the viral RNA-dependent RNA polymerase (RdRp), and a 98-kDa protein, VP3. Rotavirus transcripts are modified at the 5′ end with a cap-1 structure (17, 18). VP3 binds covalently and reversibly to GMP, which it can transfer to pyrophosphate or GDP (19–21). VP3 also has a nonspecific affinity for single-stranded RNA, with a preference for the uncapped form (22). Together, these observations suggest that the rotavirus GTase activity is contained within VP3. VP3 also has affinity for SAM, supporting a role for VP3 in methyl transfer (23). RTPase activity has not yet been observed for recombinant VP3 and is absent in hypotonically disrupted rotavirus core particles (22). High-resolution structures of rotavirus particles and VP1 are available (24–26). However, likely due to a low copy number and asymmetric positioning within the rotavirus particle, as well as difficulties in purifying the full-length recombinant protein, the structure of VP3 remains unknown.
In addition to its capping activities, VP3 may also function as a virulence determinant. Reassortant genetic analyses have revealed that the VP3-encoding rotavirus genome segment contributes to virulence in a species-specific manner in mouse and piglet models of disease (27–29). Recently, it has been reported that the C-terminal domain of VP3 antagonizes the interferon-induced oligoadenylate synthetase (OAS)-RNase L pathway by cleaving 2′-5′-oligoadenylates (2-5A) (30). This VP3 domain is predicted to resemble 2H-phosphoesterase superfamily members that form a half-barrel structure containing two characteristic catalytic HΦ(T/S)Φ motifs (where Φ is a hydrophobic residue). VP3 may contribute to virulence by antagonizing RNase L or by methylating viral RNA at the 2′-O position, a modification that subverts cellular innate antiviral responses against some viruses by permitting escape from the effects of interferon-induced proteins with tetratricopeptide repeats (31–33).
Since rotaviruses and orbiviruses belong to the same subfamily of Reoviridae, VP3 may share structural features with orbivirus capping enzymes. A structure of the capping enzyme, VP4, from the orbivirus bluetongue virus type 10 (BTV10) has been reported (7). This protein, described as a capping assembly line, contains four domains. An enzymatically inactive N-terminal kinase-like domain precedes an N7-MTase domain containing an inserted 2′-O-MTase domain. Each of the two MTase domains forms a class I, SAM-dependent MTase fold. The VP4 C-terminal domain, which covalently binds GTP, forms a six-helix bundle that is proposed to contain both GTase and RTPase activities (7). This fold is unlike that of any known GTase or RTPase, and its active site(s) remains undefined.
In the current study, we used a combination of phylogenetic analysis, sequence alignment, and homology modeling to predict the architecture and functional domain organization of the rotavirus capping enzyme and innate immune antagonist VP3. Together, our findings suggest that the capping region of VP3 structurally resembles that of BTV10 VP4. In addition to assigning putative N7-MTase, 2′-O-MTase, and GTase/RTPase functions to specific regions of VP3, our analyses permitted identification of specific residues likely involved in ligand binding and catalysis. As a proof of concept, we present biochemical evidence supporting our identification of the VP3 GTase domain. While several of our predictions remain to be tested empirically, they represent a significant advance in an area of rotavirus biology that has long remained a mystery.
MATERIALS AND METHODS
Phylogenetic analysis.
Amino acid sequences of the predicted capping enzymes of representatives from the Rotavirus, Orbivirus, and Seadornavirus genera were aligned by multiple-sequence alignment using a fast Fourier transform program (MAFFT, v7.012b) (34) with the E-INS-i strategy. On the basis of the Bayesian information criterion (BIC) ranking, the best available substitution model was selected by use of the Molecular Evolutionary Genetics Analysis (MEGA) program (v5.05) to be WAG+ Γ+I (35). PhyML software was used to construct a maximum likelihood (ML) tree evaluated with 1,000 bootstrap replicates (36). The ML tree was visualized using the FigTree program (v1.4.0; http://tree.bio.ed.ac.uk/software/figtree/). GenBank accession numbers for the aligned sequences, followed in parentheses by the rotavirus species and strain name or the virus genus and species or strain name, are Q6WVH5 (RVA WA), Q6WNV8 (RVA OSU), ADE44246 (RVA Gottfried), AEF01495 (RVA CH-1), A4ZCW6 (RVA AU-1), ACC94314 (RVA RRV), ABC66299 (RVA 30-96), ACH97430 (RVA K9), ACH97463 (RVA Cat2), YP_002302228 (RVA SA11), ABU87836 (RVA DS1), ADJ68029 (RVA GO34), ABU49762 (RVA OVR762), ACN18218 (RVA Lamb-NT), ACN86090 (RVA Chubut), Q6WAT6 (RVA UK), A7J3A0 (RVA NCDV), ACL27789 (RVA RC-18-08), AEH96570 (RVA L338), ACY95262 (RVA ETD_822), Q6WNV6 (RVA Ty-2), BAA24148 (RVA PO-13), ACN22280 (RVA 02V0002G3), AFL91885 (RVF 03V0568), AAA99239 (RVC porcine rotavirus C), P26192 (RVC Cowden), ADP76607 (RVC YNR001), ADP76606 (RVC Wu82), ADP76608 (RVC OH567), ADP76609 (RVC BK0830), ADP76604 (RVC v508), AEJ21068 (RVC CAU_10-312), ADP76605 (RVC BS347), Q82041 (RVC Bristol), Q65526 (RVC Shintoku), ADN06426 (RVD 05V0049), Q45UF7 (RVH J19), AFL91894 (RVG 03V0567), ADF57864 (RVB MMR-B1), ADF57872 (RVB IC-008), ADF57884 (RVB IDH-084), ADF57896 (RVB Bang117), ACD39823 (RVB CAL-1), AAW29086 (seadornavirus, Liao ning virus), AAF78851 (seadornavirus JKT-7075), AGK29951 (seadornavirus Balaton/2010/HUN), AAF78856 (seadornavirus JKT-6423), AY701512 (orbivirus, Yunnan virus), DQ248060 (orbivirus, Peruvian horse sickness virus [PHV]), YP_052936 (orbivirus, Palyam virus), BAA76550 (orbivirus, Chuzan virus), ACH92680 (orbivirus, African horse sickness virus 1), AAA02768 (orbivirus, bluetongue virus 10), and AF145401 (orbivirus, Saint Croix River virus).
Amino acid sequence alignment for rotavirus and orbivirus capping enzymes.
Amino acid sequences for VP3 from representative strains of each rotavirus species and for VP4 from representative orbiviruses were aligned using the PSI-COFFEE program, which aligns distantly related proteins by homology extension, on the T-COFFEE multiple-sequence-alignment server (37, 38). GenBank accession numbers for the input sequences, followed in parentheses by the strain or virus name, are ABQ59570 (RRV), Q6WVH5 (WA), Q6WNW5 (DS1), Q6WAT6 (UK), BAA24148 (PO-13), P26192 (Cowden), AAB01673 (Shintoku), YP_392516 (Bristol), YP_003896049 (05V0049), YP_008145318 (03V0568), YP_008126855 (Bang373), ACD39823 (CAL-1), YP_008136241 (03V0567), A9Q1K9 (B219), YP_392493 (J19), AAA02768 (BTV10), YP_443928 (Yunnan virus), BAA76550 (Chuzan virus), AAF61740 (African horse sickness virus serotype 3), and YP_052945 (Saint Croix River virus). Amino acid sequences for VP3 from 639 clade A and 20 clade B rotaviruses and for VP4 from 151 orbiviruses were aligned by the use of MAFFT (v7.110) (34) and the FFT-NS-i strategy. The complete alignment, which includes GenBank accession numbers, is available upon request.
Homology modeling.
A complete three-dimensional model of BTV10 VP4 was generated from the amino acid sequence (GenBank accession number AAA02768) by the I-TASSER online server (39–41). I-TASSER, which generates three-dimensional models on the basis of multiple threading alignments and iterative template fragment assembly simulations, assigns each model a confidence (C) score in the range of −5 to 2 for estimating the quality of a predicted model. The C score for the BTV10 VP4 model was 2.0, suggesting the highest confidence. An initial RVA VP3 homology model was generated by use of the Protein Homology/analogY Recognition Engine (PHYRE; v0.2) (42) from the sequence of simian RRV VP3 (GenBank accession number ABQ59570). On the basis of the predicted homology with BTV10 VP4 (Protein Data Bank [PDB] accession number 2JH8), the region of VP3 spanning amino acids 39 to 634 was modeled with an estimated precision score of 75%, suggesting a 75% chance that BTV10 VP4 is a true structural homolog. Analysis of the RRV VP3 sequence by PHYRE, v2.0 (PHYRE2) (42), resulted in models for amino acids 257 to 343 based on vaccinia virus (VV) VP39 (PDB accession numbers 1V39, 1VPT, and 1VP3) (12, 43) and for amino acids 697 to 800 based on the central domain of human protein kinase A anchoring protein 7 (AKAP7; PDB accession numbers 2VFY and 3J4R) (44). Each model was assigned >95% confidence. The PHYRE2 confidence score represents the probability (0 to 100%) that the match between an input sequence and a specific template is a true homology. To generate a threading model of RVA VP3, we employed the threading function of I-TASSER with RRV VP3 amino acids 1 to 688 (GenBank accession number ABQ59570) as the input sequence and BTV10 VP4 (PDB accession number 2JH8 or 2JHP) as the primary template. The best RRV VP3 threading model generated by I-TASSER had a C score of −1.75. Additional homology models were generated by PHYRE2 for VP3 sequences from the following different rotavirus species and strains (the amino acids included in the model, the PDB accession number for the homolog on which it was based, and the confidence score are given in parentheses): RVB CAL-1 (664 to 746, 2VFY, 35.8%), RVC Bristol (267 to 348, 1V39, 96.9%), RVD 05V0049 (238 to 401, 1V39, 98.4%), RVF 03V0568 (256 to 415, 1V39, 97.5%), RVG 03V0567 (668 to 743, 2VFY, 46%), and RVH J19 (277 to 365, 1V39, 97%).
Structure comparison and depiction.
Molecular graphics and analytical data were obtained using the UCSF Chimera package, v1.8.1 (Chimera) (45). Chimera was developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS grant P41-GM103311). Structural models and homologous structures were aligned using the MatchMaker function of Chimera. The PDB files used in the study included those with accession numbers 2JH8, 2JHP, and 2JHA (BTV10 VP4); 1V39 (VV VP39); and 2VFY and 2VFK (AKAP7 central domain). Structure-based sequence alignments were generated using the Match>Align function of Chimera. Images of BTV10 VP4 and RRV VP3 models colored by sequence conservation were generated using the Render by Conservation tool with default settings in Chimera. A corresponding color key was generated for each rendering. For Fig. 3B, all orbivirus sequences from Fig. 2 were used. For Fig. 4, all clade A rotavirus sequences from Fig. 2 were used. For Fig. 3D and 4D, the complete alignment from Fig. 2 was used.
FIG 3.

Predicted structure and conservation for BTV10 VP4. (A) Ribbon drawing of a front view of the BTV10 VP4 model homology with the ligands SAH (yellow; PDB accession number 2JHP) and GpppG (light blue; PDB accession number 2JHA) overlaid. Domains are colored gray (N terminus), green (N7-MTase), purple (2′-O-MTase), or blue (GTase/RTPase). (B) Four views of a surface representation of the BTV10 VP4 homology model colored by the degree of amino acid conservation across all orbivirus sequences from the alignment in Fig. 2, with ligands overlaid as described for panel A. The most highly conserved positions are colored maroon, and the least conserved are colored cyan, as indicated by the scale bar. (C) Ribbon drawing of the BTV10 VP4 homology model onto which residues that are identical (red) or similar (orange) for all sequences in the alignment in Fig. 2 have been mapped. The locations of sequence motifs conserved among rotavirus and orbivirus capping enzymes and identified by sequence alignment (Fig. 2) are indicated. (D) Four views of a surface representation of the BTV10 VP4 homology model colored by the degree of amino acid conservation across all sequences from the alignment in Fig. 2.
FIG 2.


Sequence conservation among rotavirus and orbivirus capping enzymes. Amino acid alignment of rotavirus (VP3) and orbivirus (VP4) capping enzymes. The rotavirus species, strain name, and clade are indicated; orbivirus (Orbi) strain names are indicated. Aligned residues are colored according to their conservation in the alignment: red, identical for all viruses; orange, similar for all viruses; green, identical for all rotaviruses; and cyan, identical for all clade A rotaviruses. The colored bar above each row of the alignment indicates a structural domain of BTV10 VP4 (gray, N terminus; green, N7-MTase; purple, 2′-O-MTase; blue, GTase/RTPase) or a predicted structural domain of RVA RRV VP3 (cyan, PDE). Asterisks indicate motifs or residues discussed in the text. Black squares highlight residues of the BTV10 VP4 2′-O-MTase KDKE motif. Predicted HΦ(T/S)Φ motifs in the PDE domain of rotavirus VP3 are outlined.
FIG 4.

Predicted structure and conservation for RRV VP3. (A to D) Ribbon drawings of front views of the RRV VP3 PHYRE homology (A and B) and I-TASSER threading (C and D) models, based on BTV10 VP4 (PDB accession number 2JH8). Predicted domains are colored as described in the legend to Fig. 2. BTV10 VP4 ligands SAH (yellow; PDB accession number 2JHP) and GpppG (light blue; PDB accession number 2JHA) have been overlaid as sphere (A and C) or stick (B and D) representations. Residues that are identical (red) or similar (orange) in structure-based alignments with BTV10 VP4 are indicated in panels B and D. Motifs identified in Fig. 2 or discussed in the text are indicated. (E and F) Four views of a surface representation of the VP3 I-TASSER threading model colored by the degree of amino acid conservation across clade A rotavirus (E) or all (F) sequences from the alignment in Fig. 2. The most highly conserved positions are colored maroon, and the least conserved are colored cyan, as indicated by the scale bar. The first nine amino acids of the model are shown as a ribbon.
Generation of baculoviruses expressing VP3 mutants.
A codon-optimized gene encoding the amino acid sequence of rotavirus strain RRV VP3 was synthesized by GenScript. The VP3 open reading frame was cloned into the Gateway vector pENTR-1A (Invitrogen) using engineered restriction sites to make plasmid RRV VP3 pENTR-1A. Point mutations were engineered in RRV VP3 pENTR-1A by around-the-horn PCR with mutagenic primers (sequences are available upon request). Mutant VP3 sequences were verified by sequencing of purified plasmid DNA. Plasmids were recombined into the baculovirus genome, and recombinant baculovirus stocks were generated, using a BaculoDirect C-Term transfection kit (Invitrogen) according to the manufacturer's guidelines. The chloramphenicol acetyltransferase (CAT)-expressing baculovirus was generated using a plasmid provided in the BaculoDirect kit. Baculovirus DNA was isolated from P3 baculovirus stocks according to protocol 7 of the Easy-DNA isolation kit (Invitrogen), and VP3 sequences were verified by DNA sequencing.
VP3 autoguanylation assays.
To generate recombinant VP3 proteins, Sf9 cells were infected with the P3 baculovirus stock for 72 h at 27°C. Infected cells were pelleted, washed in 0.5 ml phosphate-buffered saline, and resuspended in 0.25 ml 20 mM Tris, pH 8, 20 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF). Cells were lysed in a 3-inch cup horn twice for 5 s each time at 25% amplitude with a Q500 sonicator (Qsonica) and stored at 4°C. VP3 expression in Sf9 lysates was normalized by quantitation of gels stained with colloidal Coomassie using a LI-COR Odyssey system. Covalent GTP binding by recombinant VP3 was tested in a standard reaction volume of 10 μl containing a normalized amount of recombinant VP3, 2.5 mM MnCl2, 1 μM ATP to reduce background, and 10 μCi [α-32P]GTP (3,000 Ci/mmol). The reaction mixtures were incubated at room temperature for 1 min, quenched with SDS sample buffer, boiled for 2 min, loaded onto 10% polyacrylamide gels, and electrophoresed for ∼18 h at 20 mA. Gels were dried and exposed to a phosphor storage screen for 4 h. Radiolabeled VP3 was visualized and quantified using a Typhoon Trio+ variable-mode imager and ImageQuant (v5.1) software. The results of two replicate experiments from each of two batches of baculovirus-infected Sf9 lysates were quantified for statistical analysis. One-sample t tests were performed using Prism (v6.0) software (GraphPad). P values of <0.01, in comparison to the set value of 1.0 for wild-type VP3, were considered statistically significant.
Nucleotide sequence accession number.
The codon-optimized gene encoding the amino acid sequence of rotavirus strain RRV VP3 synthesized by GenScript has been submitted to GenBank and may be found under accession number KJ869109.
RESULTS
Phylogenetic analysis.
To determine the phylogenetic relationships within the Sedoreovirinae, we constructed ML trees of amino acid sequences for predicted capping enzymes from representatives of the seadornaviruses, orbiviruses, and rotaviruses (Fig. 1). Seadornavirus VP3 and orbivirus VP4 sequences clustered tightly by genus. In contrast, rotavirus VP3 amino acid sequences resolved into two distinct branches that were analogous to the two previously identified clades (clades A and B) (15, 16). The phylogenetic distances between the clade A and B rotavirus VP3 sequences were similar to the distances between the clade B rotavirus and orbivirus capping enzyme sequences (Fig. 1). Within each clade, rotavirus species occupied distinct branches, consistent with previous phylogenetic analyses (15, 16). RVA VP3 sequences, which were most highly represented in the ML tree, clustered in distinct branches containing isolates from either mammalian or avian hosts.
FIG 1.

Phylogenetic relationships among capping enzymes of the Sedoreovirinae subfamily of the Reoviridae. A tree of the amino acid sequences of rotavirus VP3, orbivirus VP4, and seadornavirus VP3 is shown. Clade A rotavirus strain names are colored red (RVA), magenta (RVC), pink (RVD), or orange (RVF). Clade B rotavirus strain names are colored royal blue (RVB), cornflower blue (RVG), or cyan (RVH). Seadornavirus and orbivirus strain names are colored green and gray, respectively. Numbers on the branches indicate bootstrap support. AHV, African horse sickness virus; StCRV, Saint Croix River virus. Bar, 0.8 change per site.
Sequence analysis.
Based on their phylogenetic and functional relatedness, we hypothesized that Sedoreovirinae capping enzymes have similar domain architectures. We further hypothesized that residues important for catalytic function are under strong selective pressure and may be revealed in sequence alignments. We began to test this hypothesis by identifying conserved amino acids in the aligned sequences of capping enzymes from representative strains of each rotavirus species and of the Orbivirus genus, including BTV10 (Fig. 2). Consistent with the phylogenetic analyses, sequence alignment revealed that the capping enzymes of rotaviruses and orbiviruses cluster into three groups that correspond well with the viral genus and clade (Fig. 1 and 2). Of the sequences in the alignment, clade A rotavirus VP3 proteins shared 25 to 90% amino acid identity (39 to 95% similarity) (Table 1). Similarly, clade B VP3 proteins shared 25 to 99% identity (38 to 100% similarity), and orbivirus VP4 proteins shared 33 to 50% identity (50 to 67% similarity). The amino acid conservation between these groups of capping enzymes was lower, with only 5 to 17% identity (11 to 30% similarity).
TABLE 1.
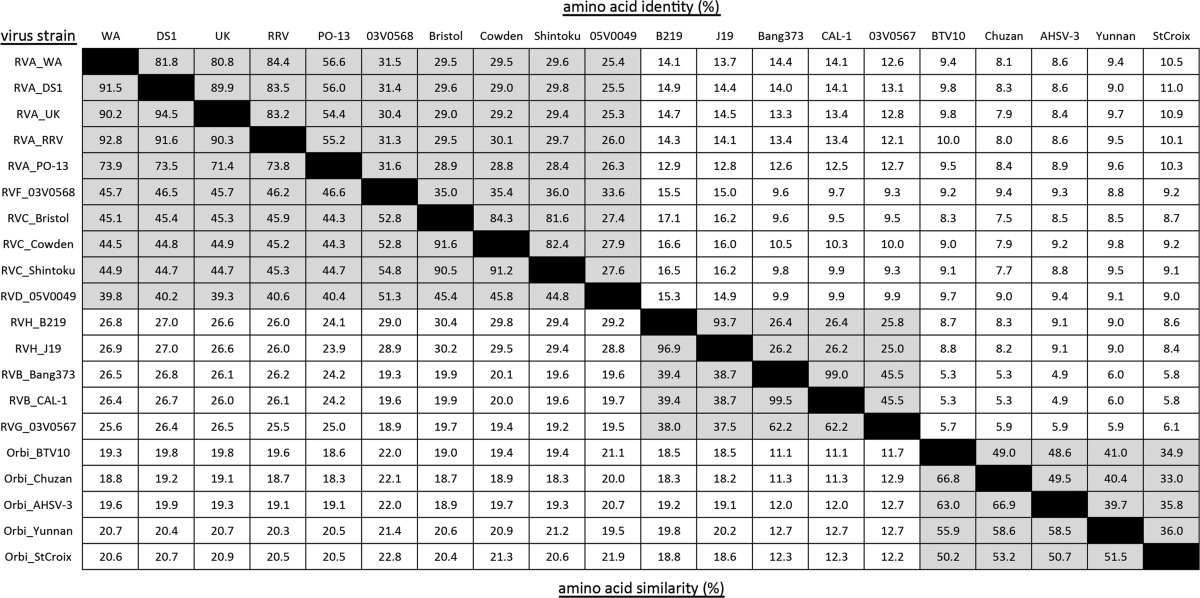
Values for intragenus (orbivirus) and intraclade (rotavirus) comparisons are shaded.
Some of the most striking differences between the capping enzymes are in regions that align with the BTV10 VP4 N-terminal and 2′-O-MTase domains (Fig. 2). The N termini of clade A rotavirus, clade B rotavirus, and orbivirus capping enzymes have modest intragroup alignment but poor intergroup alignment, with no residues being conserved across all sequences. This observation is consistent with the proposed function for the N-terminal region of BTV10 VP4 as a catalytically inactive adaptor (7). Sequences within the 2′-O-MTase domain of BTV10 VP4 differed significantly in length and composition between the clade B rotavirus VP3 sequences and the other capping enzyme sequences in the alignment, and no absolutely conserved residues were observed (Fig. 2). Consistent with an accessory function, a C-terminal domain present in RVA, RVB, and RVG rotaviruses was absent from the other capping enzymes in the alignment.
Despite many differences, several regions of conservation anchor all of the sequences in the alignment (Fig. 2). These regions align with the N7-MTase and GTase/RTPase domains of BTV10 VP4. Conserved sequences that align with the N7-MTase domain include (BTV10 VP4 numbering) Arg122, a GxxxE(S/T) motif spanning residues 134 to 139, and an LΩxL(S/T)NxxN motif (where Ω indicates an aromatic amino acid) spanning residues 409 to 417. The LΩxL(S/T)NxxN motif aligns with a previously identified region of conservation among rotavirus VP3 sequences (15). Conserved residues are found throughout the region that aligns with the GTase/RTPase domain of BTV10 VP4 and include (VP4 numbering) Ser527, Arg531, Trp571, His610, and an SGHΦ motif spanning residues 552 to 555. Notably, amino acids Arg122, Arg531, and Trp571, the SGHΦ motif, and the GxxxE(S/T) motif were absolutely conserved in an alignment of 810 rotavirus and orbivirus sequences (data not shown). Ser527 and the LΩxL(S/T)N of the LΩxL(S/T)NxxN motif were nearly completely conserved in the larger alignment, with just a few instances of substitution with similar amino acids. Although we did not choose to analyze seadornavirus capping enzymes in detail, the SGHΦ and LΩxL(S/T)NxxN motifs were conserved across all sequences in the alignment used to generate the ML tree shown in Fig. 1, but the GxxxE(S/T) motif was not conserved for the seadornavirus capping enzymes (data not shown).
A structural view of orbivirus and rotavirus capping enzyme sequence conservation.
After identifying residues and sequence motifs conserved between the rotavirus and orbivirus capping enzymes, we wanted to map them onto the BTV10 VP4 structure. Some loops were unresolved in the reported BTV10 VP4 structure, so we used I-TASSER to generate a high-confidence model of the full-length protein (Fig. 3A). Coloring of the surface of the BTV10 VP4 model on the basis of the sequence conservation among orbivirus capping enzymes (Fig. 3B) revealed that the most highly conserved amino acids cluster in discrete regions on the top and in front of the molecule, and these also correspond to predicted active sites. Conserved residues on the top of BTV10 VP4 form surface-exposed loops and line a deep depression hypothesized to be the GTase/RTPase active site (7). Conserved residues on the front of the molecule primarily line two large clefts that serve as binding sites for S-adenosyl-l-homocysteine (SAH) in the N7-MTase domain and SAH and a cap analog in the 2′-O-MTase domain (Fig. 3A and B). These observations suggest that orbivirus capping enzymes share a common domain architecture.
To identify the locations of residues and motifs conserved between the rotavirus and orbivirus capping enzymes, the surface of the BTV10 VP4 model was colored on the basis of the sequence conservation across all aligned capping enzymes (Fig. 2 and 3C and D). Although conservation in general was much lower across all sequences than across orbivirus VP4 sequences, the most highly conserved residues again mapped primarily to the top of the molecule, near the predicted GTase/RTPase active site; to the SAH-binding cleft in the N7-MTase domain; and, to a lesser extent, to the SAH-binding and cap analog-binding clefts of the 2′-O-MTase domain (Fig. 3D). The three sequence motifs identified in the alignment (Fig. 2) correspond to an α-helix that lines the deep depression in the GTase/RTPase domain (SGHΦ) and the major SAH-binding surface in the N7-MTase domain [GxxxE(S/T) and LΩxL(S/T)NxxN] (Fig. 3C). Arg122, which is conserved among all sequences in the alignment (Fig. 2), is also located in the N7-MTase domain and contributes to SAH binding by BTV10 VP4 (Fig. 3C) (7). While their contributions to activity remain unknown, residues Ser527, Arg531, Trp571, and His610, proximal to the SGHΦ motif in the BTV10 VP4 GTase/RTPase domain (Fig. 3C), are conserved among all sequences in the alignment (Fig. 2). However, a proposed RTPase active-site cysteine (Cys518) in BTV10 VP4 that is conserved among orbiviruses is not conserved among rotavirus VP3 proteins (Fig. 2). Together, these findings suggest that rotavirus and orbivirus capping enzymes share a similar GTase/RTPase domain structure and a disrupted N7-MTase domain with a conserved SAH-binding site.
Predicted structure of RVA VP3.
To gain additional insight into the RVA VP3 structure and domain organization, we inputted the sequence of simian RVA strain RRV into PHYRE (42), which uses the sequence and predicted secondary structure to search for homologs within a fold library generated from entries in the Structural Classification of Proteins (SCOP) database (46) and Protein Data Bank (PDB) (47). We obtained an extensive VP3 homology model (amino acids 39 to 634) based on the BTV10 VP4 sequence (PDB accession number 2JH8) (7) (Fig. 4A). Despite a modest confidence score (75%), the phylogenetic relatedness of RRV and BTV10 (Fig. 1) and the conservation of predicted active-site residues in sequence alignments (Fig. 2) suggest that the model is biologically relevant. Thus, to generate a more complete model of the capping region of RRV VP3, we used the threading function of I-TASSER with the BTV10 VP4 sequence (PDB accession number 2JH8) as the template and RVA RRV VP3 amino acids 1 to 688 as the input sequence (Fig. 4C). Both structural models resembled BTV10 VP4 in terms of overall architecture, and the domain boundaries suggested by each model were consistent with those predicted from the sequence alignment (Fig. 2 and 4A and C). When we generated alignments based on the structure of BTV10 VP4 and each RRV VP3 model, we observed conserved residues in each predicted domain, with several clustering in regions that align with the N7-MTase SAH-binding site and predicted GTase/RTPase active sites of BTV10 VP4 (Fig. 4B and D). For the RRV VP3 PHYRE homology model, the GxxxE(S/T) motif and LΩxL(S/T)N of the LΩxL(S/T)NxxN motif that together form the major SAH-binding surface in the N7-MTase domain of BTV10 VP4 were structurally conserved, but the SGHΦ motif in the predicted GTase/RTPase active site was shifted C terminally by one amino acid relative to the BTV10 VP4 structure, and Arg117 did not align precisely with a conserved arginine in the VP3 model (Fig. 4B and data not shown). For the RRV VP3 I-TASSER threading model, the position of the LΩxL(S/T)NxxN motif was conserved, the GxxxE(S/T) motif was shifted from the BTV10 VP4 structure toward the N terminus by one amino acid, and the SGYI sequence (residues 586 to 589) aligned structurally to SGHL (residues 552 to 555) in BTV10 VP4 (Fig. 4D and data not shown). Amino acid conservation in the predicted N-terminal and 2′-O-MTase domains was notably higher in structure-based (Fig. 4B and D) versus sequence-based (Fig. 2) alignments of the BTV10 VP4 and the RRV VP3 models.
To locate residues and motifs that are highly conserved within clade A rotavirus capping enzymes or between rotavirus and orbivirus capping enzymes, the surface of the RRV VP3 threading model was colored on the basis of sequence conservation (Fig. 4E and F). The more conserved sequences mapped primarily to regions that align with the BTV10 VP4 predicted GTase/RTPase active site at the top of the molecule, the N7-MTase SAH-binding cleft, and the 2′-O-MTase SAH- and cap analog-binding clefts. An additional region of conservation on the underside of the N-terminal domain was also observed for clade A rotavirus VP3, and the conservation on the top of the model for these viruses was more extensive than that observed for the orbiviruses (Fig. 3B and 4E). These findings support the idea that rotavirus and orbivirus capping enzymes share a similar GTase/RTPase domain structure and a disrupted N7-MTase domain with a conserved SAH-binding site.
We also searched for RRV VP3 homologs using PHYRE2 (42). Despite the low sequence identity (∼20%) with predicted homologs, high-confidence models (>95%) were generated for two distinct regions of RRV VP3. Amino acids 257 to 343 are homologous to the 2′-O-MTase of vaccinia virus (VV), VP39 (PDB accession numbers 1V39, 1VPT, and 1VP3) (12, 43) (Fig. 5A). As previously reported (30), the C-terminal region (amino acids 697 to 800) is homologous to the central domain of human AKAP7 (PDB accession number 2VFY) (44), a 2H-phosphoesterase superfamily member (Fig. 6A).
FIG 5.

Predicted 2′-O-methyltransferase domain of RRV VP3. (A to E) Overlay of ribbon drawings of VV VP39 (white; PDB accession number 1V39) and the VP39-based VP3 homology models for RVA RRV (A), RVC Bristol (B), RVD 05V0049 (C), RVF 03V0568 (D), or RVH J19 (E). The VP39 ligands SAH (yellow) and 7N-methyl-8-hydroguanosine-5′-diphosphate (light blue) are shown as stick representations. (F to H) Overlay of the 2′-O-MTase domain of the BTV10 VP4 homology model (white) with the VP4-based RVA RRV VP3 homology model (light blue; F), I-TASSER RVA RRV VP3 threading model (green; G), or both (H). The VP4 ligands SAH (yellow; PDB accession number 2JHP) and 7N-methyl-8-hydroguanosine-5′-diphosphate (blue; PDB accession number 2JH8) are shown. Side chains of residues in the KDKE motif are shown (A to H) and indicated (H). (I to J) Structure-based alignment of sequences from the VP3 models shown in panels A to E with VV VP39 (I) or from the RRV VP3 models shown in panels F and G with BTV10 VP4 (J). Identical (red) and similar (orange) residues are colored. Residues of the KDKE motif are outlined.
FIG 6.

2H-phosphodiesterase domain of rotavirus VP3. (A to C) Ribbon drawings showing the overlay of an RVA RRV (A), RVB CAL-1 (B), or RVG 03V0567 (C) VP3 homology model (cyan) onto the AKAP7 central domain structure (white; PDB accession number 2VFY). The side chains of histidine and threonine (or asparagine) residues in the HΦTΦ motifs and the AKAP7 ligand AMP (green; PDB accession number 2VFK) are shown. (D) Structure-based alignment of the AKAP7 central domain and the three models shown in panels A to C. Residues that are identical (red) or similar (orange) for all sequences in the alignment are colored. HΦTΦ motifs are outlined. (E) Ribbon drawing of the AKAP7 central domain, with conserved residues colored as in the alignment in panel D.
Predicted structure of the rotavirus VP3 2′-O-methyltransferase domain.
The region of RRV VP3 (residues 257 to 343) that was modeled on the basis of VV VP39 is contained within a region of VP3 that is homologous to the 2′-O-MTase domain of BTV10 VP4 (Fig. 4 and 5A). When VP3 sequences from representative strains of rotavirus were inputted into PHYRE2, high-confidence (>95%) homology models based on VV VP39 were also generated for a central region of the RVC, RVD, RVF, and RVH VP3 (Fig. 5B to E). Structure-based sequence alignments of these homology models with VV VP39 (PDB accession number 1V39) were generated to identify conserved amino acids (Fig. 5I). The position of residues in the VP39 KDKE catalytic tetrad was conserved for all modeled rotavirus VP3 sequences, with the exception of the first lysine for RVC VP3 and the aspartic acid for RVD VP3. Additional residues that interacted with SAM in VP39 structures and that were identical or similar for the VP3 models included Gln39, His74, and Phe115 (Fig. 5I) (12, 43). VP39 residues Gly68, Pro71, and Arg139 were also identical in the VP3 models and are located within the SAM-binding pocket. Structure-based sequence alignments of the RRV VP3 homology and threading models with BTV10 VP4 (PDB accession number 2JH8) revealed that residues in the KDKE catalytic tetrad were similarly conserved (Fig. 5J). Although the first lysine in the threading model failed to align precisely, Lys260 (Fig. 5J, RVA I-TASSER) is quite close to the first lysine in the BTV10 VP4 catalytic tetrad and may function similarly (Fig. 5H). Together, these findings suggest that, despite a lack of overall sequence conservation, clade A rotavirus VP3 and RVH VP3 contain a 2′-O-MTase domain.
Predicted structure of the rotavirus VP3 2H-PDE domain.
Two HΦ(T/S)Φ motifs are the hallmark used to identify potential 2H-phosphoesterase superfamily members, as they form the ligand-binding and catalytic site (48). Mutation of one or both histidines in these motifs resulted in the loss of 2′-5′-phosphodiesterase (2′-5′-PDE) activity for the RVA VP3 C-terminal domain (30). While the PDE domain is absent from orbivirus VP4 and RVC, RVD, RVF, and RVH VP3, it is present in RVB and RVG VP3 (Fig. 2) (30). Sequence alignments revealed two potential HΦ(T/S)Φ motifs in the RVB and RVG VP3 C terminus. However, the spacing between the motifs differed somewhat from that of RVA VP3, and the second motif was actually HΦNΦ for RVG VP3. Thus, it is unclear whether these domains function similarly to the PDE domain of RVA VP3. To gain insight into the function of this domain for RVB and RVG VP3, we inputted the C-terminal sequences into PHYRE2. In both cases, the AKAP7 central domain (PDB accession number 2VFY) was identified as a homolog, but the confidence scores were only 46% (RVG 03V0567) and 35.8% (RVB CAL-1). Nonetheless, the homology models for RVB and RVG C-terminal domains aligned the predicted HΦTΦ (or HΦNΦ) motifs with those of AKAP7 (Fig. 6B and C). On the basis of the structural alignment, residues conserved between the previously published RVA VP3 PDE model (Fig. 6A) (30), the RVB and RVG VP3 C-terminal models (Fig. 6B and C), and the AKAP7 central domain cluster primarily in the groove responsible for ligand binding and catalysis (Fig. 6D and E) (44). Residues that are identical across the four models include the catalytic histidines in both HΦTΦ motifs, the threonine in the first HΦTΦ motif, and Arg219, a component of the AKAP7 R loop that forms a cation-π interaction with the adenine moiety of its AMP and CMP ligands. While confidence in the RVB and RVG VP3 C-terminal domain models is fairly low, they provide support for the idea that these regions adopt a fold resembling 2H-phosphoesterase superfamily members and, like RVA VP3, function as 2′-5′-PDEs.
Mutational analysis of the predicted VP3 guanylyltransferase domain.
Autoguanylation, the covalent binding of GMP and release of pyrophosphate by a GTase, is the first step in the ping-pong reaction of guanylyl transfer (3, 49). Toward testing the validity of our predictions about VP3 structure and function, we engineered baculoviruses expressing RRV VP3 with mutations in the predicted GTase domain and quantified their capacity to covalently bind [32P]GMP. Lysine typically serves as the site of autoguanylation, but arginine or histidine can also form phosphoamide bonds and might theoretically serve this function (50). Although none of the lysines in the predicted GTase domain was highly conserved in sequence alignments of orbivirus and rotavirus capping enzymes (Fig. 2), we engineered individual isoleucine substitutions of each lysine in this region of RRV VP3. We also engineered isoleucine substitutions of conserved arginine and histidine residues in the predicted GTase domain of RRV VP3 (Fig. 2) and of Lys541, which is part of a KxxGNNH motif (RRV VP3 residues 541 to 545) previously proposed to be the VP3 GTase active site (51). This residue is conserved among clade A rotavirus VP3 sequences but resides in the predicted N7-MTase domain (Fig. 2). We found that several mutations in the predicted GTase region of RRV VP3, including K559I, R577I, H612I, K639I, K663I, and K687I, diminished autoguanylation, consistent with the predicted function of this domain (see Fig. 8). However, only the R591I and H650I mutations resulted in undetectable autoguanylation levels. Although lysine is capable of forming a phosphoamide bond, lysine substitutions of Arg591 and His650 failed to restore covalent GMP binding. The K541I substitution had a modest effect on VP3 autoguanylation, excluding the possibility that Lys541 is the VP3 catalytic lysine. These findings provide support for our predictions about VP3 domain architecture and identify two potential noncanonical sites of VP3 autoguanylation, Arg591 and His650.
FIG 8.

Autoguanylation of mutant VP3 proteins. Lysates of Sf9 cells infected with baculoviruses expressing CAT, wild-type RRV VP3 (VP3), or RRV VP3 point mutants were incubated with [α-32P]GTP, boiled, resolved by SDS-PAGE and autoradiography, and quantified by phosphorimage analysis. The graph shows the quantified signal from radiolabeled mutant VP3, normalized on the basis of the input protein and plotted relative to that of wild-type VP3. Point mutations in VP3 are indicated. Error bars represent SDs. *, P < 0.01.
DISCUSSION
In keeping with the description of BTV10 VP4 as a capping assembly line (7), the patterns of amino acid conservation among orbiviruses suggest an order of events and a potential pathway for newly synthesized RNA (Fig. 3B). The C-terminal domain of BTV10 VP4 has been unambiguously identified as the GTase domain and is also thought to serve as the RTPase domain (7). The poorly conserved BTV10 VP4 N-terminal domain is proposed to interact with the viral polymerase. Such an interaction could position newly synthesized viral mRNAs in proximity to the RTPase and GTase active site(s) (Fig. 2 and 3B). Winding down the front of the molecule, a viral mRNA would next encounter the conserved SAH-binding pocket of the N7-MTase domain, followed by the conserved SAH- and cap analog-binding clefts of the VP4 2′-O-MTase domain. This order of events is consistent with the canonical mRNA capping pathway (1). The pattern of VP3 amino acid conservation observed among clade A rotaviruses, when mapped onto the threading model of RVA RRV VP3, is similar to that of orbiviruses, which suggests that these enzymes also function as capping assembly lines, with viral mRNAs potentially following a similar path (Fig. 4E). It remains to be seen whether conservation of this enzymatic domain arrangement extends to other Sedoreovirinae capping enzymes.
Our findings suggest candidate catalytic regions and residues in the RRV VP3 and BTV10 VP4 GTase/RTPase domains. The SGHΦ motif is extremely well conserved among rotavirus and orbivirus capping enzymes, contains a proposed BTV10 VP4 GTase active-site histidine (His554), and is located on an α-helix in the deep depression at the top of the BTV10 VP4 GTase/RTPase domain (Fig. 2 and 3) (7). Residues in this motif may contribute to GTase, RTPase, or both activities. Our biochemical data show that His612, which is contained within the RRV VP3 SGHΦ motif, contributes to but is not required for the first step in guanylyl transfer (Fig. 2 and 7). This motif occupied divergent locations in the RRV VP3 I-TASSER model and the BTV10 VP4 structure (Fig. 4B and D), which may indicate true structural differences between BTV10 VP4 and RVA VP3 in this region or may simply reflect the quality of the models. In our biochemical studies, the three most highly conserved RRV VP3 residues that we mutated, Arg577, His612, and His650, all contributed significantly to autoguanylation; Arg591 and His650 were required for this activity (Fig. 2 and 8). RRV VP3 His612 aligns with BTV10 VP4 His554, which is part of the SGHΦ motif (Fig. 2) (7). RRV VP3 Arg591 fails to align precisely with a BTV10 VP4 residue but aligns with a region of BTV10 VP4 comprising a flexible surface loop adjacent to the α-helix containing the SGHΦ motif. The BTV10 VP4 residue corresponding to RRV VP3 His650, His610, is located in a different surface-exposed loop, adjacent to the depression containing the SGHΦ motif. The BTV10 VP4 residue corresponding to RRV VP3 Arg577 is located in an α-helix adjacent to the one containing the SGHΦ motif and preceding the flexible loop that encompasses His610. Together, these observations suggest that the deep depression containing the SGHΦ motif and the surrounding external loops of BTV10 VP4 house residues important for catalytic activity. Although Arg591 or His650 could theoretically bind GMP covalently (50), His650 is the more likely candidate for two reasons. First, His650 is more highly conserved among Sedoreovirinae capping enzymes (Fig. 2). Second, while covalent bonds with histidine occur as part of the capping process for the vesicular stomatitis virus L protein, the L-A virus Gag protein, and, likely, the bamboo mosaic virus capping enzyme, we can find no similar examples of phosphoamide bond formation with arginine (52–55). Cys518, which was proposed to form part of the RTPase catalytic site in BTV10 VP4 (7), is not conserved for rotavirus VP3 (Fig. 2). While BTV10 VP4 exhibits RTPase activity (56), this activity has not been observed for hypotonically disrupted rotavirus subviral particles (22); it is possible that another viral protein (VP1 or VP2) or its interaction with VP3 is required. Additional studies are needed to unambiguously identify the sites of autoguanylation and RTPase mechanisms for both orbivirus and rotavirus. Since the fold adopted by the BTV10 VP4 GTase/RTPase domain has not previously been observed, such studies may also reveal novel enzymatic mechanisms (1, 7).
FIG 7.

Predicted domain organization for VP3 from different rotavirus species. Amino acid domain boundaries, based on the alignments in Fig. 2, are indicated. Domains are labeled and colored by predicted function: gray, unknown (UN); green, N7-MTase; purple, 2′-O-MTase; blue, GTase/RTPase; cyan, PDE. A patterned color suggests a reduced confidence in domain identity.
Conserved sequences in the BTV10 VP4 N7-MTase domain served as anchor points to align orbivirus and clade A and B rotavirus capping enzyme sequences, which differed in some cases by more than 90% (Fig. 2 and Table 1). The strong sequence conservation of BTV10 VP4 Arg122 and the GxxxE(S/T) and LΩxL(S/T)NxxN motifs, combined with their observed interactions with SAH in VP4 structures, provides strong evidence that the N7-MTase architecture is conserved among orbivirus and rotavirus capping enzymes (Fig. 2 and 3). The positional conservation of LΩxL(S/T)N or the entire LΩxL(S/T)NxxN motif in both BTV10 VP4-based VP3 models and the GxxxE(S/T) motif in the RVA VP3 homology model, as well as the presence of amino acids conserved among clade A rotavirus VP3 proteins in the predicted catalytic cleft of the RVA VP3 threading model, provide additional evidence that the RVA VP3 N7-MTase domain architecture and the included SAH-binding site resemble those of BTV10 VP4 (Fig. 4).
Despite a lack of conservation in sequence alignments, the 2′-O-MTase domain was the VP3 capping domain most consistently modeled with high confidence (Fig. 5). The shorter models (for RVA, RVC, and RVH VP3) included four of the seven β-strands typical of the class I SAM-dependent MTase fold and conserved the two modeled residues of the KDKE catalytic tetrad, with the exception of the first lysine for RVC VP3. The more extensive models (for RVD and RVF VP3) included all seven β-strands and conserved the four residues of the KDKE catalytic tetrad, with the exception of the aspartic acid for RVD VP3. Notably, the second lysine in the tetrad, the catalytic lysine, was structurally conserved for all VP3 models that encompass this region (Fig. 5) (57). VP3 Gly68, a small residue conserved among various methyltransferases (12), was also conserved across all of the modeled structures (Fig. 5) and BTV10 VP4 (Gly197). These findings strongly support the notion that each of the RVA, RVC, RVD, RVF, and RVH VP3 proteins contains a functional 2′-O-MTase domain and reinforce the utility of homology modeling to predict the functions of proteins that share little sequence identity.
Our findings suggest an overall domain architecture for rotavirus VP3 (Fig. 7). A combination of sequence analysis, homology modeling, and mapping of conserved amino acids suggests that the VP3 N-terminal domain, whose function remains unknown, is followed by a predicted N7-MTase domain that contains an insertion, which is usually a 2′-O-MTase domain (Fig. 2 to 5 and 7). A patch of conserved amino acids on the underside of the predicted VP3 N-terminal domain and adjacent to the predicted N7-MTase SAH-binding cleft of clade A rotaviruses (Fig. 4E) is consistent with either a catalytic function for this domain or interaction with a conserved region of the viral RdRp, VP1. The first three predicted domains of VP3 are followed by a putative GTase/RTPase domain and, in some cases, a PDE domain (Fig. 2 to 7). On the basis of the sequence alignment and structure prediction, the domain architecture for the capping region of VP3 is likely well conserved among clade A rotaviruses (Fig. 2 to 5 and 7). A surprising finding for the clade B rotaviruses was the absence of a predicted 2′-O-MTase domain in RVB and RVG VP3 and its presence in RVH VP3. The insertion in the N7-MTase domain of RVB and RVG VP3 is ∼120 amino acids long, whereas it is ∼180 amino acids for the other rotavirus species. It is unknown whether RVB and RVG synthesize mRNAs with cap-0 or cap-1 structures. If they synthesize cap-1, their 2′-O-MTase domains may be too different for homologs to be identified, or a single MTase domain may perform both methylation activities, as observed for flavivirus NS5 proteins (58). The presence of a C-terminal PDE domain in some members of each rotavirus clade (Fig. 2 and 6) is consistent with the notion that it is a nonessential virulence determinant. The PDE domain maintained in a common ancestor may have been lost from some rotavirus species over time, perhaps because its innate immune antagonist function is somewhat redundant with the function of other molecules encoded in the viral genome, such as NSP1 (59), or because it is more important for infection of some hosts or cell types than of others, as is the case for coronavirus ns2 (60).
The combination of phylogenetic analysis, sequence alignment, and homology modeling provides a powerful predictive tool. While it is possible that none of the structural models generated in this study is an entirely accurate representation of VP3, they have permitted prediction of structural and functional domains and active sites. The findings reported herein provide information useful for the design of VP3 protein expression constructs and targets for mutational and biochemical analysis. An improved understanding of the enzymatic mechanisms that govern rotavirus transcription may inform strategies to recover fully recombinant rotaviruses, and a broader knowledge of the mechanisms by which rotaviruses evade innate immune defenses, including the PDE and 2′-O-MTase activities of VP3, may provide ideas for the rational design of attenuated vaccines in the future.
ACKNOWLEDGMENTS
We acknowledge Marco Morelli, Chelsea Gridley, and Elif Eren for helpful discussions and editorial comments.
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases at the National Institutes of Health.
Footnotes
Published ahead of print 4 June 2014
REFERENCES
- 1.Decroly E, Ferron F, Lescar J, Canard B. 2012. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 10:51–65. 10.1038/nrmicro2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghosh A, Lima CD. 2010. Enzymology of RNA cap synthesis. Wiley Interdiscip Rev. RNA 1:152–172. 10.1002/wrna.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shuman S, Lima CD. 2004. The polynucleotide ligase and RNA capping enzyme superfamily of covalent nucleotidyltransferases. Curr. Opin. Struct. Biol. 14:757–764. 10.1016/j.sbi.2004.10.006 [DOI] [PubMed] [Google Scholar]
- 4.Wang SP, Deng L, Ho CK, Shuman S. 1997. Phylogeny of mRNA capping enzymes. Proc. Natl. Acad. Sci. U. S. A. 94:9573–9578. 10.1073/pnas.94.18.9573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luongo CL, Reinisch KM, Harrison SC, Nibert ML. 2000. Identification of the guanylyltransferase region and active site in reovirus mRNA capping protein lambda2. J. Biol. Chem. 275:2804–2810. 10.1074/jbc.275.4.2804 [DOI] [PubMed] [Google Scholar]
- 6.Reinisch KM, Nibert ML, Harrison SC. 2000. Structure of the reovirus core at 3.6 Å resolution. Nature 404:960–967. 10.1038/35010041 [DOI] [PubMed] [Google Scholar]
- 7.Sutton G, Grimes JM, Stuart DI, Roy P. 2007. Bluetongue virus VP4 is an RNA-capping assembly line. Nat. Struct. Mol. Biol. 14:449–451. 10.1038/nsmb1225 [DOI] [PubMed] [Google Scholar]
- 8.De la Pena M, Kyrieleis OJ, Cusack S. 2007. Structural insights into the mechanism and evolution of the vaccinia virus mRNA cap N7 methyl-transferase. EMBO J. 26:4913–4925. 10.1038/sj.emboj.7601912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fabrega C, Hausmann S, Shen V, Shuman S, Lima CD. 2004. Structure and mechanism of mRNA cap (guanine-N7) methyltransferase. Mol. Cell 13:77–89. 10.1016/S1097-2765(03)00522-7 [DOI] [PubMed] [Google Scholar]
- 10.Bujnicki JM, Rychlewski L. 2001. Reassignment of specificities of two cap methyltransferase domains in the reovirus lambda 2 protein. Genome Biol. 2:RESEARCH0038. 10.1186/gb-2001-2-9-research0038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egloff MP, Benarroch D, Selisko B, Romette JL, Canard B. 2002. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 21:2757–2768. 10.1093/emboj/21.11.2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hodel AE, Gershon PD, Shi X, Quiocho FA. 1996. The 1.85 Å structure of vaccinia protein VP39: a bifunctional enzyme that participates in the modification of both mRNA ends. Cell 85:247–256. 10.1016/S0092-8674(00)81101-0 [DOI] [PubMed] [Google Scholar]
- 13.Anonymous. 2013. Rotavirus vaccines WHO position paper: January 2013—recommendations. Vaccine 31:6170–6171. 10.1016/j.vaccine.2013.05.037 [DOI] [PubMed] [Google Scholar]
- 14.Attoui H, Becnel J, Belaganahalli S, Bergoin M, Brussaard CP, Chappell JD, Ciarlet M, del Vas M, Dermody TS, Dormitzer PR, Duncan R, Fang Q, Graham R, Guglielmi KM, Harding RM, Hillman B, Makkay A, Marzachi C, Matthijnssens J, Mertens PPC, Milne RG, Mohd Jaafar F, Mori H, Noordeloos AA, Omura T, Patton JT, Rao S, Maan M, Stoltz D, Suzuki N, Upadhyaya NM, Wei C, Zhou H. 2012. Part II: the viruses—the double stranded RNA viruses—family Reoviridae, p 541–637 In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. (ed), Virus taxonomy: classification and nomenclature. Ninth report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, San Diego, CA [Google Scholar]
- 15.Kindler E, Trojnar E, Heckel G, Otto PH, Johne R. 2013. Analysis of rotavirus species diversity and evolution including the newly determined full-length genome sequences of rotavirus F and G. Infect. Genet. Evol. 14:58–67. 10.1016/j.meegid.2012.11.015 [DOI] [PubMed] [Google Scholar]
- 16.Ogden KM, Johne R, Patton JT. 2012. Rotavirus RNA polymerases resolve into two phylogenetically distinct classes that differ in their mechanism of template recognition. Virology 431:50–57. 10.1016/j.virol.2012.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imai M, Akatani K, Ikegami N, Furuichi Y. 1983. Capped and conserved terminal structures in human rotavirus genome double-stranded RNA segments. J. Virol. 47:125–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCrae MA, McCorquodale JG. 1983. Molecular biology of rotaviruses. V. Terminal structure of viral RNA species. Virology 126:204–212 [DOI] [PubMed] [Google Scholar]
- 19.Fukuhara N, Nishikawa K, Gorziglia M, Kapikian AZ. 1989. Nucleotide sequence of gene segment 1 of a porcine rotavirus strain. Virology 173:743–749. 10.1016/0042-6822(89)90590-4 [DOI] [PubMed] [Google Scholar]
- 20.Liu M, Mattion NM, Estes MK. 1992. Rotavirus VP3 expressed in insect cells possesses guanylyltransferase activity. Virology 188:77–84. 10.1016/0042-6822(92)90736-9 [DOI] [PubMed] [Google Scholar]
- 21.Pizarro JL, Sandino AM, Pizarro JM, Fernandez J, Spencer E. 1991. Characterization of rotavirus guanylyltransferase activity associated with polypeptide VP3. J. Gen. Virol. 72(Pt 2):325–332. 10.1099/0022-1317-72-2-325 [DOI] [PubMed] [Google Scholar]
- 22.Patton JT, Chen D. 1999. RNA-binding and capping activities of proteins in rotavirus open cores. J. Virol. 73:1382–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen D, Luongo CL, Nibert ML, Patton JT. 1999. Rotavirus open cores catalyze 5′-capping and methylation of exogenous RNA: evidence that VP3 is a methyltransferase. Virology 265:120–130. 10.1006/viro.1999.0029 [DOI] [PubMed] [Google Scholar]
- 24.Chen JZ, Settembre EC, Aoki ST, Zhang X, Bellamy AR, Dormitzer PR, Harrison SC, Grigorieff N. 2009. Molecular interactions in rotavirus assembly and uncoating seen by high-resolution cryo-EM. Proc. Natl. Acad. Sci. U. S. A. 106:10644–10648. 10.1073/pnas.0904024106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu X, McDonald SM, Tortorici MA, Tao YJ, Vasquez-Del Carpio R, Nibert ML, Patton JT, Harrison SC. 2008. Mechanism for coordinated RNA packaging and genome replication by rotavirus polymerase VP1. Structure 16:1678–1688. 10.1016/j.str.2008.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McClain B, Settembre E, Temple BR, Bellamy AR, Harrison SC. 2010. X-ray crystal structure of the rotavirus inner capsid particle at 3.8 Å resolution. J. Mol. Biol. 397:587–599. 10.1016/j.jmb.2010.01.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng N, Yasukawa LL, Sen A, Greenberg HB. 2013. Permissive replication of homologous murine rotavirus in the mouse intestine is primarily regulated by VP4 and NSP1. J. Virol. 87:8307–8316. 10.1128/JVI.00619-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoshino Y, Saif LJ, Kang SY, Sereno MM, Chen WK, Kapikian AZ. 1995. Identification of group A rotavirus genes associated with virulence of a porcine rotavirus and host range restriction of a human rotavirus in the gnotobiotic piglet model. Virology 209:274–280. 10.1006/viro.1995.1255 [DOI] [PubMed] [Google Scholar]
- 29.Wang W, Donnelly B, Bondoc A, Mohanty SK, McNeal M, Ward R, Sestak K, Zheng S, Tiao G. 2011. The rhesus rotavirus gene encoding VP4 is a major determinant in the pathogenesis of biliary atresia in newborn mice. J. Virol. 85:9069–9077. 10.1128/JVI.02436-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang R, Jha BK, Ogden KM, Dong B, Zhao L, Elliott R, Patton JT, Silverman RH, Weiss SR. 2013. Homologous 2′,5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc. Natl. Acad. Sci. U. S. A. 110:13114–13119. 10.1073/pnas.1306917110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M, Jr, Shi PY, Diamond MS. 2010. 2′-O Methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456. 10.1038/nature09489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szretter KJ, Daniels BP, Cho H, Gainey MD, Yokoyama WM, Gale M, Jr, Virgin HW, Klein RS, Sen GC, Diamond MS. 2012. 2′-O Methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 8:e1002698. 10.1371/journal.ppat.1002698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zust R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, Siddell SG, Ludewig B, Thiel V. 2011. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 12:137–143. 10.1038/ni.1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30:772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59:307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- 37.Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang JM, Taly JF, Notredame C. 2011. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39:W13–W17. 10.1093/nar/gkr245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kemena C, Notredame C. 2009. Upcoming challenges for multiple sequence alignment methods in the high-throughput era. Bioinformatics 25:2455–2465. 10.1093/bioinformatics/btp452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5:725–738. 10.1038/nprot.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roy A, Yang J, Zhang Y. 2012. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 40:W471–W477. 10.1093/nar/gks372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. 10.1186/1471-2105-9-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371. 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- 43.Hodel AE, Gershon PD, Shi X, Wang SM, Quiocho FA. 1997. Specific protein recognition of an mRNA cap through its alkylated base. Nat. Struct. Biol. 4:350–354. 10.1038/nsb0597-350 [DOI] [PubMed] [Google Scholar]
- 44.Gold MG, Smith FD, Scott JD, Barford D. 2008. AKAP18 contains a phosphoesterase domain that binds AMP. J. Mol. Biol. 375:1329–1343. 10.1016/j.jmb.2007.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 46.Murzin AG, Brenner SE, Hubbard T, Chothia C. 1995. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247:536–540. 10.1016/S0022-2836(05)80134-2 [DOI] [PubMed] [Google Scholar]
- 47.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mazumder R, Iyer LM, Vasudevan S, Aravind L. 2002. Detection of novel members, structure-function analysis and evolutionary classification of the 2H phosphoesterase superfamily. Nucleic Acids Res. 30:5229–5243. 10.1093/nar/gkf645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shuman S, Hurwitz J. 1981. Mechanism of mRNA capping by vaccinia virus guanylyltransferase: characterization of an enzyme–guanylate intermediate. Proc. Natl. Acad. Sci. U. S. A. 78:187–191. 10.1073/pnas.78.1.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Besant PG, Attwood PV, Piggott MJ. 2009. Focus on phosphoarginine and phospholysine. Curr. Protein Pept. Sci. 10:536–550. 10.2174/138920309789630598 [DOI] [PubMed] [Google Scholar]
- 51.Cook JP, McCrae MA. 2004. Sequence analysis of the guanylyltransferase (VP3) of group A rotaviruses. J. Gen. Virol. 85:929–932. 10.1099/vir.0.19629-0 [DOI] [PubMed] [Google Scholar]
- 52.Blanc A, Ribas JC, Wickner RB, Sonenberg N. 1994. His-154 is involved in the linkage of the Saccharomyces cerevisiae L-A double-stranded RNA virus Gag protein to the cap structure of mRNAs and is essential for M1 satellite virus expression. Mol. Cell. Biol. 14:2664–2674. 10.1128/MCB.14.4.2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang YL, Han YT, Chang YT, Hsu YH, Meng M. 2004. Critical residues for GTP methylation and formation of the covalent m7GMP-enzyme intermediate in the capping enzyme domain of bamboo mosaic virus. J. Virol. 78:1271–1280. 10.1128/JVI.78.3.1271-1280.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin HY, Yu CY, Hsu YH, Meng M. 2012. Functional analysis of the conserved histidine residue of bamboo mosaic virus capping enzyme in the activity for the formation of the covalent enzyme-m7GMP intermediate. FEBS Lett. 586:2326–2331. 10.1016/j.febslet.2012.05.024 [DOI] [PubMed] [Google Scholar]
- 55.Ogino T, Yadav SP, Banerjee AK. 2010. Histidine-mediated RNA transfer to GDP for unique mRNA capping by vesicular stomatitis virus RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 107:3463–3468. 10.1073/pnas.0913083107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martinez-Costas J, Sutton G, Ramadevi N, Roy P. 1998. Guanylyltransferase and RNA 5′-triphosphatase activities of the purified expressed VP4 protein of bluetongue virus. J. Mol. Biol. 280:859–866. 10.1006/jmbi.1998.1926 [DOI] [PubMed] [Google Scholar]
- 57.Li C, Xia Y, Gao X, Gershon PD. 2004. Mechanism of RNA 2′-O-methylation: evidence that the catalytic lysine acts to steer rather than deprotonate the target nucleophile. Biochemistry 43:5680–5687. 10.1021/bi0359980 [DOI] [PubMed] [Google Scholar]
- 58.Liu L, Dong H, Chen H, Zhang J, Ling H, Li Z, Shi PY, Li H. 2010. Flavivirus RNA cap methyltransferase: structure, function, and inhibition. Front. Biol. (Beijing) 5:286–303. 10.1007/s11515-010-0660-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arnold MM, Sen A, Greenberg HB, Patton JT. 2013. The battle between rotavirus and its host for control of the interferon signaling pathway. PLoS Pathog. 9:e1003064. 10.1371/journal.ppat.1003064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao L, Birdwell LD, Wu A, Elliott R, Rose KM, Phillips JM, Li Y, Grinspan J, Silverman RH, Weiss SR. 2013. Cell-type-specific activation of the oligoadenylate synthetase-RNase L pathway by a murine coronavirus. J. Virol. 87:8408–8418. 10.1128/JVI.00769-13 [DOI] [PMC free article] [PubMed] [Google Scholar]