

ABSTRACT
Lactococcus lactis, a Gram+ lactic acid-producing bacterium used for the manufacture of several fermented dairy products, is subject to infection by diverse virulent tailed phages, leading to industrial fermentation failures. This constant viral risk has led to a sustained interest in the study of their biology, diversity, and evolution. Lactococcal phages now constitute a wide ensemble of at least 10 distinct genotypes within the Caudovirales order, many of them belonging to the Siphoviridae family. Lactococcal siphophage 1358, currently the only member of its group, displays a noticeably high genomic similarity to some Listeria phages as well as a host range limited to a few L. lactis strains. These genomic and functional characteristics stimulated our interest in this phage. Here, we report the cryo-electron microscopy structure of the complete 1358 virion. Phage 1358 exhibits noteworthy features, such as a capsid with dextro handedness and protruding decorations on its capsid and tail. Observations of the baseplate of virion particles revealed at least two conformations, a closed and an open, activated form. Functional assays uncovered that the adsorption of phage 1358 to its host is Ca2+ independent, but this cation is necessary to complete its lytic cycle. Taken together, our results provide the complete structural picture of a unique lactococcal phage and expand our knowledge on the complex baseplate of phages of the Siphoviridae family.
IMPORTANCE Phages of Lactococcus lactis are investigated mainly because they are sources of milk fermentation failures in the dairy industry. Despite the availability of several antiphage measures, new phages keep emerging in this ecosystem. In this study, we provide the cryo-electron microscopy reconstruction of a unique lactococcal phage that possesses genomic similarity to particular Listeria phages and has a host range restricted to only a minority of L. lactis strains. The capsid of phage 1358 displays the almost unique characteristic of being dextro handed. Its capsid and tail exhibit decorations that we assigned to nonspecific sugar binding modules. We observed the baseplate of 1358 in two conformations, a closed and an open form. We also found that the adsorption to its host, but not infection, is Ca2+ independent. Overall, this study advances our understanding of the adhesion mechanisms of siphophages.
INTRODUCTION
General interest in phage biology has increased in the past decade due to their abundance in most ecosystems, their potential use as antimicrobials, and the risk they pose in bacterium-driven biotechnological processes. Phages now are predicted to be the most abundant biological entities on our planet and play key roles in the balance of bacterial populations. However, very few phages have been studied thoroughly. Thus, understanding their biology is important for ecological systems and also for industrial applications.
Over 95% of the prokaryote viruses described morphologically belong to the Caudovirales order, as they possess a double-stranded DNA (dsDNA) genome packaged into a capsid connected to a tail (1). Phages within this viral order are subdivided into three families based on their tail features: members of the Siphoviridae have a long, noncontractile tail (2, 3), Myoviridae have a contractile tail (4), and Podoviridae have a very short tail (5). The Siphoviridae family is by far the largest family, as they represent close to 60% of all characterized phages (1).
Lactococcus lactis is the most important species for the manufacture of fermented dairy products. Its phages are among the most extensively studied because they are the main cause of milk fermentation failures worldwide. The dairy industry has been dealing with this natural phenomenon for years and has relied on an array of antiphage control measures. In spite of these efforts, lactococcal phages are evolving and new variants keep emerging. Understanding this natural variation is the key to updating phage control strategies.
While hundreds of lactococcal phages have been reported, they are currently classified into only 10 genetically distinct groups (6). This classification scheme is based mainly on electron microscopy and comparative genomics (7). Members belonging to one phage group have the same general morphology, and they share a high level of nucleotide identity. Members of distinct groups share limited, if any, DNA identity, and their morphologies also are different (6). Eight of the 10 lactococcal phage groups are clustered within the Siphoviridae family (siphophages), and two are in the Podoviridae family (6). Three groups of lactococcal siphophages, namely, 936, c2, and P335, are by far the most predominant phages found in modern dairy facilities. The seven other groups are far less prevalent, and for some, only a single member has been identified, including the virulent phage 1358 (8).
In the past decade, the X-ray structures of several lactococcal phage proteins have been determined (9), particularly for the virulent phage p2 (936 group) (10–14) and the temperate phages Tuc2009 and TP901-1 (P335 group) (15–18). These structures included those of the receptor binding proteins (RBP) and of the RBP-containing host recognition device, located at the distal end of the phage tail and called the baseplate (BP). It was shown that the RBPs harbor a saccharide-binding site and that viral infection is neutralized when this site is blocked by a camelid nanobody (10, 18). It was further revealed that the BP of 936-like phages goes through a calcium-dependent conformation change (200° rotation), anchoring the RBPs to the cell surface receptors. This conformational change also led to the opening of a channel at the bottom of the BP for the exit of the phage genomic dsDNA (14, 17). On the other hand, lactococcal siphophages of the P335 group do not go through such BP activation processes (7, 17). Lastly, the complete electron microscopy (EM) structure of phage p2 (19) and phage TP901-1 (3) were newly reported.
One of the key findings of the studies mentioned above was the modular nature of the RBPs seemingly allowing lactococcal phages to shuffle their host recognition domain within the RBP to rapidly modify host specificity. It is tempting to speculate that this viral adaptation is an evolutionary response to the industrial practice of rotating numerous L. lactis strains. Another significant outcome of these studies was the discovery of the binding partner of the RBPs, a polysaccharide pellicle at the surface of the lactococcal cells (20–22). The diversity of the pellicle composition between L. lactis strains explains, at least in part, the strain specificity of most lactococcal phages (21).
Recently, we became interested in the virulent siphophage 1358 due to its unique features (8). Many of its genes share sequence identities with some Listeria siphophages (23), which led to the hypothesis that this uncommon lactococcal phage originated from a Listeria phage (8). Moreover, the tail of 1358 is significantly shorter than those of other lactococcal phages, and it is decorated by hairy appendages. We also recently reported the X-ray structure of its RBP (22). Each monomer of its trimeric RBP is formed of two domains: a “shoulder” domain linking the RBP to the rest of the phage and a jelly-roll fold “head/host recognition” domain. This domain harbors a saccharide-binding crevice located in the middle of an RBP monomer, which is notably different from phage p2, where this binding site is located between two RBP head domains. We also proposed that a trisaccharidic motif within the lactococcal pellicle hexasaccharide was a common phage receptor, while the remaining components of the pellicle are involved in strain specificity. Therefore, the study of such a unique lactococcal phage may give insights into the requirements for host recognition. We report here the complete composite cryo-EM structure of the 1358 virion and propose that the structural decorations of its capsid and tail are involved in initial host binding.
MATERIALS AND METHODS
Cryo-electron microscopy data collection. (i) Phage preparation.
Phage 1358 and its host, L. lactis SMQ-388 (HER1205), were grown in GM17, and phage 1358 was purified as described previously (8). Purified phages were conserved at 4°C in buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 8 mM MgSO4). Approximately 2.5 μl of a purified phage sample was applied to glow-discharged carbon-coated grids (Quantifoil 1.2/1.1 cryo-EM grid) and incubated for 1 min. For negative staining, excess solution was blotted, 10 μl of uranyl-acetate (2%) was added, the solution was incubated for 30 s, and excess stain was blotted. For cryo-EM, after blotting for 4 s, the grid was plunged into liquid ethane for vitrification using an FEI-Vitrobot Mark II plunge-freezing device operating at 100% humidity.
(ii) Data collection.
For negative stain, charge-coupled-device (CCD) images were collected using a Tecnai Spirit operated at 120 kV and a 2,000- by 2,000-pixel CCD camera at a magnification of 48,500×, resulting in a pixel size of 4.95 Å/pixel, and coarsened by 2 at a constant defocus value where the first zero lies around a resolution of 20 Å. For cryo-EM, CCD images were collected under low-dose conditions using a Tecnai F30-Polara microscope with a field emission gun operated at 300 kV (IGBMC, Strasbourg, France) and an FEI Eagle 4,000- by 4,000-pixel CCD camera. Images were recorded with the FEI-EPU automatic data collection software package at a dose of ∼20 electrons per Å2 at a magnification of 59,000 with a pixel size of 1.92 Å and over a range of nominal defocus values between 1.5 and 3.5 μm. Defocus estimation and contrast transfer function (CTF) correction were carried out using FINDCTF2D (Timothy Grant, Imperial College, TIGRIS).
(iii) Image processing.
Particles (see Table 2) were manually selected using the program boxer from the EMAN2 package (24), extracted into boxes of 240 by 240 pixels (capsid), 216 by 216 pixels (connector), 300 by 300 pixels (tail), and 100 by 100 pixels (baseplate), and combined into the four different data sets. To evaluate the dimension of the tail and the number of MTP rings, 1,000 isolated phage particles were manually selected from the negative staining images, extracted into boxes of 500 by 500 pixels (coarsened by 2), and combined into a data set. All five data sets were pretreated using the SPIDER package (25) and submitted to maximum likelihood (ML) classification and alignment (26) using the Xmipp package (27). Tail fragments were properly aligned as they were boxed, including either a fragment of the capsid (upper tail) or a fragment of the baseplate (lower tail). The initial models were built to form a visually selected class average representing a side view imposing the corresponding symmetry (C6 for the full phage, tail, and baseplate, C12 for the connector, and icosahedral for the capsid). The initial models were refined by three-dimensional (3D) ML and by SPIDER, with a sampling rate of 5°. For the baseplate, two different conformations (open and closed) were observed after two-dimensional (2D) ML classification. During 3D ML refinement, two different models were given as seeds, which resulted in the separation of the data set into two subsets corresponding to the two different conformations. The final models of the phage 1358 components were obtained at resolutions of 16 to 24 Å (40 Å for the open form; see Table 2) as estimated by Fourier shell correlation (FSC) and the 1/2-bit threshold criterion (28) (Fig. 1A).
TABLE 2.
Summary of data for lactococcal phage 1358 single-particle EM reconstructions
| Function | Symmetry | Resolution (Å) | No. of particles |
|---|---|---|---|
| Capsid | Icosahedral | 16 | 9,211 |
| Connector | C12 | 17 | 4,994 |
| Tail | Helicoidal | 16 | 3,641 |
| Baseplate at rest | C6 | 24 | 2,415 |
| Baseplate activated | C6 | 40 | 1,580 |
FIG 1.
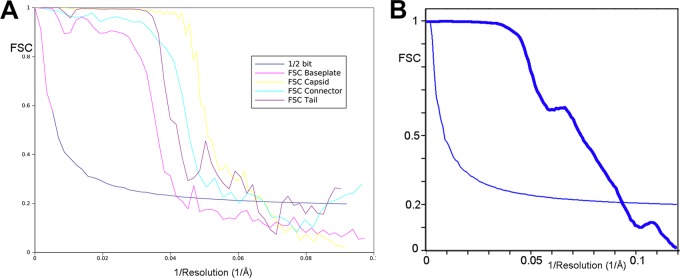
Fourier shell correlation (FSC) curves of the final 3D reconstructions. (A) These curves were obtained by correlation of two different 3D images created by splitting the particle set into two subsets. The resolution was estimated by the 1/2-bit cutoff threshold criterion as 16-Å resolution for the capsid, 17-Å resolution for the connector, 16-Å resolution for the helical tail, and 24-Å resolution for the baseplate. (B) The empty mature capsid resolution was estimated at 10 Å by the 1/2-bit cutoff threshold criterion.
Concerning the empty mature capsid, around 2,000 particles were boxed and coarsened twice, giving a final pixel size of 3.84 Å and a box size of 256 by 256 pixels. The initial structure was obtained in IMAGIC using a class average and cross-common line approach and refined using angular reconstitutions. The final resolution of the map is ∼10 Å, as estimated by FSC and the 1/2-bit threshold criterion (28) (Fig. 1B).
(iv) Tail helical processing.
The tail particles aligned as described above were submitted to helical processing. The helical map was produced using the package IHRSR++ (29). The rotational symmetry used was C6, and as the particles were already aligned, the maximum allowed in-plane rotational angle was set to 10°. The initial helical parameters were determined using the Brandeis helical package (30) to calculate the Bessel orders of the basic layer lines (6 and −6). These were later refined by IHRSR++ to a helical rise of 87.5 Å and a rotation between subunits of 27.4°.
(v) Fitting and structure visualization.
Molecular graphics and analyses were performed with the UCSF Chimera package (Resource for Biocomputing, Visualization, and Informatics at UC–San Francisco; supported by NIGMS P41-GM103311) (31). The model/EM map or EM map/EM map fitting was performed by the option fit in map of the volume register. The difference maps were calculated by the vop substract command.
Phage calcium and adsorption assays.
The phage titers were estimated with the standard double-layer method using different concentrations of CaCl2 added to both the bottom and top agars (32). Phage adsorption assays were performed as described previously (33), with the following modifications. One hundred μl of phage (104 PFU/ml) was mixed with 900 μl of bacteria (optical density at 600 nm [OD600] of 0.6 to 0.8). After incubation at 30°C for 10 min, the mixture was centrifuged at 16,000 × g for 1 min. The supernatant then was titrated. The percentage of adsorption was calculated with the formula 100 × [(phage titer in adsorption assay without bacteria − phage titer in supernatant after adsorption assay)/phage titer in adsorption assay without bacteria]. All of the assays were performed in triplicate.
EM map accession numbers.
The EM maps of capsid, connector, tail, and baseplate reconstructions have been deposited at the EMDB under accession codes EMD-12505, EMD-12533, EMD-12532, and EMD-11517, respectively.
RESULTS
Sequence analysis.
We previously identified the genes coding for the portal protein (orf3), the minor capsid protein (orf4), the major capsid protein (MCP; orf6), the tail terminator (TT; orf10), the tape measure protein (TMP; orf16), and the receptor binding protein (orf20) of phage 1358 (Table 1 and Fig. 2). In lactococcal siphophage genomes, the genes coding for components of the baseplate usually are located downstream of the gene coding for the TMP and upstream of the holin gene (14, 34–36). Using the secondary structure software HHpred (37), we identified ORF17 as the distal tail protein (Dit), because it is similar to the Dit of Bacillus siphophage SPP1 (Protein Databank [PDB] entry 2X8K; HHPred probability of 96.9%). ORF18 of phage 1358 is in a genomic position consistent with the gene encoding the tail-associated lysin (Tal) (38), which is a component of the baseplate in myo- and siphophages (39). However, ORF18 exhibits only a weak similarity (HHpred probability of 57%) with Tal of prophage 53 from Listeria welshimeri, and this remote similarity does not involve the generally conserved N terminus (residues 1 to 400). In fact, we hypothesize that ORF19 is the Tal protein. Indeed, ORF19 is similar to a short Tal protein, such as that of lactococcal phage p2 or of coliphage T4 (gp27), and differs from the long Tal proteins of lactococcal phage TP901-1 or Tuc2009 (40). As indicated above, we recently reported the X-ray structure of ORF20, the RBP of phage 1358 (22). HHpred analysis of ORF20 also revealed a striking similarity between its ∼170 first residues and the N-terminal region of the RBP of the lactococcal p2 (936 group) (10–12, 14).
TABLE 1.
Comparison of 1358 structural proteins to those of other characterized siphophages
| Function | ORF no. | No. of amino acids in ORF of phage: |
|||
|---|---|---|---|---|---|
| 1358 | SPP1 | TP901-1 | p2 | ||
| Portal | 3 | 547 | 503 | 452 | 378 |
| Capsid decoration | 5 | 210 | |||
| MCP | 6 | 297 | 324 | 272 | 293 |
| Tail terminator | 10 | 116 | 134 | 129 | 121 |
| MTP | 13 | 493 | 177/266 | 169 | 301 |
| TMP | 16 | 690 | 1,032 | 937 | 999 |
| Dit | 17 | 352 | 253 | 252 | 298 |
| Tal | 18 | 540 | 1,111 | 946 | 375 |
| RBP | 20 | 393 | NAa | 164 | 263 |
NA, not applicable.
FIG 2.
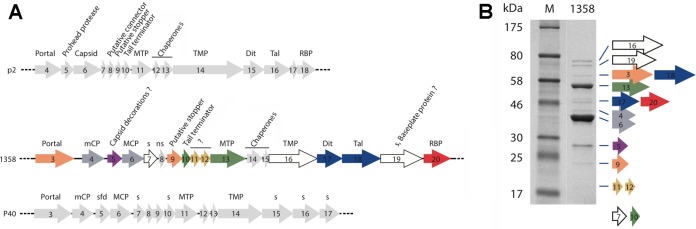
Schematic representation and assignment of the structural gene module of lactococcal phages p2 and 1358 as well as Listeria phage P40. (A) Genes coding for nonstructural ORFs are in light gray. The capsid, decorations, and connector ORFs are in dark gray, violet, and orange, respectively. The genes coding for the tail and baseplate ORFs are green, yellow, blue, and red. MTP, major tail protein; TMP, tail tape measure protein; Dit, distal tail protein; Tal, tail-associated lysine; RBP, receptor binding protein; mCP, minor capsid protein; MCP, major capsid protein; s, structural protein; ns, nonstructural protein; sfd, scaffolding protein. (B) Coomassie blue staining of a 12% SDS-polyacrylamide gel of phage 1358 structural proteins. Numbers on the left indicate the sizes (in kDa) of the proteins in the broad-range molecular mass standard (M). Each protein band corresponding to phage 1358 structural proteins was identified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analyses and are encoded by the color-coded genes shown in panel A. The small ORF7 and ORF10 were not seen in this gel but were detected by LC-MS/MS analyses of the whole phage.
Single-particle electron microscopy structure.
We next reconstructed the complete structure of the 1358 virion by assembling four overlapping regions (capsid, connector complex, tail, and baseplate) on a scaffold (3, 41) (Table 2 and Fig. 3A and B). The 1358 virion is ∼1,715 Å long (Fig. 3C) and comprises the four regions described below, with emphasis on the baseplate.
FIG 3.

Assembled structure of phage 1358. (A) Six-fold-averaged, scaffold structure reconstruction of a complete 1358 phage from a few selected virions exhibiting an almost straight tail, making it possible to obtain the number of MTPs. (B) Superposition of the virion's scaffold structure with the final reassembled structure. (C) This 1358 phage hybrid structure was assembled from separately reconstructed capsid (gray), connector (orange), tail (green), and tail tip (blue) on a low-resolution structure of the phage. The capsid decorations are in violet; 60 of such domains decorate the whole capsid. Dimensions are given in Å.
The capsid.
The full mature capsid of phage 1358 was reconstructed at 16-Å resolution using 9,211 particles with icosahedral symmetry (Table 2 and Fig. 4A to C). The dsDNA-containing mature capsid is ∼640 Å wide along its 5-fold axes and harbors 60 hexamers and 11 pentamers of the MCP (ORF6) organized with a T=7 symmetry and dextro handedness (Fig. 4D and E). A dodecamer of the portal protein (ORF3) occupies the unique vertex (see below). The connector density was averaged out by the icosahedral symmetry reconstruction. Therefore, its structure has been independently reconstructed using boxed pictures of the connector region alone. The capsid cavity is filled with the dsDNA linear genome (36,892 bp), forming concentric layers spaced at intervals of ∼25 Å (Fig. 4A), as observed for other Caudovirales phages (3, 17).
FIG 4.

Cryo-EM reconstruction of phage 1358 capsid. (A) The full mature capsid (the structure is at 16-Å resolution) measures 640 Å along its 5-fold axis. Shown is a cross-section of the capsid reconstruction (surface in blue) showing the external layer of the capsid (orange) and a few layers of the dsDNA genome organized as concentric shells (beige). (Inset) Close-up image of a decorating domain (distances in Å). (B) The icosahedral reconstruction viewed along an icosahedral 2-fold axis with HK97 capsid fit inside. (C) Transparent surface rendering of the capsid. (D) The empty mature capsid structure determined at 10-Å resolution with the dextro handedness and the HK97 MCP hexamer fitted in the map. (Inset) Close-up of the MCP hexamer. (E) The capsid with the laevo handedness. (Inset) Close-up of the MCP hexamer.
The structures of several phage MCPs have been described, and they exhibit a conserved fold, first described for the coliphage HK97 (42, 43), which is also shared by herpesviruses and some archaeal viruses (17, 39, 44). We could fit the crystal structure of the MCP from phage HK97 (PDB entry 1OHG) into the cryo-EM map of phage 1358 capsid, taking into account icosahedral symmetry (45), leading to a pseudoatomic model of the capsid which surprisingly appeared to have dextro handedness (Fig. 4C). All known phage capsids, except the enterobacteria phage P2 (46), have laevo handedness. To confirm the dextro handedness, we determined the cryo-EM structure of the mature empty capsid at higher resolution (10 Å) (Fig. 4D and E). We fitted the crystal structure of the HK97 MCP hexamer into this map, yielding a correlation coefficient (cc) value of 0.57 (Fig. 4D), while for the mirrored map the cc is only 0.4 (Fig. 4E). The h and k values of the lattice (h = 1, k = 2) positioned in a clockwise manner confirmed that the 1358 capsid has dextro handedness (Fig. 4D).
It is worth noting that 60 hook-shaped domains protrude from the capsid surface, with dimensions of ∼54 by 38 by 24 Å (Fig. 4A, inset). HHpred analysis of ORF5 (210 residues, 24 kDa) reports 89% probability of similarity with a fibrinogen-like structure (PDB entry 3GHG) for the first 110 residues. The gene orf5 is located between the genes coding for a minor capsid protein (mCP; orf4) and the MCP (orf6) on the phage 1358 genome (Fig. 2A). Supporting ORF5 as the capsid decoration is its identification as a structural protein using SDS gels and mass spectrometry and that it is the third most abundant protein in the virion (Fig. 2B). Furthermore, considering the protrusion size and average protein density, its mass fits inside.
Head-to-tail connecting region.
The connector ensures, among other things, the attachment of the phage's capsid to the tail and is located at the unique capsid vertex, replacing a penton motif. The connector often is composed of three ORFs, forming successive rings from the capsid interior to the exterior: the portal protein, the head-to-tail connector, and the stopper. The portal, a dodecameric protein involved in DNA packaging during phage assembly and DNA release at the onset of infection, shares a conserved fold in tailed phages and even in herpesviruses (17, 39). The portal of phage 1358 (ORF3; 547 residues) is comparable in length to Bacillus siphophage SPP1 portal gp6 (503 residues), and they share 44% amino acid sequence identity. We reconstructed the connector of phage 1358 at 17-Å resolution, using 4,994 particles and applying the recognized 12-fold symmetry along the connector axis (Fig. 5A to C). We then fitted the models of dodecameric portals present in the PDB into the region of the connector within the capsid of the lactococcal phage 1358. The best-fit correlation coefficient was obtained with the atomic model of SPP1 portal gp6 (PDB entry 2JES) (Fig. 5D). Using SPP1 as a model, the rest of the connector reconstruction should account for the two rings of the head-completion proteins (gp15 and gp16), but no candidates with sequence identity could be found for these proteins in the 1358 genome. However, it appears that there is not enough room for the two rings but only enough for the stopper (Fig. 5B and D).
FIG 5.

Cryo-EM reconstruction of the 1358 connector (17-Å resolution) and tail (16-Å resolution). (A) The 12-fold-averaged connector (orange) within the capsid (gray). The tail terminator is in green. (B) Side-view cross-section of the connector comprising the portal and the putative stopper (orange/light gray). The tail terminator (TT; green and medium gray), belonging to the tail, contacts the stopper. (C) Top view of the connector tube, closed by the stopper (bottom). (D) Side-view cross-section of the connector tube, with the docked structure of a dodecameric portal of phage SPP1. (E) Six-fold-averaged reconstruction of the virion tail (10 segments). Note the decorations protruding on each side of the tail central tube. A segment (yellow) is identified at position 2. (F and G) Tail segment side (F) and top (G) views.
The tail.
In order to construct the full-length structure of the tail, we applied a two-step procedure previously used with lactococcal phage TP901-1 (3) and mycobacteriophage Araucaria (41). In the first step, we produced a 6-fold-averaged reconstruction of the whole phage from a selection of virions with a straight tail (Fig. 3A). We used this reconstruction to measure the tail tube and count the number of stacked rings, comprising a tail terminator hexamer and several MTP (ORF13) hexamers forming the tube. In a second step, we boxed short tail segments that we combined in one data set. Finally, we processed this data set with the appropriate helical symmetry at 16-Å resolution using 3641 particles (Table 2 and Fig. 3B and C).
The phage 1358 tail extends over 875 Å (Fig. 3C and 4E) between the tail terminator ring (40 Å) and the baseplate, making it one of the shortest Siphoviridae tails. It counts only 10 repeat motifs and exhibits an original structure. Each motif is about twice the thickness of a classical MTP stack; hence, they might be formed by a stacking of two MTP hexameric rings. Outside the central core of ∼70 to 100 Å in diameter, large decorations of ∼80 Å (one per MTP monomer) protrude out of the tail tube (Fig. 5F and G). One branch is pointing downwards (∼30° relative to the horizontal) while a second branch is pointing upwards, almost vertically (Fig. 5E to G). The MTP hexameric stacks and decorations have a pitch angle of 27.4° and an interrepeat distance of ∼87 Å (Fig. 5E).
Finally, a 34-Å-wide central channel is present in the center of the tail tube, aligned with the connector and baseplate channels, forming the dsDNA genome ejection pathway (17, 47–51) (Fig. 5G). Only a weak density was observed inside the tail channel, which in Siphoviridae virions is presumed to be filled by the TMP (3, 41, 51, 52), probably due to a mix of TMP filled and empty tails observed in phage preparations (see below).
The baseplate.
The 2D analysis of the phage 1358 host-adsorption device in several particles (free in solution) clearly showed heterogeneity. At least two general classes could be assigned, one in which the baseplate appears to be in an open conformation and one in which it appears closed. We selected a representative class and built two initial 3D models imposing C6 symmetry corresponding to the baseplate in its open and closed conformations. Consequently, our data set included two subsets, 1,580 particles belonging to the open and 2,415 to the closed conformations. Refinement of the closed reconstruction (larger class of particles) of phage 1358 baseplate was performed to 24-Å resolution (Table 2 and Fig. 6A to E). The overall dimensions of the baseplate are 250 Å (width) by 160 Å (high) (Fig. 3C and 5A). The periphery of the baseplate displays six elongated electron densities with a quasi-3-fold symmetry, reminiscent of the phage p2 RBPs within their baseplate (14). We also performed refinement of the open form of the baseplate at ∼40-Å resolution. The low resolution perhaps is due to flexibility within this class (Table 2 and Fig. 6F).
FIG 6.

Twenty-four-Å-resolution cryo-EM reconstruction of the 1358 phage baseplate. (A) The 24-Å cryo-EM electron density map of the closed form of the 1358 baseplate together with the two last segments of the tail. (B) Six 1358 RBP trimer X-ray structures (red) and the phage p2 X-ray structure of ORF15/ORF16 complex (Dit/Tal; blue) have been fitted in the baseplate electron density map. The last tail segment is identified in green. (C) Baseplate bottom view at 90° from the structures shown in panel B. Note how the Dit arms contact the RBP shoulder domains at the center of the groove formed at the 3-fold axis (white arrow, denoted 1, at the top). (D) Close-up view of the baseplate with RBP models fitted (red and light blue), the Dit/Tal complex (dark blue), as well as the EM density of the last tail segment (green). The three contacts of each RBP are numbered 1 to 3 (white arrows). (E) Cross-section of the baseplate corresponding to the view from previous panels (RBPs are red only). Note that there is a nonmodeled zone (white arrows in the middle) between the last tail segment (green) and the Dit/Tal complex (blue). (F) EM density map of the open form of the 1358 baseplate together with the two last segments of the tail. (G) Six trimers of 1358 RBP X-ray structures (red) and the phage p2 X-ray structure of ORF15/ORF16 complex (Dit/Tal; blue) have been fitted in the baseplate electron density map. (H) Same as panel B, with the six RBPs in the closed form superimposed. The last tail segment is identified in green. Note the ∼180° rotation of the RBPs (highlighted by the white arrow).
Opening of the 1358 baseplate.
We were able to fit the trimeric RBP structure (ORF20) of phage 1358 (22) into the baseplate EM map of the closed conformation. We successively assigned six RBPs trimers (18× ORF20) to their electron densities (Fig. 6B and C). We noticed a well-defined electron density below the RBP shoulder domain, a feature suggesting the attachment to another phage structural protein as observed in the baseplate of lactococcal phage p2 (14). Together with the HHpred analysis (see above), this observation prompted us to fit 6× ORF15 as well as 3× ORF16 (Dit/Tal complex) of phage p2 baseplate into the phage 1358 EM density. We noticed that the p2 ORF15 arm/hand extensions, which attach to the p2 RBPs (ORF18), fit well into the above-mentioned electron density (Fig. 6C). The rest of the complex also fits well (cc of 0.91), aside from a symmetry mismatch of the p2 trimeric ORF16 and despite the lack of sequence identity between 1358 ORF18 and other Tal proteins. At the other ends, the C-terminal/head domains of the RBP trimers establish a contact with the lower decorations belonging to the distal tail module (Fig. 6B and D). However, taking together the distal tail module, the RBPs, and the Dit/Tal complex, the EM map of the 1358 baseplate is filled only partially, because an unassigned density is still observed between the distal tail module and the Dit/Tal complex (Fig. 6E, double white arrows).
Even though the resolution of the EM map of the baseplate in the open conformation is very low (40 Å using 1,580 particles) (Table 2), it is clearly less compact than the closed form, as large volumes of electron density protrude outside the baseplate core (Fig. 6F). We tentatively assigned these densities to the RBPs. We could fit the six RBPs satisfactorily (cc of 0.80), as well as the Dit/Tal complex, in the EM map (Fig. 6G). Superimposing the closed and open baseplate classes illustrates a large conformational change of the RBPs which have rotated by ∼180° (Fig. 6H). This RBP rotation in phage 1358 is comparable to the conformational change observed in phage p2 baseplate (14).
The conformational change of phage p2 BP was observed in the presence of Ca2+ ions (17). Therefore, we analyzed the adsorption and infectivity of phage 1358 in the absence and presence of increasing concentrations of Ca2+ ions. In the absence of Ca2+ ions, phage 1358 adsorption occurs, but no completion of its lytic cycle was observed (Fig. 7). However, new virions were produced with Ca2+ ions, up to a plateau reached at 5 mM Ca2+ (Fig. 7).
FIG 7.
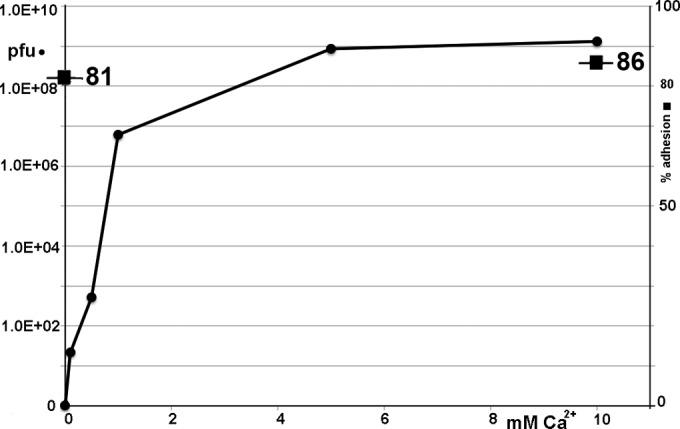
Influence of Ca2+ ions on phage 1358 adsorption and infection of its host strain. Titer of phage 1358 lysate when plated on its host strain, L. lactis SMQ-388, in the absence or presence of increasing concentrations of Ca2+ ions (●). Percent adsorption to the L. lactis host strain with or without 10 mM Ca2+ ions (◼) is given.
DISCUSSION
The Lactococcus phage 1358 genome has been shown previously to resemble those of Listeria phages P35 and P40 (8, 23). However, the gene organization of its morphogenesis module also is very similar to that of siphophage p2, a well-studied lactococcal phage (936 group). Thanks to this similarity, we readily annotated functions to several 1358 proteins. Still, the 1358 virion exhibits several peculiarities (Fig. 8). The capsid has protruding decorations of ∼54 by 38 by 24 Å, dimensions which account for a protein of 200 to 250 amino acids. The MCP (ORF6), being within the average sizes of other MCPs (Table 1), likely is not involved in this decoration. However, orf5, located next to the mcp gene and coding for a 210-residue structural protein, appears to be the best candidate. ORF5, with 60 U in the 1358 virion, is the third most abundant structural protein based on SDS gels (Fig. 2B). HHpred analyses also showed that ORF5 belongs to the fibrinogen family, and protruding decorations with an Ig-like fold have been observed in coliphage T4 (Myoviridae), extending 60 Å out of the capsid (53). Capsid decorations also have been observed by cryo-EM for Bacillus phage phi29 (Podoviridae; gp8) and Escherichia coli siphophage T5 (pb10) (54, 55). It has been postulated that these decorations are involved in nonspecific adhesion to the host (56–58), as phage Ig-like domains bind variable glycan residues. Bacillus siphophage SPP1 also possesses three small decorations (35 by 15 Å) per hexamer; however, their roles remain undocumented (59). In contrast to phage 1358, the two other lactococcal phages of known structure, p2 and TP901-1, do not exhibit such capsid decorations (Fig. 8). No equivalent of orf5 was identified in either of their genomes. Thus, we also propose that the capsid decorations of phage 1358 perform initial attachment to the host surface polysaccharides. In further support of this hypothesis, we recently showed that the cell surface of its host is covered by a polysaccharide pellicle (22).
FIG 8.

Structural comparison of the three lactococcal phages of known structures. From left to right, phages TP901-1 (3), p2 (19), and 1358. Phage p2 and 1358 activated open baseplates are displayed in the insets.
The tail of phage 1358 is 875 Å long and is significantly shorter than the tails of other lactococcal siphophages, such as p2 (1,160 Å) and TP901-1 (1,180 Å) (Fig. 8). Striking features of the tail of phage 1358 are the number and size of repeat units as well as its decorations (Fig. 5E). The size of each repeat (87.5 Å) is about twice the thickness of the MTP hexameric ring (38 Å) found in siphophage TP901-1 (3). We postulate that each repeat is formed of two hexameric MTPs stacked together, and that the decorations arise from the C terminus of the MTP (ORF13) which is formed of 493 residues compared to the 169 residues of the MTP of phage TP901-1 (3). It is worth noting that ORF13 of phage 1358 is the second most abundant protein in the virion and exhibits a unique band on SDS gels (Fig. 2B). The upper and lower MTPs exhibit different orientations, and the EM density is broken between the tail core and the upper decoration, a sign of possible static disorder (Fig. 5E). Decorations also have been identified in the tail of other phages, e.g., SPP1 (51), λ (57), Araucaria (41), and p2 (19). They have been postulated to participate in nonspecific and reversible adhesion of the phage to its host surface polysaccharides, as is the case for the capsid decorations. In favor of this adhesion hypothesis, removing the decorations of phage λ resulted in a 100-fold decrease in infectivity (57). However, the presence of tail decorations is not a general feature, as they are not observed in phage TP901-1 (Fig. 8).
The baseplate of phage 1358 gathers six trimeric RBPs that partly resemble those of phage p2, which are held by a similar Dit arm extension. The EM map of the baseplate extremity is well satisfied by the fitting of the Dit6Tal3 complex of phage p2 despite the larger size (∼40%) of these proteins in phage 1358 (Table 1). Still, two marked differences are observed. The fitting of the 6 RBPs and the Dit6Tal3 complex is not sufficient to fully account for the EM map. A residual nonassigned density is observed between the last tail module and the Dit hexamer (Fig. 6E). However, this density might be ascribed to a second Dit protein, as in the case of phage p2 (14). The second difference deals with the way in which the RBPs are held in the closed conformation of the baseplate. In lactococcal phage p2, the RBP shoulder domains are held by the lower Dit arm, and the head domains are held by the Dit arms from a second, upper Dit hexamer. Here, the head domain of the RBP is in contact with the decorations of the last tail module, which seems to block the RBP in an upward direction (toward the capsid).
Of interest, we also observed a large baseplate conformational change, the open form of phage 1358. The ∼180° rotation of the RBPs is reminiscent of the comparable phenomenon (200° rotation) observed in phage p2 baseplate activation (Fig. 8). While in p2 the baseplate conformation was provoked or stabilized by Ca2+, the open conformation form in phage 1358 is observed concomitantly with the closed form and does not require Ca2+ in vitro. This observation is in agreement with in vivo experiments that clearly indicate that Ca2+ is not necessary for phage adsorption but to complete its lytic cycle.
From our results emerges a complex putative mechanism of host recognition and infection based on a unique combination of capsid and tail decorations. Both the capsid and the tail of phage 1358 possess large numbers of decorations, 60 and 120, respectively. From our analyses and other published data, these decorations might be involved in the first, nonspecific step of host recognition, positioning the phage close to its receptor (51, 53–55, 57). A second step, allowing tight and specific binding of the phage to the host polysaccharide pellicle, likely involved the baseplate in a closed conformation. Finally, in a third step, the firm attachment to the host surface would lead to the opening of the baseplate, signaling to the connector and leading to DNA ejection. Considering the clear evolutionary link of Lactococcus phage 1358 with some Listeria phages, our results also suggest that the mechanism of baseplate conformational change is as widespread in saccharide-adhering Siphoviridae as it is in Myoviridae.
ACKNOWLEDGMENTS
This work was supported by grants from the Agence Nationale de la Recherche (grants ANR-11-BSV8-004-01 “Lactophages” and French Infrastructure for Integrated Structural Biology [FRISBI]). The IGBMC electron microscope facility is supported by the Alsace Region, the Fondation pour la Recherche Médicale, INSERM, CNRS, and the Association pour la Recherche sur le Cancer. S.M. acknowledges funding from NSERC of Canada (strategic program). S.M. holds a Tier 1 Canada Research Chair in Bacteriophages.
Footnotes
Published ahead of print 28 May 2014
REFERENCES
- 1.Ackermann HW. 2012. Bacteriophage electron microscopy. Adv. Virus Res. 82:1–32. 10.1016/B978-0-12-394621-8.00017-0 [DOI] [PubMed] [Google Scholar]
- 2.Katsura I. 1987. Determination of bacteriophage lambda tail length by a protein ruler. Nature 327:73–75. 10.1038/327073a0 [DOI] [PubMed] [Google Scholar]
- 3.Bebeacua C, Lai L, Vegge CS, Brondsted L, van Heel M, Veesler D, Cambillau C. 2013. Visualizing a complete Siphoviridae member by single-particle electron microscopy: the structure of lactococcal phage TP901-1. J. Virol. 87:1061–1068. 10.1128/JVI.02836-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kostyuchenko VA, Chipman PR, Leiman PG, Arisaka F, Mesyanzhinov VV, Rossmann MG. 2005. The tail structure of bacteriophage T4 and its mechanism of contraction. Nat. Struct. Mol. Biol. 12:810–813. 10.1038/nsmb975 [DOI] [PubMed] [Google Scholar]
- 5.Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B, Johnson JE. 2006. The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science 312:1791–1795. 10.1126/science.1127981 [DOI] [PubMed] [Google Scholar]
- 6.Deveau H, Labrie SJ, Chopin MC, Moineau S. 2006. Biodiversity and classification of lactococcal phages. Appl. Environ. Microbiol. 72:4338–4346. 10.1128/AEM.02517-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahony J, Martel B, Tremblay DM, Neve H, Heller KJ, Moineau S, van Sinderen D. 2013. Molecular analysis of lactococcal phages Q33 and BM13: identification of a new P335 subgroup. Appl. Environ. Microbiol. 79:4401–4409. 10.1128/AEM.00832-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dupuis ME, Moineau S. 2010. Genome organization and characterization of the virulent lactococcal phage 1358 and its similarities to Listeria phages. Appl. Environ. Microbiol. 76:1623–1632. 10.1128/AEM.02173-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spinelli S, Veesler D, Bebeacua C, Cambillau C. 2014. Structures and host-adhesion mechanisms of lactococcal siphophages. Front. Microbiol. 5:3. 10.3389/fmicb.2014.00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spinelli S, Desmyter A, Verrips CT, de Haard HJ, Moineau S, Cambillau C. 2006. Lactococcal bacteriophage p2 receptor-binding protein structure suggests a common ancestor gene with bacterial and mammalian viruses. Nat. Struct. Mol. Biol. 13:85–89. 10.1038/nsmb1029 [DOI] [PubMed] [Google Scholar]
- 11.Tremblay DM, Tegoni M, Spinelli S, Campanacci V, Blangy S, Huyghe C, Desmyter A, Labrie S, Moineau S, Cambillau C. 2006. Receptor-binding protein of Lactococcus lactis phages: identification and characterization of the saccharide receptor-binding site. J. Bacteriol. 188:2400–2410. 10.1128/JB.188.7.2400-2410.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siponen M, Spinelli S, Blangy S, Moineau S, Cambillau C, Campanacci V. 2009. Crystal structure of a chimeric receptor binding protein constructed from two lactococcal phages. J. Bacteriol. 191:3220–3225. 10.1128/JB.01637-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siponen M, Sciara G, Villion M, Spinelli S, Lichiere J, Cambillau C, Moineau S, Campanacci V. 2009. Crystal structure of ORF12 from Lactococcus lactis phage p2 identifies a tape measure protein chaperone. J. Bacteriol. 191:728–734. 10.1128/JB.01363-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sciara G, Bebeacua C, Bron P, Tremblay D, Ortiz-Lombardia M, Lichiere J, van Heel M, Campanacci V, Moineau S, Cambillau C. 2010. Structure of lactococcal phage p2 baseplate and its mechanism of activation. Proc. Natl. Acad. Sci. U. S. A. 107:6852–6857. 10.1073/pnas.1000232107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veesler D, Dreier B, Blangy S, Lichiere J, Tremblay D, Moineau S, Spinelli S, Tegoni M, Pluckthun A, Campanacci V, Cambillau C. 2009. Crystal structure and function of a darpin neutralizing inhibitor of lactococcal phage TP901-1: comparison of darpin and camelid VHH binding mode. J. Biol. Chem. 284:30718–30726. 10.1074/jbc.M109.037812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spinelli S, Campanacci V, Blangy S, Moineau S, Tegoni M, Cambillau C. 2006. Modular structure of the receptor binding proteins of Lactococcus lactis phages. The RBP structure of the temperate phage TP901-1. J. Biol. Chem. 281:14256–14262. 10.1074/jbc.M600666200 [DOI] [PubMed] [Google Scholar]
- 17.Veesler D, Spinelli S, Mahony J, Lichiere J, Blangy S, Bricogne G, Legrand P, Ortiz-Lombardia M, Campanacci V, van Sinderen D, Cambillau C. 2012. Structure of the phage TP901-1 1.8 MDa baseplate suggests an alternative host adhesion mechanism. Proc. Natl. Acad. Sci. U. S. A. 109:8954–8958. 10.1073/pnas.1200966109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desmyter A, Farenc C, Mahony J, Spinelli S, Bebeacua C, Blangy S, Veesler D, van Sinderen D, Cambillau C. 2013. Viral infection modulation and neutralization by camelid nanobodies. Proc. Natl. Acad. Sci. U. S. A. 110:E1371–E1379. 10.1073/pnas.1301336110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bebeacua C, Tremblay D, Farenc C, Chapot-Chartier MP, Sadovskaya I, van Heel M, Veesler D, Moineau S, Cambillau C. 2013. Structure, adsorption to host, and infection mechanism of virulent lactococcal phage p2. J. Virol. 87:12302–12312. 10.1128/JVI.02033-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chapot-Chartier MP, Vinogradov E, Sadovskaya I, Andre G, Mistou MY, Trieu-Cuot P, Furlan S, Bidnenko E, Courtin P, Pechoux C, Hols P, Dufrene YF, Kulakauskas S. 2010. Cell surface of Lactococcus lactis is covered by a protective polysaccharide pellicle. J. Biol. Chem. 285:10464–10471. 10.1074/jbc.M109.082958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ainsworth S, Sadovskaya I, Vinogradov E, Courtin P, Guerardel Y, Mahony J, Grard T, Cambillau C, Chapot-Chartier MP, van Sinderen D. 2014. Differences in lactococcal cell wall polysaccharide structure are major determining factors in bacteriophage sensitivity. mBio 5:e00880–14. 10.1128/mBio.00880-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farenc C, Spinelli S, Vinogradov E, Tremblay D, Blangy S, Sadovskaya I, Moineau S, Cambillau C. 2014. Molecular insights on the recognition of a Lactococcus lactis cell wall pellicle by a phage receptor binding protein. J. Virol. 88:7005–7015. 10.1128/JVI.00739-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorscht J, Klumpp J, Bielmann R, Schmelcher M, Born Y, Zimmer M, Calendar R, Loessner MJ. 2009. Comparative genome analysis of Listeria bacteriophages reveals extensive mosaicism, programmed translational frameshifting, and a novel prophage insertion site. J. Bacteriol. 191:7206–7215. 10.1128/JB.01041-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. 2007. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157:38–46. 10.1016/j.jsb.2006.05.009 [DOI] [PubMed] [Google Scholar]
- 25.Shaikh TR, Gao H, Baxter WT, Asturias FJ, Boisset N, Leith A, Frank J. 2008. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nat. Protoc. 3:1941–1974. 10.1038/nprot.2008.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scheres SH. 2010. Classification of structural heterogeneity by maximum-likelihood methods. Methods Enzymol. 482:295–320. 10.1016/S0076-6879(10)82012-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scheres SH, Nunez-Ramirez R, Sorzano CO, Carazo JM, Marabini R. 2008. Image processing for electron microscopy single-particle analysis using XMIPP. Nat. Protoc. 3:977–990. 10.1038/nprot.2008.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Heel M, Schatz M. 2005. Fourier shell correlation threshold criteria. J. Struct. Biol. 151:250–262. 10.1016/j.jsb.2005.05.009 [DOI] [PubMed] [Google Scholar]
- 29.Egelman EH. 2007. The iterative helical real space reconstruction method: surmounting the problems posed by real polymers. J. Struct. Biol. 157:83–94. 10.1016/j.jsb.2006.05.015 [DOI] [PubMed] [Google Scholar]
- 30.Owen CH, Morgan DG, DeRosier DJ. 1996. Image analysis of helical objects: the Brandeis helical package. J. Struct. Biol. 116:167–175. 10.1006/jsbi.1996.0027 [DOI] [PubMed] [Google Scholar]
- 31.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 32.Terzaghi BE, Sandine WE. 1975. Improved medium for lactic streptococci and their bacteriophages. Appl. Microbiol. 29:807–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanders ME, Klaenhammer TR. 1980. Restriction and modification in group N streptococci: effect of heat on development of modified lytic bacteriophage. Appl. Environ. Microbiol. 40:500–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rousseau GM, Moineau S. 2009. Evolution of Lactococcus lactis phages within a cheese factory. Appl. Environ. Microbiol. 75:5336–5344. 10.1128/AEM.00761-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahony J, van Sinderen D. 2012. Structural aspects of the interaction of dairy phages with their host bacteria. Viruses 4:1410–1424. 10.3390/v4091410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campanacci V, Veesler D, Lichiere J, Blangy S, Sciara G, Moineau S, van Sinderen D, Bron P, Cambillau C. 2010. Solution and electron microscopy characterization of lactococcal phage baseplates expressed in Escherichia coli. J. Struct. Biol. 172:75–84. 10.1016/j.jsb.2010.02.007 [DOI] [PubMed] [Google Scholar]
- 37.Soding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33:W244–W248. 10.1093/nar/gki408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kenny JG, McGrath S, Fitzgerald GF, van Sinderen D. 2004. Bacteriophage Tuc2009 encodes a tail-associated cell wall-degrading activity. J. Bacteriol. 186:3480–3491. 10.1128/JB.186.11.3480-3491.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Veesler D, Cambillau C. 2011. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol. Mol. Biol. Rev. 75:423–433. 10.1128/MMBR.00014-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stockdale SR, Mahony J, Courtin P, Chapot-Chartier MP, van Pijkeren JP, Britton RA, Neve H, Heller KJ, Aideh B, Vogensen FK, van Sinderen D. 2013. The lactococcal phages Tuc2009 and TP901-1 incorporate two alternate forms of their tail fiber into their virions for infection specialization. J. Biol. Chem. 288:5581–5590. 10.1074/jbc.M112.444901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sassi M, Bebeacua C, Drancourt M, Cambillau C. 2013. The first structure of a mycobacteriophage, the Mycobacterium abscessus subsp. bolletii phage araucaria. J. Virol. 87:8099–8109. 10.1128/JVI.01209-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wikoff WR, Conway JF, Tang J, Lee KK, Gan L, Cheng N, Duda RL, Hendrix RW, Steven AC, Johnson JE. 2006. Time-resolved molecular dynamics of bacteriophage HK97 capsid maturation interpreted by electron cryo-microscopy and X-ray crystallography. J. Struct. Biol. 153:300–306. 10.1016/j.jsb.2005.11.009 [DOI] [PubMed] [Google Scholar]
- 43.Helgstrand C, Wikoff WR, Duda RL, Hendrix RW, Johnson JE, Liljas L. 2003. The refined structure of a protein catenane: the HK97 bacteriophage capsid at 3.44 A resolution. J. Mol. Biol. 334:885–899. 10.1016/j.jmb.2003.09.035 [DOI] [PubMed] [Google Scholar]
- 44.Wikoff WR, Liljas L, Duda RL, Tsuruta H, Hendrix RW, Johnson JE. 2000. Topologically linked protein rings in the bacteriophage HK97 capsid. Science 289:2129–2133. 10.1126/science.289.5487.2129 [DOI] [PubMed] [Google Scholar]
- 45.Jaroszewski L, Li Z, Cai XH, Weber C, Godzik A. 2011. FFAS server: novel features and applications. Nucleic Acids Res. 39:W38–W44. 10.1093/nar/gkr441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dearborn AD, Laurinmaki P, Chandramouli P, Rodenburg CM, Wang S, Butcher SJ, Dokland T. 2012. Structure and size determination of bacteriophage P2 and P4 procapsids: function of size responsiveness mutations. J. Struct. Biol. 178:215–224. 10.1016/j.jsb.2012.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goulet A, Lai-Kee-Him J, Veesler D, Auzat I, Robin G, Shepherd DA, Ashcroft AE, Richard E, Lichiere J, Tavares P, Cambillau C, Bron P. 2011. The opening of the SPP1 bacteriophage tail, a prevalent mechanism in Gram-positive-infecting siphophages. J. Biol. Chem. 286:25397–25405. 10.1074/jbc.M111.243360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Veesler D, Robin G, Lichiere J, Auzat I, Tavares P, Bron P, Campanacci V, Cambillau C. 2010. Crystal structure of bacteriophage SPP1 distal tail protein (gp19.1): a baseplate hub paradigm in gram-positive infecting phages. J. Biol. Chem. 285:36666–36673. 10.1074/jbc.M110.157529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lebedev AA, Krause MH, Isidro AL, Vagin AA, Orlova EV, Turner J, Dodson EJ, Tavares P, Antson AA. 2007. Structural framework for DNA translocation via the viral portal protein. EMBO J. 26:1984–1994. 10.1038/sj.emboj.7601643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lhuillier S, Gallopin M, Gilquin B, Brasiles S, Lancelot N, Letellier G, Gilles M, Dethan G, Orlova EV, Couprie J, Tavares P, Zinn-Justin S. 2009. Structure of bacteriophage SPP1 head-to-tail connection reveals mechanism for viral DNA gating. Proc. Natl. Acad. Sci. U. S. A. 106:8507–8512. 10.1073/pnas.0812407106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Plisson C, White HE, Auzat I, Zafarani A, Sao-Jose C, Lhuillier S, Tavares P, Orlova EV. 2007. Structure of bacteriophage SPP1 tail reveals trigger for DNA ejection. EMBO J. 26:3720–3728. 10.1038/sj.emboj.7601786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pedersen M, Ostergaard S, Bresciani J, Vogensen FK. 2000. Mutational analysis of two structural genes of the temperate lactococcal bacteriophage TP901-1 involved in tail length determination and baseplate assembly. Virology 276:315–328. 10.1006/viro.2000.0497 [DOI] [PubMed] [Google Scholar]
- 53.Fokine A, Chipman PR, Leiman PG, Mesyanzhinov VV, Rao VB, Rossmann MG. 2004. Molecular architecture of the prolate head of bacteriophage T4. Proc. Natl. Acad. Sci. U. S. A. 101:6003–6008. 10.1073/pnas.0400444101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Effantin G, Boulanger P, Neumann E, Letellier L, Conway JF. 2006. Bacteriophage T5 structure reveals similarities with HK97 and T4 suggesting evolutionary relationships. J. Mol. Biol. 361:993–1002. 10.1016/j.jmb.2006.06.081 [DOI] [PubMed] [Google Scholar]
- 55.Morais MC, Choi KH, Koti JS, Chipman PR, Anderson DL, Rossmann MG. 2005. Conservation of the capsid structure in tailed dsDNA bacteriophages: the pseudoatomic structure of phi29. Mol. Cell 18:149–159. 10.1016/j.molcel.2005.03.013 [DOI] [PubMed] [Google Scholar]
- 56.Fraser JS, Yu Z, Maxwell KL, Davidson AR. 2006. Ig-like domains on bacteriophages: a tale of promiscuity and deceit. J. Mol. Biol. 359:496–507. 10.1016/j.jmb.2006.03.043 [DOI] [PubMed] [Google Scholar]
- 57.Pell LG, Gasmi-Seabrook GM, Morais M, Neudecker P, Kanelis V, Bona D, Donaldson LW, Edwards AM, Howell PL, Davidson AR, Maxwell KL. 2010. The solution structure of the C-terminal Ig-like domain of the bacteriophage lambda tail tube protein. J. Mol. Biol. 403:468–479. 10.1016/j.jmb.2010.08.044 [DOI] [PubMed] [Google Scholar]
- 58.Davidson AR, Cardarelli L, Pell LG, Radford DR, Maxwell KL. 2012. Long noncontractile tail machines of bacteriophages. Adv. Exp. Med. Biol. 726:115–142. 10.1007/978-1-4614-0980-9_6 [DOI] [PubMed] [Google Scholar]
- 59.White HE, Sherman MB, Brasiles S, Jacquet E, Seavers P, Tavares P, Orlova EV. 2012. Capsid structure and its stability at the late stages of bacteriophage SPP1 assembly. J. Virol. 86:6768–6777. 10.1128/JVI.00412-12 [DOI] [PMC free article] [PubMed] [Google Scholar]