

ABSTRACT
Bracoviruses (BVs) from the Polydnaviridae family are symbiotic viruses used as biological weapons by parasitoid wasps to manipulate lepidopteran host physiology and induce parasitism success. BV particles are produced by wasp ovaries and injected along with the eggs into the caterpillar host body, where viral gene expression is necessary for wasp development. Recent sequencing of the proviral genome of Cotesia congregata BV (CcBV) identified 222 predicted virulence genes present on 35 proviral segments integrated into the wasp genome. To date, the expressions of only a few selected candidate virulence genes have been studied in the caterpillar host, and we lacked a global vision of viral gene expression. In this study, a large-scale transcriptomic analysis by 454 sequencing of two immune tissues (fat body and hemocytes) of parasitized Manduca sexta caterpillar hosts allowed the detection of expression of 88 CcBV genes expressed 24 h after the onset of parasitism. We linked the expression profiles of these genes to several factors, showing that different regulatory mechanisms control viral gene expression in the host. These factors include the presence of signal peptides in encoded proteins, diversification of promoter regions, and, more surprisingly, gene position on the proviral genome. Indeed, most genes for which expression could be detected are localized in particular proviral regions globally producing higher numbers of circles. Moreover, this polydnavirus (PDV) transcriptomic analysis also reveals that a majority of CcBV genes possess at least one intron and an arthropod transcription start site, consistent with an insect origin of these virulence genes.
IMPORTANCE Bracoviruses (BVs) are symbiotic polydnaviruses used by parasitoid wasps to manipulate lepidopteran host physiology, ensuring wasp offspring survival. To date, the expressions of only a few selected candidate BV virulence genes have been studied in caterpillar hosts. We performed a large-scale analysis of BV gene expression in two immune tissues of Manduca sexta caterpillars parasitized by Cotesia congregata wasps. Genes for which expression could be detected corresponded to genes localized in particular regions of the viral genome globally producing higher numbers of circles. Our study thus brings an original global vision of viral gene expression and paves the way to the determination of the regulatory mechanisms enabling the expression of BV genes in targeted organisms, such as major insect pests. In addition, we identify sequence features suggesting that most BV virulence genes were acquired from insect genomes.
INTRODUCTION
Polydnaviruses (PDVs) are symbiotic viruses produced by parasitoid wasps (Hymenoptera) that are essential for the parasitic success of these insects (1–3). Viral replication and particle production occur in the wasp ovaries. The particles constitute the major component of the fluid injected with the eggs into the parasitized caterpillar host during wasp oviposition. These particles enter lepidopteran host cells, and genes harbored by the double-stranded DNA (dsDNA) contained in the particles are expressed by the host cellular machinery. Viral products ensure wasp survival in the lepidopteran larvae by interfering with caterpillar host immune responses and development (4–7).
PDVs are associated with over 30,000 parasitoid wasp species from two families, Braconidae and Ichneumonidae. These wasp-PDV associations derive from independent ancestral associations between wasps and viruses in a remarkable example of convergent evolution, enabling wasps to face the specific constraints of living within a developing host insect larva (8, 9). In particular, PDVs associated with braconid wasps, named bracoviruses (BVs), were shown to originate from nudiviruses, a sister group of baculoviruses (10–12). Braconid wasps harboring BVs constitute a monophyletic group, and the proposed evolutionary scenario is that the genome of an ancestral nudivirus was captured and integrated into the genome of the ancestor of these wasps approximately 100 million years ago. Since then, the virus has been chromosomally transmitted and is thought to have contributed to the diversity observed today in these particularly species-rich hymenopteran families (13). PDVs associated with ichneumonid wasps, named ichnoviruses (IVs), originate from a different ancestral virus, belonging to a new virus entity having its own characteristic gene set (9).
PDV genomes are thus constituted of two different parts that are both integrated into wasp genomes. The first part is composed of genes involved in particle production (e.g., nudivirus genes in BV) but not packaged in the particles, leading to the injection of a nonreplicative virus in the caterpillar hosts (8, 9). The second part corresponds to proviral segments used to produce multiple dsDNA circles, which are packaged in the particles and contain virulence genes involved in the regulation of caterpillar host physiology (7, 14). Both parts of PDV genomes are amplified during PDV particle production (9, 15). For example, in Cotesia congregata wasps, the 35 Cotesia congregata BV (CcBV) proviral segments are amplified within 12 different molecules, each constituting a replication unit (RU), that are further resolved to give the circles packaged within the particles. Proviral segments are named segment 1 (S1) to S36. After replication and amplification, segments are circularized and become circle 1 (C1) to C36, respectively (except for S34, which does not contain signals for excision) (14). A genomic region containing half of the nudiviral genes is also amplified during viral replication and constitutes a 13th replication unit (15). DNA amplification therefore allows the massive production of both particle protein components and circular DNA molecules that will be injected into the caterpillar.
In the last few years, many BV packaged genomes have been sequenced (16–19), and chromosomal forms (proviral segments) have been obtained for three BVs (14, 20). All sequenced BV genomes share common structural features: they consist of highly segmented large genomes (15 to 35 segments with a total size of 189 to 730 kb) with low coding densities (17 to 33%) harboring numerous genes organized into gene families (14, 21). The recent sequencing of the integrated form of CcBV has revealed that this proviral genome is composed of 35 segments organized in a macrolocus comprising over two-thirds of the proviral segments and seven smaller dispersed loci (14). The macrolocus itself comprises two proviral loci, PL1 and PL2, that are separated by a region containing wasp genes and a nudiviral gene encoding a particle envelope protein. Large duplications contributing to the expansion of gene families have been identified within this macrolocus (14). Bioinformatic analysis of proviral segments combined with expert annotation predicted 222 CDSs (coding DNA sequences) and 29 pseudogenes. An overall total of 183 CcBV genes and 26 pseudogenes belonging to 37 multigenic families and 11 remnants from mobile elements were identified in CcBV segments (14). Only seven of these gene families encode proteins containing eukaryotic conserved domains (PTP, VANK, Cystatin, RNaseT2, BEN, CRP, and C-type lectin), and 29 families are specific to BVs (EP1-like, EP2-like, SRP, and BV family 1 [BV1]to BV26) (14). In contrast to nudiviral genes, which do not contain introns, 60% of the genes present in proviral segments were predicted to contain introns, like cellular genes. This abundance of introns suggests that most of the genes present in proviral segments originated from insect genomes. However, phylogenetic analyses suggesting viral genes could be of hymenopteran origin have so far been performed for only a few genes (20). Indeed, most genes appear to have diverged considerably in their sequence from insect genes (22). The encapsidated genomes of BVs from closely related wasps share many conserved genes (20), whereas distantly related wasps share very few genes (17). These observations led to the hypothesis that the gene content of the injected genome is very much shaped by the physiology that it is confronted with in the lepidopteran host (7, 23). However, due to its large size, the proviral form of BV is also the target of mobile element insertions as any part of the wasp genome. Remnants of known mobile elements (retroelements and Maverick/Polinton) that encode proteins interrupted by stop codons have been detected within CcBV (24, 25), and it is not known whether these elements are expressed in parasitized host tissues.
Transcriptomic analyses of these particular host-parasitoid interactions have so far focused on host genes (26, 27) or on specific virulence genes or multigenic families and have rarely given a global vision of PDV transcription during these interactions (3, 5, 28). Recently, an extensive BV transcriptome profile was reported, which concerned Microplitis demolitor BV (MdBV) expression in Pseudoplusia includens, where the spatial and temporal expressions of most predicted open reading frames (ORFs) of this BV were analyzed by reverse transcription-PCR (RT-PCR) (29). In addition, deep sequencing approaches at the transcriptome level concerned the expression of the Diadegma semiclausum ichnovirus (DsIV) in Plutella xylostella larvae and the expression of Cotesia chilonis bracovirus (CchBV) in the Chilo suppressalis host. In these de novo transcriptomic approaches applied to new model species, the relative contributions of IV and BV gene expression could not be evaluated because the genomes of these PDVs have not yet been sequenced (30, 31).
Here we present a large-scale transcriptome analysis of a BV using a deep sequencing approach on two distinct immune tissues of the lepidopteran host Manduca sexta parasitized by Cotesia congregata wasps harboring CcBV. Both the circular and proviral forms of this BV genome have been sequenced (14, 16). We used the proviral virus genome as a reference, thereby giving a picture of viral genes actually expressed without the bias of studying computationally predicted genes. Expressions of only a limited number of the predicted CDSs (81 out of 222), corresponding to genes localized in particular regions of the proviral genome, were detected in the analyzed tissues with this technique. These results show that genes that are highly expressed are contained in certain proviral regions. We also identified seven new CcBV genes. Moreover, we identified different properties possibly influencing CcBV gene expression, such as (i) the presence or absence of signal peptides in encoded proteins, (ii) the gene position on the proviral genome, (iii) circle abundance in viral particles, and (iv) diversification of promoter regions, illustrating the complexity of BV gene regulation in the host. Finally, this BV transcriptomic analysis also reveals that a majority of CcBV genes possess at least one intron and an arthropod transcription start site, which is consistent with an insect origin of these virulence genes.
MATERIALS AND METHODS
Insects, parasitization, and sample collection.
C. congregata wasps (Hymenoptera: Braconidae) were reared under laboratory conditions on their natural host, the tobacco hornworm, M. sexta (Lepidoptera: Sphingidae). M. sexta larvae were reared on an artificial diet at 27°C under a 16-h-light/8-h-dark photoperiod and 70% ± 5% relative humidity, as previously described (32). Five fourth-instar M. sexta larvae were exposed to C. congregata females until at least two ovipositions were observed on each larva. They were then maintained under the same rearing conditions for 24 h, as were five M. sexta larvae from the same cohort, which were used as controls. The time point of 24 h postoviposition was chosen for expression analysis for consistency with previous studies (33–35) and also because physiological effects on hemocytes are visible at this time point (28). Caterpillars were anesthetized for 10 min on ice before dissection. Two hundred microliters of hemolymph was then collected for each caterpillar. To isolate hemocytes, hemolymph was centrifuged at 1,000 × g for 10 min at 4°C. The supernatant was discarded, and the cell pellet was washed in 100 μl of sterile 1× phosphate-buffered saline (PBS). After centrifugation at 1,000 × g for 10 min at 4°C, the cell pellet was resuspended in 150 μl of NucleoSpin RNA II purification kit RA1 buffer (Macherey-Nagel) containing 1.5 μl β-mercaptoethanol, according to the manufacturer's instructions, and stored at −80°C until RNA isolation. To isolate fat body tissues, 3 mg of fat body was dissected from each individual after hemolymph collection, rinsed in 1× PBS, and then resuspended in 350 μl of RA1 buffer containing 3.5 μl β-mercaptoethanol and stored at −80°C until RNA isolation.
Sample preparation and deep sequencing.
Total RNA was extracted from each sample by using the NucleoSpin RNA II Purification kit according to the user's manual. Sample RNA concentrations were measured by using a spectrophotometer (Varian Cary 50 Scan), and RNA integrity was assessed on a 1% (wt/vol) agarose gel. The absence of DNA contamination in RNA samples was checked by controlling for the lack of amplification of CcBV and M. sexta genes by PCR (cystatin-1 [cyst1] and actin, respectively). The parasitized status of hosts exposed to oviposition by parasitoid females was verified by amplifying PDV cystatin-1 gene transcript sequences from the corresponding RNA samples by RT-PCR (Omniscrip RT kit; Qiagen). Finally, RNA samples corresponding to the same tissues (fat body or hemocytes) and the same conditions (parasitized or control larvae) were pooled.
cDNA libraries were prepared by using a SMARTer PCR cDNA synthesis kit (Clontech). First-strand cDNA synthesis was performed with 3′ SMART CDS Primer II A according to the supplier's protocols and by using 995 and 70 ng of total RNA from fat body and hemocytes, respectively. Double-strand cDNAs were prepared by long-distance PCR (LD-PCR) from 1 and 10 μl (according to the manufacturer's instructions) of first-strand cDNA from fat body and hemocytes, respectively. A pool of three independent LD-PCRs was used for deep sequencing of each cDNA library. Sequencing was performed by using 454 GS FLX Titanium technology (Roche) at the French National Sequencing Institute (Genoscope, France).
Read mapping and assembly.
Reads generated by 454 sequencing were trimmed for poly(A), 454 adaptors (A and B), and PCR primers. The origin of reads was established by mapping onto two reference genomes, M. sexta (Agripestbase database [http://www.agripestbase.org/]) and the CcBV chromosomal form (EMBL accession numbers HF586472 to HF586480) (14). Mapping was performed by using GS Reference Mapper from the Newbler software package (version 2.5.6; Roche) with the following parameters: project, cdna; seed step, 1; seed length, 16; minimum overlap length, 80%; minimum overlap identity, 95%; minimum reads depth, 5. The minimum read depth was set to 5 to exclude genomic DNA contamination. The 454 expression detection limit was therefore set to a minimum read depth of 5. To distinguish and annotate genes belonging to gene families and sharing high sequence similarity, CcBV reads were aligned to the CcBV genome by using Exonerate (36) with the following parameters: model, cdna2genome; gene seed, 100; codon word limit, 1; refine, region. For each cluster of reads aligned with a given genomic region, a consensus sequence was generated by de novo assembly of reads using GS De Novo Assembler from the Newbler software package with the following parameters: project, cdna; maximum number of isotigs in an isogroup, 1; maximum number of contigs in an isogroup, 1; maximum number of contigs in an isotig, 1; seed step, 1; seed length, 16; minimum overlap length, 90%; minimum overlap identity, 80. Whenever overlapping consensus sequences encompassed an entire CDS predicted by FGENESH+ software detection (SoftBerry) on the CcBV integrated genome (14), the corresponding gene was considered to produce full-length mRNAs under our experimental conditions. However, for certain genes, only partial mRNA could be obtained. We used custom-written Perl and Bash scripts to localize and count read abundances for each gene. Expression levels were normalized and are given in reads per kilobase per million (RPKM) according to a previously described method (37). RPKM values were also calculated for the M. sexta rpl3 reference gene. Five pairs of genes (bv1-1/bv1-2, bv5-1/bv5-2, bv7-1/bv7-6, bv7-2/bv7-5, and bv11-1/bv11-2) have two copies with identical nucleotide sequences due to their localization in a recently duplicated region (14), and it was therefore not possible to determine which transcript corresponded to the expression of one gene copy or the other. All assembly outputs were visualized by using Tablet software (38).
Sequence analyses and motif prediction.
All sequences used in this study were manipulated with Geneious version 6.0.3, created by Biomatters, and with the Artemis genome browser and annotation tool (39).
Full-length de novo-assembled cDNA sequences or CDSs predicted from the CcBV integrated genome reference (when incomplete cDNAs were obtained) were translated by using ORF Finder (40). The deduced amino acid sequence of the main ORF encoded by each nucleotide sequence was submitted to the SignalP 4.1 server (41) for signal peptide prediction. Statistical analysis of the relation between expression level and the presence of a signal peptide was performed by using a Wilcoxon-Mann-Whitney test (P value of <0.001) with R statistical software (http://www.r-project.org/).
The region upstream of the 5′ untranslated region (UTR) from each full-length cDNA sequence was submitted to the Multiple Em for Motif Elicitation (MEME) 4.9.0 Web server (42) for the detection of transcription start sites (TSSs). The following parameters were used: distribution of motif occurrences, 0 or 1 per sequence; number of different motifs, 1; minimum motif width, 3; maximum motif width, 7.
Amino acid sequence alignments of the gene products of the BV5 CcBV gene family were performed by using the ClustalOmega program (43). They were converted into codon-based sequence alignments, and phylogenetic analyses were then performed by using the RAxML program (44) with the following substitution models and parameters: general time reversible + gamma + proportion of invariant sites (GTR + G + I). Support for nodes was obtained from 100 bootstrap iterations. Potential transcription factor binding sites of the same genes were searched for within the 1,000 bp upstream of the ATG start codon by using Matinspector from the Genomatix software suite (45). Previous studies on a PDV promoter had indeed suggested that a 1-kb region upstream of the transcription start site contained most regulatory sequences (46). An insect matrix library and standard parameters were used.
Quantitative RT-PCR validation.
Quantitative analysis of gene expression was performed by quantitative RT-PCR (qRT-PCR) on the same fat body RNA samples as those used for 454 sequencing to determine whether the differences observed in the number of reads by transcriptome profiling reflected differences in transcript abundance. We also performed qRT-PCR on 11 CcBV genes (cyst1, cyst2, C-type lectin CcV3 and CcV3-like, viral ankyrin 4 [vank-4], vank-9, protein tyrosine phosphatase p [ptpp], protein tyrosine phosphatase h [ptph], CrV1-like, DUFFY-like, and a unique gene, CcBV_13.2) using RNA samples (hemocytes and fat body) from five newly parasitized larvae (maintained as described above) to determine whether the differences observed were reproducible.
RNA was extracted by using a NucleoSpin RNA II purification kit, concentrations were measured by using a Qubit 2.0 fluorometer (Invitrogen), and RNA integrity was assessed by analysis on an Agilent 2100 Bioanalyzer (Agilent Technologies).
First-strand cDNA was synthesized from either 1 μg of total RNA from 454 sequencing libraries or 250 ng of total RNA extracted from newly parasitized larvae for qRT-PCR. Fifty picograms of kanamycin RNA was added to the reaction mix to serve as an external control and reference. Reverse transcription was performed by using a QuantiTect reverse transcription kit (Qiagen). The qRT-PCR mixture consisted of 2 μl of diluted cDNA, 0.96 μl of primer mix (300 mM), 5 μl of qPCR MasterMix Plus for SYBR assay (Eurogentec), and H2O for a final volume of 10 μl. Reactions were performed in triplicates with an ABI Prism 7000 sequence detection system (Life Technologies) under the following conditions: 50°C for 2 min, 95°C for 10 min, and 40 cycles at 95°C for 15 s and 60°C for 1 min, followed by a dissociation step (60°C to 95°C). Melting curves for each sample were analyzed to check the specificity of amplification and primer efficiency. Expression was measured by using the 2−ΔCT method with efficiency correction for each gene. Normalization was performed by using two reference genes, the internal reference gene rpl3 (endogenous expression) and the external reference gene kanamycin. The significance levels of differential gene expressions were tested by using pairwise Wilcoxon rank sum tests (47) with Benjamini-Hochberg correction (48) in R statistical software. Statistical differences in expression were then compared to 454 RPKM results. On the basis of this comparison, we established different levels of expression in the 454 analysis, with high expression levels corresponding to values equal to and above 25 RPKM, intermediate expression levels corresponding to values of between 5 and 25 RPKM (5 ≤ RPKM < 25), and low expression levels corresponding to values below 5 RPKM.
Detection of expression of genes below the 454 detection limit in different tissues.
Analysis of expression of genes below the 454 detection limit was performed on RNA samples from five tissues: hemocytes, fat body, nervous system, malpighian tubules, and midgut from five newly parasitized larvae (maintained as described above).
RNA was extracted by using the NucleoSpin RNA II purification kit (Macherey-Nagel, France), concentrations were measured by using a Qubit 2.0 fluorometer (Invitrogen), and RNA integrity was assessed by analysis on a 1% agarose gel.
First-strand cDNA was synthesized from 100 ng of total RNA, and reverse transcription was performed by using the QuantiTect reverse transcription kit (Qiagen). RT-PCRs were performed with eight CcBV genes (cystatin-1, bv8-3, ben-2, crp-4, crp-2, ptph, DNApolB2, and CcBV_4.4), using 1 μl of diluted cDNA (1:10) in a 25-μl reaction mixture volume. In order to assess genomic DNA contamination, negative controls, corresponding to RNA samples to which no RT enzyme was added, were generated for each tissue and were tested for each gene primer pair. In all cases, no genomic DNA contamination was detected.
Reaction cycling conditions were as follows: an initial denaturation step at 94°C for 1 min followed by 35 cycles at 94°C for 1 min, annealing at 60°C (all genes) for 1 min, extension at 72°C for 1 min, and a final extension step at 72°C for 7 min. The rpl3 gene from M. sexta was used as an endogenous control.
Determination of CcBV circle abundance.
Virus particles were purified from 200 pairs of freshly dissected ovaries from C. congregata female wasps by filtration using SpinX columns (Costar, France), and the PDV DNA packaged in the particles was extracted as previously described (49). Briefly, ovaries were dissected in 400 μl Tris-acetate-EDTA (TAE), and viral particles were then homogenized with a syringe with 18- to 23-gauge needles. Debris and tissue fragments were eliminated by spinning at 3,000 × g for 3 min at 4°C and discarding the pellet. Supernatants were filtrated on a SpinX filter at 15,000 × g for 15 min at 4°C, and filter-purified particles were diluted in 500 μl TAE. Viral particles were then treated with proteinase K for 1 h at 55°C, 25 μl of 20% SDS was added, and samples were incubated overnight at 55°C. Viral DNA was then extracted with phenol-chloroform and precipitated by using ethyl alcohol (EtOH). The concentration of viral DNA solubilized in aqueous solution was measured by using a Varian Cary 50 Scan spectrophotometer. The presence of wasp genomic contamination was assessed by attempting to amplify the C. congregata actin and ef1α genes by PCR. Circle abundance was measured by qPCR on 15 different CcBV circles corresponding to 8 different RUs (C19, C30, C23, and C5 in RU1; C9, C31, and C13 in RU2.2; C28, C35, and C18 in RU2.3; C1 in RU5; C14 in RU6.2; C4 in RU7; and C26 in RU8). Circle quantity was measured by using the 2−ΔCT method for each circle and relative to the less abundant circle.
Contact the corresponding authors for a list of primer sequences used in this paper.
Nucleotide sequence accession numbers.
454 raw data corresponding to CcBV cDNA were deposited in the Sequence Read Archive (SRA) under accession number SRP035265.
RESULTS
Global transcriptome statistics obtained from nonparasitized and parasitized tissues.
In order to obtain a global vision of CcBV expression in two immune tissues (fat body and hemocytes) of the parasitized host M. sexta, we performed a large-scale transcriptomic analysis of these two tissues, 24 h after oviposition, using 454 pyrosequencing. A total of 380,670 reads were obtained from the sequencing of the four cDNA libraries (fat body and hemocytes from parasitized and control larvae) (Table 1). Out of the 236,173 reads that could be assembled, 124,214 corresponded to the nontreated control conditions, whereas 111,959 corresponded to cDNA obtained from tissues of parasitized caterpillars. The fact that less concentrated RNA could be extracted from hemocytes impacted sequencing depth: 5,449 reads corresponding to the CcBV genome were obtained from the hemocyte library, compared to 55,427 reads from the fat body library.
TABLE 1.
Read statistics resulting from 454 transcriptomic deep sequencing of parasitized and nonparasitized M. sexta larvae
| Condition and tissue | No. of reads |
|||
|---|---|---|---|---|
| Before assembly | After assembly |
|||
| CcBV | M. sexta | Total | ||
| Parasitized | ||||
| Fat body | 143,297 | 55,427 | 47,575 | |
| Hemocytes | 17,135 | 5,449 | 3,508 | |
| Total | 160,432 | 60,876 | 51,083 | 111,959 |
| No treatment | ||||
| Fat body | 178,301 | 0 | 105,173 | |
| Hemocytes | 41,937 | 0 | 19,041 | |
| Total | 220,238 | 0 | 124,214 | 124,214 |
| Total expt | 380,670 | 236,173 | ||
In parasitized tissues, more than half of the reads obtained could be mapped to the CcBV genome (60,876 reads), and less than half could be mapped to the M. sexta genome (51,083 reads), indicating that an important part of the global transcriptional activity is devoted to virus genes.
Identification of CcBV genes expressed in fat body and hemocytes.
By 454 sequencing, we identified 88 CcBV genes expressed in the fat body and/or hemocytes of M. sexta 24 h after parasitization (Table 2). Among these 88 genes, five pairs of genes correspond to duplicate genes for which expression levels could not be distinguished. Out of the 88 expressed genes, 72 genes belonged to 23 multigenic families, while the remaining genes were single-copy genes. A set of 81 genes figure among the 222 predicted genes of the CcBV integrated genome, and this approach allowed us to support the assembly of this genome (14) and to validate and refine the predicted annotation of these genes (data not shown). This approach also allowed the identification of 7 new CcBV genes that were not previously identified. Three of these new genes could be mapped to the integrated genome (CcBV_3.9, CcBV_20.6, and CcBV_28.22), whereas the remaining four genes (bv3-4, bv5-9, bv7-8, and ep1-like7) belonged to known BV multigenic families but were not present in the integrated or packaged genome previously sequenced, suggesting that either certain segments were not perfectly assembled or a few proviral segments remain unidentified.
TABLE 2.
Complete list of CcBV genes expressed in two tissues (fat body and hemocytes) in M. sexta 24 h after parasitization by C. congregata detected by 454 transcriptomic analysisa
| Gene | Gene family | No. of introns | Segment position | Region position | Signal peptide | mRNA size (bp) | RPKM for fat body | RPKM for hemocytes | mRNA sequence type | TSS motif |
|---|---|---|---|---|---|---|---|---|---|---|
| ben-8 | Ben | 3 | 20 | PL2 | No | 1,034 | 3.48 | 0.00 | Partial | |
| bv1-1/bv1-2 | BV1 | 1 | 9/33 | PL2 | Yes | 755 | 21.75 | 0.00 | Full length | CACAGT |
| bv2-2 | BV2 | 1 | 19 | PL1 | Yes | 1,265 | 17.74 | 81.37 | Full length | CGCAGT |
| bv2-3 | BV2 | 1 | 19 | PL1 | Yes | 1,104 | 38.98 | 117.56 | Full length | CGCAGT |
| bv2-5 | BV2 | 1 | 25 | PL1 | Yes | 1,272 | 49.49 | 202.30 | Full length | CGCAGT |
| bv2-6 | BV2 | 1 | 30 | PL1 | Yes | 1,038 | 6.08 | 8.62 | Full length | CGTAGT |
| bv2-8 | BV2 | 1 | 23 | PL1 | Yes | 537 | 8.86 | 22.92 | Partial | |
| bv3-1 | BV3 | 1 | 30 | PL1 | No | 664 | 63.79 | 11.79 | Full length | GTCAGT |
| bv3-2 | BV3 | 1 | 18 | PL2 | Yes | 1,383 | 15.38 | 23.46 | Full length | CACAGC |
| bv3-3 | BV3 | 1 | 31 | PL2 | Yes | 817 | 83.59 | 0.00 | Full length | ATCATT |
| bv3-4* | BV3 | Yes | 749 | 42.80 | 8.96 | Full length | ||||
| bv5-2 | BV5 | 1 | 9 | PL2 | Yes | 610 | 205.93 | 78.87 | Full length | AGCAGT |
| bv5-3 | BV5 | 1 | 9 | PL2 | Yes | 714 | 350.64 | 393.31 | Full length | CTCAGT |
| bv5-4 | BV5 | 1 | 23 | PL1 | Yes | 479 | 1.62 | 0.00 | Full length | AGCAGT |
| bv5-5 | BV5 | 1 | 25 | PL1 | No | 575 | 3.55 | 0.00 | Partial | |
| bv5-6 | BV5 | 1 | 33 | PL2 | Yes | 713 | 251.39 | 84.74 | Full length | CTCAGT |
| bv5-7 | BV5 | 1 | 33 | PL2 | Yes | 1,098 | 14.78 | 29.55 | Full length | GACAGT |
| bv5-8/bv5-1 | BV5 | 1 | 33/9 | PL2 | Yes | 1,061 | 194.02 | 138.14 | Full length | AGCAGT |
| bv5-9* | BV5 | Yes | 725 | 54.14 | 24.69 | Full length | ||||
| bv6-8 | BV6 | 0 | 29 | PL2 | No | 48 | 20.24 | 0.00 | Partial | |
| bv7-2/bv7-5 | BV7 | 1 | 22/36 | PL2 | Yes | 780 | 48.08 | 0.00 | Full length | CACAGT |
| bv7-6/bv7-1 | BV7 | 1 | 36/22 | PL2 | Yes | 762 | 306.11 | 214.37 | Full length | CGCAGT |
| bv7-7 | BV7 | 1 | 36 | PL2 | Yes | 595 | 465.83 | 534.02 | Full length | CACAGT |
| bv7-8* | BV7 | Yes | 690 | 185.15 | 163.77 | Full length | ||||
| bv8-3 | BV8 | 1 | 28 | PL2 | No | 409 | 1.66 | 0.00 | Partial | |
| bv8-12 | BV8 | 2 | 12 | PL6 | No | 373 | 3.13 | 0.00 | Partial | |
| bv8-13 | BV8 | 1 | 11 | PL6 | No | 153 | 5.08 | 0.00 | Partial | |
| bv9-4 | BV9 | 1 | 29 | PL2 | No | 657 | 3.55 | 0.00 | Partial | |
| bv9-7 | BV9 | 1 | 28 | PL2 | No | 435 | 4.47 | 0.00 | Partial | |
| bv11-1 | BV11 | 1 | 30 | PL1 | Yes | 2,269 | 82.93 | 181.95 | Full length | CACAGT |
| bv11-2/bv11-4 | BV11 | 2 | 22/36 | PL2 | Yes | 1,106 | 5.36 | 7.08 | Partial | |
| bv11-3 | BV11 | 1 | 2 | PL2 | Yes | 1,209 | 135.72 | 15.73 | Full length | TTCAGT |
| bv12-1 | BV12 | 5 | 17 | PL3 | No | 858 | 13.36 | 18.26 | Full length | TACAGT |
| bv12-2 | BV12 | 4 | 10 | PL3 | No | 674 | 14.41 | 0.00 | Partial | |
| bv15-1 | BV15 | 1 | 31 | PL2 | No | 613 | 73.69 | 16.43 | Full length | TGCAGT |
| bv15-2 | BV15 | 1 | 2 | PL2 | Yes | 615 | 296.03 | 138.26 | Full length | TTCAGT |
| bv16-1 | BV16 | 1 | 5 | PL1 | Yes | 575 | 33.79 | 40.86 | Full length | CACAGT |
| bv19-1 | BV19 | 1 | 19 | PL1 | Yes | 904 | 44.92 | 0.00 | Full length | AGCATT |
| bv19-2 | BV19 | 1 | 13 | PL2 | Yes | 777 | 8.75 | 0.00 | Full length | AACAAT |
| bv19-3 | BV19 | 1 | 30 | PL1 | Yes | 689 | 20.45 | 0.00 | Full length | TGCATT |
| bv24-1 | BV24 | 1 | 29 | PL2 | No | 338 | 3.16 | 0.00 | Full length | TCCAGT |
| bv25-1 | BV25 | 1 | 31 | PL2 | Yes | 832 | 65.97 | 32.27 | Full length | AACAGT |
| CcV3 | C-type lectin | 1 | 13 | PL2 | Yes | 691 | 349.65 | 367.54 | Full length | GACAGT |
| CcV3-like | C-type lectin | 1 | 30 | PL1 | Yes | 591 | 109.97 | 253.67 | Full length | GACAGT |
| CrV1-like | CrV1 unique gene | 1 | 13 | PL2 | Yes | 1,657 | 33.77 | 29.71 | Full length | GGCATT |
| cystatin-1 | Cystatin | 0 | 19 | PL1 | Yes | 660 | 271.14 | 184.77 | Full length | GACAGT |
| cystatin-2 | Cystatin | 0 | 19 | PL1 | Yes | 664 | 324.51 | 471.79 | Full length | GACAGT |
| DUFFY-like | Duffy unique gene | 1 | 5 | PL1 | Yes | 2,450 | 5.99 | 0.00 | Full length | GTCAGT |
| ep1 | EP1 | 1 | 8 | PL9 | Yes | 1,940 | 257.00 | 388.13 | Full length | CACAGT |
| ep1-like1 | EP1 | 1 | 1 | PL5 | Yes | 1,216 | 26.68 | 155.49 | Full length | CGCATT |
| ep1-like2 | EP1 | 2 | 1 | PL5 | Yes | 1,171 | 132.66 | 389.82 | Full length | CGCATT |
| ep1-like3 | EP1 | 2 | 1 | PL5 | Yes | 1,472 | 40.85 | 104.89 | Full length | CGCATT |
| ep1-like4 | EP1 | 1 | 5 | PL1 | Yes | 1,710 | 114.02 | 138.71 | Full length | CACAGT |
| ep1-like5 | EP1 | 1 | 7 | PL4 | No | 1,140 | 15.25 | 0.00 | Full length | CACAGT |
| ep1-like6 | EP1 | 1 | 28 | PL2 | Yes | 941 | 11.15 | 16.65 | Full length | CACAGT |
| ep1-like7* | EP1 | Yes | 967 | 502.93 | 528.75 | Full length | ||||
| ep2 | EP2 | 1 | 2 | PL2 | Yes | 1,068 | 140.36 | 97.43 | Full length | GACAAT |
| ep2-like1 | EP2 | 1 | 31 | PL2 | Yes | 2,024 | 49.39 | 30.40 | Full length | GACAGT |
| ep2-like2 | EP2 | 1 | 13 | PL2 | Yes | 604 | 50.02 | 14.82 | Partial | |
| P494-like | P494 unique gene | 1 | 22 | PL2 | Yes | 2,055 | 60.13 | 58.25 | Full length | AACAGT |
| ptpi | PTP | 0 | 1 | PL5 | No | 733 | 1.19 | 12.21 | Partial | |
| ptpl | PTP | 0 | 1 | PL5 | No | 309 | 2.20 | 0.00 | Partial | |
| ptpm | PTP | 0 | 1 | PL5 | No | 500 | 2.53 | 0.00 | Partial | |
| ptpp | PTP | 0 | 1 | PL5 | No | 426 | 0.00 | 13.13 | Partial | |
| ptpr | PTP | 0 | 7 | PL4 | No | 631 | 2.77 | 0.00 | Partial | |
| ptpy | PTP | 0 | 17 | PL3 | No | 426 | 0.00 | 1.82 | Partial | |
| RNAseT2-like1 | RNAseT2 | 2 | 23 | PL1 | Yes | 567 | 1.20 | 25.65 | Partial | |
| vank-4 | Vank | 0 | 14 | PL6 | No | 548 | 7.45 | 0.00 | Partial | |
| vank-5 | Vank | 0 | 26 | PL8 | No | 871 | 2.68 | 0.00 | Full length | CGCAGT |
| vank-9 | Vank | 0 | 26 | PL8 | No | 1,042 | 21.26 | 46.17 | Full length | CACAGT |
| CcBV_12.6 | Unique gene | 0 | 12 | PL6 | No | 192 | 3.04 | 0.00 | Partial | |
| CcBV_13.2 | Unique gene | 1 | 13 | PL2 | No | 967 | 1.00 | 9.26 | Full length | GGCAGT |
| CcBV_13.6 | Unique gene | 1 | 13 | PL2 | Yes | 831 | 40.68 | 12.12 | Full length | GACAGT |
| CcBV_18.13 | Unique gene | 1 | 18 | PL2 | No | 476 | 4.29 | 0.00 | Partial | |
| CcBV_18.2 | Unique gene | 1 | 18 | PL2 | Yes | 177 | 6.04 | 0.00 | Partial | |
| CcBV_20.4 | Unique gene | 1 | 20 | PL2 | No | 899 | 7.46 | 0.00 | Full length | CACAGT |
| CcBV_20.6* | Unique gene | 0 | 20 | PL2 | 356 | 1.64 | 0.00 | |||
| CcBV_21.2 | Unique gene | 1 | 21 | PL9 | Yes | 497 | 129.40 | 177.84 | Full length | TACAGT |
| CcBV_28.22* | Unique gene | 0 | 28 | PL2 | No | 176 | 3.86 | 0.00 | Partial | |
| CcBV_6.1b | Unique gene | 1 | 6 | PL1 | Yes | 543 | 44.01 | 0.00 | Full length | CACAGT |
| CcBV_6.2b | Unique gene | 1 | 6 | PL1 | No | 646 | 2.11 | 0.00 | Partial | |
| CcBV_8.1b | Unique gene | 1 | 8 | PL9 | Yes | 452 | 25.36 | 59.41 | Full length | TACAGT |
| CcBV_3.9* | Unique gene | 1 | 3 | PL2 | No | 323 | 5.71 | 0.00 | Full length | ATCAGT |
Asterisks indicate new genes. Boldface type in sequences corresponds to arthropod TSS.
Organization of CcBV genes expressed in immune tissues.
These transcriptomic data provided large-scale experimental evidence on the organization of expressed CcBV genes. Comparison of CcBV integrated genome and transcriptomic data allowed us to identify introns and UTRs of CcBV expressed genes.
Our data show that 92.9% of full-length CcBV mRNAs have at least 1 intron (Fig. 1A and Table 2). Concerning UTRs, sequence comparison of 5′ UTRs of 57 full-length CcBV mRNAs using the MEME Web server (42) revealed the presence of a CAGT motif in 46 sequences (Fig. 1B), which corresponded to the arthropod TSS motif (50). An additional 3′-UTR analysis performed on 50 full-length CcBV mRNAs revealed the presence of a hexamer motif, “AAUAAA” or “AUUAAA,” corresponding to the polyadenylation signal (PAS) identified in eukaryote and baculovirus mRNAs (51, 52). As in eukaryotic transcripts (53), this motif is accompanied in 21 CcBV mRNAs by a U-rich motif, “UUUUAU,” named the cytoplasmic polyadenylation element (CPE). We also identified in 36 full-length CcBV mRNAs an “AUUUA” motif corresponding to the AU-rich element (ARE) described as being involved in mRNA destabilization (51). However, no correlation was established between the presence of this ARE motif and the transcription level recorded by 454 sequencing. We might explain this observation by the fact that wasp genomes are enriched in AT nucleotides, leading to an increase in the false-positive detection of U-rich motifs (54–56).
FIG 1.
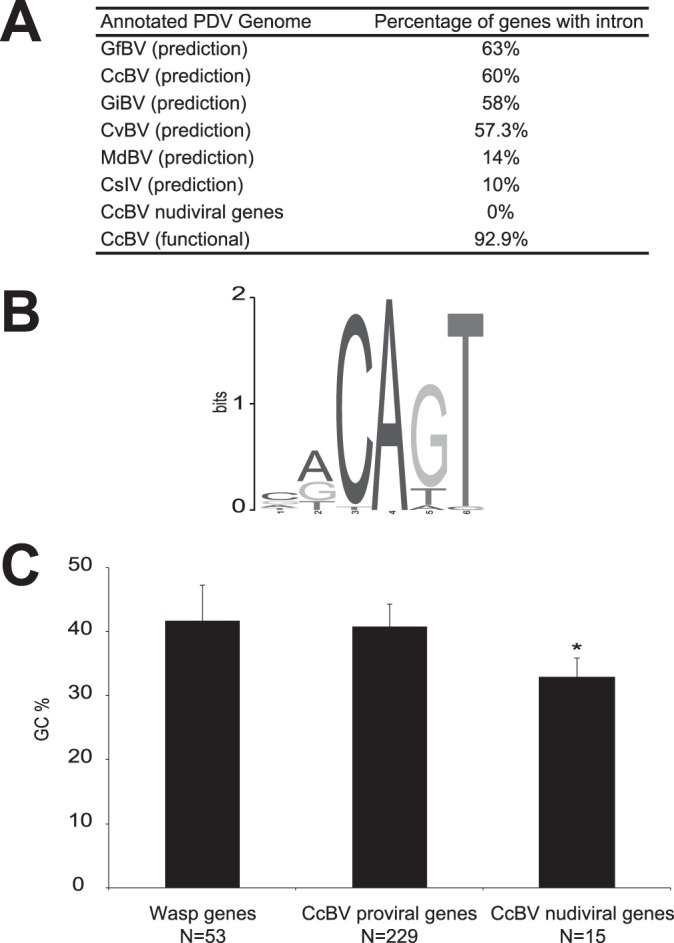
Eukaryotic features of CcBV genes. (A) Percentages of genes predicted to contain at least one intron among PDV genomes. Data for Glyptapanteles flavicoxis bracovirus (GfBV), Glyptapanteles indiensis bracovirus (GiBV) (20), Cotesia congregata bracovirus (CcBV) (14), Cotesia vestalis bracovirus (CvBV) (18), Microplitis demolitor bracovirus (MdBV), and Campoletis sonorensis ichnovirus (CsIV) (17) are shown. (B) CcBV gene transcription start site (TSS) predictions. Shown is a pictogram representation of a motif detected by MEME in the 5′ UTR of the 52 full-length mRNAs detected by 454 sequencing. A total of 10 bp of genomic sequence upstream of the mRNA was also included in the analysis. The height of the letters corresponds to their frequencies relative to the single-nucleotide background used when running MEME. (C) GC content among wasp genes, CcBV segment genes, and CcBV nudiviral genes. GC content (percent) was monitored in (i) 53 C. congregata genes present in the vicinity of the proviral form of CcBV or the nudiviral genes, (ii) 227 CcBV genes present in the CcBV integrated proviral form (222 predicted genes and 5 new genes), and (iii) 15 CcBV nudiviral genes. Error bars indicate standard deviations, and the asterisk indicates that the GC content differs significantly (P < 0.05 by pairwise t test).
Analysis of GC content was performed on 53 wasp genes (14), 229 CcBV proviral segment genes, and 15 CcBV nudiviral genes that are involved in particle production (7, 8, 11). No differences in GC content between wasp and CcBV proviral genes were identified (41.7% and 40.8%, respectively), whereas a significant difference was observed regarding CcBV nudiviral genes (32.8%), which are slightly more AT rich (P < 0.05 by pairwise t test) (Fig. 1C).
In conclusion, in both their organization and GC content, CcBV expressed genes resemble wasp genes.
CcBV gene expression levels (454 and qRT-PCR analyses).
Expression was detected for a total of 88 CcBV genes and was compared to that of the host rpl3 gene. Most genes (51 genes) were expressed in both tissues, 86 genes were expressed in the fat body, and 53 genes were expressed in hemocytes. The expression of 35 genes was detected exclusively in the fat body; among these genes, 5 and 13 genes are expressed at high and intermediate levels, respectively, and 17 genes are expressed at low levels. The protein tyrosine phosphatase y (ptpy) and ptpp genes were the only two genes for which transcripts were detected only in hemocytes. The difference in library size between tissues did not allow comparison of the levels of expression between these tissues (57).
Expression levels of these 88 CcBV genes deduced from deep sequencing revealed a heterogeneous distribution in fat body and in hemocytes (Fig. 2A and 3A). To validate these observations, 11 genes were selected across the expression range for qRT-PCR analysis of the cDNA sample from fat body used for 454 sequencing (Fig. 2B) and independently on fat body and hemocyte cDNAs from five newly parasitized M. sexta larvae (Fig. 2C and 3B). The selected genes were cyst1, cyst2, CcV3, CcV3-like, vank-9, CrV1-like, DUFFY-like, and a unique gene, CcBV_13.2. Expression was also examined for ptpp, detected by 454 sequencing only in hemocytes; vank-4, detected only in fat body; and ptph, for which expression was not detected by 454 sequencing in both tissues. All three genes had previously been reported to be expressed by using a multiplex RT-PCR analysis (33, 35).
FIG 2.

Expression levels of CcBV genes in fat body of M. sexta larvae 24 h after parasitization by C. congregata. (A) RPKM values deduced from transcriptomic 454 sequencing for each CcBV gene detected in M. sexta fat body 24 h after parasitization. The M. sexta rpl3 gene was used as a reference (in gray). Genes encoding proteins with predicted signal peptides are symbolized by black boxes, whereas genes encoding potential intracellular proteins are indicated by white boxes. (B) Validation of expression levels by qRT-PCR analysis of 11 selected CcBV genes on cDNA used for 454 analysis. Gene expression was measured in triplicate on the same cDNA as that used for 454 library construction. The M. sexta rpl3 gene was used as a reference (in gray). (C) Validation of expression levels by qRT-PCR analysis of 11 selected CcBV genes in new samples. Gene expression was measured in five new independently parasitized individuals in triplicate. The M. sexta rpl3 gene was used as a reference (in gray). Error bars indicate standard deviations, and different letters indicate significant differences in transcript abundance. Gene expression levels were separated into three categories based on the statistical differences between gene expression levels deduced from qRT-PCR analysis on new cDNA samples: high expression levels (red bars) (≥25 RPKM), intermediate expression levels (green bars) (5 ≤ RPKM < 25), and low expression levels (blue bars) (<5 RPKM).
FIG 3.

Expression levels of CcBV genes in hemocytes of M. sexta larvae 24 h after parasitization by C. congregata. (A) RPKM values deduced from transcriptomic 454 sequencing for each CcBV gene detected in M. sexta hemocytes 24 h after parasitization. The M. sexta rpl3 gene was used as a reference (in gray). Genes encoding proteins with predicted signal peptides are symbolized by black boxes, whereas genes encoding potential intracellular proteins are indicated by white boxes. (B) Validation of expression levels by qRT-PCR analysis of 11 selected CcBV genes in new samples. Gene expression was measured in triplicate on samples from five independently parasitized individuals. The M. sexta rpl3 gene was used as a reference (in gray). Error bars indicate standard deviations, and different letters indicate significant differences in transcript abundance. Gene expression levels were separated into two categories based on the statistical differences between gene expression levels deduced from qRT-PCR analysis of new cDNA samples: high expression levels (red bars) (≥25 RPKM) and low expression levels (green bars) (<5 RPKM).
Taken together, these qRT-PCR analyses confirmed the trend observed by transcriptome analysis (Fig. 2 and 3). Cystatin-2 and CcV3-like showed high expression levels, whereas CcBV_13.2, vank-4, and ptpp showed low expression levels in both tissues. ptph revealed barely detectable expression by qRT-PCR. Based on the statistical differences between gene expression levels deduced from the qRT-PCR analysis for individual samples in both tissues, we classified gene expression levels into 3 categories in fat body, high (≥25 RPKM), intermediate (5 ≤ RPKM < 25), and low (<5 RPKM), and into 2 categories in hemocytes, high (≥25 RPKM) and low (<25 RPKM).
Genes with expression levels below the limit of detection.
This relative CcBV gene expression analysis by 454 sequencing did not allow the detection of expression of certain CcBV genes that had previously been reported to be expressed by using RT-PCR (13 PTP genes and 3 VANK genes) (33, 35). The expression levels of these genes were therefore below the limit of detection of the 454 analysis, which had been set to a minimum depth of 5 reads. Genes at this limit of detection are expressed at levels 20 times lower than the level of the rpl3 gene in fat body and in hemocytes (Fig. 2 and 3).
We therefore investigated expression levels of genes below the 454 detection limit. We found that 44 genes, 12 pseudogenes, and 4 transposons (Table 3) presented read depths either in fat body or in hemocytes that were between 1 and 4 (Table 3) and that 97 genes, 17 pseudogenes, and 7 transposons had no reads by 454 analysis. We selected a subset of genes below the 454 detection limit (ben-2, crp-4, crp-2, ptph, DNApolB2, and CcBV_4.4) to determine whether expression levels could be detected by using a more sensitive gene-specific amplification. Expression of these genes was analyzed by RT-PCR not only in hemocytes and fat body but also in the nervous system, malpighian tubules, and midgut at 24 h postoviposition. A low level of amplification was observed for all genes in at least one tissue (Fig. 4), indicating that genes for which relative expression could not be detected by the 454 analysis are weakly expressed.
TABLE 3.
Complete list of CcBV genes presenting depths of 1 to 4 reads by 454 transcriptomic analysis in two tissues (fat body and hemocytes) in M. sexta 24 h after parasitization by C. congregataa
| Gene | Region position | Signal peptide | No. of reads for fat body | No. of reads for hemocytes |
|---|---|---|---|---|
| ben-13 pseudogene | S27/RU2.3 | 4 | 0 | |
| ben-14 | S24/RU2.3 | No | 2 | 0 |
| ben-2 | S3/RU2.2 | No | 1 | 2 |
| ben-3 | S6/RU1 | No | 5 | 3 |
| ben-5 | S12/RU6.1 | No | 1 | 0 |
| ben-6 pseudogene | S18/RU2.3 | 1 | 0 | |
| ben-7 | S18/RU2.3 | No | 5 | 0 |
| bv2-1 | S2/RU2.1 | Yes | 4 | 2 |
| bv2-4 pseudogene | S19/RU1 | 1 | 0 | |
| bv2-7/Maverick capsid-like p31.10* | S31/RU2.1 | Yes | 4 | 0 |
| bv6-7 | S29/RU2.2 | No | 1 | 0 |
| bv6-9 pseudogene | S28/RU2.3 | 1 | 0 | |
| bv6-14 pseudogene | S28/RU2.3 | 2 | 0 | |
| bv6-15 pseudogene | S32/RU2.3 | 1 | 0 | |
| bv6-20 | S32/RU2.3 | No | 0 | 1 |
| bv6-24 | S35/RU2.3 | No | 1 | 0 |
| bv6-27 | S16/RU2.3 | No | 2 | 0 |
| bv8-5 | S27/RU2.3 | No | 2 | 0 |
| bv8-7 | S32/RU2.3 | No | 1 | 0 |
| bv8-10 | S35/RU2.3 | No | 2 | 0 |
| bv9-1 pseudogene | S29/RU2.2 | 1 | 0 | |
| bv9-3 | S29/RU2.2 | No | 1 | 0 |
| bv9-5 | S28/RU2.3 | No | 2 | 0 |
| bv12-3 | S4/RU7 | No | 3 | 0 |
| bv14-2 | S18/RU2.3 | Yes | 1 | 1 |
| bv16-2 | S5/RU1 | Yes | 4 | 1 |
| bv17-2 pseudogene | S27/RU2.3 | 1 | 0 | |
| bv21-2 | S32/RU9 | No | 2 | 0 |
| bv22-2 | S28/RU2.3 | No | 1 | 0 |
| bv24-2 pseudogene | S28/RU2.3 | 3 | 0 | |
| bv25-2 pseudogene | S31/RU2.1 | 1 | 0 | |
| bv26-2 pseudogene | S33/RU2.1 | 3 | 0 | |
| h4 | S7/RU4 | No | 2 | 0 |
| p94-like1 | S7/RU4 | No | 2 | 0 |
| p94-like2 | S7/RU4 | No | 1 | 0 |
| ptpa | S26/RU8 | No | 0 | 1 |
| ptpc | S10/RU3 | No | 3 | 0 |
| ptpdelta | S26/RU3 | No | 0 | 1 |
| ptpe | S10/RU3 | No | 6 | 3 |
| ptpepsilon pseudogene | S26/RU3 | 0 | 1 | |
| ptpk | S1/RU5 | No | 4 | 0 |
| ptpq | S1/RU5 | No | 0 | 3 |
| ptps | S10/RU3 | No | 0 | 1 |
| ptpu | S14/RU6.2 | No | 2 | 0 |
| ptpx | S17/RU3 | No | 4 | 0 |
| RNAseT2-like3 | S25/RU1 | Yes | 5 | 0 |
| srp1 | S29/RU2.3 | No | 2 | 0 |
| srp2 | S29/RU2.3 | No | 3 | 0 |
| vank-1 | S11/RU6.1 | No | 4 | 0 |
| vank-2 | S15/RU2.3 | No | 5 | 0 |
| vank-7 | S16/RU2.3 | No | 2 | 0 |
| CcBV_12.1 | S12/RU6.1 | No | 1 | 0 |
| CcBV_15.5 | S15/RU2.3 | Yes | 4 | 0 |
| CcBV_20.1b | S20/RU2.1 | No | 1 | 0 |
| CcBV_24.2 | S24/RU2.3 | No | 5 | 0 |
| CcBV_32.6 | S32/RU2.3 | No | 1 | 4 |
| Dong-like1 pseudogene | S31/RU2.1 | 6 | 2 | |
| DIRS gag p31.7/DIRS RT RNaseH p31.6 pseudogene* | S31/RU2.1 | 1 | 0 |
The expression levels of these genes are below the limit of detection of the 454 analysis, which was set to 5 reads. Asterisks correspond to genes sharing identical reads.
FIG 4.
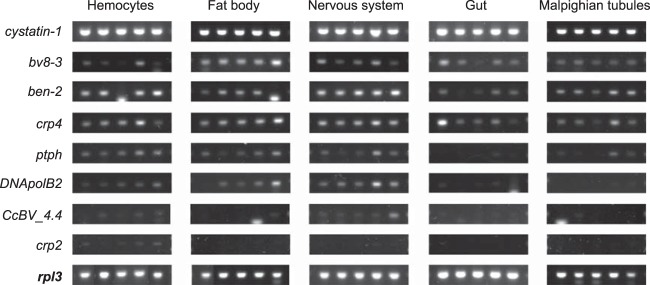
RT-PCR expression analysis of genes below the limit of detection by 454 analysis. Genes tested were cystatin-1, showing high expression levels by 454 analysis; bv8-3, showing low expression levels by 454 analysis; and genes below the 454 limit of detection: ben-2, crp4, ptph, DNApolB2, CcBV_4.4, and crp2. Host tissues examined were from five independently parasitized M. sexta larvae. Expression of the host rpl3 gene was used as an internal control.
Analysis of PDV gene expression and targeting of encoded proteins.
High-level expression in both tissues appeared to be strongly correlated with the presence of a sequence coding for a predicted signal peptide in the CDS of the gene (P value of <0.001) (Fig. 2A and 3A). Accordingly, the genes encoding predicted intracellular proteins have no signal peptide and were expressed at a low level. Moreover, among the 141 genes below the limit of detection, 129 have no signal peptide (Table 3) (14).
We next investigated whether other factors could contribute to the different expression levels observed for CcBV genes, such as gene membership in a given circle, replication unit, and gene family or localization of the gene in the proviral genome.
Gene expression and circle position.
To investigate whether BV gene expression levels could be correlated to the presence of these genes on particular segments (or, by extension, to a particular circle), we mapped gene expression levels based on the above-described qRT-PCR analyses onto the packaged CcBV genome (Fig. 5 and 6).
FIG 5.

Localization in CcBV segments of genes expressed in fat body. Each segment is represented by its predicted genes but is not to scale in terms of segment or gene length. Segment names and RU numbers are positioned to the right of each segment. Gene names are indicated below each bar. Expression levels are log normalized to facilitate visualization and are represented by colored bars.
FIG 6.

Localization in CcBV segments of genes expressed in hemocytes. Each segment is represented by its predicted genes but is not to scale in terms of segment or gene length. Segment names and RU numbers are positioned to the right of each segment. Gene names are indicated below each bar. Expression levels are log normalized to facilitate visualization and are represented by colored bars.
The results indicate that levels of expression of BV genes were not simply linked to their belonging to particular circles, as genes with different levels of expression were found on the same circle. However, the distribution of expressed genes appeared to clearly occur on a nonrandom basis. Interestingly, 15 circles had at least one highly expressed gene in both tissues, and 2 of them (C8 and C13) had all their genes expressed. Conversely, eight circles exhibited gene expression below the detection limit in both tissues (Fig. 5 and 6), and five additional circles displayed no detectable gene expression in hemocytes.
Gene expression and replication units.
We then assessed whether BV gene expression levels could be linked to the position of the proviral segments within the wasp genome (Fig. 7). During particle production, the segments clustered in the wasp genome are not amplified separately but within several molecules (RU) comprising several contiguous segments (15). The circles are produced later by resolution of the amplified molecule. Therefore, DNAs amplified together might be present in a similar amount in the particles. Louis et al. (15) showed previously that CcBV proviral segments are amplified within 12 RUs, which have been mapped onto the CcBV integrated genome. Remarkably, the majority of segments containing genes expressed at high levels are localized in five replication units (RU1, RU2.1, RU5, RU8, and RU9), which are the same for hemocytes and fat body (Fig. 7). In contrast, the seven other RUs contain genes with no detectable expression or only a few genes expressed at low levels (RU2.2, RU2.3, RU3, RU4, RU6.1, RU6.2, and RU7). These results indicate that belonging to a particular RU may strongly influence the gene expression profile of a given CcBV gene.
FIG 7.

Relationships between gene localization in the proviral genome of CcBV, circle abundance, and gene expression levels in fat body (A) and hemocytes (B) of M. sexta larvae 24 h after parasitization by C. congregata. CcBV DNA segments are represented by boxes, with the segment numbers indicated below. The 35 segments are organized into nine proviral loci (PL) that correspond to wasp genomic regions containing integrated CcBV segments. Segments are coamplified during particle production in the wasp ovaries in 12 different molecules constituting replication units (RUs) (15). The position of expressed CcBV genes is represented in each segment by either a red, green, or blue bar, which corresponds to the level of expression in host tissues (high, intermediate, or low expression levels, respectively). Only positions of genes detected by 454 analysis are indicated. Genes with identical sequences are indicated by dashed bars. Segments corresponding to high-abundance circles are shown in black, and those corresponding to low-abundance circles are shown in gray.
Gene expression and circle abundance.
This contrast in numbers of genes expressed and levels of expression between RUs led us to assess whether circles harboring highly expressed genes were produced at higher abundances in wasp ovaries. Indeed, the relative abundance of PDV circles in female wasps was shown in other models to be the same as in the parasitized host (58, 59). The abundances of 15 circles (corresponding to 8 different RUs) were examined by using qPCR on DNA extracted from filter-purified CcBV particles (Fig. 8). Most of the tested circles produced from RU1, RU2.1, and RU5 were present in high abundances in viral particles and harbored highly expressed genes in host tissues (Fig. 8). Conversely, circles corresponding to proviral segments belonging to RU2.2, RU2.3, RU6.2, and RU7 had low abundances, and these circles contained genes expressed either at low levels or at levels below the detection limit. These results suggest that certain RUs produced less abundant circles, leading to low levels of gene expression in the host, while others produced abundant circles, with at least one gene being expressed at a high level. There were, however, a few exceptions to this trend: circles 26 and 23 were produced at high levels but showed low BV gene expression levels, whereas circle 5 was produced at low levels but contained two highly expressed genes (S5) (Fig. 8).
FIG 8.
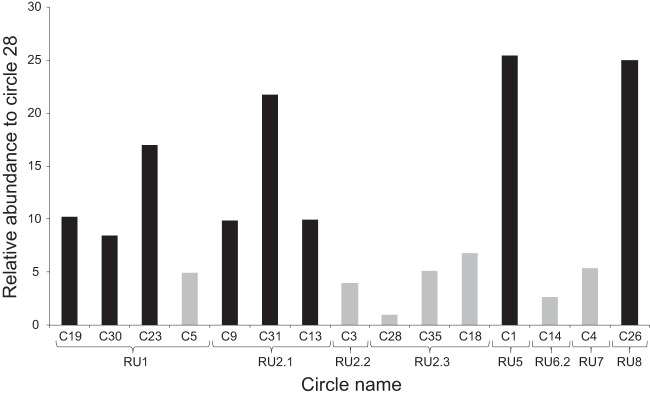
Relative abundance of CcBV circles in viral particles produced by C. congregata female wasps. Circle abundance in a purified CcBV preparation was monitored by qPCR. Circle abundance is represented as a factor of the less represented circle (C28). Segments corresponding to high-abundance circles are shown in black, and those corresponding to low-abundance circles are shown in gray. Braces indicate circles that belong to the same replication unit (RU).
Gene expression and gene family membership.
To identify other factors that could explain the differential expression levels, we performed an inter- and intragene family expression analysis. Deep sequencing showed disparities in gene expression levels between CcBV gene families. Indeed, 14 multigenic families (BV4, BV10, BV13, BV14, BV17, BV18, BV20, BV21, BV22, BV23, BV26, P94-like, CRP, and SRP) displayed expression levels below the limit of detection, whereas in other multigenic families, expression was detected for at least some gene members (BV1, BV2, BV5, BV6, BV7, BV8, BV9, BV11, BV12, BV16, BV24, BV25, Cystatin, RNAseT2, VANK, BEN, and PTP). Only six multigenic families (BV3, BV15, BV19, C-type lectin, EP1-like, and EP2-like) had all of their genes expressed at levels above the 454 detection limit (Table 2).
Most genes with expression levels below the limit of detection were located at RU2.2 and RU2.3, which produced less abundant circles, but some were also present on abundantly produced circles, such as bv14-1 on C30 (RU1) and bv20-1 and bv20-2 on C26 (RU8).
Within families with visibly expressed genes, different levels of expression were observed and were investigated by combining analyses of promoter sequence comparisons, gene positions, and phylogeny.
In the case of gene families exclusively present in RU1 and RU2.1 producing abundant circles, variations in gene expression levels might be linked to promoter sequences. For example, within the BV5 gene family (Fig. 9A and B), low, intermediate, and high levels of gene expression were observed. As shown in the combined phylogenetic and expression analysis, certain closely related genes that are potentially recent gene duplicates showed similar expression levels and harbored identical (bv5–1/bv5-8) or nearly identical (bv5–6/bv5-3) organizations of promoter regions, as reflected by the respective positions of the sequences described as recognition sites of transcription factors in Drosophila melanogaster. An exception was the recent bv5-7 and bv5-2 duplicates, which showed contrasting levels of expression. In this case, a 500-bp insertion upstream of the bv5-7 ATG start site displaced the bv5-2-like promoter region, which might contribute to the lower levels of expression observed for bv5-7. In the case of the bv5-4 and bv5-5 genes expressed at low levels, the promoters were completely different from those of the other BV5 genes, which might be an explanation for their different expression levels. Furthermore, a gene encoding RNAseT2 is present within the 1,000 bp upstream of the bv5-4 ATG start site, which may also be involved in the perturbation of gene expression.
FIG 9.

Phylogenetic analyses, expression profiles in fat body, and transcription factor binding site predictions for the CcBV BV5 multigenic family. (A) Phylogenetic trees of genes from the BV5 gene family, with their respective log-normalized RPKM expression levels represented by colored circles. Bootstrap values of >70 are represented by asterisks. Long branches are interrupted by dashed lines for clarity. (B) Transcription factor binding site predictions for the BV5 gene family using Matinspector from the Genomatix software suite. Analyzed sequences correspond to 1,000 bp upstream of the ATG start codon of each gene. Colored motifs represent motif family predictions. A complete description of motif names is available (http://www.genomatix.de/). Sequences within a black rectangle possess a majority of identical motifs, represented by asterisks. Red lines correspond to regions with predicted CDSs or known expressed mRNAs.
Taken together, the data show that expression levels within CcBV gene families can vary, and this variation can be explained by the localization of genes in nonabundant or abundantly produced circles and also possibly by differences in promoter regions, for which activities need to be assessed experimentally.
DISCUSSION
This study provides a global vision of CcBV genes expressed 24 h after parasitism by the braconid wasp C. congregata in two tissues (fat body and hemocytes) involved in the immune responses of the host M. sexta.
The availability of the reference genomes of the host M. sexta and the CcBV integrated genome allowed the identification of transcript origins in parasitized and control larvae (Table 1). Few studies dedicated to parasite-host or pathogen-host biological systems have evaluated the transcript proportion produced by each protagonist during the course of the interaction. In a plant-fungal pathogen interaction, pathogenic fungal transcripts were estimated to represent 0.5% to 28% of the total transcripts analyzed (60, 61). During the course of a baculovirus infection, viral mRNA in host cells increased from 3% to >80% of the total mRNA within the first 48 h after infection (52). This was correlated with a tremendous increase in the number of viral genome copies in infected cells (ms42 cells from the lepidopteran host Trichoplusia ni). Our data show that despite the fact that the viral genome does not replicate in infected cells, more than half of the total transcriptional activity of the host immune tissues is dedicated to the expression of PDV gene products.
High-throughput 454 analysis identified expression levels above the limit of detection for only 36.5% of predicted genes 24 h after wasp oviposition. Eighty-one genes figured among the 222 predicted genes present in the integrated CcBV genome. Further analysis revealed that CcBV genes for which expression was not detected by 454 sequencing are probably weakly expressed. Indeed, the expression levels of a subset of genes below the 454 detection limit were analyzed in a series of tissues (nervous system, midgut, and malpighian tubules) by RT-PCR. The results show that these genes are expressed at very low levels. However, it is interesting to note that this category of genes includes a series of pseudogenes and transposable elements that are not likely to play a role in the host-parasite interaction. In summary, at 24 h postoviposition, 36.5% of CcBV genes are expressed at levels that can be detected by 454 analysis, whereas the other genes are expressed at much lower levels, below the 454 limit of detection.
To date, only two other high-throughput transcriptomic approaches have been conducted on similar host-parasitoid systems involving DsIV and CchBV; in each case, the expressions of only 19 viral genes from 5 and 6 gene families, respectively, were detected (30, 31). Even if the reference genomes are not available for these viruses, the expression of a larger number of genes might have been expected, considering the number of genes predicted in related viruses (19, 23). These results could be explained by the fact that de novo transcriptome assembly of short reads is very difficult for these models because of the presence of many multigenic families. Conversely, longer reads produced by 454 sequencing allowed a better assembly sensitivity but did not allow the detection of genes with very low expression levels.
In contrast, high levels of expression of certain well-characterized genes (ep1, cystatin-1, and CrV1-like) (62–64) were confirmed in our analysis. All EP1-like family members were found to be expressed, and notably, six of them were expressed at a high level. It is noteworthy that gene families that have so far been neglected because of their lack of similarity to known genes in databanks also reveal high levels of expression (BV2, BV3, BV5, BV7, BV11, BV15, BV16, BV19, and BV25) (Fig. 2 and 3 and Table 2). These genes may represent new potential candidates involved in this host-parasite interaction. Concerning Cystatin genes, although previous studies characterized three copies in the CcBV genome (11, 16, 34), 454 sequencing detected the expression of only two genes. The coding sequence of cystatin-3 could not be amplified from parasitized host mRNA or from recent genomic DNA samples of C. congregata (data not shown). We hypothesize that the cystatin-3 gene has been lost, possibly because of successive genetic bottlenecks imposed on our laboratory population of parasitoid wasps.
Our study allowed the identification of seven new CcBV genes. The sequences of the new CcBV_3.9, CcBV_20.6, and CcBV_28.22 genes were known to be part of the integrated genome sequence, but automated annotation tools used so far failed to predict them. The remaining four new genes (bv3-4, bv5-9, bv7-8, and ep1-like7) had original sequences sharing similarities with members of known BV multigenic families. The initial sequencing of the CcBV genome (16) missed some circles produced in low abundance that were later identified in the proviral sequence of the virus (14). It is therefore possible that these new genes belong to low-abundance and provisionally elusive CcBV circles. Altogether, these transcriptomic data allowed us to improve the assembly of the CcBV integrated genome along with a refinement of the annotation of some software-predicted genes, arguing for the need to have biological data to correct automatic genome assembly and annotation.
This BV transcriptomic analysis yielded new information about CcBV gene properties. So far, intron detection has been performed in silico with the assistance of prediction software. An important heterogeneity in intron proportions predicted with this method was reported among PDV genomes depending on the software and criterion used, with 10% of genes in Campoletis sonorensis ichnovirus (CsIV), 14% in MdBV (17), 57.3% in Cotesia vestalis bracovirus (CvBV) (18), 58% in Glyptapanteles indiensis bracovirus (GiBV), 63% in Glyptapanteles flavicoxis bracovirus (GfBV) (20), and 60% in CcBV (14) predicted to contain introns. It was proposed that gene identification criteria were sources of this heterogeneity. Applied to the CcBV genome, the criteria used on CsIV led Webb et al. (17) to estimate that only 6.8% of CcBV genes may contain introns. Here functional annotation based on transcriptomic data shows that 92.9% of the assembled full-length CcBV mRNAs have at least one intron. All genes previously predicted to contain introns were validated experimentally, indicating that the criteria used by Espagne (16) and Bézier et al. (14) were correct.
This transcriptomic study also allowed us to perform a 5′-UTR analysis, with the identification of a CAGT motif corresponding to the arthropod TSS (50) initially identified in Drosophila melanogaster (65) and in early-expressed baculovirus genes (52). Moreover, 3′-UTR analysis allowed us to identify eukaryotic features such as PAS, CPE, and ARE involved in poly(A) tail fixation and mRNA stability (51, 53). Taken together, our results indicate that potential virulence genes that are expressed in the caterpillar have an insect gene structure (with introns, arthropod TSS, and GC content similar to that of wasp genes), which is different from that of the nudiviral genes involved in particle production. This structure is compatible with a wasp (or lepidopteran) origin for these viral genes, in accordance with the gene transfer scenario that we recently proposed (23) and with the results of BV genome sequencing of two Glyptapanteles species showing a phylogenetic link between Nasonia vitripennis wasp and BV sugar transporter genes (20). However, it is also possible that these insect features may have been acquired after the integration of genes originating from different sources (e.g., bacteria and other viruses) into the provirus. Indeed, these genes are involved in parasitism success, and strong selection pressure is likely operating to promote their expression in lepidopteran cells.
Expression analyses (RPKM) from 454 sequencing gave us an estimation of the relative expression levels of 88 CcBV genes in fat body and hemocytes. These levels were confirmed by performing qRT-PCR analyses on the same cDNA samples, for a set of 11 selected genes with various expression levels. Three categories of BV gene expression levels in fat body and two categories in hemocytes were statistically defined and used for delineating factors covarying with BV gene expression. Globally, the diversity of CcBV gene expression levels that we observed is in accordance with the various PDV gene expression levels described recently for MdBV (29) and DsIV (30) genes. The current hypotheses to explain this diversity are membership of genes to a segment (66), relative abundance of the circles in the particles, promoter strength, and mRNA stability (58). In this analysis, we have identified a correlation between CcBV gene expression levels and (i) the presence of a predicted signal peptide in the gene product, (ii) the replication unit to which they belong and circle abundance, and (iii) the promoter structure.
First, genes coding for proteins harboring predicted signal peptides appeared to be clearly overexpressed. This observation suggests that viral proteins directed to the secretory system of host cells are produced in large quantities to be effective against distant or diffuse host targets, whereas viral proteins that act locally on intracellular host pathways could be efficient at lower concentrations. For instance, the ep1 and cystatin-1 genes, which are known to code for proteins possessing a signal peptide sequence and to be abundantly secreted in the host hemolymph (63, 67), displayed high levels of expression in both tissues investigated. These results also confirm observations made for certain MdBV and CsIV genes encoding secreted proteins, which represent abundant viral gene products in the parasitized host (17).
Strikingly, our data show that certain parts of the integrated CcBV genome are characterized by the abundance/scarcity of genes expressed at detectable levels. Segments located in RU2.2, RU2.3, RU6, and RU7 were shown to produce less abundant circles. Therefore, the lack of detectable gene expression or low-level expression characterizing the segments of these RUs could be linked to low circle abundance. RU2.2 and RU2.3 correspond to a large triplicated region consisting of triplicate 1 (Tr1) (S29 and S3), Tr2 (S28, S27, and S15), and Tr3 (S32, S24, and S35) (14). While Tr1 was detected only in C. congregata, Tr2 and Tr3 are conserved in species that separated from the Cotesia lineage approximately 17 million years ago, suggesting that although the circles from this region are produced in lower abundances, the genes that they encode might, however, play an important role in the host-parasite interaction.
These results show that certain RUs produce less abundant circles, leading to very low levels of gene expression in the host, while others produce abundant circles, with at least one gene being highly expressed. There were, however, some exceptions to this trend: two circles (C26 and C23) that are abundantly produced possess PTP and VANK genes that are expressed at low levels by M. sexta fat body and hemocytes. Interestingly, the proteins encoded by these genes lack peptide signals and potentially act intracellularly (35, 68). Selection could be acting to maintain high-level production and encapsidation of these circles by the ovaries of female wasps, thereby enabling the widespread intracellular production of CcBV virulence genes expressed at low levels. Circle abundance is not the only determinant of CcBV gene expression, since genes expressed at low levels are also found on abundant circles, but it clearly plays an important role as a prerequisite to have a gene highly expressed on a circle.
Promoter efficiency could also play a role in CcBV gene expression. Indeed, the analysis of the promoter region of genes belonging to the BV5 multigenic family allowed us to detect important sequence differences, insertions, and deletions that might explain the different expression levels observed. These results are consistent with previous studies of other PDV gene families that suggested that promoters play an important role in gene regulation (59, 69). However, in-depth promoter analyses are now required to delineate the most important regulatory elements in PDV promoters.
Gene expression within multigenic families revealed different patterns of expression. Certain multigenic families displayed expression levels below the detection limit in fat body and hemocytes at 24 h postoviposition. Most genes from these families are localized in RU2.2 and RU2.3, which produce less abundant circles, which could explain in part their very low levels of expression. Exceptions to this rule were bv14-1 and bv20-1/bv20-2, which showed no detectable levels of expression despite being on abundant circles (C30 and C26, respectively); in the case of bv20-1/bv20-2, this might be explained by the intracellular localization of their protein products. Four gene families, BV1, BV15, C-type lectin, and EP2-like, have all their genes expressed at the same high level. The other gene families showed variations in expression depending on the gene family members.
Gene duplications are recognized to play an important role in evolution, as duplicated genes can be sources of new functions (70, 71). Gene duplications can potentially lead to different fates: one of the duplicated copies can be silenced by degenerative mutations (nonfunctionalization) or can evolve to give a new function (neofunctionalization). Alternatively, duplicated copies can persist and share a function (subfunctionalization) (71). The different patterns of expression that we observed suggest that these processes may be ongoing in the CcBV genome. Gene families showing the same expression levels could constitute examples of genes undergoing subfunctionalization, whereas gene families with variable gene expression levels could be in the process of eliminating genes and/or creating new genes with new functions.
Conclusion.
In summary, this study has allowed the experimental identification of genes actually expressed in host immune tissues, including seven new genes probably involved in the interaction with the host. The insect features of CcBV genes were confirmed by an extensive analysis. This work also highlights that certain genes, so far neglected because of their lack of similarity to known genes in databanks, reveal impressive levels of expression, suggesting that they may be involved in host regulation. Conversely, large regions of the CcBV genome appear to produce low-abundance circles with genes that are poorly expressed in the tissues analyzed, which questions the past and present roles of these sequences.
To go beyond this landmark step, it will now be necessary to obtain a dynamic and spatial vision of expressed CcBV genes throughout the course of parasitism, from very early to very late time points. It will then be possible to link CcBV gene expression to M. sexta transcriptomic responses to begin to delineate the consequences of viral expression during parasitism on the numerous alterations of host physiology previously described in this model of interaction.
ACKNOWLEDGMENTS
This work was supported by the ANR project Paratoxose and was done by using the facilities of the sequencing platform PPF Genome at the Université François-Rabelais. G.C. was supported by a Ph.D. grant from the French MENESR (Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche). J.T. was supported by a Ph.D. grant from the CNRS and the Région Centre.
We thank the GenOuest BioInformatics Platform (http://www.genouest.org/), which allowed the use of a computing cluster for bioinformatic analyses. We thank Cindy Menoret for insect rearing. We thank the three anonymous reviewers for constructive comments, which helped to improve the manuscript.
Footnotes
Published ahead of print 28 May 2014
REFERENCES
- 1.Beckage NE, Drezen J-M. (ed). 2011. Parasitoid viruses. Academic Press, San Diego, CA [Google Scholar]
- 2.Strand MR, Burke GR. 2013. Polydnavirus-wasp associations: evolution, genome organization, and function. Curr. Opin. Virol. 3:587–594. 10.1016/j.coviro.2013.06.004 [DOI] [PubMed] [Google Scholar]
- 3.Gundersen-Rindal D, Dupuy C, Huguet E, Drezen J-M. 2013. Parasitoid polydnaviruses: evolution, pathology and applications. Biocontrol Sci. Technol. 23:1–61. 10.1080/09583157.2012.731497 [DOI] [Google Scholar]
- 4.Dupuy C, Huguet E, Drezen J-M. 2006. Unfolding the evolutionary story of polydnaviruses. Virus Res. 117:81–89. 10.1016/j.virusres.2006.01.001 [DOI] [PubMed] [Google Scholar]
- 5.Strand MR. 2012. Polydnavirus gene products that interact with the host immune system, p 149–161 In Beckage NE, Drezen J-M. (ed), Parasitoid viruses. Academic Press, San Diego, CA [Google Scholar]
- 6.Beckage NE. 2012. Polydnaviruses as endocrine regulators, p 163–168 In Beckage NE, Drezen J-M. (ed), Parasitoid viruses. Academic Press, San Diego, CA [Google Scholar]
- 7.Herniou EA, Huguet E, Theze J, Bézier A, Periquet G, Drezen J-M. 2013. When parasitic wasps hijacked viruses: genomic and functional evolution of polydnaviruses. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368:20130051. 10.1098/rstb.2013.0051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bézier A, Annaheim M, Herbinière J, Wetterwald C, Gyapay G, Bernard-Samain S, Wincker P, Roditi I, Heller M, Belghazi M, Pfister-Wilhem R, Periquet G, Dupuy C, Huguet E, Volkoff A-N, Lanzrein B, Drezen J-M. 2009. Polydnaviruses of braconid wasps derive from an ancestral nudivirus. Science 323:926–930. 10.1126/science.1166788 [DOI] [PubMed] [Google Scholar]
- 9.Volkoff A-N, Jouan V, Urbach S, Samain S, Bergoin M, Wincker P, Demettre E, Cousserans F, Provost B, Coulibaly F, Legeai F, Béliveau C, Cusson M, Gyapay G, Drezen J-M. 2010. Analysis of virion structural components reveals vestiges of the ancestral ichnovirus genome. PLoS Pathog. 6:e1000923. 10.1371/journal.ppat.1000923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stoltz DB, Whitfield JB. 2009. Virology. Making nice with viruses. Science 323:884–885. 10.1126/science.1169808 [DOI] [PubMed] [Google Scholar]
- 11.Bézier A, Herbinière J, Lanzrein B, Drezen J-M. 2009. Polydnavirus hidden face: the genes producing virus particles of parasitic wasps. J. Invertebr. Pathol. 101:194–203. 10.1016/j.jip.2009.04.006 [DOI] [PubMed] [Google Scholar]
- 12.Thézé J, Bézier A, Periquet G, Drezen J-M, Herniou EA. 2011. Paleozoic origin of insect large dsDNA viruses. Proc. Natl. Acad. Sci. U. S. A. 108:15931–15935. 10.1073/pnas.1105580108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitfield JB, O'Connor JM. 2012. Molecular systematics of wasp and polydnavirus genomes and their coevolution, p 89–97 In Beckage NE, Drezen J-M. (ed), Parasitoid viruses. Academic Press, San Diego, CA [Google Scholar]
- 14.Bézier A, Louis F, Jancek S, Periquet G, Thézé J, Gyapay G, Musset K, Lesobre J, Lenoble P, Dupuy C, Gundersen-Rindal D, Herniou EA, Drezen J-M. 2013. Functional endogenous viral elements in the genome of the parasitoid wasp Cotesia congregata: insights into the evolutionary dynamics of bracoviruses. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368:20130047. 10.1098/rstb.2013.0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Louis F, Bézier A, Periquet G, Ferras C, Drezen J-M, Dupuy C. 2013. The bracovirus genome of the parasitoid wasp Cotesia congregata is amplified within 13 replication units, including sequences not packaged in the particles. J. Virol. 87:9649–9660. 10.1128/JVI.00886-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Espagne E. 2004. Genome sequence of a polydnavirus: insights into symbiotic virus evolution. Science 306:286–289. 10.1126/science.1103066 [DOI] [PubMed] [Google Scholar]
- 17.Webb BA, Strand MR, Dickey SE, Beck MH, Hilgarth RS, Barney WE, Kadash K, Kroemer JA, Lindstrom KG, Rattanadechakul W, Shelby KS, Thoetkiattikul H, Turnbull MW, Witherell RA. 2006. Polydnavirus genomes reflect their dual roles as mutualists and pathogens. Virology 347:160–174. 10.1016/j.virol.2005.11.010 [DOI] [PubMed] [Google Scholar]
- 18.Chen Y-F, Gao F, Ye X-Q, Wei S-J, Shi M, Zheng H-J, Chen X-X. 2011. Deep sequencing of Cotesia vestalis bracovirus reveals the complexity of a polydnavirus genome. Virology 414:42–50. 10.1016/j.virol.2011.03.009 [DOI] [PubMed] [Google Scholar]
- 19.Jancek S, Bézier A, Gayral P, Paillusson C, Kaiser L, Dupas S, Le Ru BP, Barbe V, Periquet G, Drezen J-M, Herniou EA. 2013. Adaptive selection on bracovirus genomes drives the specialization of Cotesia parasitoid wasps. PLoS One 8:e64432. 10.1371/journal.pone.0064432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desjardins CA, Gundersen-Rindal DE, Hostetler JB, Tallon LJ, Fadrosh DW, Fuester RW, Pedroni MJ, Haas BJ, Schatz MC, Jones KM, Crabtree J, Forberger H, Nene V. 2008. Comparative genomics of mutualistic viruses of Glyptapanteles parasitic wasps. Genome Biol. 9:R183. 10.1186/gb-2008-9-12-r183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dupuy C, Gundersen-Rindal DE, Drezen J-M. 2012. Genomics and replication of polydnaviruses, p 47–61 In Beckage NE, Drezen J-M. (ed), Parasitoid viruses. Academic Press, San Diego, CA [Google Scholar]
- 22.Bézier A, Herbinière J, Serbielle C, Lesobre J, Wincker P, Huguet E, Drezen J-M. 2008. Bracovirus gene products are highly divergent from insect proteins. Arch. Insect Biochem. Physiol. 67:172–187. 10.1002/arch.20219 [DOI] [PubMed] [Google Scholar]
- 23.Huguet E, Serbielle C, Moreau SJM. 2012. Evolution and origin of polydnavirus virulence genes, p 63–78 In Beckage NE, Drezen J-M. (ed), Parasitoid viruses. Academic Press, San Diego, CA [Google Scholar]
- 24.Drezen J-M, Bézier A, Lesobre J, Huguet E, Cattolico L, Periquet G, Dupuy C. 2006. The few virus-like genes of Cotesia congregata bracovirus. Arch. Insect Biochem. Physiol. 61:110–122. 10.1002/arch.20108 [DOI] [PubMed] [Google Scholar]
- 25.Dupuy C, Periquet G, Serbielle C, Bézier A, Louis F, Drezen J-M. 2011. Transfer of a chromosomal Maverick to endogenous bracovirus in a parasitoid wasp. Genetica 139:489–496. 10.1007/s10709-011-9569-x [DOI] [PubMed] [Google Scholar]
- 26.Barat-Houari M, Hilliou F, Jousset F-X, Sofer L, Deleury E, Rocher J, Ravallec M, Galibert L, Delobel P, Feyereisen R, Fournier P, Volkoff A-N. 2006. Gene expression profiling of Spodoptera frugiperda hemocytes and fat body using cDNA microarray reveals polydnavirus-associated variations in lepidopteran host genes transcript levels. BMC Genomics 7:160. 10.1186/1471-2164-7-160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Provost B, Jouan V, Hilliou F, Delobel P, Bernardo P, Ravallec M, Cousserans F, Wajnberg E, Darboux I, Fournier P, Strand MR, Volkoff A-N. 2011. Lepidopteran transcriptome analysis following infection by phylogenetically unrelated polydnaviruses highlights differential and common responses. Insect Biochem. Mol. Biol. 41:582–591. 10.1016/j.ibmb.2011.03.010 [DOI] [PubMed] [Google Scholar]
- 28.Moreau SJM, Huguet E, Drezen J-M. 2009. Polydnaviruses as tools to deliver wasp virulence factors to impair lepidopteran host immunity, p 137–158 In Rolff J, Reynolds SE. (ed), Insect infection and immunity. Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 29.Bitra K, Zhang S, Strand MR. 2011. Transcriptomic profiling of Microplitis demolitor bracovirus reveals host, tissue and stage-specific patterns of activity. J. Gen. Virol. 92:2060–2071. 10.1099/vir.0.032680-0 [DOI] [PubMed] [Google Scholar]
- 30.Etebari K, Palfreyman RW, Schlipalius D, Nielsen LK, Glatz RV, Asgari S. 2011. Deep sequencing-based transcriptome analysis of Plutella xylostella larvae parasitized by Diadegma semiclausum. BMC Genomics 12:446. 10.1186/1471-2164-12-446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu S-F, Sun F-D, Qi Y-X, Yao Y, Fang Q, Huang J, Stanley D, Ye G-Y. 2013. Parasitization by Cotesia chilonis influences gene expression in fatbody and hemocytes of Chilo suppressalis. PLoS One 8:e74309. 10.1371/journal.pone.0074309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harwood SH, McElfresh JS, Nguyen A, Conlan CA, Beckage NE. 1998. Production of early expressed parasitism-specific proteins in alternate sphingid hosts of the braconid wasp Cotesia congregata. J. Invertebr. Pathol. 71:271–279. 10.1006/jipa.1997.4745 [DOI] [PubMed] [Google Scholar]
- 33.Provost B, Varricchio P, Arana E, Espagne E, Falabella P, Huguet E, La Scaleia R, Cattolico L, Poirié M, Malva C, Olszewski JA, Pennacchio F, Drezen J-M. 2004. Bracoviruses contain a large multigene family coding for protein tyrosine phosphatases. J. Virol. 78:13090–13103. 10.1128/JVI.78.23.13090-13103.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Espagne E, Douris V, Lalmanach G, Provost B, Cattolico L, Lesobre J, Kurata S, Iatrou K, Drezen J-M, Huguet E. 2005. A virus essential for insect host-parasite interactions encodes cystatins. J. Virol. 79:9765–9776. 10.1128/JVI.79.15.9765-9776.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Falabella P, Varricchio P, Provost B, Espagne E, Ferrarese R, Grimaldi A, de Eguileor M, Fimiani G, Ursini MV, Malva C, Drezen J-M, Pennacchio F. 2007. Characterization of the IkappaB-like gene family in polydnaviruses associated with wasps belonging to different braconid subfamilies. J. Gen. Virol. 88:92–104. 10.1099/vir.0.82306-0 [DOI] [PubMed] [Google Scholar]
- 36.Slater GSC, Birney E. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6:31. 10.1186/1471-2105-6-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5:621–628. 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 38.Milne I, Stephen G, Bayer M, Cock PJA, Pritchard L, Cardle L, Shaw PD, Marshall D. 2013. Using Tablet for visual exploration of second-generation sequencing data. Brief. Bioinformatics 14:193–202. 10.1093/bib/bbs012 [DOI] [PubMed] [Google Scholar]
- 39.Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA. 2012. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28:464–469. 10.1093/bioinformatics/btr703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rombel IT, Sykes KF, Rayner S, Johnston SA. 2002. ORF-FINDER: a vector for high-throughput gene identification. Gene 282:33–41. 10.1016/S0378-1119(01)00819-8 [DOI] [PubMed] [Google Scholar]
- 41.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8:785–786. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 42.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37:W202–W208. 10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. 10.1093/bioinformatics/btl446 [DOI] [PubMed] [Google Scholar]
- 45.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. 2005. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21:2933–2942. 10.1093/bioinformatics/bti473 [DOI] [PubMed] [Google Scholar]
- 46.Cuit L, Webb BA. 1997. Promoter analysis of a cysteine-rich Campoletis sonorensis polydnavirus gene. J. Gen. Virol. 78(Part 7):1807–1817 [DOI] [PubMed] [Google Scholar]
- 47.Yuan JS, Reed A, Chen F, Stewart CN. 2006. Statistical analysis of real-time PCR data. BMC Bioinformatics 7:85. 10.1186/1471-2105-7-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57:289–300 [Google Scholar]
- 49.Beckage NE, Tan F, Schleifer KW, Lane RD, Cherubin LL. 1994. Characterization and biological effects of Cotesia congregata polydnavirus on host larvae of the tobacco hornworm, Manduca sexta. Arch. Insect Biochem. Physiol. 26:165–195. 10.1002/arch.940260209 [DOI] [Google Scholar]
- 50.Cherbas L, Cherbas P. 1993. The arthropod initiator—the capsite consensus plays an important role in transcription. Insect Biochem. Mol. Biol. 23:81–90. 10.1016/0965-1748(93)90085-7 [DOI] [PubMed] [Google Scholar]
- 51.Matoulkova E, Michalova E, Vojtesek B, Hrstka R. 2012. The role of the 3′ untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol. 9:563–576. 10.4161/rna.20231 [DOI] [PubMed] [Google Scholar]
- 52.Chen YR, Zhong S, Fei Z, Hashimoto Y, Xiang JZ, Zhang S, Blissard GW. 2013. The transcriptome of the baculovirus Autographa californica multiple nucleopolyhedrovirus in Trichoplusia ni cells. J. Virol. 87:6391–6405. 10.1128/JVI.00194-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mendez R, Richter JD. 2001. Translational control by CPEB: a means to the end. Nat. Rev. Mol. Cell Biol. 2:521–529. 10.1038/35080081 [DOI] [PubMed] [Google Scholar]
- 54.Honeybee Genome Sequencing Consortium. 2006. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 443:931–949. 10.1038/nature05260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Werren JH, Richards S, Desjardins CA, Niehuis O, Gadau J, Colbourne JK, Nasonia Genome Working Group 2010. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 327:343–348. 10.1126/science.1178028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nygaard S, Zhang G, Schiøtt M, Li C, Wurm Y, Hu H, Zhou J, Ji L, Qiu F, Rasmussen M, Pan H, Hauser F, Krogh A, Grimmelikhuijzen CJP, Wang J, Boomsma JJ. 2011. The genome of the leaf-cutting ant Acromyrmex echinatior suggests key adaptations to advanced social life and fungus farming. Genome Res. 21:1339–1348. 10.1101/gr.121392.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tarazona S, García-Alcalde F, Dopazo J, Ferrer A, Conesa A. 2011. Differential expression in RNA-seq: a matter of depth. Genome Res. 21:2213–2223. 10.1101/gr.124321.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beck MH, Inman RB, Strand MR. 2007. Microplitis demolitor bracovirus genome segments vary in abundance and are individually packaged in virions. Virology 359:179–189. 10.1016/j.virol.2006.09.002 [DOI] [PubMed] [Google Scholar]
- 59.Djoumad A, Dallaire F, Lucarotti CJ, Cusson M. 2013. Characterization of the polydnaviral “T. rostrale virus” (TrV) gene family: TrV1 expression inhibits in vitro cell proliferation. J. Gen. Virol. 94:1134–1144. 10.1099/vir.0.049817-0 [DOI] [PubMed] [Google Scholar]
- 60.Kawahara Y, Oono Y, Kanamori H, Matsumoto T, Itoh T, Minami E. 2012. Simultaneous RNA-seq analysis of a mixed transcriptome of rice and blast fungus interaction. PLoS One 7:e49423. 10.1371/journal.pone.0049423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fernandez D, Tisserant E, Talhinhas P, Azinheira H, Vieira A, Petitot A-S, Loureiro A, Poulain J, Da Silva C, Silva MDC, Duplessis S. 2012. 454-pyrosequencing of Coffea arabica leaves infected by the rust fungus Hemileia vastatrix reveals in planta-expressed pathogen-secreted proteins and plant functions in a late compatible plant-rust interaction. Mol. Plant Pathol. 13:17–37. 10.1111/j.1364-3703.2011.00723.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harwood SH, Grosovsky AJ, Cowles EA, Davis JW, Beckage NE. 1994. An abundantly expressed hemolymph glycoprotein isolated from newly parasitized Manduca sexta larvae is a polydnavirus gene product. Virology 205:381–392. 10.1006/viro.1994.1659 [DOI] [PubMed] [Google Scholar]
- 63.Serbielle C, Moreau S, Veillard F, Voldoire E, Bézier A, Mannucci M-A, Volkoff A-N, Drezen J-M, Lalmanach G, Huguet E. 2009. Identification of parasite-responsive cysteine proteases in Manduca sexta. Biol. Chem. 390:493-502. 10.1515/BC.2009.061 [DOI] [PubMed] [Google Scholar]
- 64.Le NT, Asgari S, Amaya K, Tan FF, Beckage NE. 2003. Persistence and expression of Cotesia congregata polydnavirus in host larvae of the tobacco hornworm, Manduca sexta. J. Insect Physiol. 49:533–543. 10.1016/S0022-1910(03)00052-0 [DOI] [PubMed] [Google Scholar]
- 65.Ohler U, Liao G-C, Niemann H, Rubin GM. 2002. Computational analysis of core promoters in the Drosophila genome. Genome Biol. 3:RESEARCH0087. 10.1186/gb-2002-3-12-research0087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weber B, Annaheim M, Lanzrein B. 2007. Transcriptional analysis of polydnaviral genes in the course of parasitization reveals segment-specific patterns. Arch. Insect Biochem. Physiol. 66:9–22. 10.1002/arch.20190 [DOI] [PubMed] [Google Scholar]
- 67.Harwood SH, Beckage NE. 1994. Purification and characterization of an early-expressed polydnavirus-induced protein from the hemolymph of Manduca sexta larvae parasitized by Cotesia congregata. Insect Biochem. Mol. Biol. 24:685–698. 10.1016/0965-1748(94)90056-6 [DOI] [Google Scholar]
- 68.Pruijssers AJ, Strand MR. 2007. PTP-H2 and PTP-H3 from Microplitis demolitor bracovirus localize to focal adhesions and are antiphagocytic in insect immune cells. J. Virol. 81:1209–1219. 10.1128/JVI.02189-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bae S, Kim Y. 2009. IkB genes encoded in Cotesia plutellae bracovirus suppress an antiviral response and enhance baculovirus pathogenicity against the diamondback moth, Plutella xylostella. J. Invertebr. Pathol. 102:79–87. 10.1016/j.jip.2009.06.007 [DOI] [PubMed] [Google Scholar]
- 70.Ohno S. 1970. The enormous diversity in genome sizes of fish as a reflection of nature's extensive experiments with gene duplication. Trans. Am. Fish. Soc. 99:120–130 [Google Scholar]
- 71.Innan H, Kondrashov F. 2010. The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 11:97–108. 10.1038/nrg2689 [DOI] [PubMed] [Google Scholar]