

ABSTRACT
Occasional transmission of highly pathogenic avian H5N1 influenza viruses to humans causes severe pneumonia with high mortality. To better understand the mechanisms via which H5N1 viruses induce severe disease in humans, we infected cynomolgus macaques with six different H5N1 strains isolated from human patients and compared their pathogenicity and the global host responses to the virus infection. Although all H5N1 viruses replicated in the respiratory tract, there was substantial heterogeneity in their replicative ability and in the disease severity induced, which ranged from asymptomatic to fatal. A comparison of global gene expression between severe and mild disease cases indicated that interferon-induced upregulation of genes related to innate immunity, apoptosis, and antigen processing/presentation in the early phase of infection was limited in severe disease cases, although interferon expression was upregulated in both severe and mild cases. Furthermore, coexpression analysis of microarray data, which reveals the dynamics of host responses during the infection, demonstrated that the limited expression of these genes early in infection led to a failure to suppress virus replication and to the hyperinduction of genes related to immunity, inflammation, coagulation, and homeostasis in the late phase of infection, resulting in a more severe disease. Our data suggest that the attenuated interferon-induced activation of innate immunity, apoptosis, and antigen presentation in the early phase of H5N1 virus infection leads to subsequent severe disease outcome.
IMPORTANCE Highly pathogenic avian H5N1 influenza viruses sometimes transmit to humans and cause severe pneumonia with ca. 60% lethality. The continued circulation of these viruses poses a pandemic threat; however, their pathogenesis in mammals is not fully understood. We, therefore, investigated the pathogenicity of six H5N1 viruses and compared the host responses of cynomolgus macaques to the virus infection. We identified differences in the viral replicative ability of and in disease severity caused by these H5N1 viruses. A comparison of global host responses between severe and mild disease cases identified the limited upregulation of interferon-stimulated genes early in infection in severe cases. The dynamics of the host responses indicated that the limited response early in infection failed to suppress virus replication and led to hyperinduction of pathological condition-related genes late in infection. These findings provide insight into the pathogenesis of H5N1 viruses in mammals.
INTRODUCTION
Since late 2003, H5N1 highly pathogenic avian influenza viruses have spread among poultry and wild birds in Asia, Africa, and Europe. These H5N1 influenza viruses sometimes transmit to humans and cause severe diseases, ca. 60% of which have been fatal (1). Continued circulation of the H5N1 viruses among poultry allows for sporadic transmission to humans, which raises the possibility of the avian virus acquiring the ability to transmit efficiently from human to human. Therefore, a comprehensive understanding of the pathogenesis of H5N1 virus infection is needed to develop effective prevention and intervention strategies.
Typically, human patients infected with H5N1 virus develop progressive pneumonia accompanied by diffuse alveolar damage and acute respiratory distress syndrome (ARDS) (2–8). Severe H5N1 virus infection induces high serum levels of inflammatory molecules such as interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), gamma interferon (IFN-γ), chemokine (C-X-C motif) ligand 10 (CXCL10; IP-10), chemokine (C-C motif) ligand 2 (CCL2; MCP-1), and IL-8 (5, 9, 10). These findings suggest that hypercytokinemia is involved in the pathogenicity of H5N1 virus in humans; however, the mechanisms by which the H5N1 virus infection induces hypercytokinemia and high virulence are not yet fully understood.
Several comprehensive analyses of the global host responses to highly pathogenic influenza A virus infections have been performed using nonhuman primate models (11). These studies have shown that genes related to type I interferons, innate immunity, inflammation, and cell death, expressed in the acute phase of infection, are responsible for the high pathogenicity of H5N1 viruses (12–14). However, because these studies examined a single H5N1 virus strain (14) or compared an H5N1 strain with an H1N1 “Spanish flu” 1918 virus (13) or with a seasonal human influenza virus and reassortant 1918 viruses (12), it remains unknown whether differences in host responses to different H5N1 virus strains exist and cause different outcomes.
Therefore, we first examined the growth properties and pathogenicity in macaques of H5N1 viruses isolated from lethal and nonlethal human cases and then compared the host responses to severe and mild H5N1 virus infections. The results suggest that the differences in severity of the disease outcome upon infection with different H5N1 viruses originate from differential expression of host genes related to specific biologic events.
MATERIALS AND METHODS
Viruses and cells.
Six Vietnamese H5N1 influenza viruses (Table 1) were isolated from human samples and passaged in Madin-Darby canine kidney (MDCK) cells to produce viral stocks (15). All strains except for A/Vietnam/UT3028II/2004 clone3 (VN3028IIcl3) and A/Vietnam/30408/2005 clone7 (VN30408cl7) were passaged twice in MDCK cells; VN3028IIcl3 and VN30408cl7 were previously plaque purified by use of twice-passaged samples in MDCK cells (16), that is, they were passaged four times. VN3040 was isolated in our laboratory from the same human specimen from which A/Viet Nam/1203/2004 (H5N1) was isolated. There are no amino acid differences between VN3040 and A/Viet Nam/1203/04, although there are two nucleotide differences in the NA gene at position 143 (the former having adenine and the latter guanine) and the M gene at position 250 (the former having adenine and the latter guanine). All experiments with these H5N1 viruses were performed in approved enhanced biosafety level 3 (BSL3) containment laboratories.
TABLE 1.
The six highly pathogenic avian influenza H5N1 viruses analyzed in this studya
| Virus strain | Abbreviation | Clinical outcome of patient | HA clade | Receptor-binding specificityb |
PB1-F2 | PB2 |
NS1 |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| α2,3 | α2,6 | 66 | 591 | 627 | 701 | 92 | C terminus | ||||
| A/Vietnam/UT3028II/2004 clone3 | VN3028IIcl3 | Died | 1 | ++++ | ++++ | N | Q | K | D | D | ESEV |
| A/Vietnam/UT30259/2004 | VN30259 | Died | 1 | ++++ | ++ | N | Q | K | D | D | ESEV |
| A/Vietnam/UT3040/2004 | VN3040 | Died | 1 | ++++ | – | N | Q | K | D | D | 10-aa deletion |
| A/Vietnam/UT3062/2004 | VN3062 | Died | 1 | ++++ | – | N | Q | K | D | D | ESEV |
| A/Vietnam/30408/2005 clone7 | VN30408cl7 | Recovered | 1 | ++++ | +++ | N | Q | K | D | D | ESEV |
| A/Vietnam/UT30850/2005 | VN30850 | Died | 2.3.4 | ++++ | – | N | Q | K | D | D | ESEV |
For PB1-F2, PB2, and NS1, the amino acid at the position(s) indicated in the subheadings is specified.
The level of receptor-binding affinity is indicated using a “+/−” scale, where “−” indicates no receptor binding affinity, “+” indicates affinity, and the number of “+” symbols indicates the intensity of the receptor-binding affinity (21).
MDCK cells were maintained in Eagle minimal essential medium (MEM) containing 5% newborn calf serum and antibiotics. After infection with influenza virus, the MDCK cells were maintained in MEM containing 0.3% bovine serum albumin.
Experimental infection of cynomolgus macaques.
We used 4- to 7-year-old female cynomolgus macaques (Macaca fascicularis) from Vietnam (obtained from Ina Research, Inc.; Keari Co., Ltd.; or Japan Laboratory Animals, Inc.), weighing 2.5 to 3.5 kg and serologically negative by AniGen AIV antibody enzyme-linked immunosorbent assay, which detects antibody to nucleoprotein of all influenza A virus subtypes (Animal Genetics, Inc.). While the animals were under anesthesia, telemetry probes (TA10CTA-D70; Data Sciences International) were implanted in their peritoneal cavities to monitor body temperature. Three macaques per group were intramuscularly anesthetized with ketamine (5 mg/kg) and xylazine (1 mg/kg) and inoculated with a suspension containing 107 PFU of each H5N1 virus/ml through a combination of the intratracheal (4.5 ml), intranasal (0.5 ml per nostril), ocular (0.25 ml per eye), and oral (1 ml) routes, resulting in a total infectious dose of 107.8 PFU. Macaques were monitored every 15 min for changes in body temperature. For one of the macaques (macaque 18), the telemetry probes did not work, and therefore its temperature was rectally determined. On days 1, 3, 5, 7, 9, 11, and 13 postinfection (p.i.), nasal swab, bronchial brush, and rectal swab samples were collected and suspended in 500 μl of phosphate-buffered saline. One macaque died on day 9 (macaque 9) and underwent pathological and virological analyses; the other macaques were euthanized on day 14 p.i. for pathological analysis. Virus titers in swabs, bronchial brushes, and various organs were determined by plaque assays in MDCK cells. All experiments were carried out in accordance with the Guidelines for the Husbandry and Management of Laboratory Animals of the Research Center for Animal Life Science at Shiga University of Medical Science, Shiga, Japan, and approved by the Shiga University of Medical Science Animal Experiment Committee and Biosafety Committee.
Pathological examination.
Excised macaque lung tissue was preserved in 10% phosphate-buffered formalin. The tissue was then processed for paraffin embedding and cut into 5-μm-thick sections. Each tissue sample was stained using a standard hematoxylin-and-eosin procedure (17).
Bacterial identification.
Frozen blood or tracheal swabs from the dead macaques were plated on Trypticase soy agar (TSA) with 5% sheep blood (Becton Dickinson) and incubated at 37°C in aerobic condition for 24 h. Individual bacterial colonies from the TSA plates were identified by using an API test system (SYSMEX bioMérieux Co., Ltd.) according to the manufacturer's instructions.
Microarray analysis.
Total RNA was extracted from bronchial brush samples by using the RNeasy minikit (Qiagen), according to the manufacturer's protocol. Fluorescently labeled probes were generated by using a one-color QuickAmp labeling kit (Agilent Technologies), and each sample was hybridized to a rhesus macaque gene expression microarray (Agilent microarray design identification number 015421; Agilent Technologies), as previously described (18). Individual microarrays were performed for each bronchial brush sample that was collected from animals prior to infection and on days 1, 3, 5, and 7 p.i.
Statistical analysis was performed using the LIMMA package. The log2 of the intensity of each probe was background corrected and normalized between arrays (using the quantile method). Duplicate probes were averaged. Hierarchical clustering was used to review biological replicate quality. Contrasts comparing the expression of each infection-day group to its respective preinfection data were constructed and the moderated t-statistic was calculated to determine the significance between the means. Lastly, the P values were Benjamini-Hochberg corrected to control the false discovery rate (FDR). Probes were considered differentially expressed (DE) if they had a fold change of ≥1.5 and an FDR of <0.01. Primary gene expression data are available in Gene Expression Omnibus (series number GSE57970) in accordance with proposed Minimum Information About a Microarray Experiment (MIAME) guidelines.
Coexpression analysis.
Probes that were not DE at least once were removed to ensure confident variation in the coexpressed genes. Signed gene coexpression networks were constructed by using the WGCNA package in R, a statistics software environment. A detailed explanation of the WGCNA package is available elsewhere (19). Briefly, the pairwise correlation matrix of the probe expression for all probe combinations is computed and then, to ensure a scale free network topology, the correlation matrix is raised to a power, n, and referred to as the adjacency matrix. Next, hierarchical clustering is performed on the adjacency matrix to identify modules of coexpressed genes/probes. We used the blockwiseModules function with the default parameters. A check of the scale free characteristics showed that n = 12 was a reasonable choice based on the scale free topology criteria.
To relate the gene expression to secondary measurements, for example, virus titer, we summarized the gene expression of each gene module by using the module eigengene (the first principal component of the module's gene expression matrix) and then calculated the pairwise correlation between each module eigengene and virus titer in R. A module eigengene was defined as being significantly correlated with virus titer if they had a correlation value of ≥0.6.
Gene functional enrichment analysis.
Each module of coexpressed probes was analyzed for enrichment of gene function annotations by using ToppCluster (20). ToppCluster uses the Fisher exact test to determine annotations that are enriched in a set of genes. The resulting P values were Benjamini-Hochberg-corrected to control the FDR. The enrichment scores reported are the −log10 of the FDR. Only annotations with scores of >2 (corresponding to an FDR of <0.01) are reported and ToppCluster puts an upper bound of 10 on the enrichment score.
RESULTS
Clinical observations of macaques infected with different H5N1 viruses isolated from humans.
To evaluate the differences in pathogenicity among H5N1 highly pathogenic avian influenza viruses, we infected groups of three cynomolgus macaques with one of six H5N1 influenza virus strains (Table 1). These viruses were chosen because their history is well known; we isolated them from the original samples and therefore know their exact passage history. These viruses were also chosen because their receptor specificity was known, having been previously tested (21). These strains were isolated from humans in Vietnam in 2004 or 2005, and all except for VN30408cl7 resulted in fatal infections. All of these strains contain lysine at amino acid position 627 of PB2, which is a key virulence marker for mammalian hosts (22). All strains, except for VN3040, possess the PDZ domain-binding motif, ESEV, at the C terminus of NS1 (23), which is associated with virulence in mice (24); VN3040 has a 10-amino-acid truncation at the C terminus of its NS1 (15). Three of the viruses—VN3028IIcl3, VN30259, and VN30408cl7—recognize α-2,6-linked sialic acid (human-type receptor) in addition to α-2,3-linked sialic acid (avian-type receptor), whereas the other viruses recognize only α-2,3-linked sialic acid (21).
To compare the pathogenicity among the six H5N1 viruses, body temperature was monitored, body weight was measured every 2 days (i.e., at the time of every sample collection), and clinical symptoms (i.e., posture, respiration, recumbency, attitude, and skin condition) were observed twice daily. Within 24 h of inoculation, the body temperature had increased in most of the animals (Fig. 1). All animals infected with VN3040 and one infected with VN3028IIcl3 exhibited decreased activity and mild depression (Table 2). Some animals huddled slightly or exhibited coughing, nasal discharge, and piloerection of body hair; two animals infected with VN30850 exhibited these symptoms for several days. Macaque 9, which was infected with VN3040, succumbed to its infection on day 9 p.i. The weight loss experienced by this animal was <5%, and it did not exhibit any clinical symptoms that would have prompted euthanization according to the Guidelines for the Husbandry and Management of Laboratory Animals. On the other hand, VN30408cl7-infected animals showed little change in body temperature and did not show any clinical symptoms except for nasal discharge. These results demonstrate that there are appreciable variations in the disease severity caused by these H5N1 highly pathogenic avian influenza viruses and that VN3040 is the most virulent strain among these viruses in this animal model.
FIG 1.
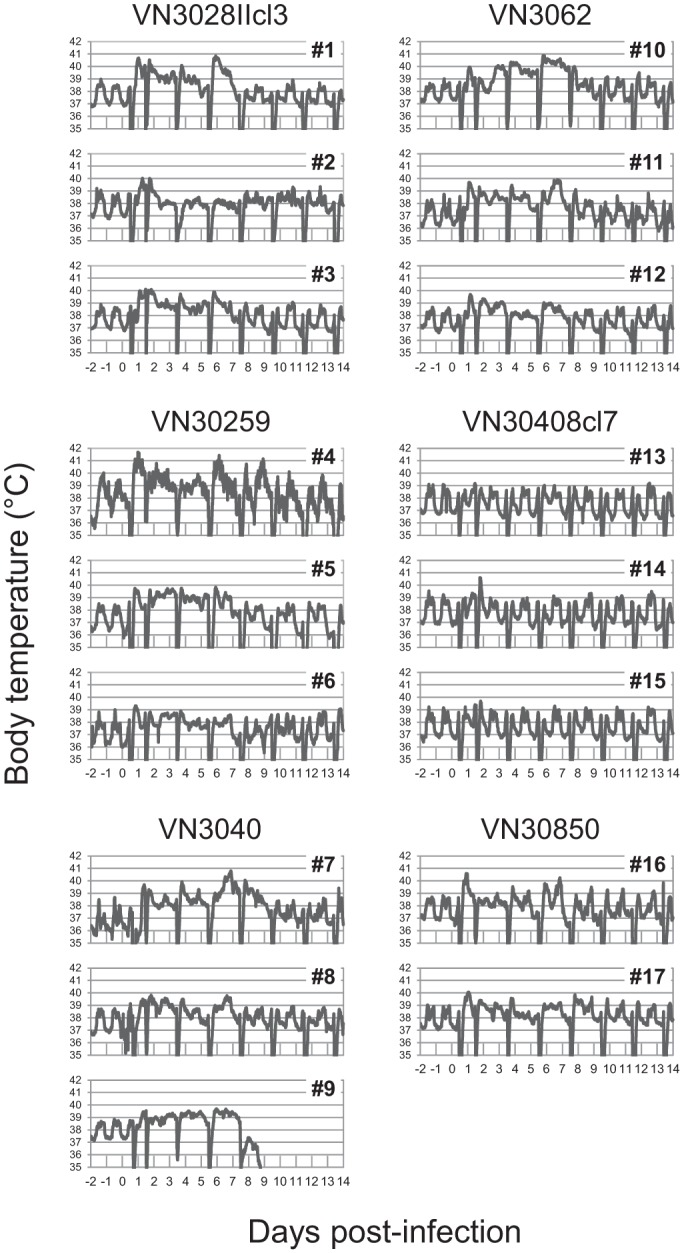
Body temperature of H5N1 virus-infected cynomolgus macaques. Three macaques per group were inoculated with 107.8 PFU (total volume, 7.0 ml) of each H5N1 virus through multiple routes. Temperatures were monitored every 15 min by telemetry probes implanted in the peritoneal cavities. The periodic sharp reductions in body temperature on days 0, 1, 3, 5, 7, 9, 11, and 13 were caused by anesthesia for sample collection. Numbers 1 to 17 indicate animal identification numbers (IDs). No data were obtained for macaque 18 because of a problem with the telemetry probe.
TABLE 2.
Clinical observations in H5N1 virus-infected animalsa
| Virus strain | Animal ID | Clinical symptom(s)b |
|---|---|---|
| VN3028IIcl3 | 1 | Decreased activity and mild depression on day 2 |
| 2 | – | |
| 3 | Mild coughing on day 10 | |
| VN30259 | 4 | – |
| 5 | Mild huddling on day 5; nasal discharge on days 5 and 6 | |
| 6 | – | |
| VN3040 | 7 | Decreased activity on days 2, 3, 4, 5, 7, and 8; mild depression on days 3, 5, 7, and 8; mild coughing on day 8 |
| 8 | Decreased activity on days 2 and 5; mild depression on day 5 | |
| 9 | Decreased activity and lying down on day 8; dying on day 9 | |
| VN3062 | 10 | – |
| 11 | Mild huddling on day 6 | |
| 12 | Piloerection of body hair on days 1, 2, 4, 6, and 7 | |
| VN30408cl7 | 13 | Nasal discharge on day 5 |
| 14 | – | |
| 15 | – | |
| VN30850 | 16 | – |
| 17 | Mild coughing on days 2 and 3; mild huddling on day 7 | |
| 18 | Piloerection of body hair on day 0; mild huddling on days 1, 2, and 3 |
Cynomolgus macaques were inoculated with 107.8 PFU thorough multiple routes and monitored twice daily throughout the study for each clinical parameter, i.e., posture, respiration, recumbency, attitude, and skin condition.
–, No clinical symptom observed.
Comparison of viral replication in the organs of infected macaques.
To evaluate virus replication in the organs of infected animals, bronchial brushes, nasal swabs, and rectal swabs were collected on every second day after infection. Although all virus strains were detected in the nasal swabs and bronchial brushes, there were differences among these strains in both the periods when the virus was detected and in the virus titers (Table 3). VN3040 and VN30850 were detected in bronchial samples through days 7 or 9, whereas VN3028IIc3, VN30259, and VN30408cl7 were cleared soon after the infection by day 5 or 7. VN3062 was detected in the bronchus through day 7, but in nasal swabs it was only detected in two of three macaques through day 3, suggesting that this strain may not replicate efficiently in the upper respiratory tract. No virus was detected in rectal swabs throughout the study. Given that VN3040 and VN30850 showed efficient virus replication in the respiratory tracts and were associated with more severe clinical symptoms (Table 2), efficient virus replication appears to contribute to disease severity in macaques. This relationship was also observed in human patients infected with H5N1 viruses (10).
TABLE 3.
Virus titers in respiratory swabs collected from H5N1 virus-infected animalsa
| Virus strain | Animal ID | Virus titer (log10 PFU/ml)b in: |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bronchial brush at: |
Nasal swab at: |
||||||||||
| 1 dpi | 3 dpi | 5 dpi | 7 dpi | 9 dpi | 1 dpi | 3 dpi | 5 dpi | 7 dpi | 9 dpi | ||
| VN3028IIcl3 | 1 | 3.1 | 1.0 | – | – | – | – | – | 1.0 | – | – |
| 2 | – | 3.5 | – | – | – | – | – | – | – | – | |
| 3 | 1.3 | 1.3 | – | – | – | – | – | – | – | – | |
| VN30259 | 4 | 2.6 | 2.1 | 1.6 | – | – | 1.0 | 1.0 | 2.8 | 3.1 | – |
| 5 | 2.7 | 1.6 | – | – | – | 1.9 | – | – | 1.0 | – | |
| 6 | 2.5 | – | – | – | – | – | – | – | – | – | |
| VN3040 | 7 | 3.3 | 3.8 | 5.4 | 3.7 | 1.3 | 3.1 | 3.7 | – | – | 2.5 |
| 8 | 1.9 | 2.5 | 1.0 | 1.0 | – | 4.0 | 2.8 | – | 2.7 | – | |
| 9 | 3.7 | 3.8 | 5.2 | 3.9 | Died | 3.6 | 1.0 | 1.0 | 2.5 | Died | |
| VN3062 | 10 | 3.8 | 4.1 | 3.6 | – | – | 2.2 | 2.5 | – | – | – |
| 11 | 3.6 | 3.6 | 1.7 | 1.5 | – | 1.0 | 1.0 | – | – | – | |
| 12 | 4.2 | 4.3 | 3.3 | – | – | – | – | – | – | – | |
| VN30408cl7 | 13 | 2.0 | 1.0 | – | – | – | 1.5 | 1.7 | 1.6 | – | – |
| 14 | 4.3 | – | 2.2 | – | – | 1.8 | 2.7 | – | – | – | |
| 15 | – | – | – | – | – | – | – | – | – | – | |
| VN30850 | 16 | 3.2 | 3.3 | 2.0 | – | – | 1.0 | 1.0 | 3.4 | 2.5 | 1.0 |
| 17 | 4.8 | 1.9 | 1.6 | 2.2 | – | 4.0 | 1.5 | 1.5 | 1.9 | – | |
| 18 | 2.0 | 2.7 | 2.6 | 1.0 | – | 2.6 | 1.6 | 2.8 | 2.8 | 2.0 | |
Bronchial brushes and nasal swabs were collected every other day for virus titration after virus infection. Virus titers in the samples were determined by plaque assays in MDCK cells.
−, Virus not detected (detection limit = 1.0 log10 PFU/ml). dpi, days p.i.
To evaluate whether certain variants in the inoculum became dominant during replication in animals, we sequenced the genes of all of the viruses that were isolated from respiratory swabs on days 7 and 9 (i.e., VN30259, VN3040, VN3062, and VN30850). Twenty-eight amino acid substitutions compared to the consensus virus sequences of the inoculated viruses were detected among 14 virus isolates in total. Although four amino acid substitutions were common to two of three animals infected with the same virus, the other substitutions were specific to the virus in each sample. In addition, we did not detect any substitutions that are known to be important for mammalian adaptation and pathogenicity in mammals among these 28 substitutions. Therefore, it is unlikely that the pathogenicity shown by the inoculated viruses was due to any of these substitutions that became dominant during replication in animals.
Analysis of the VN3040-infected macaque that died.
Some previous studies have reported detection of H5N1 viruses outside the respiratory tract, that is, in the brains, cerebrospinal fluid, intestines, and feces of human patients (8, 25, 26). To investigate the cause of death for VN3040-infected macaque 9, virus distribution in its organs was analyzed (Table 4). Virus was detected not only in the lower respiratory tract but also in the upper respiratory tract, including the nasal mucosa, nasal turbinates, and trachea. The virus titer of the tonsils was also high, which is consistent with previous reports in macaques (12, 14, 27, 28) and in ex vivo culture of human tissue (29). In addition, a high virus titer was also detected in the conjunctiva. No virus was detected in the heart, spleen, kidney, liver, small intestines, colon, or brain of this animal, indicating that VN3040 did not cause a systemic infection.
TABLE 4.
Virus titers in organs of macaque 9
| Organ | Virus titer (log10 PFU/g of tissue)a |
|---|---|
| Brain | – |
| Conjunctiva | 4.4 |
| Nasal mucosa | 3.0 |
| Nasal turbinates | 2.5 |
| Oro/nasopharynx | – |
| Tonsil | 4.8 |
| Trachea | 3.2 |
| Bronchus, right | 4.4 |
| Bronchus, left | 4.8 |
| Lung, right upper | 4.3 |
| Lung, right middle | 3.0 |
| Lung, right lower | 3.2 |
| Lung, left upper | 2.0 |
| Lung, left middle | 4.6 |
| Lung, left lower | 3.8 |
| Lymph node, chest | – |
| Heart | – |
| Spleen | – |
| Kidney | – |
| Liver | – |
| Duodenum | – |
| Ileum | – |
| Jejunum | – |
| Transverse colon | – |
| Descending colon | – |
–, Virus not detected (detection limit = 2.0 log10 PFU/g of tissue).
Fifteen amino acid substitutions compared to the consensus sequence of the inoculum were detected among 13 isolates from the tissues of the animal 9. One substitution detected in a lung lobe was also found in a nasal swab from animal 8, which was infected with the same virus (VN3040): however, the other substitutions were specific to each sample. In addition, we did not detect any substitutions known to be important for mammalian adaptation or pathogenicity in mammals. Therefore, these substitutions are unlikely to be responsible for the lethal outcome of the animal.
To evaluate the possibility of bacterial superinfection in the dead animal, we examined a frozen tracheal swab and a frozen blood sample collected during the dissection of the animal. Some bacteria were detected from the tracheal swab (Staphylococcus chonii subsp. urealyticus and Enterococcus faecalis), as well as from the blood sample (S. chonii subsp. urealyticus, E. faecalis, Pseudomonas putida, Streptococcus hyointestinalis, P. aeruginosa, and Escherichia coli). However, these are indigenous bacteria, which are commonly found in intestines, on the skin, or in soil, and are not associated with influenza virus infection. In addition, we did not detect any histological evidence of bacterial infection in the organs listed in Table 4. Therefore, the bacteria found in these samples likely invaded tracheal and blood vessels from intestines and skin between the death and the dissection of the animal (about 12 h). Accordingly, the virus infection of the respiratory tract was likely the cause of the death.
Pathological changes in lungs after H5N1 virus infection.
Next, we performed a pathological examination of lungs after H5N1 virus infection (Fig. 2). Lungs collected from all surviving animals at 14 days p.i. showed thickened alveolar walls with granulation tissues composed of alveolar epithelial cells and fibroblasts, although the extent of the lesion differed among the individual animals (Fig. 2A to F), suggesting that these lungs on day 14 was undergoing tissue remodeling. In contrast, the lungs of macaque 9, which died on day 9, exhibited severe lesions with diffuse alveolar damage in some lobes (Fig. 2G). These observations are identical to those in human patients who died as a result of H5N1 virus infection (6–8), suggesting that these histological lesions induced by VN3040 infection would reflect respiratory failure, leading to death.
FIG 2.

Pathological changes in the lungs of animals infected with H5N1 viruses. Lungs recovered from animals on day 14 p.i. (A) Lung of macaque 3 infected with VN3028IIcl3; (B) lung of macaque 5 infected with VN30259; (C) lung of macaque 7 infected with VN3040; (D) lung of macaque 11 infected with VN3062; (E) lung of macaque 14 infected with VN30408cl7; (F) lung of macaque 16 infected with VN30850; (G) lung of macaque 9, which was infected with VN3040 and died on day 9. The lumina of the alveoli and bronchioles were filled with edema fluid, fibrin, and cell debris from neutrophils and lymphocytes. The epithelia of the alveolar and bronchiolar walls were disrupted, and hyaline membranes were apposed to the alveolar walls (arrowheads). The alveolar and bronchiolar walls were thickened due to infiltration of lymphocytes, alveolar epithelial cells, and neutrophils. (H) Normal lung of a mock-infected macaque.
Different host responses elicited by different H5N1 virus strains in the early phase of infection.
To elucidate the differences in the host responses to H5N1 virus infection between severe and mild disease cases, we performed global gene expression profiling by expression microarray. We analyzed animals infected with VN3040, which replicated efficiently in the lower respiratory tract (Table 3) and induced more severe disease (Table 2), as well as animals infected with VN3028IIcl3 or VN30259, which were cleared from the lower respiratory tract soon after infection and induced milder disease. Total RNA was extracted from each bronchial brush sample collected from each animal prior to infection and on days 1, 3, 5, and 7 and was used for the microarray analysis.
Initially, we compared the genes that were DE in severe and mild disease cases on each day (Fig. 3A). A gene probe was defined as being DE in the infected groups if it had a fold change in expression relative to preinfection of 1.5 and an FDR of <0.01 at each of the four time points sampled. In VN3028IIcl3- and VN30259-infected animals, 894 (549 upregulated and 345 downregulated) and 409 (315 upregulated and 94 downregulated) gene probes, respectively, were DE on day 1, and the number of DE gene probes subsequently decreased with time. In contrast, in VN3040-infected animals, fewer gene probes (202 gene probes; 180 upregulated and 22 downregulated) were DE on day 1, but the number of DE gene probes gradually increased with time, culminating in 3,676 gene probes on day 5. These results suggest that the host transcriptional response in the early phase of infection (on day 1) may contribute to the control of the virus infection and that the differences in the DE gene probes in the early phase of infection, as well as the subsequent regulation of these genes (i.e., attenuation or excessive activation of expression) may cause the different outcomes in H5N1 virus infection.
FIG 3.

Differential gene expression during VN3028IIcl3, VN30259, or VN3040 infection. (A) Total RNAs were extracted from each bronchial brush sample collected from each animal infected with VN3040, VN3028IIcl3, or VN30259 at preinfection and on days 1, 3, 5, and 7 p.i. and were used for the microarray analysis. Within each infection group, postinfection samples were compared to preinfection samples from the respective animal to identify DE genes on each day of the infection (DE was defined as having a fold change of at least 1.5 and an FDR < 0.01). The upregulated and downregulated genes are indicated by darker- and lighter-shaded portions of the stacked bars, respectively, and the numbers of upregulated and downregulated genes are indicated in black and in gray numbers, respectively, above each time point bar. (B) Venn diagram representing the overlap of DE genes on day 1 p.i. in a direct comparison among the respective virus infections. The numbers in red and black indicate upregulated and downregulated gene numbers, respectively. As shown, genes detected by 135 probes were commonly upregulated in all three virus infections (darker gray) and genes detected by 125 probes were commonly upregulated in only VN3028IIcl3 and VN30259 infections (lighter gray). (C) Heat map of ISG expression in each of the virus-infected animals on day 1 p.i. Each column represents the log2-fold change in gene expression in each animal after each virus infection, which is shown in red if it was upregulated relative to preinfection. For several genes, multiple probes are present on the microarray slide.
The observations described above prompted us to analyze the similarities and the differences of the host transcriptional responses on day 1 between severe and mild outcomes (Fig. 3B). In the Venn diagram representing the DE gene probes of day 1, we focused on 125 gene probes that were upregulated only by VN3028IIcl3 and VN30259 infection (mild cases), and 135 gene probes that were commonly upregulated by infection with all three viruses. All of the genes belonging to the 125- and the 135- probe sets are listed in Tables S1 and S2 in the supplemental material. Interestingly, the 125-probe set, which was upregulated only in mild cases, contained many interferon-stimulated genes (ISGs; 74 gene probes, 59.2%), including SOCS2, HLAs, GBP2, DDX58 (RIG-I), CXCL11, FAS, TLR2, MYD88, and JAK2 according to the Interferome (30) (Fig. 3C and see Table S1 in the supplemental material). Gene Ontology (GO) functional enrichment analysis by ToppCluster (20) showed that the 125-probe set was associated with “immune response” (score 5.1; enrichment scores are the −log10 of the FDR and significant scores ranged from 2 to 10; see Materials and Methods), “regulation of apoptotic process” (score 3.7), and “innate immune response” (score 2.2), which would contribute to the control of the virus infection. In addition, the subdivided biological processes “activation of cysteine-type endopeptidase activity involved in apoptosis” (score 3.7) and “antigen processing and presentation of exogenous antigen via major histocompatibility complex (MHC) class, TAP dependent” (score 2.0) were enriched only for the 125-probe set. Thus, innate immune response, apoptosis, and antigen presentation may be important for the control of the infection and the disease.
Similarly, 105 of the 135 gene probes upregulated in all three infections were ISGs, including ISGs with anti (influenza) viral functions (31–54) (Fig. 3C and see Table S2 in the supplemental material). Functional enrichment analysis of these 135 gene probes revealed that a large proportion of these genes was associated with “innate immune response” (score 10), indicating that upregulation of these 135 gene probes was largely involved in the interferon responses to control the virus infection. Interestingly, the fold change values in the expression of ISGs, especially IFIT1/2/3, OAS2, GBP1, RSAD2 (viperin), IFITM1/2, APOBEC3G, and TRIM22 among the antiviral ISGs in the VN3040 infection were lower than those in the VN3028IIcl3 and VN30259 infections (Fig. 3C and see Table S2 in the supplemental material), suggesting that VN3040 induced weaker interferon-mediated antiviral responses than did VN3028IIcl3 and VN30259 in the early phase of infection. In contrast, the expression levels of type I interferon genes (IFNAs and IFNB1) in VN3040-infected animals on day 1 were similar to those in VN3028IIcl3- and VN30259-infected animals, and the expression levels of type III interferon genes (IL-28A, IL-28B, and IL-29) induced by VN3040 were higher than those by VN3028IIcl3 and VN30259 infections (see Table S3 in the supplemental material). These findings suggest that limited induction of ISGs in the early phase of infection, which was observed only in VN3040-infected animals, is related to disease severity.
Differences in the dynamics of host responses during severe and mild diseases.
Next, to further identify the differences in the dynamics of transcriptional responses between severe and mild outcomes and to identify potential molecular modulators of gene expression, we performed a coexpression analysis, known as a weighted gene correlation network analysis (WGCNA). Within the WGCNA framework, we can identify modules of genes that have a highly correlated gene (19). We can then use the eigengene of each module (the first principal component of the gene expression matrix), which is a scaled representation of the expression of the genes assigned to each module to explore for potentially strain-dependent responses (55). This coexpression analysis is not biased by existing knowledge of pathways or interactions and can identify systemic gene expression patterns based on underlying correlation structures.
By using WGCNA, we identified 11 modules of coexpressed genes (M1 [module 1] to M11) (Fig. 4). All of the gene probes belonging to each module are listed in Table S4 in the supplemental material. We performed GO functional enrichment analysis and identified the biological functions of the individual modules; representative GO biological processes (or GO molecular functions for M11) are indicated in Table 5, and all GO biological processes are shown in Table S5 in the supplemental material. In addition, we analyzed the correlation between the virus titers and the module eigengenes (i.e., the gene expression levels) (Fig. 5) and found a significant correlation in several modules. In particular, the M2, M6, and M7 genes had the greatest correlation (correlation value of ≥0.6), suggesting that virus replication induced the expression of those genes.
FIG 4.

Expression dynamics of 11 modules of genes representing host responses to H5N1 virus infection for each animal (M1 to M11). WGCNA was performed to identify modules of coexpressed genes. The 11 gene modules identified here were further analyzed for enriched GO categories as shown in Tables 5 and see Table S5 in the supplemental material. For each gene module, the expression levels of the eigengene for each sample are indicated by a colored bar: blue, VN3028IIcl3-infected animals; green, VN30259-infected animals; pink, and VN3040-infected animals. The time points for sample collection and the animal IDs (1 to 9) are indicated above and below the graph, respectively.
TABLE 5.
Functional enrichment analysis of genes identified as being coexpressed by WGCNA
| Gene module | No. of probes | Representative enriched gene functiona (enrichment score) | ISG content (%)b |
|---|---|---|---|
| M1 | 1,140 | Microtubule-based process (10), cell projection assembly (10) | 6.0 |
| M2 | 922 | Immune response (10), innate immune response (10), adaptive immune response (10), inflammatory response (10), cytokine production (10), lymphocyte activation (10), lymphocyte migration (10), positive regulation of apoptotic process (10), coagulation (10), hemopoiesis (10), homeostatic process (10), IFN-γ production (3.7) | 18.4 |
| M3 | 859 | Antigen processing and presentation of peptide antigen via MHC class I (10), macromolecular complex subunit organization (10), intracellular protein transport (10), RNA catabolic process (10), translation (10), viral infectious cycle (10), G1/S transition checkpoint (10), regulation of apoptotic process (2.6) | 8.1 |
| M4 | 858 | – | 3.3 |
| M5 | 377 | – | 4.2 |
| M6 | 291 | Immune response (10), innate immune response (10), cytoplasmic pattern recognition receptor signaling pathway in response to virus (10), type I interferon production (10), type I interferon-mediated signaling pathway (10), response to virus (10), cytokine production (10), cytokine-mediated signaling pathway (10), I-κB kinase/NF-κB cascade (10), regulation of apoptotic process (10), IFN-γ-mediated signaling pathway (5.8), RIG-I signaling pathway (3.4), JAK-STAT cascade (2.8), ISG15-protein conjugation (2.5) | 62.9 |
| M7 | 138 | Immune response (10), innate immune response (5.1), pattern recognition receptor signaling pathway (3.4), cytokine-mediated signaling pathway (3.3), regulation of sequence-specific DNA binding transcription factor activity (3.0) | 36.2 |
| M8 | 133 | Microtubule-based process (10), mitosis (10), mitotic cell cycle (10) | 6.8 |
| M9 | 128 | Antigen processing and presentation of peptide antigen via MHC class I (10) | 35.9 |
| M10 | 75 | – | 26.7 |
| M11 | 38 | Neurotransmitter receptor activity (GO molecular function) (2.7), cation transmembrane transporter activity (GO molecular function) (2.6) | 2.6 |
Genes in each module were analyzed using ToppCluster to identify enriched GO biological processes and GO molecular functions. ToppCluster used the Fisher exact test for enrichment and reports an “enrichment score” that is the −log10 of the BH-adjusted P value. Because many GO annotations are related, representative terms were selected to summarize the enrichment results. M11 was not enriched by any GO biological process but was enriched by GO molecular functions. –, No enriched GO function was identified.
The ISGs in each module were identified by using Interferome.
FIG 5.

Relationship between virus titers in bronchial brushes and the expression levels of genes for each module (M1 to M11). The x axis represents the virus titer and the y axis represents the expression level of the eigengene of the gene module. Each marker indicates each sample; virus strains are indicated by different colors. Correlation values, which are given in each graph, were calculated by use of the pairwise correlation. Significant correlations between the virus titers and the expression levels of genes were observed for M2, M6, and M7.
To elucidate host responses that were potentially involved in the control of the infection, we focused on the early host responses (on day 1) of VN3028IIcl3- and VN30259-infected animals (mild disease). Interestingly, a remarkable upregulation of M6, M7, M9, and M10 on day 1 was observed in VN3028IIcl3- and VN30259-infected animals (Fig. 4). In contrast, VN3040-infected animals exhibited attenuated responses for these modules. All of these gene modules contained many ISGs based on the Interferome (30) (Table 5), implying that strong expression of the M6, M7, M9, and M10 genes in the early phase of infection may be important for virus clearance.
M6 was strongly enriched in functions involved in “innate immune response” and more specific biological processes such as “cytoplasmic pattern recognition receptor signaling pathway,” “type I interferon production,” “type I interferon-mediated signaling pathway,” and “apoptosis” (Table 5). M6 contained a number of genes that play roles in initiating and regulating the innate responses to influenza virus infection (see Table S4 in the supplemental material): DDX58 (RIG-I) and TLR3, which recognize influenza virus infection (56–60), type I (IFNA5) and type III (IL-28B and IL-29) interferons (61, 62), and ISGs with anti-influenza virus functions (MX1, EIF2AK2 [PKR], OAS1/2/3/L, ISG15, ISG20, RSAD2 [viperin], BST2 [tetherin], IFITM1/2, IFIT1/2/3, GBP1, TRIM22, CASP1, and ZC3HAV1 [ZAP]) (31–54). The chemokine genes CXCL10 (IP-10), which is related to disease severity in influenza (63), CXCL2 (MIP-2), and CXCL11 were also found in M6. In addition, M6 contained the RelA gene (RELA), a subunit of the pleiotropic transcription factor NF-κB, which is involved in many biological processes, including inflammation, immunity, and apoptosis. Similarly, M7 contained type I interferon (IFNA8, IFNA16, and IFNA21) genes (all type I and type III interferon genes belonged to M6 or M7) and another subunit of NF-κB (NFKB1). M7 was also functionally enriched as “innate immune response,” “pattern recognition receptor signaling pathway,” and “regulation of sequence-specific DNA binding transcription factor activity” (Table 5). Therefore, M6 and M7 genes probably contribute to the inhibition of virus replication via the interferon response in the early phase of infection.
Although VN3028IIcl3- and VN30259-infected animals showed dramatic upregulation of M6 and M7 genes in the early phase of infection (on day 1), their expression levels largely declined with time (Fig. 4 and 6A). In contrast, although VN3040-infected animals exhibited weaker expression of these genes on day 1, their gene expression was sustained or accelerated in the late phase of infection (days 3 to 7). In addition, correlation analysis revealed that there were significant correlations between virus titers and the expression levels of genes in these gene modules (Fig. 5), suggesting that insufficient expression of M6 and M7 genes early in infection allows continuous virus replication, resulting in excessive upregulation of M6 and M7 later in the infection.
FIG 6.

Expression profiles of M6, M7, and M2 genes for each virus infection. (A) Type I and III interferon genes, the NF-κB genes, and representative ISGs, which belong to M6 or M7. (B) Representative M2 genes involved in the indicated biological process. Each column represents the mean log2-fold change among three animals for each virus infection, which is shown in red if it was upregulated relative to preinfection.
M9, which was functionally enriched as “antigen processing and presentation of peptide antigen via MHC class I,” was also upregulated in mild cases on day 1, although there was no clear difference between severe and mild cases in the expression of these genes in the late phase of infection (Fig. 4). The comparative analysis of the DE gene probes on day 1 also revealed that VN3040 failed to activate this biological process on day 1 (see Table S1 in the supplemental material). Antigen presentation is essential for initiating the adaptive immune response; therefore, the strength of activation of antigen processing and presentation in the early phase of infection in mild cases would influence the outcome of the infection. M10 was not functionally enriched and expression was strongly attenuated on day 3; its importance for virus clearance is therefore unclear.
Dysregulation of immune responses, inflammation, coagulation, and homeostasis in the late phase of infection in severe outcome cases.
Finally, to elucidate how the differences in early gene expression influence disease severity, we focused on the gene expression pattern in the late phase of infection, which showed different patterns for severe and mild cases (Fig. 4). There was a remarkable difference in M2 expression in the late phase of infection between severe and mild cases; VN3040-infected animals exhibited a dramatic upregulation of M2 genes on days 3 to 7, whereas VN3028IIcl3- and VN30259-infected animals showed weaker upregulation of M2 (Fig. 4 and 6B). M2 was GO functionally enriched as “innate immune response,” “adaptive immune response,” “inflammatory response,” “lymphocyte activation/migration,” “gamma interferon production,” “coagulation,” “homeostasis,” and “apoptosis” (Table 5). Since some of these biological functions, such as homeostasis and coagulation as well as inflammation, are directly related to the pathological condition of the host, the dramatic upregulation of M2 genes in VN3040-infected animals would represent the severe pathological condition of these animals.
Importantly, M2 contains genes of proinflammatory cytokines (TNF [TNF-α], TNFSFs, and IFNG [IFN-γ]), anti-inflammatory cytokine IL-10, which is an indicator of inflammation (64), TGF-β1, and many chemokines that are chemotactic for monocytes/macrophages, neutrophils, natural killer (NK) cells, dendritic cells, T cells, and B cells (see Table S4 in the supplemental material). The expression of these cytokines/chemokines resembles the hyperinduction of cytokines/chemokines that has been observed in human patients with fatal outcomes (9, 10).
Expression of M2 as well as M6 and M7 was significantly correlated with virus titers (Fig. 5). The M2 gene module also contained many ISGs (170 gene probes, 18.4%) (Table 5). As for the biological processes enriched for M2, it is well known that adaptive immune response, inflammation, and coagulation are promoted by the innate immune response (65–67). In addition, coagulation and homeostatic processes are also linked to inflammation; inflammation activates coagulation, which in turn activates inflammation (67, 68), and the expression and function of ion channels that control homeostasis are influenced by cytokines/chemokines and other inflammatory mediators (69). Taken together, our data suggest that excessive upregulation of M2 genes, which were probably activated by sustained strong expression of innate immune genes in M6 and M7, including type I and III interferons, in the late phase of infection is likely responsible for the severe disease outcome observed in VN3040-infected animals.
DISCUSSION
Here, we compared the pathogenicity of six highly pathogenic H5N1 viruses that were isolated from humans in a nonhuman primate model. Interestingly, there were substantial variations among these H5N1 viruses in terms of replicative ability and the severity of the disease they caused from asymptomatic to fatal. We used plaque-purified VN3028IIcl3 and VN30408cl7 viruses due to their receptor preferences. Although we cannot tell whether limited sequence diversity due to plaque purification had an effect on the mild outcome of infection, we do know that non-plaque-purified VN30259 also caused asymptomatic disease. Coexpression analysis of microarray data revealed the dynamics of host responses in severe outcome cases of H5N1 virus infections. In addition, there were significant correlations between virus titer and the gene expression levels of M2, M6, and M7 genes. These findings suggest that the attenuated activation of innate immunity, apoptosis, and antigen processing/presentation in the early phase of infection, which are usually induced by interferons, led to an inability to suppress virus replication. The subsequent continuous virus replication in additional cells in respiratory organs induced sustained activation of the innate immune response and excessive activation of inflammation, coagulation, and homeostatic process in the late phase of infection, leading to the severe outcome; however, it is not clear why the virus was able to replicate under the strong upregulation of interferons and ISGs at the later time in the infection. Nonetheless, our findings demonstrate the importance of the early strong ISG response to control highly pathogenic H5N1 virus infection.
We previously reported that macaques infected with a seasonal H1N1 virus showed clear upregulation of interferons and ISGs early in infection (day 3) (70); therefore, it is possible for host responses in animals with mild symptoms to be similar between animals infected with seasonal human influenza viruses and those infected with H5N1 viruses that cause only mild symptoms.
Type I and III interferons are the principal molecules of innate immunity against virus infection (71). These interferons show their effects through interactions with their cognate receptors, which activate the JAK/STAT cascade and additional signaling pathways to induce ISGs; a subset of ISGs are classified based on their putative functions as antiviral, host defense, apoptosis, antigen processing/presentation, cell cycle, and transcription factors (72). In our study, many ISGs were expressed weakly in severe cases in the early phase, suggesting that the severe outcome may be caused by attenuated interferon responses to H5N1 virus infection in the early phase. However, expression levels of type I and III interferon genes in severe cases on day 1 were comparable to or higher than those in mild cases (see Table S3 in the supplemental material and Fig. 6A). Therefore, the attenuated interferon-mediated signaling to induce ISGs may be involved in the severe and lethal outcome following VN3040 infection.
What causes the differences in ISG induction among VN3028IIcl3, VN30259 (mild cases), and VN3040 (severe case)? Among the amino acid differences between the viruses causing severe and mild outcomes (Table 6), differences in NS1 and HA are potentially important, because NS1 inhibits host antiviral responses elicited by type I and III interferons via multiple mechanisms (61, 73), and the receptor binding specificity of HA influences the target cells (74, 75). NS1 limits interferon production pretranscriptionally (by inhibiting RIG-I activation) and posttranscriptionally (by inhibiting the nuclear posttranscriptional processing and the export of cellular mRNAs to the cytoplasm). The latter also prevents efficient production of ISGs. A previous study showed that the NS1 of 1918 virus is more efficient in blocking ISG expression than is the NS1 of WSN virus in a human lung cell line (76). It may be that the NS1 of VN3040 blocks ISG expression more efficiently than does that of VN3028IIcl3 and VN30259.
TABLE 6.
Amino acid differences among VN3040, VN3028IIcl3, and VN30259
| Virus protein | Position | Amino acid |
||
|---|---|---|---|---|
| VN3040a | VN3028IIcl3 | VN30259 | ||
| PB2 | 391 | Q | E | E |
| 717 | A | A | T | |
| PB1 | 178 | E | D | E |
| 375 | N | N | T | |
| 384 | L | V | L | |
| 640 | V | V | I | |
| PB1-F2 | 51 | T | M | M |
| 84 | N | N | S | |
| PA | 142 | E | K | K |
| 270 | L | L | I | |
| 421 | I | S | S | |
| HA1b | 36 | K | T | T |
| 123 | S | S | P | |
| 192 | Q | R | Q | |
| 223 | S | N | S | |
| HA2 | 128 | D | D | N |
| 167 | R | R | K | |
| NP | 452 | R | R | K |
| NA | 54 | F | L | F |
| 130 | K | K | N | |
| 186 | L | L | V | |
| 264 | N | D | D | |
| 324 | Y | C | Y | |
| M1 | 248 | M | V | M |
| NS1c | 71 | G | E | E |
| 171 | G | G | D | |
| 205 | N | N | S | |
| 221 | Stop codon | K | K | |
| NS2 | 14 | V | V | M |
| 48 | T | T | A | |
Amino acid differences, which are unique to VN3040, are indicated in boldface type.
Amino acid positions are indicated in the H5 numbering system.
NS1 of these three viruses contained a deletion of amino acids 80 to 84.
The NS1 of VN3040 possesses a 10-amino-acid truncation in its C terminus. The truncated C-terminal region of NS1 possesses some functional sites: a poly(A)-binding protein II (PABII) binding site at amino acid positions 223 to 230 (73), the PDZ-binding motif “ESEV” at the C-terminal 4 amino acids (22), and a sumoylation and ISGylation site at the lysine at position 221 (77, 78). NS1 binds to PABII and blocks the export of cellular mRNAs to the cytoplasm; the truncation of the PABII binding site in VN3040-NS1 may be disadvantageous for virus replication. The PDZ-binding motif “ESEV” at the C-terminal 4 amino acids, which is a known virulence factor for mice (23), contributes to pathogenicity in a host species-specific manner (for example, a virus with the ESEV motif exhibited more efficient replication in mouse cells and yet showed attenuated replication in human cells compared to a virus that lacked the ESEV motif [79]). Thus, it remains unclear whether the ESEV motif enhances virus replication in macaques. Lysine at position 221, which is highly conserved among H5N1 viruses, is both a sumoylation site as well as an ISGylation site; the sumoylation of NS1 enhances NS1 stability and virus growth (78), whereas ISGylation inhibits NS1 functions and reduces virus replication (77, 80), suggesting that the C terminus of NS1 possesses conflicting functions for virus replication. However, ca. 20% of NS1 proteins from different subtypes of influenza A viruses have C-terminal truncations of the same, smaller, or even a larger size (81), implying that these truncations may provide an advantage to the viruses.
The HA of VN3040 recognizes only α2,3-linked sialic acid, whereas that of VN3028IIcl3 and VN30259 also recognizes α2,6-linked sialic acid as a result of amino acid substitutions at positions 123, 192, and 223 (H5 numbering) (21). In addition, all of the viruses that recognized both α2,6-linked and α2,3-linked sialic acids (VN3028IIcl3, VN30259, and VN30408cl7) caused mild disease and showed limited replicative ability in macaques (Tables 2 and 3). On the other hand, viruses that recognized α2,3-linked but not α2,6-linked sialic acid (VN3040, VN3062, and VN30850) replicated efficiently in macaques. Although it is not well understood how the difference in receptor specificity influences the pathogenicity of H5N1 viruses, it has been reported that the 2009 pandemic H1N1 virus with enhanced α2,3-linked sialic acid binding ability shows increased infection of type II pneumocytes and higher pathogenicity in macaques than the related virus that does not recognize α2,3-linked sialic acid (75). A seasonal H1N1 virus that preferentially recognizes α2,6-linked sialic acid infects type I but not type II pneumocytes in macaques, resulting in less lung damage (82). In addition, a highly pathogenic H5N1 virus that recognizes α2,3-linked sialic acid infects type II pneumocytes, Clara cells, and bronchial epithelial cells in macaques in the early phase of infection, resulting in severe alveolar damage (14). Type II pneumocytes express cytokines/chemokines and participate in the innate immune responses of the lung. However, it was recently reported that type I pneumocytes in rodent models mount innate immune responses (83) and produce higher levels of proinflammatory cytokines on lipopolysaccharide stimulation than do type II pneumocytes (84), suggesting that type I pneumocyte targeting of H5N1 viruses by α2,6-linked sialic acid recognition would cause the stronger innate immune responses at the early phase of infection, leading to early virus clearance.
Compared to the World Health Organization-documented fatality rate of H5N1 virus infections in humans (which is close to 60%), the virus lethality in cynomolgus macaques (one out of 18 animals) in the present study was low. Other studies on H5N1 virus infections in macaques have reported similar findings of only one fatal outcome (12). Such findings have brought into question the suitability of macaques as an animal model for H5N1 virus infection. To address this question, one must carefully consider the reported fatality rate of H5N1 infections in humans. The reported ∼60% fatality rate in humans is a case fatality rate; some humans with no history of influenza-like illness but with confirmed seropositivity for H5N1 virus infection are not included in the confirmed cases of H5N1 virus infections (85–92). Therefore, the true fatality rate for H5N1 virus-infected humans is less than the reported rate. Also, although ferrets and mice are used to study influenza, highly pathogenic H5N1 viruses cause systemic infection in these animals, whereas systemic infections rarely occur with H5N1 viruses in humans (8, 93). Moreover, because laboratory mice lack the Mx1 gene, they are very susceptible to highly pathogenic H5N1 viruses. Therefore, the pathobiologies of H5N1 virus infections in ferrets and laboratory mice do not always represent all of the features of human infections with these viruses. In contrast, in macaques, highly pathogenic H5N1 viruses replicate mainly in respiratory organs and induce severe pneumonia as is the case in human patients infected with H5N1 viruses. The lung lesions of macaques infected with highly pathogenic influenza viruses, including H5N1 viruses, are similar to those seen in human patients (12–14, 27, 28, 70, 75, 82, 94–100). Therefore, macaques are in fact excellent animal models for the study of severe influenza.
In summary, our results suggest that early ISG induction determines the pathogenicity of H5N1 virus infection. Since virus infection can be viewed as competitive antagonism between virus replication and host antiviral responses, it appears that even a slight advantage on either side can influence the outcome for the infection. A better understanding of this antagonism will aid in the development of immunomodulatory therapeutics against highly pathogenic influenza.
Supplementary Material
ACKNOWLEDGMENTS
We thank Fusayo Adachi for technical support and Susan Watson for editing the manuscript.
This study was supported by a Grant-in-Aid for Specially Promoted Research, by the Japan Initiative for Global Research Network on Infectious Diseases from the Ministry of Education, Culture, Sports, Science, and Technology, by grants-in-aid from the Ministry of Health, Labor, and Welfare of Japan, and by ERATO (Japan Science and Technology Agency).
Footnotes
Published ahead of print 4 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00907-14.
REFERENCES
- 1.WHO. 2013. Cumulative number of confirmed human cases of avian influenza A/(H5N1) reported to WHO. Accessed 17 December 2013 World Health Organization, Geneva, Switzerland: http://www.who.int/influenza/human_animal_interface/EN_GIP_20131008CumulativeNumberH5N1cases.pdf [Google Scholar]
- 2.Tran TH, Nguyen TL, Nguyen TD, Luong TS, Pham PM, Nguyen v V, Pham TS, Vo CD, Le TQ, Ngo TT, Dao BK, Le PP, Nguyen TT, Hoang TL, Cao VT, Le TG, Nguyen DT, Le HN, Nguyen KT, Le HS, Le VT, Christiane D, Tran TT, Menno de J, Schultz C, Cheng P, Lim W, Horby P, Farrar J; World Health Organization International Avian Influenza Investigative Team 2004. Avian influenza A (H5N1) in 10 patients in Vietnam. N. Engl. J. Med. 350:1179–1188. 10.1056/NEJMoa040419 [DOI] [PubMed] [Google Scholar]
- 3.Grose C, Chokephaibulkit K. 2004. Avian influenza virus infection of children in Vietnam and Thailand. Pediatr. Infect. Dis. J. 23:793–794. 10.1097/00006454-200408000-00024 [DOI] [PubMed] [Google Scholar]
- 4.Chotpitayasunondh T, Ungchusak K, Hanshaoworakul W, Chunsuthiwat S, Sawanpanyalert P, Kijphati R, Lochindarat S, Srisan P, Suwan P, Osotthanakorn Y, Anantasetagoon T, Kanjanawasri S, Tanupattarachai S, Weerakul J, Chaiwirattana R, Maneerattanaporn M, Poolsavathitikool R, Chokephaibulkit K, Apisarnthanarak A, Dowell SF. 2005. Human disease from influenza A (H5N1), Thailand, 2004. Emerg. Infect. Dis. 11:201–209. 10.3201/eid1102.041061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beigel JH, Farrar J, Han AM, Hayden FG, Hyer R, de Jong MD, Lochindarat S, Nguyen TK, Nguyen TH, Tran TH, Nicoll A, Touch S, Yuen KY, Writing Committee of the World Health Organization (WHO) Consultation on Human Influenza A/H5 2005. Avian influenza A (H5N1) infection in humans. N. Engl. J. Med. 353:1374–1385. 10.1056/NEJMra052211 [DOI] [PubMed] [Google Scholar]
- 6.Ng WF, To KF, Lam WW, Ng TK, Lee KC. 2006. The comparative pathology of severe acute respiratory syndrome and avian influenza A subtype H5N1–a review. Hum. Pathol. 37:381–390. 10.1016/j.humpath.2006.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uiprasertkul M, Kitphati R, Puthavathana P, Kriwong R, Kongchanagul A, Ungchusak K, Angkasekwinai S, Chokephaibulkit K, Srisook K, Vanprapar N, Auewarakul P. 2007. Apoptosis and pathogenesis of avian influenza A (H5N1) virus in humans. Emerg. Infect. Dis. 13:708–712. 10.3201/eid1305.060572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu J, Xie Z, Gao Z, Liu J, Korteweg C, Ye J, Lau LT, Lu J, Gao Z, Zhang B, McNutt MA, Lu M, Anderson VM, Gong E, Yu AC, Lipkin WI. 2007. H5N1 infection of the respiratory tract and beyond: a molecular pathology study. Lancet 370:1137–1145. 10.1016/S0140-6736(07)61515-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peiris JS, Yu WC, Leung CW, Cheung CY, Ng WF, Nicholls JM, Ng TK, Chan KH, Lai ST, Lim WL, Yuen KY, Guan Y. 2004. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet 363:617–619. 10.1016/S0140-6736(04)15595-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, Cam BV, Ha DO, Guan QY, Peiris JS, Chinh NT, Hien TT, Farrar J. 2006. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 12:1203–1207. 10.1038/nm1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnard DL. 2009. Animal models for the study of influenza pathogenesis and therapy. Antivir. Res. 82:A110–A122. 10.1016/j.antiviral.2008.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baskin CR, Bielefeldt-Ohmann H, Tumpey TM, Sabourin PJ, Long JP, García-Sastre A, Tolnay AE, Albrecht R, Pyles JA, Olson PH, Aicher LD, Rosenzweig ER, Murali-Krishna K, Clark EA, Kotur MS, Fornek JL, Proll S, Palermo RE, Sabourin CL, Katze MG. 2009. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc. Natl. Acad. Sci. U. S. A. 106:3455–3460. 10.1073/pnas.0813234106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cillóniz C, Shinya K, Peng X, Korth MJ, Proll SC, Aicher LD, Carter VS, Chang JH, Kobasa D, Feldmann F, Strong JE, Feldmann H, Kawaoka Y, Katze MG. 2009. Lethal influenza virus infection in macaques is associated with early dysregulation of inflammatory related genes. PLoS Pathog. 5:e1000604. 10.1371/journal.ppat.1000604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shinya K, Gao Y, Cilloniz C, Suzuki Y, Fujie M, Deng G, Zhu Q, Fan S, Makino A, Muramoto Y, Fukuyama S, Tamura D, Noda T, Eisfeld AJ, Katze MG, Chen H, Kawaoka Y. 2012. Integrated clinical, pathologic, virologic, and transcriptomic analysis of H5N1 influenza virus-induced viral pneumonia in the rhesus macaque. J. Virol. 86:6055–6066. 10.1128/JVI.00365-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le QM, Ito M, Muramoto Y, Hoang PV, Vuong CD, Sakai-Tagawa Y, Kiso M, Ozawa M, Takano R, Kawaoka Y. 2010. Pathogenicity of highly pathogenic avian H5N1 influenza A viruses isolated from humans between 2003 and 2008 in northern Vietnam. J. Gen. Virol. 91:2485–2490. 10.1099/vir.0.021659-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le QM, Kiso M, Someya K, Sakai YT, Nguyen TH, Nguyen KH, Pham ND, Ngyen HH, Yamada S, Muramoto Y, Horimoto T, Takada A, Goto H, Suzuki T, Suzuki Y, Kawaoka Y. 2005. Avian flu: isolation of drug-resistant H5N1 virus. Nature 437:1108. 10.1038/4371108a [DOI] [PubMed] [Google Scholar]
- 17.Itoh Y, Ozaki H, Tsuchiya H, Okamoto K, Torii R, Sakoda Y, Kawaoka Y, Ogasawara K, Kida H. 2008. A vaccine prepared from a non-pathogenic H5N1 avian influenza virus strain confers protective immunity against highly pathogenic avian influenza virus infection in cynomolgus macaques. Vaccine 26:562–572. 10.1016/j.vaccine.2007.11.031 [DOI] [PubMed] [Google Scholar]
- 18.Shoemaker JE, Fukuyama S, Eisfeld AJ, Muramoto Y, Watanabe S, Watanabe T, Matsuoka Y, Kitano H, Kawaoka Y. 2012. Integrated network analysis reveals a novel role for the cell cycle in 2009 pandemic influenza virus-induced inflammation in macaque lungs. BMC Syst. Biol. 6:117. 10.1186/1752-0509-6-117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang B, Horvath S. 2005. A general framework for weighted gene coexpression network analysis. Stat. Appl. Genet. Mol. Biol. 4:Article17. 10.2202/1544-6115.1128 [DOI] [PubMed] [Google Scholar]
- 20.Kaimal V, Bardes EE, Tabar SC, Jegga AG, Aronow BJ. 2010. ToppCluster: a multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic Acids Res. 38:W96–W102. 10.1093/nar/gkq418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamada S, Suzuki Y, Suzuki T, Le MQ, Nidom CA, Sakai-Tagawa Y, Muramoto Y, Ito M, Kiso M, Horimoto T, Shinya K, Sawada T, Kiso M, Usui T, Murata T, Lin Y, Hay A, Haire LF, Stevens DJ, Russell RJ, Gamblin SJ, Skehel JJ, Kawaoka Y. 2006. Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature 444:378–382. 10.1038/nature05264 [DOI] [PubMed] [Google Scholar]
- 22.Hatta M, Gao P, Halfmann P, Kawaoka Y. 2001. Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science 293:1840–1842. 10.1126/science.1062882 [DOI] [PubMed] [Google Scholar]
- 23.Obenauer JC, Denson J, Mehta PK, Su X, Mukatira S, Finkelstein DB, Xu X, Wang J, Ma J, Fan Y, Rakestraw KM, Webster RG, Hoffmann E, Krauss S, Zheng J, Zhang Z, Naeve CW. 2006. Large-scale sequence analysis of avian influenza isolates. Science 311:1576–1580. 10.1126/science.1121586 [DOI] [PubMed] [Google Scholar]
- 24.Jackson D, Hossain MJ, Hickman D, Perez DR, Lamb RA. 2008. A new influenza virus virulence determinant: the NS1 protein four C-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. U. S. A. 105:4381–4386. 10.1073/pnas.0800482105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Jong MD, Bach VC, Phan TQ, Vo MH, Tran TT, Nguyen BH, Beld M, Le TP, Truong HK, Nguyen VV, Tran TH, Do QH, Farrar J. 2005. Fatal avian influenza A (H5N1) in a child presenting with diarrhea followed by coma. N. Engl. J. Med. 352:686–691. 10.1056/NEJMoa044307 [DOI] [PubMed] [Google Scholar]
- 26.Uiprasertkul M, Puthavathana P, Sangsiriwut K, Pooruk P, Srisook K, Peiris M, Nicholls JM, Chokephaibulkit K, Vanprapar N, Auewarakul P. 2005. Influenza A H5N1 replication sites in humans. Emerg. Infect. Dis. 11:1036–1041. 10.3201/eid1107.041313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rimmelzwaan GF, Kuiken T, van Amerongen G, Bestebroer TM, Fouchier RA, Osterhaus AD. 2001. Pathogenesis of influenza A (H5N1) virus infection in a primate model. J. Virol. 75:6687–6691. 10.1128/JVI.75.14.6687-6691.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuiken T, Rimmelzwaan GF, Van Amerongen G, Osterhaus AD. 2003. Pathology of human influenza A (H5N1) virus infection in cynomolgus macaques (Macaca fascicularis). Vet. Pathol. 40:304–310. 10.1354/vp.40-3-304 [DOI] [PubMed] [Google Scholar]
- 29.Nicholls JM, Chan MC, Chan WY, Wong HK, Cheung CY, Kwong DL, Wong MP, Chui WH, Poon LL, Tsao SW, Guan Y, Peiris JS. 2007. Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat. Med. 13:147–149. 10.1038/nm1529 [DOI] [PubMed] [Google Scholar]
- 30.Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, Hertzog PJ. 2013. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41:D1040–D1046. 10.1093/nar/gks1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pichlmair A, Lassnig C, Eberle CA, Górna MW, Baumann CL, Burkard TR, Bürckstümmer T, Stefanovic A, Krieger S, Bennett KL, Rülicke T, Weber F, Colinge J, Müller M, Superti-Furga G. 2011. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 12:624–630. 10.1038/ni.2048 [DOI] [PubMed] [Google Scholar]
- 32.Haller O, Arnheiter H, Lindenmann J, Gresser I. 1980. Host gene influences sensitivity to interferon action selectively for influenza virus. Nature 283:660–662. 10.1038/283660a0 [DOI] [PubMed] [Google Scholar]
- 33.Turan K, Mibayashi M, Sugiyama K, Saito S, Numajiri A, Nagata K. 2004. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res. 32:643–652. 10.1093/nar/gkh192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, Wolff T, Osiak A, Levine B, Schmidt RE, García-Sastre A, Leib DA, Pekosz A, Knobeloch KP, Horak I, Virgin HW., 4th 2007. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. U. S. A. 104:1371–1376. 10.1073/pnas.0607038104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai C, Struckhoff JJ, Schneider J, Martinez-Sobrido L, Wolff T, García-Sastre A, Zhang DE, Lenschow DJ. 2009. Mice lacking the ISG15 E1 enzyme UbE1L demonstrate increased susceptibility to both mouse-adapted and non-mouse-adapted influenza B virus infection. J. Virol. 83:1147–1151. 10.1128/JVI.00105-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Min JY, Krug RM. 2006. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: inhibiting the 2′-5′ oligo(A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. U. S. A. 103:7100–7105. 10.1073/pnas.0602184103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malathi K, Dong B, Gale M, Jr, Silverman RH. 2007. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448:816–819. 10.1038/nature06042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melchjorsen J, Kristiansen H, Christiansen R, Rintahaka J, Matikainen S, Paludan SR, Hartmann R. 2009. Differential regulation of the OASL and OAS1 genes in response to viral infections. J. Interferon Cytokine Res. 29:199–207. 10.1089/jir.2008.0050 [DOI] [PubMed] [Google Scholar]
- 39.Chakrabarti A, Jha BK, Silverman RH. 2011. New insights into the role of RNase L in innate immunity. J. Interferon Cytokine Res. 31:49–57. 10.1089/jir.2010.0120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nordmann A, Wixler L, Boergeling Y, Wixler V, Ludwig S. 2012. A new splice variant of the human guanylate-binding protein 3 mediates anti-influenza activity through inhibition of viral transcription and replication. FASEB J. 26:1290–1300. 10.1096/fj.11-189886 [DOI] [PubMed] [Google Scholar]
- 41.Zhu Z, Shi Z, Yan W, Wei J, Shao D, Deng X, Wang S, Li B, Tong G, Ma Z. 2013. Nonstructural protein 1 of influenza A virus interacts with human guanylate-binding protein 1 to antagonize antiviral activity. PLoS One 8:e55920. 10.1371/journal.pone.0055920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stasakova J, Ferko B, Kittel C, Sereinig S, Romanova J, Katinger H, Egorov A. 2005. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1β and 18. J. Gen. Virol. 86:185–195. 10.1099/vir.0.80422-0 [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Hinson ER, Cresswell P. 2007. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2:96–105. 10.1016/j.chom.2007.06.009 [DOI] [PubMed] [Google Scholar]
- 44.Watanabe R, Leser GP, Lamb RA. 2011. Influenza virus is not restricted by tetherin whereas influenza VLP production is restricted by tetherin. Virology 417:50–56. 10.1016/j.virol.2011.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yondola MA, Fernandes F, Belicha-Villanueva A, Uccelini M, Gao Q, Carter C, Palese P. 2011. Budding capability of the influenza virus neuraminidase can be modulated by tetherin. J. Virol. 85:2480–2491. 10.1128/JVI.02188-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254. 10.1016/j.cell.2009.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang IC, Bailey CC, Weyer JL, Radoshitzky SR, Becker MM, Chiang JJ, Brass AL, Ahmed AA, Chi X, Dong L, Longobardi LE, Boltz D, Kuhn JH, Elledge SJ, Bavari S, Denison MR, Choe H, Farzan M. 2011. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 7:e1001258. 10.1371/journal.ppat.1001258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Everitt AR, Clare S, Pertel T, John SP, Wash RS, Smith SE, Chin CR, Feeley EM, Sims JS, Adams DJ, Wise HM, Kane L, Goulding D, Digard P, Anttila V, Baillie JK, Walsh TS, Hume DA, Palotie A, Xue Y, Colonna V, Tyler-Smith C, Dunning J, Gordon SB, GenISIS Investigators. Investigators MOSAIC. Smyth RL, Openshaw PJ, Dougan G, Brass AL, Kellam P. 2012. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484:519–523. 10.1038/nature10921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.García-Sastre A, Durbin RK, Zheng H, Palese P, Gertner R, Levy DE, Durbin JE. 1998. The role of interferon in influenza virus tissue tropism. J. Virol. 72:8550–8558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolff T, Ludwig S. 2009. Influenza viruses control the vertebrate type I interferon system: factors, mechanisms, and consequences. J. Interferon Cytokine Res. 29:549–557. 10.1089/jir.2009.0066 [DOI] [PubMed] [Google Scholar]
- 51.Di Pietro A, Kajaste-Rudnitski A, Oteiza A, Nicora L, Towers GJ, Mechti N, Vicenzi E. 2013. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 87:4523–4533. 10.1128/JVI.02548-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bergmann M, Garcia-Sastre A, Carnero E, Pehamberger H, Wolff K, Palese P, Muster T. 2000. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J. Virol. 74:6203–6206. 10.1128/JVI.74.13.6203-6206.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Espert L, Degols G, Gongora C, Blondel D, Williams BR, Silverman RH, Mechti N. 2003. ISG20, a new interferon-induced RNase specific for single-stranded RNA, defines an alternative antiviral pathway against RNA genomic viruses. J. Biol. Chem. 278:16151–16158. 10.1074/jbc.M209628200 [DOI] [PubMed] [Google Scholar]
- 54.Hayakawa S, Shiratori S, Yamato H, Kameyama T, Kitatsuji C, Kashigi F, Goto S, Kameoka S, Fujikura D, Yamada T, Mizutani T, Kazumata M, Sato M, Tanaka J, Asaka M, Ohba Y, Miyazaki T, Imamura M, Takaoka A. 2011. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat. Immunol. 12:37–44. 10.1038/ni.1963 [DOI] [PubMed] [Google Scholar]
- 55.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. 2011. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474:380–384. 10.1038/nature10110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. 10.1038/nature04734 [DOI] [PubMed] [Google Scholar]
- 57.Guo Z, Chen LM, Zeng H, Gomez JA, Plowden J, Fujita T, Katz JM, Donis RO, Sambhara S. 2007. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 36:263–269. 10.1165/rcmb.2006-0283RC [DOI] [PubMed] [Google Scholar]
- 58.Matikainen S, Sirén J, Tissari J, Veckman V, Pirhonen J, Severa M, Sun Q, Lin R, Meri S, Uzé G, Hiscott J, Julkunen I. 2006. Tumor necrosis factor alpha enhances influenza A virus-induced expression of antiviral cytokines by activating RIG-I gene expression. J. Virol. 80:3515–3522. 10.1128/JVI.80.7.3515-3522.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sirén J, Imaizumi T, Sarkar D, Pietilä T, Noah DL, Lin R, Hiscott J, Krug RM, Fisher PB, Julkunen I, Matikainen S. 2006. Retinoic acid inducible gene-I and mda-5 are involved in influenza A virus-induced expression of antiviral cytokines. Microbes Infect. 8:2013–2020. 10.1016/j.micinf.2006.02.028 [DOI] [PubMed] [Google Scholar]
- 60.Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, Si-Tahar M. 2007. Cutting edge: influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J. Immunol. 178:3368–3372. 10.4049/jimmunol.178.6.3368 [DOI] [PubMed] [Google Scholar]
- 61.Mordstein M, Kochs G, Dumoutier L, Renauld JC, Paludan SR, Klucher K, Staeheli P. 2008. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 4:e1000151. 10.1371/journal.ppat.1000151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang J, Oberley-Deegan R, Wang S, Nikrad M, Funk CJ, Hartshorn KL, Mason RJ. 2009. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-λ1) in response to influenza A infection. J. Immunol. 182:1296–1304. 10.4049/jimmunol.182.3.1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ichikawa A, Kuba K, Morita M, Chida S, Tezuka H, Hara H, Sasaki T, Ohteki T, Ranieri VM, Dos Santos CC, Kawaoka Y, Akira S, Luster AD, Lu B, Penninger JM, Uhlig S, Slutsky AS, Imai Y. 2013. CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am. J. Respir. Crit. Care Med. 187:65–77. 10.1164/rccm.201203-0508OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. 2001. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19:683–765. 10.1146/annurev.immunol.19.1.683 [DOI] [PubMed] [Google Scholar]
- 65.Li M, Liu X, Zhou Y, Su SB. 2009. Interferon-lambdas: the modulators of antivirus, antitumor, and immune responses. J. Leukoc. Biol. 86:23–32. 10.1189/jlb.1208761 [DOI] [PubMed] [Google Scholar]
- 66.Tough DF. 2012. Modulation of T-cell function by type I interferon. Immunol. Cell Biol. 90:492–497. 10.1038/icb.2012.7 [DOI] [PubMed] [Google Scholar]
- 67.Engelmann B, Massberg S. 2013. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 13:34–45. 10.1038/nri3345 [DOI] [PubMed] [Google Scholar]
- 68.Esmon CT, Xu J, Lupu F. 2011. Innate immunity and coagulation. J. Thromb. Haemost. 9:182–188. 10.1111/j.1538-7836.2011.04323.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eisenhut M, Wallace H. 2011. Ion channels in inflammation. Pflugers Arch. 461:401–421. 10.1007/s00424-010-0917-y [DOI] [PubMed] [Google Scholar]
- 70.Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, Hatta Y, Kim JH, Halfmann P, Hatta M, Feldmann F, Alimonti JB, Fernando L, Li Y, Katze MG, Feldmann H, Kawaoka Y. 2007. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445:319–323. 10.1038/nature05495 [DOI] [PubMed] [Google Scholar]
- 71.Levy DE, Marié IJ, Durbin JE. 2011. Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Virol. 1:476–486. 10.1016/j.coviro.2011.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, Silverman RH, Williams BR. 2001. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 69:912–920 [PubMed] [Google Scholar]
- 73.Hale BG, Randall RE, Ortín J, Jackson D. 2008. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89:2359–2376. 10.1099/vir.0.2008/004606-0 [DOI] [PubMed] [Google Scholar]
- 74.Shinya K, Ebina M, Yamada S, Ono M, Kasai N, Kawaoka Y. 2006. Avian flu: influenza virus receptors in the human airway. Nature 440:435–436. 10.1038/440435a [DOI] [PubMed] [Google Scholar]
- 75.Watanabe T, Shinya K, Watanabe S, Imai M, Hatta M, Li C, Wolter BF, Neumann G, Hanson A, Ozawa M, Yamada S, Imai H, Sakabe S, Takano R, Iwatsuki-Horimoto K, Kiso M, Ito M, Fukuyama S, Kawakami E, Gorai T, Simmons HA, Schenkman D, Brunner K, Capuano SV, III, Weinfurter JT, Nishio W, Maniwa Y, Igarashi T, Makino A, Travanty EA, Wang J, Kilander A, Dudman SG, Suresh M, Mason RJ, Hungnes O, Friedrich TC, Kawaoka Y. 2011. Avian-type receptor-binding ability can increase influenza virus pathogenicity in macaques. J. Virol. 85:13195–13203. 10.1128/JVI.00859-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Geiss GK, Salvatore M, Tumpey TM, Carter VS, Wang X, Basler CF, Taubenberger JK, Bumgarner RE, Palese P, Katze MG, García-Sastre A. 2002. Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc. Natl. Acad. Sci. U. S. A. 99:10736–10741. 10.1073/pnas.112338099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang Y, Zhong G, Zhu L, Liu X, Shan Y, Feng H, Bu Z, Chen H, Wang C. 2010. Herc5 attenuates influenza A virus by catalyzing ISGylation of viral NS1 protein. J. Immunol. 184:5777–5790. 10.4049/jimmunol.0903588 [DOI] [PubMed] [Google Scholar]
- 78.Xu K, Klenk C, Liu B, Keiner B, Cheng J, Zheng BJ, Li L, Han Q, Wang C, Li T, Chen Z, Shu Y, Liu J, Klenk HD, Sun B. 2011. Modification of nonstructural protein 1 of influenza A virus by SUMO1. J. Virol. 85:1086–1098. 10.1128/JVI.00877-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Soubies SM, Volmer C, Croville G, Loupias J, Peralta B, Costes P, Lacroux C, Guérin JL, Volmer R. 2010. Species-specific contribution of the four C-terminal amino acids of influenza A virus NS1 protein to virulence. J. Virol. 84:6733–6747. 10.1128/JVI.02427-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao C, Hsiang TY, Kuo RL, Krug RM. 2010. ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proc. Natl. Acad. Sci. U. S. A. 107:2253–2258. 10.1073/pnas.0909144107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dundon WG, Capua I. 2009. A closer look at the NS1 of influenza virus. Viruses 1:1057–1072. 10.3390/v1031057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Itoh Y, Shinya K, Kiso M, Watanabe T, Sakoda Y, Hatta M, Muramoto Y, Tamura D, Sakai-Tagawa Y, Noda T, Sakabe S, Imai M, Hatta Y, Watanabe S, Li C, Yamada S, Fujii K, Murakami S, Imai H, Kakugawa S, Ito M, Takano R, Iwatsuki-Horimoto K, Shimojima M, Horimoto T, Goto H, Takahashi K, Makino A, Ishigaki H, Nakayama M, Okamatsu M, Takahashi K, Warshauer D, Shult PA, Saito R, Suzuki H, Furuta Y, Yamashita M, Mitamura K, Nakano K, Nakamura M, Brockman-Schneider R, Mitamura H, Yamazaki M, Sugaya N, Suresh M, Ozawa M, Neumann G, Gern J, Kida H, Ogasawara K, Kawaoka Y. 2009. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 460:1021–1025. 10.1038/nature08260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yamamoto K, Ferrari JD, Cao Y, Ramirez MI, Jones MR, Quinton LJ, Mizgerd JP. 2012. Type I alveolar epithelial cells mount innate immune responses during pneumococcal pneumonia. J. Immunol. 189:2450–2459. 10.4049/jimmunol.1200634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wong MH, Johnson MD. 2013. Differential response of primary alveolar type I and type II cells to LPS stimulation. PLoS One 8:e55545. 10.1371/journal.pone.0055545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Katz JM, Lim W, Bridges CB, Rowe T, Hu-Primmer J, Lu X, Abernathy RA, Clarke M, Conn L, Kwong H, Lee M, Au G, Ho YY, Mak KH, Cox NJ, Fukuda K. 1999. Antibody response in individuals infected with avian influenza A (H5N1) viruses and detection of anti-H5 antibody among household and social contacts. J. Infect. Dis. 180:1763–1770. 10.1086/315137 [DOI] [PubMed] [Google Scholar]
- 86.Buxton Bridges C, Katz JM, Seto WH, Chan PK, Tsang D, Ho W, Mak KH, Lim W, Tam JS, Clarke M, Williams SG, Mounts AW, Bresee JS, Conn LA, Rowe T, Hu-Primmer J, Abernathy RA, Lu X, Cox NJ, Fukuda K. 2000. Risk of influenza A (H5N1) infection among health care workers exposed to patients with influenza A (H5N1), Hong Kong. J. Infect. Dis. 181:344–348. 10.1086/315213 [DOI] [PubMed] [Google Scholar]
- 87.Bridges CB, Lim W, Hu-Primmer J, Sims L, Fukuda K, Mak KH, Rowe T, Thompson WW, Conn L, Lu X, Cox NJ, Katz JM. 2002. Risk of influenza A (H5N1) infection among poultry workers, Hong Kong, 1997-1998. J. Infect. Dis. 185:1005–1010. 10.1086/340044 [DOI] [PubMed] [Google Scholar]
- 88.Cavailler P, Chu S, Ly S, Garcia JM, Ha do Q, Bergeri I, Som L, Ly S, Sok T, Vong S, Buchy P. 2010. Seroprevalence of anti-H5 antibody in rural Cambodia, 2007. J. Clin. Virol. 48:123–126. 10.1016/j.jcv.2010.02.021 [DOI] [PubMed] [Google Scholar]
- 89.Buchy P, Vong S, Chu S, Garcia JM, Hien TT, Hien VM, Channa M, Ha do Q, Chau NV, Simmons C, Farrar JJ, Peiris M, de Jong MD. 2010. Kinetics of neutralizing antibodies in patients naturally infected by H5N1 virus. PLoS One 5:e10864. 10.1371/journal.pone.0010864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.World Health Organization. 2008. Human cases of avian influenza A (H5N1) in North-West Frontier Province, Pakistan, October-November 2007. Wkly. Epidemiol. Rec. 83:359–364 [PubMed] [Google Scholar]
- 91.Wang TT, Parides MK, Palese P. 2012. Seroevidence for H5N1 influenza infections in humans: meta-analysis. Science 335:1463. 10.1126/science.1218888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Le MQ, Horby P, Fox A, Nguyen HT, Le Nguyen HK, Hoang PM, Nguyen KC, de Jong MD, Jeeninga RE, Rogier van Doorn H, Farrar J, Wertheim HF. 2013. Subclinical avian influenza A(H5N1) virus infection in human, Vietnam. Emerg. Infect. Dis. 19:1674–1677. 10.3201/eid1910.130730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang Z, Zhang J, Huang K, Li KS, Yuen KY, Guan Y, Chen H, Ng WF. 2009. Systemic infection of avian influenza A virus H5N1 subtype in humans. Hum. Pathol. 40:735–739. 10.1016/j.humpath.2008.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rimmelzwaan GF, Kuiken T, van Amerongen G, Bestebroer TM, Fouchier RA, Osterhaus AD. 2003. A primate model to study the pathogenesis of influenza A (H5N1) virus infection. Avian Dis. 47:931–933. 10.1637/0005-2086-47.s3.931 [DOI] [PubMed] [Google Scholar]
- 95.Chen Y, Deng W, Jia C, Dai X, Zhu H, Kong Q, Huang L, Liu Y, Ma C, Li J, Xiao C, Liu Y, Wei Q, Qin C. 2009. Pathological lesions and viral localization of influenza A (H5N1) virus in experimentally infected Chinese rhesus macaques: implications for pathogenesis and viral transmission. Arch. Virol. 154:227–233. 10.1007/s00705-008-0277-5 [DOI] [PubMed] [Google Scholar]
- 96.Herfst S, van den Brand JM, Schrauwen EJ, de Wit E, Munster VJ, van Amerongen G, Linster M, Zaaraoui F, van Ijcken WF, Rimmelzwaan GF, Osterhaus AD, Fouchier RA, Andeweg AC, Kuiken T. 2010. Pandemic 2009 H1N1 influenza virus causes diffuse alveolar damage in cynomolgus macaques. Vet. Pathol. 47:1040–1047. 10.1177/0300985810374836 [DOI] [PubMed] [Google Scholar]
- 97.Safronetz D, Rockx B, Feldmann F, Belisle SE, Palermo RE, Brining D, Gardner D, Proll SC, Marzi A, Tsuda Y, Lacasse RA, Kercher L, York A, Korth MJ, Long D, Rosenke R, Shupert WL, Aranda CA, Mattoon JS, Kobasa D, Kobinger G, Li Y, Taubenberger JK, Richt JA, Parnell M, Ebihara H, Kawaoka Y, Katze MG, Feldmann H. 2011. Pandemic swine-origin H1N1 influenza A virus isolates show heterogeneous virulence in macaques. J. Virol. 85:1214–1223. 10.1128/JVI.01848-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Richt JA, Rockx B, Ma W, Feldmann F, Safronetz D, Marzi A, Kobasa D, Strong JE, Kercher L, Long D, Gardner D, Brining D, Feldmann H. 2012. Recently emerged swine influenza A virus (H2N3) causes severe pneumonia in cynomolgus macaques. PLoS One 7:e39990. 10.1371/journal.pone.0039990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Watanabe T, Imai M, Watanabe S, Shinya K, Hatta M, Li C, Neumann G, Ozawa M, Hanson A, Zhong G, Fukuyama S, Kawakami E, Simmons HA, Schenkman D, Brunner K, Capuano SV, III, Weinfurter JT, Kilander A, Dudman SG, Suresh M, Hungnes O, Friedrich TC, Kawaoka Y. 2012. Characterization in vitro and in vivo of pandemic (H1N1) 2009 influenza viruses isolated from patients. J. Virol. 86:9361–9368. 10.1128/JVI.01214-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang K, Xu W, Zhang Z, Wang T, Sang X, Cheng K, Yu Z, Zheng X, Wang H, Zhao Y, Huang G, Yang S, Qin C, Gao Y, Xia X. 2013. Experimental infection of non-human primates with avian influenza virus (H9N2). Arch. Virol. 158:2127–2134. 10.1007/s00705-013-1721-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.