

ABSTRACT
Asthma was the most common comorbidity observed among patients hospitalized with influenza A virus during the 2009 pandemic. However, little remains known about how the asthmatic phenotype influences protective immune responses against respiratory viral pathogens. Using the ovalbumin-induced allergic lung inflammation model, we found that asthmatic mice, unlike nonasthmatic mice, were highly susceptible to secondary heterologous virus challenge. While primary virus infection generated protective memory immune responses against homologous secondary virus challenge in both asthmatic and nonasthmatic mice, full protection against heterologous A/California/04/2009 (CA04) viral infection was observed only in nonasthmatic mice. Significant reductions in CA04-specific IgA, IgG, and IgM levels and in CA04-neutralizing activity of bronchoalveolar lavage fluid (BALF) was observed following secondary CA04 challenge of PR8-immunized asthmatic mice. Furthermore, transfer of immune BALF obtained from nonasthmatic, but not asthmatic, donors following secondary viral infection generated protection against CA04 in naive recipients. Nonspecific B-cell activation by CpG inoculation restored protection in PR8-immunized, CA04-challenged asthmatic mice. These results demonstrate a causal link between defective mucosal antibody responses and the heightened susceptibility of asthmatic mice to influenza infection and provide a mechanistic explanation for the observation that asthma was a major risk factor during the 2009 influenza pandemic.
IMPORTANCE The prevalence of asthma worldwide is increasing each year. Unfortunately, there is no cure for asthma. Asthmatic individuals not only suffer from consistent wheezing and coughing but are also believed to be more prone to serious lung infections that result in bronchitis and pneumonia. However, little is known about the influence of asthma on host mucosal immunity. Here we show that antibody responses during secondary heterologous influenza infections are suboptimal and that this is responsible for the increased mortality in asthmatic mice from viral infections. Understanding the mechanism of increased susceptibility will aid in developing new antiviral therapies for asthmatic patients.
INTRODUCTION
Asthma is an incurable disease afflicting 300 million people worldwide and causing 250,000 asthma-associated deaths per year (1). Its prevalence is increasing each year for unknown reasons, especially in developed countries. Patients with asthma typically suffer from chronic bronchial hyperresponsiveness, overproduction of mucus, allergen-specific IgE expression, and airway remodeling (2, 3). Allergic airway inflammation is characterized by an infiltrate of eosinophils, neutrophils, and Th2 and Th17 lymphocytes expressing interleukin-4 (IL-4), IL-5 IL-13, and IL-17 (4).
Asthmatic individuals are thought to be more susceptible to respiratory viral infections, but evidence that actually supports a causal relationship is weak and the mechanisms are poorly understood. It is important to note that severely asthmatic individuals are typically treated with inhaled corticosteroids, which are highly immunosuppressive; this thereby complicates an understanding of the reason for the apparent susceptibility of asthmatic patients to influenza infection. Nevertheless, recent studies have reported detrimental effects of asthma on host antiviral immunity. Papadopoulos and colleagues (5) showed that peripheral blood mononuclear cells isolated from asthmatic individuals and stimulated with rhinovirus ex vivo produced significantly lower levels of gamma interferon (IFN-γ) and IL-12 but higher levels of IL-4 and IL-10 than those cells isolated from nonasthmatic control groups. Furthermore, asthma severity has been associated with reduced rhinovirus-induced IFN-γ responses (6). A recent report by Message et al. (7) has confirmed the presence of Th2-skewed responses among asthmatics in response to rhinovirus infection or ex vivo virus stimulation. Taken together, these studies indicate that asthmatic patients have deficient Th1 immunity, which is known to be important for protection against influenza infection.
During the influenza pandemic of 2009, asthma was found to be the most common comorbidity among patients hospitalized with influenza (8). However, although asthma was associated with higher hospital admission rates, hospitalized asthmatics were less likely to develop severe disease or die than nonasthmatics (9, 10). These contradictory observations prompted us to initiate an in-depth investigation into the role of asthma in susceptibility to influenza infection. The majority of human adults possess preexisting immunity against influenza virus due to yearly exposure to seasonal influenza A viruses (11, 12); therefore, only relatively low increases in mortality were reported during the 2009 pandemic (11–16). To recapitulate the 2009 pandemic scenario in an asthmatic mouse model, we developed a comorbidity model of ovalbumin (OVA)-induced allergic lung inflammation and influenza reinfection. In humans (also mirrored in animal models), natural infection with a seasonal influenza virus confers protection not only against that same virus but also against similar viruses such as the 2009 H1N1 strain (12–16). In the present study, we show that nonasthmatic mice acquire strong cross-protective immunity against the pandemic H1N1 A/California/04/2009 (CA04) strain following natural immunization with PR8. However, asthmatic mice fail to develop protective anti-CA04 immune responses. We also observed that asthmatic mice have defective airway antibody responses during reinfection. These findings provide a mechanistic explanation for the observation that asthma was a major risk factor during the 2009 influenza pandemic.
MATERIALS AND METHODS
Ethics statement.
All animal procedures were approved by the Institutional Animal Care and Use Committee at Albany Medical College (protocol number 11-04004).
Mice.
Six- to seven-week-old female BALB/c mice were used in these studies. Mice were purchased from Charles River Laboratories under a contract with the National Cancer Institute (Frederick, MD) and infected in biosecurity level 2 containment facilities at the Albany Medical College. For immunizations and infections, mice were anesthetized by intraperitoneal (i.p.) injection with 100 μl xylazine (20 mg/ml) and ketamine (1 mg/ml) in phosphate-buffered saline (PBS).
Mouse model of allergic airway inflammation.
A well-established mouse model of OVA-induced allergic airway inflammation was employed in this study (17). BALB/c mice were sensitized to OVA by i.p injection of 10 μg of OVA (Sigma-Aldrich, St. Louis, MO) and 4 mg of aluminum hydroxide (General Chemical, Berkeley Heights, NJ) at weekly intervals for 2 weeks. One week after the second i.p. sensitization, the mice were lightly anesthetized using isoflurane and challenged intranasally (i.n.) with 100 μg of OVA in 50 μl of PBS for 5 consecutive days. Control, nonasthmatic mice were sensitized and challenged with PBS only.
Mouse model of influenza immunization and challenge.
Nonasthmatic and asthmatic mice were i.n. immunized with a sublethal dose (15 PFU) of either PR8 or CA04 1 day after the last OVA or PBS i.n. inoculation. Five weeks later, the mice were challenged i.n. with a lethal dose of CA04 (3 × 105 PFU). The viral doses required for immunization and for lethal infection were established in previous studies (18, 19). Survival and weight loss were monitored daily. Viral titers were determined by a plaque assay on Madin-Darby canine kidney cell monolayers.
The nomenclature that is used to denote the various experimental groups is shown in Table 1. Briefly, each group is designated first as asthmatic or nonasthmatic, then as immunized through sublethal infection with PR8 or CA04, and finally, as challenged with a lethal dose of CA04. For example, PR8-immunized and CA04-challenged asthmatic mice are referred to as asthma:PR8/CA04 mice; unimmunized (received PBS instead of PR8) and CA04-challenged asthmatic mice are referred to as asthma:PBS/CA04 mice.
TABLE 1.
Overview of experimental groups
| Exptl group | Treatment of mice |
|||
|---|---|---|---|---|
| OVA treatment | Vaccination with PR8 | Vaccination with CA04 | Infection with CA04 | |
| Homologous challenge | ||||
| Nonasthma: PBS/CA04 | − | − | − | + |
| Nonasthma: CA04/CA04 | − | − | + | + |
| Asthma: PBS/CA04 | + | − | − | + |
| Asthma: CA04/CA04 | + | − | + | + |
| Heterologous challenge | ||||
| Nonasthma: PBS/CA04 | − | − | − | + |
| Nonasthma: PR8/CA04 | − | + | − | + |
| Asthma: PBS/CA04 | + | − | − | + |
| Asthma: PR8/CA04 | + | + | − | + |
Cytokine, chemokine, and granzyme B analysis.
Bronchoalveolar lavage fluid (BALF) samples were harvested by delivering 1 ml of PBS through a tracheal cannula. The collected BALF was centrifuged at 4°C for 5 min at 500 × g to remove cell debris and stored at −80°C. Cytokine and chemokine protein levels were measured using mouse Bio-Plex Luminex assays in accordance with the manufacturer's instructions (Bio-Rad). Amphiregulin and granzyme B (gzmB) levels in BALF were determined by a DuoSet enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN) and a mouse gzmB ELISA kit (eBioscience), respectively.
BALF total protein and nitrite levels.
The concentration of total protein in the BALF was determined using a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific Pierce). Nitric oxide levels in BALF were indirectly quantified by measuring nitrites using a commercial Griess reagent kit (Invitrogen).
Flow cytometric analysis.
The lungs were harvested, and single-cell suspensions were obtained by incubation with 2 mg/ml collagenase D (Roche Diagnostics), 0.25 mg/ml DNase I (Roche Diagnostics), and 10 mM MgCl2 for 1 h at 37°C. Following red blood cell depletion by incubation in ammonium chloride-potassium lysis buffer, the cells were washed with PBS in 2% fetal calf serum (FCS). Fc receptors were blocked by incubation with the 2.4G2 (anti-mouse FcγIII/II receptor) monoclonal antibody (MAb) for 20 min at 4°C. The cells were further incubated with mixtures of phycoerythrin (PE)-conjugated anti-F4/80 (eBioscience), fluorescein isothiocyanate (FITC)-conjugated anti-DX5 (eBioscience), allophycocyanin (APC)-conjugated anti-CD11c (BD Pharmingen), PE-conjugated anti-Siglec F (BD Pharmingen), FITC-conjugated anti-CD11b (eBioscience), and PE-Cy7-conjugated anti-Ly6G (Biolegend) MAbs for macrophage, NK cell, dendritic cell (DC), monocyte, eosinophil, and neutrophil identification, respectively. Dead cells were identified using 7-aminoactinomycin D (eBioscience). For B- and T-cell analysis, cells were stained with PE-Cy7-conjugated anti-B220 (BD Pharmingen), FITC-conjugated anti-CD3 (BD Pharmingen), FITC-conjugated anti-CD4 (BD Pharmingen), PE-Cy7-conjugated anti-CD8 (BD Pharmingen), FITC-conjugated anti-CD62L (Biolegend), and PE-conjugated anti-CD127 (Biolegend) MAbs. For analysis of influenza virus nucleoprotein (NP)-specific CD8+ T lymphocytes, cells were stained with APC-conjugated major histocompatibility complex (MHC) class I H-2Kd tetramers loaded with the NP peptide (amino acids [aa] 147 to 155; TYQRTRALV). Tetramers were provided by the NIH Tetramer Facility. For intracellular granzyme B (gzmB) staining, cells were incubated for 20 min in BD fixation/permeabilization solution and then washed twice with BD Perm/Wash buffer and stained for 30 min with FITC-conjugated anti-gzmB MAb (eBioscience). After the final wash, the cells were resuspended in 200 μl of PBS–2% bovine serum albumin (BSA). Stained cells were quantitated using a FACSCanto flow cytometer.
CD3+ T-cell transfer.
Pulmonary CD3+ T cells were isolated from asthmatic:PR8/CA04 or nonasthmatic:PR8/CA04 mice on day 5 following CA04 secondary infection using an EasySep-negative mouse T-cell isolation kit (StemCell Technologies, Vancouver, British Columbia, Canada). The isolated CD3+ T cells were determined to be approximately 93% pure by flow cytometry. A total of 107 cells were transferred intravenously (i.v.) into unimmunized mice on day 2 postinfection with CA04. The lungs were then harvested to assess viral burden by plaque assay.
Airway responsiveness.
Airway responsiveness to aerosolized methacholine was measured on day 6 postinfection, as previously described (20). Infected or mock-treated mice were anesthetized, tracheostomized, paralyzed with pancuronium bromide, and then connected to a computer-controlled small-animal ventilator (flexiVent; SCIREQ). Increasing doses of methacholine were used to measure airway resistance and tissue damping.
HI titer.
The hemagglutination inhibition (HI) assay was performed as previously described (21). Briefly, serially diluted sera were mixed with 4 hemagglutination (HA) units of PR8 or CA04 influenza virus in V-bottom 96-well plates. Following 30 min of incubation at room temperature, 0.5% of chicken red blood cells were added and incubated for an additional hour. The HI titer was defined as the reciprocal of the last dilution that prevented hemagglutination activity.
Serum/BALF passive transfer.
Sera and BALF samples from PR8-immunized mice were collected at 5 weeks postimmunization. BALF from PR8/CA04-challenged mice were collected on day 6 after secondary infection. The pooled immune sera or BALF samples were heated for 1 h at 60°C to inactivate complement. Recipient mice were then inoculated i.n. with either PR8 or CA04 influenza virus mixed with 10% sera or 20% BALF. Total Ig in the pooled BALF samples was depleted using goat anti-mouse Ig-coated Sepharose beads as previously described (22). Complete depletion of CA04-specific antibody was confirmed by ELISA. Mice were monitored for body weight and mortality until day 20 postchallenge.
Plaque reduction assay.
Heat-treated BALF was mixed with 5,000 PFU of CA04 virus. Residual virus infectivity was then measured by plaque assay on Madin-Darby canine kidney cells.
Antibody analysis.
Influenza virus-specific antibody levels in BALF were determined by ELISA. Briefly, Nunc-Immuno 96-microwell plates were coated with monovalent subunit vaccine (2009 formulation: Sanofi Pasteur Inc.). The plates were then incubated with serial dilutions of BALF for 2 h at room temperature. Biotin-conjugated goat anti-mouse antibodies specific for IgA, IgG, or IgM (Caltag Laboratories, Burlingame, CA) were incubated for 1 h, and then horseradish peroxidase-conjugated streptavidin was added (Biosource). After 5 to 20 min of incubation, 3,3′,5,5′-tetramethylbenzidine (TMB) peroxidase substrate (BD Biosciences) was added, and optical density was measured at 450 nm. The antibody titer is expressed as the reciprocal dilution that gave 50% of the maximum optical density.
PCR array/quantitative RT-PCR.
Total RNA was extracted from whole mouse lung using TRIzol (Invitrogen, CA) in accordance with the manufacturer's recommended procedures. RNA quality was determined at 260/280 nm using a Nanodrop spectrophotometer (Thermo Scientific). Reverse transcription was performed using the RT2 first-strand synthesis kit (SABiosciences) in accordance with the manufacturer's instructions. For PCR array, a mouse T-cell and B-cell activation array (SABiosciences) was used, and PCR was performed using the Bio-Rad iQ5 real-time thermal cycler. For quantitative reverse transcription (RT)-PCR, reverse transcription was performed using a SuperScript III first-strand synthesis kit (Invitrogen) according to the manufacturer's instructions. The primers used to quantify IgM, IgA, IgG2a, IgG2b, IgG3, and activation-induced deaminase (AID) transcripts were obtained from Invitrogen and are listed in Table 2. Quantitative real-time PCR was performed using BioRadiQ SYBR green Supermix (Bio-Rad Laboratories), and PCR was performed in a Bio-Rad iQ5 real-time thermal cycler using the recommended cycle conditions. All relative expression quantities were determined using the ΔCT method with the ß2-microglobulin gene as the internal reference gene.
TABLE 2.
List of primers used for qRT-PCR
| Transcript | Sequence (5′ to 3′) |
|
|---|---|---|
| Forward primer | Reverse primer | |
| IgM | CAGGGGTCTCACCTTCTTGA | CACTTTGAGAAGCCCAGGAG |
| IgA | GAATGTGACCTGGGGAAAGA | GGAGGAGTAGGACCAGAGCA |
| IgG2a | CAGTCCATCACCTGCAATGT | TGGTCCACCCAAGAGGTTAG |
| IgG2b | AAATGCCCAGCTCCTAACCT | TTGGGTGTCAGGGAGATCAT |
| IgG3 | CTTCATGCCCACCTGGTAAC | TTGTCCACAAACCAGCTGAC |
| AID | TGCTACGTGGTGAAGAGGAG | TCCCAGTCTGAGATGTAGCG |
Prophylactic immunomodulation.
PR8-immunized asthmatic mice were treated twice i.n. with 30 μg of CpG oligonucleotide (Invitrogen) in a 50-μl volume of PBS. Control nonasthmatic and asthmatic mice were treated with the same volume of PBS. One day after the last administration, mice were infected with the CA04 virus.
In vivo CD4+ and CD8+ T-cell depletion.
PR8-immunized nonasthmatic and asthmatic mice were treated i.p. with 500 μg of GK1.5 anti-CD4 MAb (Bio X Cell) or 2.43 anti-CD8 MAb (Bio X Cell) on days −5, −4, −3, −2, −1, +1, +4, and +7. Control mice were injected i.p. with rat IgG. The treated mice were infected with CA04 on day 0, and effective cell depletion was confirmed in the lungs by flow cytometry analysis on day +6.
Statistical analyses.
Student's t test (two-tailed distribution, two-sample unequal variance) was used to analyze statistical significance between two groups. Multiple groups were compared using a one-way analysis of variance (ANOVA). Survival data were analyzed with the Kaplan-Meier log rank test using GraphPad Prism 5 software (San Diego, CA). A P value of <0.05 was considered to be statistically significant.
RESULTS
Asthmatic mice have increased susceptibility to secondary heterologous, but not homologous, influenza infection.
To determine the susceptibility of asthmatic mice to influenza infection, we developed a comorbidity model of allergic asthma and influenza infection. The established murine model of OVA-induced allergic lung inflammation was used to induce acute pulmonary hyperresponsiveness in mice (Fig. 1A) (17). We then examined homologous or heterologous immunity against the pandemic CA04 influenza virus strain after natural immunization by sublethal viral infection, which was designed to mimic the human scenario. On day 1 after the last OVA challenge, treated and nontreated mice were immunized i.n. by low-dose infection with either influenza A H1N1 seasonal virus (PR8) or the 2009 pandemic virus (CA04). Five weeks later, the mice were infected with lethal doses of the CA04 virus. All unimmunized, nonasthmatic mice (nonasthma:PBS/CA04 mice; see Table 1 for nomenclature) and unimmunized, asthmatic mice (asthma:PBS/CA04 mice) suffered significant weight loss and succumbed to infection (Fig. 1B to E). Both CA04-immunized, nonasthmatic mice (nonasthma:CA04/CA04 mice) and asthmatic mice (asthma:CA04/CA04 mice) survived a lethal homologous challenge with negligible weight loss (Fig. 1B and C). Similarly, all PR8-immunized mice survived a lethal PR8 challenge (data not shown). However, PR8-immunized, asthmatic mice challenged with the CA04 virus strain (asthma:PR8/CA04 mice) exhibited a more severe weight loss than PR8-immunized, nonasthmatic mice (nonasthma:PR8/CA04 mice) (Fig. 1D); asthma:PR8/CA04 mice also had significantly reduced survival rates (Fig. 1E). Significant decreases in lung viral titers were detected in both nonasthma:PR8/CA04 and asthma:PR8/CA04 mice compared to the unimmunized asthma or nonasthma:PBS/CA04 mice (Fig. 1F). However, viral titers were 10-fold higher in the asthma:PR8/CA04 mice than in nonasthma:PR8/CA04 mice on both days 3 and 6 post-CA04 challenge. Taken together, these results indicate that asthma increases susceptibility to heterologous but not homologous infection.
FIG 1.

Asthmatic mice are more susceptible to secondary heterologous but not homologous influenza A virus challenge. (A) Schematic of OVA-induced asthma and influenza virus immunization and challenge. BALB/c mice were sensitized twice, via the i.p. route, to OVA and OVA challenged i.n. for 5 consecutive days. One day following the final OVA challenge, the mice were inoculated i.n. with a sublethal dose of either PR8 or CA04 influenza virus for immunization; nonimmunized mice were given PBS. Five weeks after immunization, the mice were then challenged with lethal doses of CA04. (B to E) Weight loss (B and D) and survival (C and E) were monitored after challenge with the homologous virus strain (B and C) or the heterologous strain (D and E) (8 to 17 mice/group). (F) On days 3 and 6 post-secondary challenge, virus lung titers were determined by plaque assay (4 to 9 mice/group). The data are represented as mean viral titers ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Survival data were combined from two independent experiments.
Heterologous secondary infection results in severe lung inflammation in asthmatic mice.
Influenza infections cause lung immunopathology through the production of proinflammatory cytokines and chemokines that attract inflammatory cells (23). PR8-immunized, nonasthmatic and asthmatic mice were challenged with a lethal dose of CA04, and 6 days later, BALF cytokine and chemokine levels were analyzed. Consistent with the ability of asthma to induce increased production of Th2 type cytokines, IL-4, IL-5, and IL-13 levels were significantly increased in asthma:PR8/CA04 mice compared to nonasthmatic, infected mice (Fig. 2A). Interestingly, IL-6 levels were comparable between nonasthma:PR8/CA04 and asthma:PR8/CA04 mice. The Th1 cytokines, IL-12 and IFN-γ, were also significantly upregulated in asthma:PR8/CA04 mice. Increased levels of fever- and inflammation-associated mediators (e.g., IL-1α and IL-1β) were also detected in asthma:PR8/CA04 mice. Typical inflammatory chemokines, which are involved in the recruitment and activation of both innate and adaptive immune cells, were similarly expressed at significantly higher levels in asthma:PR8/CA04 mice than in nonasthma:PR8/CA04 mice (Fig. 2B). Severe lung injury often results in elevated BALF protein levels, which are indicative of edema and alveolar leakage. Indeed, increased total protein levels were detected in cell-free BALF collected from asthma:PR8/CA04 mice (Fig. 2C). A recently identified cell population, the innate lymphoid cell subset, can restore airway epithelial integrity in response to influenza-induced lung pathology through production of the epidermal growth factor amphiregulin (24). In fact, asthma:PR8/CA04 mice had increased levels of amphiregulin on day 3 post-CA04 influenza virus challenge, suggesting ongoing lung tissue repair (Fig. 2D). Nitric oxide also mediates the resolution of lung inflammation (25). Total nitrite/nitrate levels, which indicate nitric oxide production, were measured using the Griess reaction. BALF nitric oxide levels were significantly reduced in asthma:PR8/CA04 mice relative to the nonasthmatic controls on both days 3 and 6 post-CA04 influenza virus challenge (Fig. 2E). Collectively, these data demonstrate that asthmatic mice exhibit more severe lung pathology than nonasthmatic mice after secondary heterologous influenza challenge.
FIG 2.

Asthmatic mice exhibit severe pulmonary inflammation. Levels of cytokines (A) and chemokines (B) in BALF of nonasthmatic:PR8/CA04 and asthmatic:PR8/CA04 mice on day 6 post-CA04 challenge. Total BALF protein (C) and amphiregulin (D) levels on days 3 and 6 post-CA04 challenge. (E) Nitrite/nitrate levels, an indicator of NO, in the BALF were measured with Griess reagent. The data represent means ± SD of results for 7 to 9 mice/group from two independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Altered recruitment of inflammatory cells during reinfection in asthmatic mice.
To further characterize the inflammatory responses of asthma:PR8/CA04 mice, cell infiltration was analyzed at days 3 and 6 after CA04 challenge. Comparable numbers of neutrophils (CD11c− CD11b+ F4/80− Ly6G+), monocytes (CD11c− CD11b+ F4/80+ Ly6G−), NK cells (Dx5+ CD3−), and dendritic cells (CD11c+ CD11b+ F4/80− Ly6G−) were observed in nonasthma:PR8/CA04 and asthma:PR8/CA04 mice (Fig. 3A to D). However, numbers of alveolar macrophages (CD11c+ CD11b− F4/80+ Ly6G−) were reduced in both groups on days 3 and 6 postchallenge (Fig. 3E). These results are in agreement with previous studies demonstrating that alveolar macrophages are susceptible to influenza virus infection and undergo apoptosis to limit viral spread (26, 27). Thus, the decrease in numbers of alveolar macrophages in asthma:PR8/CA04 mice was not unexpected, because of the higher viral burden in these mice. Eosinophils (Siglec F+ CD3−) are considered to be a terminal effector cell associated with airway disease, and this population was increased in asthma:PR8/CA04 mice (Fig. 3F).
FIG 3.

Analysis of inflammatory cell responses following secondary heterologous influenza virus challenge. Levels of neutrophils (A), monocytes (B), NK cells (C), dendritic cells (D), alveolar macrophages (E), and eosinophils (F) in the lungs of nonasthmatic:PR8/CA04 and asthmatic:PR8/CA04 mice on days 3 and 6 post-CA04 challenge. The data represent means ± SD of results for 8 to 9 mice/group from two independent experiments.
These results suggest that secondary CA04 infection causes asthma exacerbation. To examine this, lung function was measured by oscillatory mechanics before and after reinfection. Immunized nonasthmatic or asthmatic mice, in the absence of secondary viral challenge, exhibited comparable baseline levels of airway resistance (RN) (Fig. 4A) and tissue damping (G) (Fig. 4B). On day 6 after challenge with CA04 virus, airway resistance and tissue damping increased significantly above baseline levels in both nonasthma:PR8/CA04 and asthma:PR8/CA04 mice; however, the levels were comparable in the two groups. Weight loss as a measure of morbidity indicated that asthma:PR8/CA04 mice suffered more severe influenza infection despite having airway hyperresponsiveness similar to that of nonasthma:PR8/CA04 mice (Fig. 4C). Therefore, these results exclude the possibility that asthma exacerbation was the cause of increased mortality.
FIG 4.
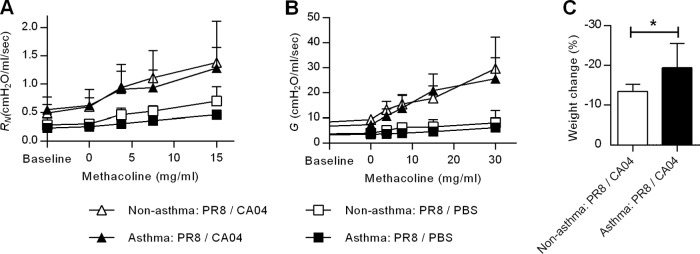
Airway hyperresponsiveness during secondary CA04 reinfection. (A) Newtonian resistance (RN) and (B) tissue damping (G) in response to increasing doses of methacholine were measured on day 6 post-CAO4 challenge using a mechanical animal ventilator. (C) Weight loss was also determined on day 6 post-CA04 challenge. The data represent the means ± SD of results for 3 to 8 mice/group from two independent experiments. *, P < 0.05.
Asthmatic mice generate equivalent cross-reactive recall T-cell responses.
Cross-protection against pandemic influenza virus typically requires both humoral and cell-mediated immunity. T cells have been shown to recognize internal conserved regions of influenza viruses, which can be cross-protective against pandemic influenza strains (15, 16, 28, 29). Therefore, we next evaluated pulmonary T-cell responses in nonasthma:PR8/CA04 and asthma:PR8/CA04 mice. On days 3 and 6 post-CA04 challenge, the numbers of lung CD4+ and CD8+ T cells were comparable between nonasthmatic and asthmatic mice (Fig. 5A and B). Since the influenza nucleoprotein (NP) contains immunodominant T-cell epitopes (30, 31), we next assessed the magnitude of pulmonary NP-specific T-cell responses. Lungs were harvested at different time points and stained with an NP-specific tetramer. NP-specific CD8+ T cells were rapidly recruited to the lungs after CA04 challenge, as shown by approximately 10- and 100-fold increases on days 3 and 6 postchallenge, respectively; similar increases were observed in both nonasthma:PR8/CA04 and asthma:PR8/CA04 mice (Fig. 5C). NP-specific T cells were further characterized based on cell surface expression of CD62L and CD127; expression of these markers distinguishes effector memory T cells (CD62L− CD127+), activated effector T cells (CD62L− CD127−), and central memory T cells (CD62L+ CD127+) (32, 33). The levels of effector memory T cells expanded over time in the lungs of both nonasthmatic and asthmatic mice, whereas the percentages of central memory T cells tended to decrease (Fig. 5D). Taken together, these results show that PR8 immunization efficiently primes T-cell immunity in both asthmatic and nonasthmatic mice and that equivalent memory T-cell responses are elicited upon CA04 challenge.
FIG 5.

Comparable numbers of influenza-specific T cells are present in the lungs of nonasthmatic and asthmatic mice following secondary heterologous CA04 virus challenge. Numbers of CD4+ (A), CD8+ (B), and NP+ CD8+ (C) T cells were determined in the lungs of nonasthmatic:PR8/CA04 and asthmatic:PR8/CA04 mice on days 3 and 6 post-CA04 challenge. (D) Percentages of CD62L− CD127−, CD62L+ CD127+, and CD62L− CD127+ cells within the CD8+ T-cell subset on days 3 and 6 post-CA04 challenge. A total of 3 to 9 mice/group were analyzed at each time point. The experiments were repeated at least twice.
Because no differences in the percentages or numbers of influenza virus-specific CD8+ T cell were detected between nonasthmatic and asthmatic mice, we next tested whether asthma affected the antiviral effector function of T cells. Granzyme B (gzmB) is the most abundant member of the granzyme family involved in T-cell-mediated cytolytic killing of virally infected cells (reviewed in reference 34). The percentages of gzmB+ CD8+ T cells (Fig. 6A) and gzmB+ CD4+ T cells (data not shown) were slightly higher in asthmatic mice. However, no differences were found in the numbers of gzmB+ CD8+ and gzmB+ CD4+ T cells between asthmatic and nonasthmatic mice (Fig. 6B and C). Interestingly, the mean fluorescence intensity (MFI) for gzmB staining of CD8+ cells was significantly higher in asthma:PR8/CA04 mice than in nonasthma:PR8/CA04 mice (Fig. 6D and E). The greater gzmB+ expression in asthmatic mice was associated with increased levels of gzmB in the BALF on days 3 and 6 post-CA04 challenge (Fig. 6F). To examine the protective function of T cells during heterologous influenza virus infection, activated lung T cells were adoptively transferred from either nonasthma:PR8/CA04 or asthma:PR8/CA04 mice into CA04-infected mice. Similar 10-fold reductions in lung viral titers at day 5 postinfection were detected in recipients of either nonasthma:PR8/CA04 or asthma:PR8/CA04 donor T cells compared to CA04-infected mice that did not receive T cells (Fig. 6G). Collectively, these data show that asthmatic mice do not exhibit any functional T-cell impairment in the pulmonary tract.
FIG 6.

Antiviral T-cell effector function is not compromised in asthmatic mice. (A) Representative dot plots showing the mean percentages ± SD of gzmB+ CD8+ T cells on days 0, 3, and 6 following CA04 challenge (3 or 4 mice/group). Numbers of gzmB+ CD4+ (B) and gzmB+ CD8+ (C) pulmonary T cells in nonasthmatic:PR8/CA04 and asthmatic:PR8/CA04 mice as assessed by intracellular flow cytometry on days 0, 3, and 6 post-CA04 challenge (3 or 4 mice/group). MFI of gzmB+ CD4+ (D) and CD8+ (E) lung T cells (3 or 4 mice/group). (F) gzmB levels in BALF on days 0, 3, and 6 post-CA04 infection (7 or 8 mice/group from two independent experiments). (G) Five days after CA04 challenge, CD3+ T cells were isolated from the lungs of nonasthmatic:PR8/CA04 and asthmatic:PR8/CA04 mice and transferred into day 2 CA04 (5,000 PFU)-infected recipients. Viral lung titers in the recipients 5 days after T-cell transfer are shown (4 or 5 mice/group). *, P < 0.05; ***, P < 0.001.
To further delineate the potential role of T cells, nonasthmatic:PR8/CA04 and asthmatic: PR8/CA04 mice were depleted of CD4+ or CD8+ T cells prior to the challenge. However, depletion of either CD4+ or CD8+ T cells had an insignificant effect on the survival of nonasthma:PR8/CA04 and asthma:PR8/CA04 mice (Fig. 7). This observation agrees with other studies showing that the function of either CD4+ or CD8+ T cells is sufficient for T-cell-mediated cross-protection (35, 36). Thus, both CD4+ and CD8+ T cells of asthmatic mice are capable of compensating for the lack of the other.
FIG 7.
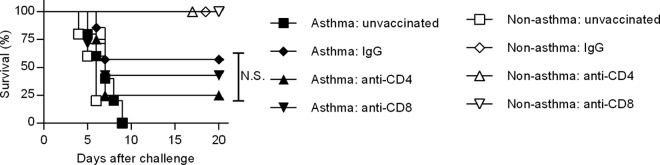
Depletion of either CD4+ or CD8+ T cells does not diminish cross-protective immunity. T cells were depleted by i.p. injection of either GK1.5 MAb (for CD4+ T-cell depletion) or 2.43 MAb (for CD8+ T-cell depletion). Control mice were treated with rat IgG. Successful T-cell depletion was confirmed by flow cytometry using fluorescent conjugated anti-CD4 (clone RM4.4) and anti-CD8 (clone 53-6.72) MAbs. Control and T-cell-depleted mice were infected with CA04 virus and monitored for survival (4 to 7 mice/group). N.S., P > 0.05.
PR8-specific systemic antibodies do not confer protection against CA04 virus.
Previous exposure to seasonal influenza A viruses affords some cross-protection against pandemic strains (12–14, 37–39), but the mechanism responsible for protection is not fully understood. Generally, antibody-mediated immunity is highly strain specific and is targeted against the mutable viral surface epitopes on hemagglutinin and neuraminidase. To investigate how asthma affects humoral immune responses against influenza virus, nonasthmatic and asthmatic mice were immunized i.n. with PR8 and bled 5 weeks later. Sera from immunized mice were then tested for HI titers. Consistent with other reports (15), this functional antibody assay showed that PR8 immunization generated strain-specific HI antibody in both nonasthmatic and asthmatic mice that was not cross-reactive with CA04 (Fig. 8A). Hence, CA04 cross-reactive HI antibodies were not elicited during priming with the PR8 virus.
FIG 8.

Humoral immune responses do not play a role in cross-protective immunity in either nonasthmatic or asthmatic mice. (A) Five weeks post-PR8 immunization, serum HI titers were determined against PR8 and CA04. (B to E) Pooled serum samples, collected at 5 weeks postimmunization, were diluted to 10% (vol/vol) and inoculated i.n. into naive mice together with 2,000 PFU of PR8 or CA04. Weight loss (B and D) and survival (C and E) were monitored for 20 days (8 mice/group). **, P < 0.01.
We next tested the protective activity of antibodies after PR8 immunization. Sera were harvested from PR8-immunized nonasthmatic and asthmatic mice and then administered i.n. to naive mice together with lethal doses of either PR8 or CA04 virus. Mice that received either nonimmune sera or PR8-immune sera lost similar amounts of weight upon CA04 challenge (Fig. 8B), and all of the mice succumbed to infection (Fig. 8C). In contrast, regardless of the asthmatic phenotype, naive recipient mice that received PR8-immune sera were fully protected from challenge with PR8 (Fig. 8E); no weight loss or other clinical signs were observed (Fig. 8D). These data indicate that asthmatic mice are capable of producing protective, strain-specific antibodies following PR8 immunization; however, as expected, those antibodies are not cross-reactive with CA04 virus.
Increased susceptibility of asthmatic mice to secondary CA04 influenza virus is due to impaired mucosal antibody responses following CA04 challenge.
We next examined the CA04-specific humoral immune response in the lungs of nonasthmatic and asthmatic mice that were challenged with CA04. BALF was harvested on day 6 post-CA04 influenza virus challenge from nonasthma:PR8/CA04 and asthma:PR8/CA04 mice. A plaque reduction assay revealed that BALF collected from asthma:PR8/CA04 mice exhibited significantly lower neutralizing activity against CA04 influenza virus than BALF collected from nonasthma:PR8/CA04 mice (Fig. 9A). As expected, CA04-specific IgA, IgG, or IgM antibodies were undetectable in the BALF after PR8 immunization of either nonasthmatic or asthmatic mice. In contrast, CA04-specific IgA, IgG, and IgM antibodies were induced by CA04 infection, but preexisting asthma caused an approximate 2-fold decrease in levels of these antibodies (Fig. 9, B-D). These results were further supported by data obtained by quantitative PCR analysis of lung Ig gene transcripts 3 days post-CA04 challenge. Asthma caused significant reductions in IgA, IgG2a, IgG2b, and IgG3 H chain transcript levels, whereas no differences in IgM H chain transcript levels were observed (Fig. 9E). These results suggest that asthma impairs the production of isotype-switched antibodies during subsequent influenza virus infection. Indeed, expression of AID, an enzyme required for class switch recombination in B cells, was also reduced in asthmatic mice (Fig. 9F).
FIG 9.

Impaired mucosal antibody responses in asthmatic mice during secondary heterologous challenge. (A) On day 6 post-CA04 challenge, BALF was collected from PR8-immunized nonasthmatic and asthmatic mice and tested for virus-neutralizing activity against CA04 (4 mice/group). (B to D) IgA (B), IgG (C), and IgM (D) anti-CA04 BALF antibody levels were determined by ELISA (4 to 12 mice/group from two independent experiments). (E) On day 3 post-CA04 challenge, IgM, IgA, IgG2a, IgG2b, and IgG3 H chain transcript levels were determined by quantitative RT-PCR (5 mice/group). (F) Transcript levels of AID were also determined on day 3 post-CA04 challenge (5 mice/group). (G and H) Immune BALF was collected from the indicated mice before or after CA04 challenge and inoculated i.n. into naive recipients together with 5,000 PFU of CA04 virus. Weight loss (G) and survival (H) were then monitored (5 to 10 mice/group). (I) Viral lung burden in the recipient mice on day 1 following CA04 challenge (5 mice/group). **, P < 0.01; ***, P < 0.001.
To directly measure the functional impairment of antibody responses in asthma:PR8/CA04 mice, BALF was passively transferred into naive mice. BALF was collected on day 6 after secondary CA04 infection and inoculated i.n. into naive mice together with lethal doses of CA04 virus. BALF collected from PR8-immunized, but not CA04-challenged, mice showed no protection against CA04 in naive mice regardless of donor asthmatic phenotype. All recipient mice lost significant weight (Fig. 9G) and succumbed to CA04 infection by day 7 (Fig. 9H). In contrast, immune BALF from donor mice that were PR8 immunized and CA04 challenged was protective against CA04 challenge. However, analysis of weight loss (Fig. 9G) and survival (Fig. 9H) showed that BALF from asthma:PR8/CA04 mice was significantly less protective in the recipient mice than BALF from nonasthma:PR8/CA04 mice. The enhanced survival mediated by BALF from nonasthma:PR8/CA04 mice is consistent with a 10-fold reduction in lung viral titers (Fig. 9I). To determine whether there was a role for virus-specific antibody in mediating passive protection, total mouse Ig was depleted prior to BALF transfer. Total Ig depletion completely abrogated protection, suggesting that CA04-specific antibody was responsible for passive protection (Fig. 9H). None of the mice that received BALF alone without virus challenge exhibited weight loss or any signs of morbidity. This rules out the possibility that heat-resistant cytokines in the BALF of asthma:PR8/CA04 mice contributed to the increased weight loss and mortality.
Susceptibility of asthmatic mice to secondary CA04 challenge is not due to impaired expression of B cells.
The above results showed that asthma reduces lung AID expression, thereby impairing the production of influenza-specific isotype-switched antibodies. Therefore, we determined whether asthma affects pulmonary B-cell abundance. PR8-immunized nonasthmatic and asthmatic mice were challenged with CA04 virus, and the percentage of B220+ cells in the lung was quantified. Comparable percentages of B220+ B cells were present in nonasthma:PR8/CA04 and asthma:PR8/CA04 mice (Fig. 10A). Furthermore, the percentages of necrotic and apoptotic B cells were similar in both groups of mice (Fig. 10B and C).
FIG 10.
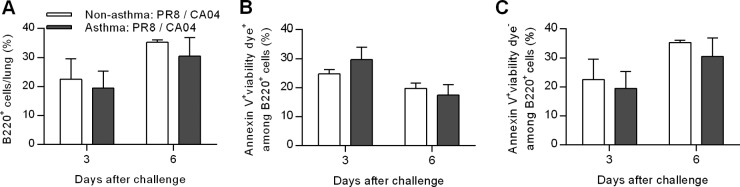
Asthma does not impact B-cell apoptosis/necrosis after heterologous influenza reinfection. (A) Total percentages of B220+ B cells in the lungs. (B and C) Percentages of necrotic (B) and apoptotic (C) cells within the B220+ B-cell subset on days 3 and 6 post-CA04 influenza challenge. Data are the means ± SD of results for 4 mice/group.
CpG confers protection in asthmatic mice against secondary CA04 challenge.
Having identified humoral immunity as the defective immune response in asthma:PR8/CA04 mice, we sought to determine if the B-cell-stimulating agent CpG could overcome this defect. CpG is a synthetic oligonucleotide that initiates signaling through TLR9 (40). Following PR8 immunization, asthmatic mice were treated i.n. with CpG before secondary CA04 infection (Fig. 11A). It was found that CpG inoculation significantly reduced weight loss in asthmatic mice compared to non-CpG-treated asthmatic mice (Fig. 11B). CpG also significantly improved the survival rates of asthma:PR8/CA04 mice (Fig. 11C). These results indicate that CpG could be useful for prophylactic treatment of asthmatics to help prevent influenza.
FIG 11.
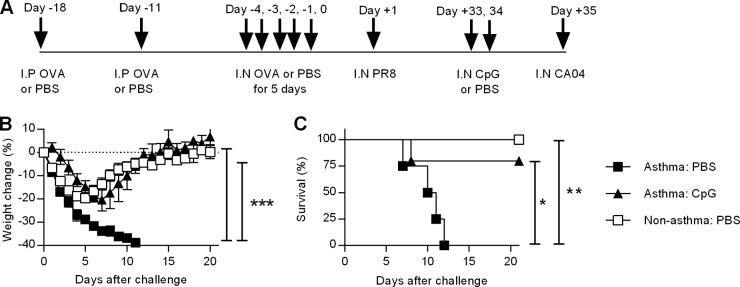
CpG provides protection against CA04 reinfection in asthmatic mice. (A) Schematic of CpG treatment of asthmatic mice challenged with a heterologous influenza infection. PR8-immunized mice were treated i.n. with 30 μg CpG or PBS for 2 days and then challenged with CA04 virus the following day. Infected mice were monitored for weight loss (B) and survival (C) for 20 days (4 or 5 mice/group). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
Epidemiological evidence indicates that asthma is a significant risk factor for influenza virus infection. However, this hypothesis has never been experimentally tested. In the current study, we linked asthma to adverse effects on cross-protective immunity against secondary infection with the 2009 H1N1 pandemic (CA04) virus. Using a novel comorbidity model of asthma and influenza reinfection, we found that asthmatic mice immunized against seasonal influenza A virus (PR8) through natural infection remained highly vulnerable to the pandemic CA04 virus while nonasthmatic mice were protected. Increased susceptibility was attributable to inadequate activation of influenza-specific respiratory B cells, but not T cells, upon reinfection. These novel findings may help explain the increased rate of hospitalization among asthmatics during the 2009 H1N1 pandemic.
Asthmatic mice showed uncontrolled viral burden, excessive cytokine/chemokine production, and increased total protein levels in BALF following CA04 infection, indicative of edema and severe inflammatory responses. However, although asthmatic mice had increased numbers of eosinophils, pulmonary lung functions following secondary influenza infection were equivalent to those of nonasthmatic mice, ruling out the possibility of infection-induced asthma exacerbation as a cause of mortality. In addition, we confirmed that the increased mortality rate was not a result of an already inflamed pulmonary environment, because baseline cytokine and chemokine levels were comparable in nonasthmatic and asthmatic mice immediately prior to secondary infection (data not shown). Thus, the severe cytokine storm seen in asthmatic mice following secondary challenge was likely due to uncontrolled viral replication. It is important to note that the heightened susceptibility of asthmatic mice was apparent only in the secondary heterologous virus challenge model. Asthmatic mice were fully protected against primary virus infection as well as secondary homologous challenge. This led us to initially hypothesize that strain-specific antibody responses were intact but that cross-reactive T-cell immunity was negatively impacted by asthma, leading to decreased resistance against heterologous challenge. This hypothesis was based on numerous publications showing that cross-protection is predominantly mediated by T-cell immunity due to cross-reactivity with influenza A virus internal proteins (28–31, 39). In line with this, PR8 immunization induced comparable serum HI titers in nonasthmatic and asthmatic mice, which protected naive mice against PR8 infection following passive serum transfer. However, the antisera did not protect against CA04 challenge, confirming the findings of others (15) that PR8 immunization-induced antibodies are not cross-reactive with the CA04 strain and therefore do not contribute to protection against secondary heterologous challenge.
Surprisingly, however, we did not observe any differences in cellular immunity that could account for the severe illness that was observed in CA04-infected asthmatic mice. Absolute numbers of recall CD4+, CD8+, and influenza NP-specific T cells were comparable between nonasthmatic and asthmatic mice during secondary CA04 infection. In addition, the memory/activation phenotype of influenza-specific CD8+ T cells was normal in asthmatic mice. Interestingly, gzmB expression by CD8+ T cells in asthmatic mice was found to be even slightly greater. Studies of asthma in human patients (41, 42) and in animal models (43) have demonstrated upregulation of gzmB during asthma attacks, thereby implicating gzmB in asthma pathogenesis. Consistent with these studies, we have also observed greater expression of gzmB in the airways of asthmatic mice during infection, mostly due to increased gzmB expression by CD8+ T cells. Thus, although the significance of high CD8+ T cell gzmB expression in asthmatic mice in relation to the outcome of influenza infection was not investigated in this study, it is clear that the cytolytic function of T cells was not compromised. This conclusion was further supported by T-cell transfer experiments in which T cells from reinfected mice were harvested and transferred into unimmunized CA04-infected mice. In this case, T cells from asthmatic mice were fully capable of controlling viral replication in the recipient mice. Thus, memory T-cell responses are not compromised in asthmatic mice and unlikely to be responsible for the heightened sensitivity against pandemic H1N1 challenge.
Humoral immunity in asthmatic mice upon reinfection was characterized by significantly lower levels of CA04-neutralizing antibodies as well as decreased CA04-specific IgA, IgG, and IgM titers. This is consistent with the reduced protective effect of BALF from asthmatic mice. Thus, we have identified a defective immune response in asthmatic mice, namely, pulmonary humoral immunity, which is responsible for reduced resistance. However, the precise mechanism by which mucosal antibody responses are downregulated in asthmatic mice remains unknown. Because levels of apoptotic gzmB were upregulated in the BALF of asthmatic mice, we determined if the suppressed antibody responses were due to higher rates of B-cell necrosis or apoptosis. However, the percentages of total, necrotic, and apoptotic B cells were equivalent between nonasthmatic and asthmatic mice, ruling out this possibility. Instead, significant reduction in AID expression was observed in asthmatic mice and corresponded to diminished Ig class switching in these animals, suggesting that B-cell activation was suboptimal. Indeed, inoculation of the B-cell TLR9 agonist CpG provided significant protection against secondary CA04 infection in asthma:PR8/CA04 mice, to the levels seen in nonasthma:PR8/CA04 mice.
The impact of impaired antibody responses on the survival of asthmatic mice may be compounded by the significantly lower number of alveolar macrophages in asthma:PR8/CA04 mice. Phagocytes such as alveolar macrophages directly contribute to the control of influenza virus replication (44) via ingestion of antibody-opsonized virus (22) and to the elimination of infected cells in an Fc receptor-dependent manner (45). Furthermore, the phagocytic activities of alveolar macrophages in asthmatic mice have been found to be impaired (46). Thus, it is likely that a reduced number of alveolar macrophages also contribute to the heightened susceptibility of asthmatic mice.
Antibodies elicited by influenza infections are generally T-cell-dependent responses and CD4+ helper T cells are needed to provide signals important for B-cell antibody class switching. During reinfection, the numbers of CD4+ T cells found in the BALF were equivalent between nonasthmatic and asthmatic groups. Furthermore, in vivo depletion of CD4+ T cells during reinfection did not alter the survival rates in nonasthmatic or asthmatic mice (25% survival in asthmatic mice versus 100% survival in nonasthmatic mice), suggesting that the observed reduced resistance and defective B-cell responsiveness of asthma:PR8/CA04 mice is independent of CD4+ helper T cells. Alternatively, the defects could lie in CD4-independent antibody response mechanisms. T-cell-independent antibody responses have been reported for influenza infections (47); induction of such antibody responses typically involves TLR signaling and/or B-cell-stimulating cytokines such as B-cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL) (48–50). An important mechanism that promotes the production of BAFF and APRIL by DCs and enhances T-independent antibody responses is mediated by nitric oxide (51). Nitric oxide levels in BALF were significantly reduced in asthmatic mice. Thus, additional studies are under way to determine whether BAFF/APRIL and TLR signaling pathways are defective in asthmatic mice. An impairment in TLR responses in asthmatic mice that leads to defective bacterial clearance has been recently reported (52). It is therefore plausible that a similar TLR signaling impairment exists in our asthma/influenza model and is responsible for diminished mucosal antibody responses. This would be consistent with the ability of CpG to overcome the observed antibody defect.
It is important to note that our findings contrast with a recent report by Ishikawa et al. (53) showing that asthmatic mice are more resistant to influenza infection than are nonasthmatic mice and that this resistance is mediated by NK cells. Several major differences can be noted between this previous study and our current results, including the use of the PR8 strain for lethal challenges of C57BL/6 mice in the previous study compared to utilization of the 2009 pandemic virus and BALB/c mice in the present study. BALB/c mice are classically used for mouse models of allergic asthma, and asthma pathology differs significantly between BALB/c and C57BL/6 mice (54–56). Thus, it is not surprising that the effect of asthma on host immunity is also different. Perhaps most importantly, the time intervals between asthma and influenza infection were different between the two studies. The study by Ishikawa et al. used a primary influenza challenge model in which OVA-sensitized mice were lethally infected 1 day after the final i.n OVA challenge. Day 1 after the final OVA challenge is the peak of the acute asthma attack during which pulmonary inflammation is severe and a number of various immune cells, including NK cells, have infiltrated the lungs. In contrast, we immunized with a sublethal dose of PR8 after OVA challenge, followed by a lethal inoculation of CA04 virus 5 weeks later, at a time when the mice no longer exhibited any asthmatic symptoms. Thus, the contexts in which the susceptibility of asthmatic mice was evaluated were vastly different and the nature of the immune responses would also be expected to be different. In our study, a 5-week interval for lethal influenza challenge was chosen for two reasons. First, in clinical settings, viral infections typically precede asthma attacks. In fact, it has been estimated that 70% of asthma exacerbations are due to viral infections (57, 58). Hence, it is clinically more relevant to induce asthma and then infect with influenza virus after lung homeostasis has been restored. Second, the mouse model of successive influenza virus challenges (seasonal PR8 followed by pandemic CA04) better resembles human influenza infection among asthmatic adults. Together, it is likely that the differing results are due to differences in a number of experimental conditions. However, a definitive link between asthma and antiviral immunity is a common finding between our study and the study by Ishikawa and colleagues. These two studies suggest a complexity of interactions between asthma and host mucosal immunity, with the influence of asthma on antiviral responses different depending upon genetic background, the immunological history of the host, and the timing of viral infection in relation to allergen exposure. Furthermore, the types of virus used may also have a significant impact on the outcome of the lung allergic/infection model. For example, OVA-sensitized/challenged mice exhibit enhanced viral clearance against parainfluenza (59) and rhinovirus (60, 61) infections. The reduction of viral burden has been attributed to increased recruitment of eosinophils that may help clear viral infection (61). Consistent with this hypothesis, the ability of eosinophils to directly inhibit parainfluenza virus infectivity has been demonstrated in vitro (62). Similarly, we detected an increased influx of eosinophils in OVA-sensitized/challenged mice infected with influenza virus; however, enhanced viral replication was observed. This suggests that eosinophils may not possess an antiviral effect against influenza virus infection or that eosinophils and other inflammatory cells are required at the site of infection before the viral challenge in order to exert an effective antiviral response.
Although the acute asthma mouse model used here mimics many aspects of human asthma, a number of limitations do exist. The short-term nature of the acute allergic lung inflammation lacks many of the characteristics of human asthma, such as chronic inflammation of the airway wall and airway remodeling (63). Indeed, airway inflammation and airway hyperresponsiveness in the OVA mouse model resolve within a few weeks after the last allergen challenge (64). Thus, a variety of chronic allergen challenge models have been recently developed. It will be of great interest to investigate in future studies whether the negative effect of acute allergic lung inflammation on mucosal humoral immunity is also recapitulated in a chronic model.
It is important to note that the observed immune defects in asthmatic mice may also exist to some degree during homologous viral challenge but not to the degree that would result in reduced survival. The defect appears to be specific to mucosal humoral immunity, since systemic antibody levels in immunized mice at later time points were comparable between nonasthmatic and asthmatic mice, as shown in this study. Thus, it is likely that systemic humoral immunity together with effector T cells compensates for a possible reduction in B-cell activation in the airways during homologous challenge. Alternatively, the kinetics of antibody responses may be delayed in asthmatic mice, but the peak of antibody titers, which occurs around 4 to 7 weeks after immunization, could be intact. This scenario is also consistent with our observation that immunity was not impaired at week 5 postimmunization, when the mice were either challenged with a homologous viral strain or bled to measure antibody levels. In both scenarios, the heightened susceptibility of asthmatic mice in the immunization/challenge model became apparent when memory T cells were present but effective memory B cells were absent.
Our observation that asthmatic mice are more susceptible than nonasthmatic mice to secondary influenza virus infection, despite having intact T-cell memory, suggests that the presence of cross-reactive T-cell immunity alone cannot compensate for suboptimal primary antibody responses that occur during reinfection. The clinical significance of this finding is that even though the live attenuated influenza vaccine (FluMist; MedImmune) is thought to be superior to the inactivated subunit vaccine due to its ability to stimulate cross-reactive T-cell immunity (65, 66), it may be less effective in asthmatic individuals in the event of a pandemic, when strain-specific B-cell memory is absent. In addition, natural infection-induced immunity, which is considered to be the gold standard in vaccinology, is established in humans by yearly exposure to seasonal influenza virus and thought to provide significant cross-protection. However, our findings suggest that the asthmatic patient will still remain vulnerable, even after natural immunization, against not only pandemic strains but also drifted strains. This explains why hospital admission rates were higher among asthmatics during the 2009 pandemic despite preexisting immunity against influenza virus in the human population. Our results pose a unique problem for asthmatics during influenza pandemics, since the manufacture of strain-specific subunit vaccines can take up to 6 months from the time of the virus isolation to delivery to the human population. Thus, we would advocate that patients with asthma should be prioritized for vaccination in the event of vaccine shortages during a pandemic.
Since predictions of seasonal influenza virus strains are based on educated guesses and the appearance of a new pandemic influenza virus is unpredictable, the current vaccination strategy that is focused on strain-specific antibodies is inadequate for both asthmatics and the general population and, hence, the current interest in “universal” influenza vaccines. Nevertheless, our findings that asthmatics have a unique defect in mucosal humoral immunity and that memory T cells failed to protect asthmatic mice imply that the development of universal flu vaccines should focus on induction of cross-reactive antibodies, rather than T cells, to be fully effective in asthmatics. In addition, non-antigen-specific prophylactic treatment that broadly stimulates B cells may represent an effective alternative to vaccination for asthmatics. Indeed, we demonstrated in this study that survival rates of asthmatic mice can be significantly improved by i.n. administration of CpG. Based on our current understanding of CpG, the likely mechanism responsible for protection is enhancement of mucosal B-cell responses through direct activation of B cells (67), increased survival of B cells via preventing apoptosis and promoting cell proliferation (68), and activation of antigen-presenting cells that facilitate class switch recombination (69).
Due to the absence of effective vaccines against a number of respiratory pathogens and the increasing prevalence of asthma, a better understanding of the link between asthma and mucosal immunity is critical. The impact of asthma in downregulation of mucosal antibody responses is clear; however, the molecular mechanisms involved remain to be fully elucidated. We are currently investigating specific pathways that exist in asthmatic mice and that render B cells less responsive during influenza reinfection. Inhibition of mucosal humoral responses may have an impact on susceptibility to a variety of respiratory pathogens. Indeed, asthmatic patients are also known to be more vulnerable to other viral infections, such as rhinovirus (70, 71), and bacterial infections, including pneumococcal pneumonia (72, 73). The therapeutic potential of agents that enhance mucosal humoral immunity ultimately might be exploited to improve respiratory disease outcomes in asthmatic patients.
ACKNOWLEDGMENTS
We thank the Center for Immunology and Microbial Disease Immunology Core Laboratory for their technical assistance.
This work was supported by NIH grant RO1 AI75312 to D.W.M. and by American Lung Association Senior Research Training Fellowship RT-226959-N to Y.F.
Footnotes
Published ahead of print 4 June 2014
REFERENCES
- 1.Akinbami LJ, Moorman JE, Liu X. 2011. Asthma prevalence, health care use, and mortality: United States, 2005-2009. Natl. Health Stat. Report 12:1–14 [PubMed] [Google Scholar]
- 2.Wills-Karp M. 1999. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu. Rev. Immunol. 17:255–281. 10.1146/annurev.immunol.17.1.255 [DOI] [PubMed] [Google Scholar]
- 3.Cohn L, Elias JA, Chupp GL. 2004. Asthma: mechanisms of disease persistence and progression. Annu. Rev. Immunol. 22:789–815. 10.1146/annurev.immunol.22.012703.104716 [DOI] [PubMed] [Google Scholar]
- 4.Durrant DM, Metzger DW. 2010. Emerging roles of T helper subsets in the pathogenesis of asthma. Immunol. Invest. 39:526–549. 10.3109/08820131003615498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papadopoulos NG, Stanciu LA, Papi A, Holgate ST, Johnston SL. 2002. A defective type 1 response to rhinovirus in atopic asthma. Thorax 57:328–332. 10.1136/thorax.57.4.328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brooks GD, Buchta KA, Swenson CA, Gern JE, Busse WW. 2003. Rhinovirus-induced interferon-gamma and airway responsiveness in asthma. Am. J. Respir. Crit. Care Med. 168:1091–1094. 10.1164/rccm.200306-737OC [DOI] [PubMed] [Google Scholar]
- 7.Message SD, Laza-Stanca V, Mallia P, Parker HL, Zhu J, Kebadze T, Contoli M, Sanderson G, Kon OM, Papi A, Jeffery PK, Stanciu LA, Johnston SL. 2008. Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proc. Natl. Acad. Sci. U. S. A. 105:13562–13567. 10.1073/pnas.0804181105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilca R, De Serres G, Boulianne N, Ouhoummane N, Papenburg J, Douville-Fradet M, Fortin E, Dionne M, Boivin G, Skowronski DM. 2011. Risk factors for hospitalization and severe outcomes of 2009 pandemic H1N1 influenza in Quebec, Canada. Influenza Other Respir. Viruses 5:247–255. 10.1111/j.1750-2659.2011.00204.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Myles P, Nguyen-Van-Tam JS, Semple MG, Brett SJ, Bannister B, Read RC, Taylor BL, McMenamin J, Enstone JE, Nicholson KG, Openshaw PJ, Lim WS. 2013. Differences between asthmatics and nonasthmatics hospitalised with influenza A infection. Eur. Respir. J. 41:824–831. 10.1183/09031936.00015512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Louie JK, Acosta M, Samuel MC, Schechter R, Vugia DJ, Harriman K, Matyas BT. 2011. A novel risk factor for a novel virus: obesity and 2009 pandemic influenza A (H1N1). Clin. Infect. Dis. 52:301–312. 10.1093/cid/ciq152 [DOI] [PubMed] [Google Scholar]
- 11.Hancock K, Veguilla V, Lu X, Zhong W, Butler EN, Sun H, Liu F, Dong L, DeVos JR, Gargiullo PM, Brammer TL, Cox NJ, Tumpey TM, Katz JM. 2009. Cross-reactive antibody responses to the 2009 pandemic H1N1 influenza virus. N. Engl. J. Med. 361:1945–1952. 10.1056/NEJMoa0906453 [DOI] [PubMed] [Google Scholar]
- 12.Tu W, Mao H, Zheng J, Liu Y, Chiu SS, Qin G, Chan PL, Lam KT, Guan J, Zhang L, Guan Y, Yuen KY, Peiris JS, Lau YL. 2010. Cytotoxic T lymphocytes established by seasonal human influenza cross-react against 2009 pandemic H1N1 influenza virus. J. Virol. 84:6527–6535. 10.1128/JVI.00519-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alam S, Sant AJ. 2011. Infection with seasonal influenza virus elicits CD4 T cells specific for genetically conserved epitopes that can be rapidly mobilized for protective immunity to pandemic H1N1 influenza virus. J. Virol. 85:13310–13321. 10.1128/JVI.05728-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen GL, Lau YF, Lamirande EW, McCall AW, Subbarao K. 2011. Seasonal influenza infection and live vaccine prime for a response to the 2009 pandemic H1N1 vaccine. Proc. Natl. Acad. Sci. U. S. A. 108:1140–1145. 10.1073/pnas.1009908108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo H, Santiago F, Lambert K, Takimoto T, Topham DJ. 2011. T cell-mediated protection against lethal 2009 pandemic H1N1 influenza virus infection in a mouse model. J. Virol. 85:448–455. 10.1128/JVI.01812-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hillaire ML, van Trierum SE, Kreijtz JH, Bodewes R, Geelhoed-Mieras MM, Nieuwkoop NJ, Fouchier RA, Kuiken T, Osterhaus AD, Rimmelzwaan GF. 2011. Cross-protective immunity against influenza pH1N1 2009 viruses induced by seasonal influenza A (H3N2) virus is mediated by virus-specific T-cells. J. Gen. Virol. 92:2339–2349. 10.1099/vir.0.033076-0 [DOI] [PubMed] [Google Scholar]
- 17.Alouani S, Juillard P, Chvatchko Y. 2000. Murine model of allergic lung inflammation. Methods Mol. Biol. 138:285–293. 10.1385/1-59259-058-6:285 [DOI] [PubMed] [Google Scholar]
- 18.Ilyushina NA, Khalenkov AM, Seiler JP, Forrest HL, Bovin NV, Marjuki H, Barman S, Webster RG, Webby RJ. 2010. Adaptation of pandemic H1N1 influenza viruses in mice. J. Virol. 84:8607–8616. 10.1128/JVI.00159-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. 2009. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 206:79–87. 10.1084/jem.20081667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durrant DM, Gaffen SL, Riesenfeld EP, Irvin CG, Metzger DW. 2009. Development of allergen-induced airway inflammation in the absence of T-bet regulation is dependent on IL-17. J. Immunol. 183:5293–5300. 10.4049/jimmunol.0803109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cottey R, Rowe CA, Bender BS. 2001. Influenza virus. Curr. Protoc. Immunol. Chapter 19:Unit 19.11. 10.1002/0471142735.im1911s42 [DOI] [PubMed] [Google Scholar]
- 22.Huber VC, Lynch JM, Bucher DJ, Le J, Metzger DW. 2001. Fc receptor-mediated phagocytosis makes a significant contribution to clearance of influenza virus infections. J. Immunol. 166:7381–7388. 10.4049/jimmunol.166.12.7381 [DOI] [PubMed] [Google Scholar]
- 23.La Gruta NL, Kedzierska K, Stambas J, Doherty PC. 2007. A question of self-preservation: immunopathology in influenza virus infection. Immunology and cell biology 85:85–92. 10.1038/sj.icb.7100026 [DOI] [PubMed] [Google Scholar]
- 24.Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, Kubota M, Turner D, Diamond JM, Goldrath AW, Farber DL, Collman RG, Wherry EJ, Artis D. 2011. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 12:1045–1054. 10.1038/ni.2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D'Alessio FR, Tsushima K, Aggarwal NR, Mock JR, Eto Y, Garibaldi BT, Files DC, Avalos CR, Rodriguez JV, Waickman AT, Reddy SP, Pearse DB, Sidhaye VK, Hassoun PM, Crow MT, King LS. 2012. Resolution of experimental lung injury by monocyte-derived inducible nitric oxide synthase. J. Immunol. 189:2234–2245. 10.4049/jimmunol.1102606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghoneim HE, Thomas PG, McCullers JA. 2013. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J. Immunol. 191:1250–1259. 10.4049/jimmunol.1300014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mok CK, Lee DC, Cheung CY, Peiris M, Lau AS. 2007. Differential onset of apoptosis in influenza A virus H5N1- and H1N1-infected human blood macrophages. J. Gen. Virol. 88:1275–1280. 10.1099/vir.0.82423-0 [DOI] [PubMed] [Google Scholar]
- 28.Furuya Y, Chan J, Regner M, Lobigs M, Koskinen A, Kok T, Manavis J, Li P, Mullbacher A, Alsharifi M. 2010. Cytotoxic T cells are the predominant players providing cross-protective immunity induced by γ-irradiated influenza A viruses. J. Virol. 84:4212–4221. 10.1128/JVI.02508-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slutter B, Pewe LL, Lauer P, Harty JT. 2013. Cutting edge: rapid boosting of cross-reactive memory CD8 T cells broadens the protective capacity of the Flumist vaccine. J. Immunol. 190:3854–3858. 10.4049/jimmunol.1202790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grant E, Wu C, Chan KF, Eckle S, Bharadwaj M, Zou QM, Kedzierska K, Chen W. 2013. Nucleoprotein of influenza A virus is a major target of immunodominant CD8(+) T-cell responses. Immunol. Cell Biol. 91:184–194. 10.1038/icb.2012.78 [DOI] [PubMed] [Google Scholar]
- 31.Lee LY, Ha do LA, Simmons C, de Jong MD, Chau NV, Schumacher R, Peng YC, McMichael AJ, Farrar JJ, Smith GL, Townsend AR, Askonas BA, Rowland-Jones S, Dong T. 2008. Memory T cells established by seasonal human influenza A infection cross-react with avian influenza A (H5N1) in healthy individuals. J. Clin. Invest. 118:3478–3490. 10.1172/JCI32460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bachmann MF, Wolint P, Schwarz K, Jager P, Oxenius A. 2005. Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of IL-7 receptor alpha and CD62L. J. Immunol. 175:4686–4696. 10.4049/jimmunol.175.7.4686 [DOI] [PubMed] [Google Scholar]
- 33.Huster KM, Busch V, Schiemann M, Linkemann K, Kerksiek KM, Wagner H, Busch DH. 2004. Selective expression of IL-7 receptor on memory T cells identifies early CD40L-dependent generation of distinct CD8+ memory T cell subsets. Proc. Natl. Acad. Sci. U. S. A. 101:5610–5615. 10.1073/pnas.0308054101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kägi D, Ledermann B, Burki K, Zinkernagel RM, Hengartner H. 1996. Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu. Rev. Immunol. 14:207–232. 10.1146/annurev.immunol.14.1.207 [DOI] [PubMed] [Google Scholar]
- 35.Liang S, Mozdzanowska K, Palladino G, Gerhard W. 1994. Heterosubtypic immunity to influenza type A virus in mice. Effector mechanisms and their longevity. J. Immunol. 152:1653–1661 [PubMed] [Google Scholar]
- 36.Laidlaw BJ, Decman V, Ali MA, Abt MC, Wolf AI, Monticelli LA, Mozdzanowska K, Angelosanto JM, Artis D, Erikson J, Wherry EJ. 2013. Cooperativity between CD8+ T cells, non-neutralizing antibodies, and alveolar macrophages is important for heterosubtypic influenza virus immunity. PLoS Pathog. 9:e1003207. 10.1371/journal.ppat.1003207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alsharifi M, Furuya Y, Bowden TR, Lobigs M, Koskinen A, Regner M, Trinidad L, Boyle DB, Mullbacher A. 2009. Intranasal flu vaccine protective against seasonal and H5N1 avian influenza infections. PLoS One 4:e5336. 10.1371/journal.pone.0005336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jegaskanda S, Weinfurter JT, Friedrich TC, Kent SJ. 2013. Antibody-dependent cellular cytotoxicity is associated with control of pandemic H1N1 influenza virus infection of macaques. J. Virol. 87:5512–5522. 10.1128/JVI.03030-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weinfurter JT, Brunner K, Capuano SV, III, Li C, Broman KW, Kawaoka Y, Friedrich TC. 2011. Cross-reactive T cells are involved in rapid clearance of 2009 pandemic H1N1 influenza virus in nonhuman primates. PLoS Pathog. 7:e1002381. 10.1371/journal.ppat.1002381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408:740–745. 10.1038/35047123 [DOI] [PubMed] [Google Scholar]
- 41.Tschopp CM, Spiegl N, Didichenko S, Lutmann W, Julius P, Virchow JC, Hack CE, Dahinden CA. 2006. Granzyme B, a novel mediator of allergic inflammation: its induction and release in blood basophils and human asthma. Blood 108:2290–2299. 10.1182/blood-2006-03-010348 [DOI] [PubMed] [Google Scholar]
- 42.Bratke K, Bottcher B, Leeder K, Schmidt S, Kupper M, Virchow JC, Jr, Luttmann W. 2004. Increase in granzyme B+ lymphocytes and soluble granzyme B in bronchoalveolar lavage of allergen challenged patients with atopic asthma. Clin. Exp. Immunol. 136:542–548. 10.1111/j.1365-2249.2004.02468.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen KD, Vanichsarn C, Nadeau KC. 2008. Increased cytotoxicity of CD4+ invariant NKT cells against CD4+CD25hiCD127lo/− regulatory T cells in allergic asthma. Eur. J. Immunol. 38:2034–2045. 10.1002/eji.200738082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin-Jackwood MJ, Schultz-Cherry S, Solorzano A, Van Rooijen N, Katz JM, Basler CF. 2005. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J. Virol. 79:14933–14944. 10.1128/JVI.79.23.14933-14944.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.El Bakkouri K, Descamps F, De Filette M, Smet A, Festjens E, Birkett A, Van Rooijen N, Verbeek S, Fiers W, Saelens X. 2011. Universal vaccine based on ectodomain of matrix protein 2 of influenza A: Fc receptors and alveolar macrophages mediate protection. J. Immunol. 186:1022–1031. 10.4049/jimmunol.0902147 [DOI] [PubMed] [Google Scholar]
- 46.Naessens T, Vander Beken S, Bogaert P, Van Rooijen N, Lienenklaus S, Weiss S, De Koker S, Grooten J. 2012. Innate imprinting of murine resident alveolar macrophages by allergic bronchial inflammation causes a switch from hypoinflammatory to hyperinflammatory reactivity. Am. J. Pathol. 181:174–184. 10.1016/j.ajpath.2012.03.015 [DOI] [PubMed] [Google Scholar]
- 47.Lee BO, Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Makris M, Sprague F, Lund FE, Randall TD. 2005. CD4 T cell-independent antibody response promotes resolution of primary influenza infection and helps to prevent reinfection. J. Immunol. 175:5827–5838. 10.4049/jimmunol.175.9.5827 [DOI] [PubMed] [Google Scholar]
- 48.Xu W, Santini PA, Matthews AJ, Chiu A, Plebani A, He B, Chen K, Cerutti A. 2008. Viral double-stranded RNA triggers Ig class switching by activating upper respiratory mucosa B cells through an innate TLR3 pathway involving BAFF. J. Immunol. 181:276–287. 10.4049/jimmunol.181.1.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolf AI, Mozdzanowska K, Quinn WJ, III, Metzgar M, Williams KL, Caton AJ, Meffre E, Bram RJ, Erickson LD, Allman D, Cancro MP, Erikson J. 2011. Protective antiviral antibody responses in a mouse model of influenza virus infection require TACI. J. Clin. Invest. 121:3954–3964. 10.1172/JCI57362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kang SM, Yoo DG, Kim MC, Song JM, Park MK, Quan O EFS, Akira S, Compans RW. 2011. MyD88 plays an essential role in inducing B cells capable of differentiating into antibody-secreting cells after vaccination. J. Virol. 85:11391–11400. 10.1128/JVI.00080-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tezuka H, Abe Y, Iwata M, Takeuchi H, Ishikawa H, Matsushita M, Shiohara T, Akira S, Ohteki T. 2007. Regulation of IgA production by naturally occurring TNF/iNOS-producing dendritic cells. Nature 448:929–933. 10.1038/nature06033 [DOI] [PubMed] [Google Scholar]
- 52.Habibzay M, Saldana JI, Goulding J, Lloyd CM, Hussell T. 2012. Altered regulation of Toll-like receptor responses impairs antibacterial immunity in the allergic lung. Mucosal Immunol. 5:524–534. 10.1038/mi.2012.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishikawa H, Sasaki H, Fukui T, Fujita K, Kutsukake E, Matsumoto T. 2012. Mice with asthma are more resistant to influenza virus infection and NK cells activated by the induction of asthma have potentially protective effects. J. Clin. Immunol. 32:256–267. 10.1007/s10875-011-9619-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morokata T, Ishikawa J, Ida K, Yamada T. 1999. C57BL/6 mice are more susceptible to antigen-induced pulmonary eosinophilia than BALB/c mice, irrespective of systemic T helper 1/T helper 2 responses. Immunology 98:345–351. 10.1046/j.1365-2567.1999.00890.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takeda K, Hamelmann E, Joetham A, Shultz LD, Larsen GL, Irvin CG, Gelfand EW. 1997. Development of eosinophilic airway inflammation and airway hyperresponsiveness in mast cell-deficient mice. J. Exp. Med. 186:449–454. 10.1084/jem.186.3.449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brewer JP, Kisselgof AB, Martin TR. 1999. Genetic variability in pulmonary physiological, cellular, and antibody responses to antigen in mice. Am. J. Respir. Crit. Care Med. 160:1150–1156. 10.1164/ajrccm.160.4.9806034 [DOI] [PubMed] [Google Scholar]
- 57.Heymann PW, Carper HT, Murphy DD, Platts-Mills TA, Patrie J, McLaughlin AP, Erwin EA, Shaker MS, Hellems M, Peerzada J, Hayden FG, Hatley TK, Chamberlain R. 2004. Viral infections in relation to age, atopy, and season of admission among children hospitalized for wheezing. J. Allergy Clin. Immunol. 114:239–247. 10.1016/j.jaci.2004.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murray CS, Poletti G, Kebadze T, Morris J, Woodcock A, Johnston SL, Custovic A. 2006. Study of modifiable risk factors for asthma exacerbations: virus infection and allergen exposure increase the risk of asthma hospital admissions in children. Thorax 61:376–382. 10.1136/thx.2005.042523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adamko DJ, Yost BL, Gleich GJ, Fryer AD, Jacoby DB. 1999. Ovalbumin sensitization changes the inflammatory response to subsequent parainfluenza infection. Eosinophils mediate airway hyperresponsiveness, m(2) muscarinic receptor dysfunction, and antiviral effects. J. Exp. Med. 190:1465–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagarkar DR, Bowman ER, Schneider D, Wang Q, Shim J, Zhao Y, Linn MJ, McHenry CL, Gosangi B, Bentley JK, Tsai WC, Sajjan US, Lukacs NW, Hershenson MB. 2010. Rhinovirus infection of allergen-sensitized and -challenged mice induces eotaxin release from functionally polarized macrophages. J. Immunol. 185:2525–2535. 10.4049/jimmunol.1000286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Q, Miller DJ, Bowman ER, Nagarkar DR, Schneider D, Zhao Y, Linn MJ, Goldsmith AM, Bentley JK, Sajjan US, Hershenson MB. 2011. MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness. PLoS Pathog. 7:e1002070. 10.1371/journal.ppat.1002070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Domachowske JB, Dyer KD, Bonville CA, Rosenberg HF. 1998. Recombinant human eosinophil-derived neurotoxin/RNase 2 functions as an effective antiviral agent against respiratory syncytial virus. J. Infect. Dis. 177:1458–1464. 10.1086/515322 [DOI] [PubMed] [Google Scholar]
- 63.Kumar RK, Foster PS. 2002. Modeling allergic asthma in mice: pitfalls and opportunities. Am. J. Respir. Cell Mol. Biol. 27:267–272. 10.1165/rcmb.F248 [DOI] [PubMed] [Google Scholar]
- 64.McMillan SJ, Lloyd CM. 2004. Prolonged allergen challenge in mice leads to persistent airway remodelling. Clin. Exp. Allergy 34:497–507. 10.1111/j.1365-2222.2004.01895.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pearce MB, Belser JA, Houser KV, Katz JM, Tumpey TM. 2011. Efficacy of seasonal live attenuated influenza vaccine against virus replication and transmission of a pandemic 2009 H1N1 virus in ferrets. Vaccine 29:2887–2894. 10.1016/j.vaccine.2011.02.014 [DOI] [PubMed] [Google Scholar]
- 66.Sun K, Ye J, Perez DR, Metzger DW. 2011. Seasonal FluMist vaccination induces cross-reactive T cell immunity against H1N1 (2009) influenza and secondary bacterial infections. J. Immunol. 186:987–993. 10.4049/jimmunol.1002664 [DOI] [PubMed] [Google Scholar]
- 67.Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. 1995. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 374:546–549. 10.1038/374546a0 [DOI] [PubMed] [Google Scholar]
- 68.Yi AK, Chang M, Peckham DW, Krieg AM, Ashman RF. 1998. CpG oligodeoxyribonucleotides rescue mature spleen B cells from spontaneous apoptosis and promote cell cycle entry. J. Immunol. 160:5898–5906 [PubMed] [Google Scholar]
- 69.Davis HL, Weeratna R, Waldschmidt TJ, Tygrett L, Schorr J, Krieg AM. 1998. CpG DNA is a potent enhancer of specific immunity in mice immunized with recombinant hepatitis B surface antigen. J. Immunol. 160:870–876 [PubMed] [Google Scholar]
- 70.Corne JM, Marshall C, Smith S, Schreiber J, Sanderson G, Holgate ST, Johnston SL. 2002. Frequency, severity, and duration of rhinovirus infections in asthmatic and non-asthmatic individuals: a longitudinal cohort study. Lancet 359:831–834. 10.1016/S0140-6736(02)07953-9 [DOI] [PubMed] [Google Scholar]
- 71.van Elden LJ, Sachs AP, van Loon AM, Haarman M, van de Vijver DA, Kimman TG, Zuithoff P, Schipper PJ, Verheij TJ, Nijhuis M. 2008. Enhanced severity of virus associated lower respiratory tract disease in asthma patients may not be associated with delayed viral clearance and increased viral load in the upper respiratory tract. J. Clin. Virol. 41:116–121. 10.1016/j.jcv.2007.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Juhn YJ, Kita H, Yawn BP, Boyce TG, Yoo KH, McGree ME, Weaver AL, Wollan P, Jacobson RM. 2008. Increased risk of serious pneumococcal disease in patients with asthma. J. Allergy Clin. Immunol. 122:719–723. 10.1016/j.jaci.2008.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Talbot TR, Hartert TV, Mitchel E, Halasa NB, Arbogast PG, Poehling KA, Schaffner W, Craig AS, Griffin MR. 2005. Asthma as a risk factor for invasive pneumococcal disease. N. Engl. J. Med. 352:2082–2090. 10.1056/NEJMoa044113 [DOI] [PubMed] [Google Scholar]