

ABSTRACT
Diacylglycerol acyltransferase-1 (DGAT1) is involved in the assembly of hepatitis C virus (HCV) by facilitating the trafficking of the HCV core protein to the lipid droplet. Here, we abrogated DGAT1 expression in Huh-7.5 cells by using either the transcription activator-like effector nuclease (TALEN) or lentivirus vector short hairpin RNA (shRNA) and achieved complete long-term silencing of DGAT1. HCV entry was severely impaired in DGAT1-silenced Huh-7.5 cell lines, which showed markedly diminished claudin-1 (CLDN1) expression. In DGAT1-silenced cell lines, the forced expression of CLDN1 restored HCV entry, implying that the downregulation of CLDN1 is a critical factor underlying defective HCV entry. The expression of the gene coding for hepatocyte nuclear factor 4α (HNF4α) and other hepatocyte-specific genes was also reduced in DGAT1-silenced cell lines. After DGAT1 gene rescue, CLDN1 expression was preserved, and HCV entry was restored. Strikingly, after DGAT1 silencing, CLDN1 expression and HCV entry were also restored by low-dose palmitic acid treatment, indicating that the downregulation of CLDN1 was associated with altered fatty acid homeostasis in the absence of DGAT1. Our findings provide novel insight into the role of DGAT1 in the life cycle of HCV.
IMPORTANCE In this study, we report the novel effect of complete silencing of DGAT1 on the entry of HCV. DGAT1 was recently reported as a host factor of HCV, involved in the assembly of HCV by facilitating the trafficking of the HCV core protein to lipid droplets. We achieved complete and long-term silencing of DGAT1 by either TALEN or repeated transduction of lentivirus shRNA. We found that HCV entry was severely impaired in DGAT1-silenced cell lines. The impairment of HCV entry was caused by CLDN1 downregulation, and the expression of HNF4α and other hepatocyte-specific genes was also downregulated in DGAT1-silenced cell lines. Our results suggest new roles of DGAT1 in human liver-derived cells: maintaining intracellular lipid homeostasis and affecting HCV entry by modulating CLDN1 expression.
INTRODUCTION
Hepatitis C virus (HCV) infection poses a major threat to human health, with a prevalence of more than 160 million people worldwide (1). In addition to a combination regimen of pegylated interferon alpha (IFN-α) and ribavirin, drugs acting directly on HCV have now been developed, although these direct-acting antivirals may prompt the emergence of resistant strains (2, 3). Our increasing understanding of the HCV-host interactions is allowing novel therapeutic approaches to be developed that modulate host factors that are required for viral entry, replication, and egress; these factors may have a higher genetic barrier to viral resistance (4).
Diacylglycerol acyltransferase-1 (DGAT1) is one of two known DGAT enzymes catalyzing the final step in triglyceride biosynthesis (5, 6). The expression of DGAT1 and its physiologic role differ in humans and mice. In mice, DGAT1 is expressed in many organs, including the skeletal muscle, heart, and intestines, but it is barely expressed in the liver (5). Because DGAT1-deficient mice are resistant to diet-induced obesity (7), pharmacological DGAT1 inhibitors are being developed for the treatment of metabolic diseases (8). In contrast to mice, human DGAT1 is mainly expressed in the small intestine and liver (9). Human DGAT1 reesterifies partial glycerides to triglycerides using exogenous fatty acids. In addition, intracellular lipid homeostasis is dysregulated in human hepatocytes in the absence of DGAT1 (10).
As a host factor interacting with HCV, DGAT1 has drawn attention for its role in trafficking the HCV core protein to lipid droplets (11). In addition, the same study reported that treatment with a DGAT1 inhibitor blocked the association of the HCV core protein with lipid droplets and decreased the production of infectious HCV virions (11). Further research has demonstrated that DGAT1 is involved in the localization of the HCV NS5A protein to lipid droplets and promotes NS5A interaction with the HCV core protein (12). However, in these reports, DGAT1 inhibitors were mainly used to block DGAT1 activity, and the observation was limited to as late as 72 h after treatment with DGAT1 inhibitors.
In the present study, we investigated the effects of complete, long-term silencing of DGAT1 on the whole life cycle of HCV. We established DGAT1 knockdown cell lines and a DGAT1 knockout (KO) cell line and observed that the entry of HCV into DGAT1-silenced cells was impaired. Furthermore, we identified the underlying mechanism of defective HCV entry into these cell lines.
MATERIALS AND METHODS
Cell culture.
Huh-7.5 cells (Apath, LLC, Brooklyn, NY) (13), HepG2 cells (ATCC, Manassas, VA), Caco-2 cells (ATCC, Manassas, VA), and 293TN cells (System Biosciences, Mountain View, CA) were maintained at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (WelGENE, Daegu, South Korea), 4.5 g/liter glucose, l-glutamine, and 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA). Short hairpin RNA (shRNA)-transduced cells were maintained in complete medium containing 1 μg/ml puromycin (Sigma-Aldrich, St. Louis, MO), and shRNA-resistant DGAT1-transfected cells were cultured in complete medium that was supplemented with 1 μg/ml puromycin and 1 mg/ml G418 (A.G. Scientific, San Diego, CA). All of the transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA).
Generation and infection of JFH-1 HCVcc.
The Japanese fulminant hepatitis 1 (JFH-1) strain (genotype 2a) (14) of cell-culture-derived HCV (HCVcc) (15–17) was generated and propagated as previously described (18). For the titration of HCV infectivity, a colorimetric focus-forming assay was performed as previously described (19). Cell cultures were inoculated with JFH-1 HCVcc at a multiplicity of infection (MOI) of 0.1 to 0.5.
HCVpp entry assay.
For the evaluation of HCV entry, HCV pseudoparticles (HCVpp) with E1 and E2 glycoproteins of various HCV genotypes (H77, genotype 1a; TH, genotype 1b; JFH-1, genotype 2a; and J6CF, genotype 2a) were generated, and cell cultures were inoculated as previously described (20, 21). Virus pseudoparticles with vesicular stomatitis virus glycoproteins (VSV-G) were used as positive controls.
Preparation and transduction of shRNA-DGAT1-expressing lentiviruses.
Three bacterial glycerol stocks with lentiviral shRNA constructs expressing validated sequences of DGAT1 (shRNA-DGAT1) and one stock with lentiviral constructs expressing validated sequences of DGA2 (shRNA-DGAT2) were purchased from Sigma-Aldrich. The sequences that are recognized by these shRNAs are presented in Fig. S1 in the supplemental material. A control shRNA vector (shRNA-control) encoding a scrambled sequence was also prepared. Lentiviral vectors comprising VSV-G plasmid, gag-pol plasmid, and a MISSION shRNA plasmid were transfected into 293TN cells using Lipofectamine 2000 (Invitrogen). After 48 h of transfection, lentiviral particles were harvested, passed through a 0.45-μm-pore syringe filter, concentrated with WelProt virus concentration reagent (WelGENE), and stored at −70°C. To maximize transduction efficiency, cells were repeatedly transduced with concentrated lentivirus. After 48 h of transduction, the cells were treated with 1 μg/ml puromycin and expanded over 3 to 6 weeks in puromycin-containing selection medium until they showed a stable phenotype. For the forced expression of CLDN1, shRNA-DGAT1-transduced cell lines were transfected with a plasmid-encoding CLDN1 gene cloned from Huh-7 cells and regulated by an EF-1α promoter.
Generation of DGAT1-KO cell line using TALEN.
The target sequence of the DGAT1-targeting transcription activator-like effector nuclease (TALEN) was TGCCATCGCCTGCAGGATTC (left)-TTTATTCAGCTCT (spacer)-GACAGTGGCTTCAGCAACTA (right). The plasmid encoding DGAT1-targeting TALEN was prepared as recently described (22) and provided by Toolgen, Seoul, Republic of Korea (http://www.talenlibrary.net/) (order no. H157465). Huh-7.5 cells were cotransfected with a total of 8 μg DNA comprising 2 μg of plasmid encoding one member of the DGAT1-targeting TALEN, 2 μg of plasmid encoding another member of the TALENs, and 4 μg of the reporter encoding H-2Kk (23) via electroporation using the Invitrogen Neon transfection system (at a voltage of 1,100 V for 30 ms, using a 100-ml tip and one pulse). The transfected cells were cultured for 1 day at 37°C and for 2 days at 30°C (cold shock) (24) and subjected to magnetic separation as previously described (23). Briefly, trypsinized cells were mixed with magnetic bead-conjugated antibodies against H-2Kk (MACSelect Kk microbeads; Miltenyi Biotech, Germany) and incubated for 20 min at 4°C. Labeled cells were separated using a MACS LS column (Miltenyi Biotech). The DGAT1 mutant cells that were enriched by this magnetic separation were plated into 96-well plates at a density of 0.25 cell/well. Two weeks after plating, only single-cell-derived colonies of a circular shape were manually selected under a microscope. Genomic DNA was isolated using the DNeasy blood and tissue kit (Qiagen, Valencia, CA). We analyzed 62 single-cell-derived colonies using the T7E1 enzyme digestion assay, which selectively detects heteroduplex DNA that results from gene editing by TALEN. We also analyzed these colonies by sequencing. The region of the DGAT1 gene containing the TALEN target site was amplified by nested PCR (primer sequence, 1st PCR, forward, 5′-CAAGCTCCATGTAGGTCCAG-3′, and reverse, 5′-CTCTCCTGGGATCCAATG-3′; 2nd PCR, forward, 5′-TCTGACCCTGACATGCTCGT-3′, and reverse, 5′-CCAATGGGAAGCAGCAAGTA-3′), cloned into the T-Blunt vector, and sequenced.
Flow cytometry.
For the detection of HCV receptors on the cell surface, the following antibodies were used: phycoerythrin (PE)-conjugated mouse monoclonal anti-CD81 IgG1 (clone JS-81; BD Biosciences, San Jose, CA), mouse monoclonal anti-CLDN1 (clone 7A5; developed by DNA immunization of the CLDN1 expression construct), rabbit polyclonal anti-SR-B1 (clone H-180; BD Biosciences), and rabbit polyclonal anti-occludin (anti-OCLN) (Sigma-Aldrich). For the staining of OCLN, the cells were fixed and permeabilized using the Foxp3 staining kit (eBioscience, San Diego, CA). PE-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was used as a secondary antibody for the SR-B1 and OCLN staining. For the intracellular staining of DGAT1, mouse monoclonal anti-DGAT1 IgG1 (A-5; Santa Cruz Biotechnology, Inc.) was used as a primary antibody, and allophycocyanin (APC)-conjugated anti-mouse IgG (BD Biosciences) was used as a secondary antibody. For the intracellular staining of the HCV core protein, mouse monoclonal anti-HCV core IgG1 (clone C7-50; Thermo Scientific/Affinity BioReagents, Rockford, IL) was used as a primary antibody, and fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG1 (clone A85-1; BD Biosciences) was used as a secondary antibody. For the intracellular staining, the cells were fixed and permeabilized using the Foxp3 staining kit (eBioscience) and then stained with the primary antibody for 20 min at 4°C. After a washing step with permeabilization buffer (eBioscience), the cells were stained with the appropriate secondary antibodies. The LSR II instrument (BD Biosciences) was used for the flow cytometry. The FlowJo software (TreeStar, Ashland, OR) was used for data analysis.
Immunoblots.
Cell lysates were prepared, and 20 μg of each lysate was loaded onto SDS-PAGE gels and analyzed by immunoblotting. The antibodies that were used for the immunoblots were as follows: mouse monoclonal anti-DGAT1 IgG1 (clone A-5; Santa Cruz Biotechnology, Inc.), rabbit polyclonal anti-DGAT1 (H-255; Santa Cruz Biotechnology, Inc.), rabbit polyclonal anti-DGAT2 (Sigma-Aldrich), rabbit polyclonal anti-CLDN1 (Abcam, Cambridge, United Kingdom), rabbit polyclonal anti-hepatocyte nuclear factor 4α (anti-HNF4α) (H-171; Santa Cruz Biotechnology, Inc.), and mouse monoclonal anti-tubulin (clone B-5-1-2; Sigma-Aldrich). Following the blotting, the membrane was incubated with primary antibodies (diluted at 1:200 to 1:1,000) overnight at 4°C or for 1 h at room temperature. The signal was detected using the horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) with enhanced chemiluminescence reagents (GE Healthcare/Amersham, Buckinghamshire, United Kingdom).
Confocal microscopy.
The antibodies that were used for the confocal microscopy imaging were as follows: mouse monoclonal anti-claudin-1 (anti-CLDN1) (clone 1C5-D9; Abcam), rabbit polyclonal anti-occludin (Invitrogen), Cy3-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA), and Alexa Fluor 488-conjugated donkey anti-rabbit IgG (BioLegend, San Diego, CA). The cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.5% (vol/vol) Triton X-100 for 10 min. The slides were incubated with primary antibodies (diluted at 1:100) for 3 h at room temperature. After being washed with phosphate-buffered saline three times, they were incubated with secondary antibodies (diluted at 1:1,000). Following the addition of mounting solution, images were acquired using an LSM-510 confocal laser-scanning microscope (Carl Zeiss, Jena, Germany).
Intracellular lipid droplet quantification.
Cells were fixed with 2% paraformaldehyde for 20 min on slides and permeabilized with 0.5% (vol/vol) Triton X-100 for 10 min. The slides were incubated in phosphate-buffered saline (PBS) containing 10 μM BODIPY (boron-dipyrromethene) lipid probe 493/503 (Invitrogen) for 1 h at room temperature. After the addition of mounting solution, images were acquired using an LSM-510 confocal laser-scanning microscope (Carl Zeiss). For the flow cytometry analysis, control and DGAT1-silenced cells were pelleted and resuspended in fluorescence-activated cell sorter (FACS) buffer (PBS with 2% fetal bovine serum [FBS], 2 mM EDTA). Then, cells were fixed and permeabilized using a Foxp3 staining kit (eBioscience). After permeabilization, the cells were resuspended with FACS buffer containing 10 μM BODIPY lipid probe 493/503 and incubated for 30 min in the dark. Next, the cells were washed in FACS buffer, pelleted, and resuspended in FACS buffer. Up to 50,000 events were detected using the LSR II instrument (BD Biosciences) for flow cytometry. FlowJo software (TreeStar) was used for the data analysis.
RNA extraction, cDNA synthesis, and real-time qPCR.
Total RNA isolation, cDNA synthesis, and TaqMan real-time quantitative PCR (qPCR) were performed as previously described (18). In brief, total RNA was isolated with the RNeasy minikit (Qiagen), and cDNA was synthesized using an Applied Biosystems high-capacity cDNA synthesis kit (Applied Biosystems, Foster City, CA). TaqMan gene expression assays (Applied Biosystems) were used to determine the mRNA levels of the target genes. For the titration of intracellular HCV RNA copies, the sequences of the primers and probes that were used are as follows (25): forward, CGG GAG AGC CAT AGT GG-3; reverse, AGT ACC ACA AGG CCT TTC G; and probe, 6-carboxyfluorescein (FAM)-CTG CGG AAC CGG TGA GTA CAC-6-carboxytetramethylrhodamine (TAMRA). The results were standardized to an endogenous control, β-actin.
Palmitic acid treatment and cell culture.
Cells were incubated in medium containing 10% charcoal-stripped fetal bovine serum (WelGENE) that was supplemented with 50 μM palmitic acid (Sigma-Aldrich) or dimethyl sulfoxide (DMSO) in complexes with 1% bovine serum albumin (BSA). The medium was changed every 2 days for the continuous treatment.
Cell proliferation and viability assay.
The cell proliferation activities were assessed using the BD Biosciences apoptosis, DNA damage, and cell proliferation kit according to the manufacturer's protocol. A 1 mM concentration of bromodeoxyuridine (BrdU) was incorporated into the control and DGAT1-silenced cell lines for 3 h. For the flow cytometry analysis, control and DGAT1-silenced cells were pelleted and resuspended in FACS buffer. Then, the cells were fixed and permeabilized, treated with DNase, stained with peridinin chlorophyll protein (PerCP)-Cy5.5-conjugated mouse anti-BrdU (BD Biosciences), and incubated for 30 min in the dark. Next, cells were washed in FACS buffer, pelleted, and resuspended in FACS buffer. Up to 50,000 events were detected using the LSR II instrument (BD Biosciences). FlowJo software (TreeStar) was used for the data analysis. For the cell viability assay, the cells were fixed and permeabilized, stained with PE-conjugated mouse anti-cleaved poly(ADP-ribose) polymerase (PARP) (Asp214) antibody, and incubated for 30 min in the dark. Data were obtained using the LSR II instrument (BD Biosciences).
ChIP.
Chromatin immunoprecipitation (ChIP) assays were performed according to the manufacturer's instructions (Pierce agarose ChIP kit; Pierce, Rockford, IL). Approximately 1 × 106 cells were used for each immunoprecipitation. Whole-cell lysates were immunoprecipitated using 1 μg of rabbit anti-HNF4α (H171) (Santa Cruz Biotechnology, Inc.) or IgG. Real-time qPCR was used to amplify the region near the HNF4α binding site in the promoter of the CLDN1 gene (26, 27). Reactions were performed in triplicate in three independent experiments, and the means were normalized to 1% of the chromatin input.
Statistical analyses.
Data are presented as means ± standard errors of the means (SEM). Unpaired t tests or two-tailed Mann-Whitney U tests were performed. All of the statistical analyses were conducted using GraphPad Prism version 5.01 (GraphPad Software, San Diego, CA). A P value of <0.05 was considered statistically significant.
RESULTS
Establishment of DGAT1-silenced Huh-7.5 cell lines using either TALEN or lentivirus vector shRNA.
To identify the effects of complete, long-term silencing of DGAT1 on the whole life cycle of HCV in Huh-7.5 cells, we knocked down DGAT1 expression by lentivirus vector shRNA transduction or knocked out the DGAT1 gene using a pair of TALENs in these cells. Three stable cell lines (shRNA-DGAT1-1666, shRNA-DGAT1-1183, and shRNA-DGAT1-1402) were established by the repeated transduction of three different lentiviruses carrying shRNA (see Fig. S1 in the supplemental material) and expanded in puromycin-containing selection medium for more than 2 months. For the more complete silencing of DGAT1 without residual expression, we established a DGAT1-knockout cell line (DGAT1-TALEN) using TALENs. To isolate cells in which the TALEN-knocked out DGAT1, we transfected Huh-7.5 cells with plasmids encoding a pair of DGAT1-targeting TALENs in combination with a reporter plasmid. The reporter plasmid expressed H-2Kk when a pair of DGAT1-targeting TALENs functioned successfully in the transfected cells. After transfection, we selected H-2Kk-expressing cells using magnetic bead-conjugated anti-H-2Kk antibodies. We analyzed 62 single-cell-derived colonies using a T7E1 enzyme digestion assay, which selectively detects heteroduplex DNA resulting from gene editing by TALEN (Fig. 1A), and then we chose a DGAT1-knockout cell line harboring a shifted open reading frame or premature stop codon in the DGAT1 gene (Fig. 1B). In the cell lines with DGAT1 that was silenced by either shRNA or TALEN, we verified the loss of DGAT1 expression at both the mRNA and protein levels. The expression of DGAT1 mRNA in the shRNA-DGAT1-transduced cell lines was less than 20% of that in a control cell line (shRNA-control), and DGAT1 mRNA was not detected in the DGAT1-TALEN cell line (Fig. 1C). The DGAT1 protein was deficient in all of the DGAT1-silenced cell lines (Fig. 1D).
FIG 1.

Establishment of DGAT1-silenced cell lines using DGAT1-TALEN or lentivirus vector shRNA DGAT1. (A) Huh-7.5 cells were transfected with plasmids encoding a pair of DGAT1-targeting TALENs in combination with the reporter encoding H-2Kk. After the magnetic sorting of cells with TALEN-driven mutations, we analyzed 62 single-cell-derived colonies using the T7E1 assay. The region of the DGAT1 gene containing the TALEN target site was amplified by nested PCR and visualized by agarose gel electrophoresis. Representative data from the T7E1 assay are presented here. Circled numbers indicate clones that were selected for DGAT1 gene sequencing. (B) DNA sequences of the DGAT1-TALEN cell line. The sequence presented here is part of the DGAT1 gene of clone 13, one of the clones that are presented in panel A. Zinc finger nuclease recognition sites are underlined, and deleted bases are indicated by dashes. TGA, the premature stop codon, is shaded in gray. Clone 13 has a shifted open reading frame (ORF) or premature stop codon in the DGAT1 gene. (C) DGAT1 mRNA levels were determined by real-time quantitative PCR in DGAT1-silenced cell lines and standardized to β-actin mRNA levels (n = 3). N.D., not detected. (D) DGAT1 protein expression in DGAT1-silenced cell lines was assessed by immunoblotting. (E) Intracellular lipid droplet staining was performed using the BODIPY lipid probe 493/503 in the control and DGAT1-silenced cell lines. The scale bar represents 20 μm. The right panel represents flow cytometry analyses of the same cell lines.
We compared several basic characteristics of the DGAT1-silenced cell lines with those of the control cell line. For example, triglyceride-rich intracellular lipid droplets were barely detectable in the DGAT1-silenced cell lines, whereas they were abundant in the control cell line (Fig. 1E). Cell proliferation and apoptosis were also analyzed by BrdU incorporation and cleaved PARP staining, respectively. Cell proliferation rates were higher in the DGAT1-silenced cell lines than in the control cell line, whereas apoptotic death was not altered (data not shown).
HCV entry is impaired in DGAT1-silenced Huh-7.5 cell lines.
To identify the effects of DGAT1 silencing on the whole life cycle of HCV, we inoculated DGAT1-silenced Huh-7.5 cell lines with JFH-1 cell culture-produced HCV (HCVcc) and measured infectious virions in the culture supernatants. In accordance with the role of DGAT1 in the assembly of HCV, infectious virions were not detected in the culture supernatants of the DGAT1-silenced cells (Fig. 2A). We also quantified intracellular HCV RNA copies and unexpectedly discovered that the intracellular HCV RNA titer was very low in the DGAT1-silenced cell lines (Fig. 2B). Following inoculation of the DGAT1-silenced cell lines with a higher dose of JFH-1, we detected no intracellular HCV core protein by flow cytometry (Fig. 2C).
FIG 2.

HCV entry into DGAT1-silenced Huh-7.5 cell lines is impaired. (A and B) DGAT1-silenced cell lines were inoculated with JFH-1 HCVcc at a multiplicity of infection (MOI) of 0.1. After 72 h, infectious HCV virions were quantified in culture supernatants by a colorimetric focus-forming assay (19). The data are presented as focus-forming units (FFU) per ml of culture supernatant (n = 3) (A). Intracellular HCV RNA levels were determined by real-time quantitative PCR and standardized to β-actin mRNA levels (n = 3) (B). (C) DGAT1-silenced cell lines were inoculated with JFH-1 HCVcc at an MOI of 0.5. After 60 h, intracellular HCV core proteins were detected by flow cytometry. Data are representative of two independent experiments. (D and E) DGAT1-silenced cell lines were transfected with 5 μg in vitro-transcribed JFH1 RNA. After 72 h, infectious HCV virions were quantified in culture supernatants by a colorimetric focus-forming assay (19). The data are presented as focus-forming units (FFU) per ml of culture supernatant (n = 3) (D). Intracellular HCV RNA levels were determined by real-time quantitative PCR and standardized to β-actin mRNA levels (n = 3) (E). (F) HCV entry into DGAT1-silenced cell lines was assessed using HCVpp harboring E1 and E2 glycoproteins of various HCV genotypes. Virus pseudoparticles harboring the vesicular stomatitis virus G (VSV-G) envelope glycoprotein were used as a positive control. HCVpp entry was determined by luciferase activity. Data are expressed as percentages of the shRNA-control cell line (n = 3). Bar graphs represent means ± SEM. *, P < 0.001 compared to the control. N.D., not detected.
Next, we transfected the control and DGAT1-silenced cell lines with JFH-1 RNA. As expected from the previous study (11) and loss of intracellular lipid droplets in the DGAT1-silenced cell lines, the production of infectious virions was severely impaired in the DGAT1-silenced cell lines (Fig. 2D). The intracellular HCV RNA copy number was also lower in the DGAT1-silenced cell lines than in the control cell line, with a difference of less than 1 log (Fig. 2E), indicating that HCV RNA replication is slightly impaired in DGAT1-silenced cells. However, this minor decrease is in contrast to the enormous decrease that was observed in the HCVcc inoculation study (Fig. 2B). This finding indicates that another factor is the major cause of the very low intracellular HCV RNA levels in HCVcc-inoculated, DGAT1-silenced cells.
With the hypothesis that HCV entry is defective in DGAT1-silenced cell lines, we evaluated HCV entry using HCVpp of various genotypes. No HCVpp entered the DGAT1-silenced cell lines (Fig. 2F), whereas the control virus pseudoparticles with VSV-G did enter these lines, indicating that HCV entry is impaired in the absence of DGAT1.
Claudin-1 expression is downregulated in DGAT1-silenced cell lines.
Because of the impaired HCV entry into DGAT1-silenced cell lines, we investigated the expression of host proteins participating in HCV entry, including tetraspanin CD81 (28), the high-density lipoprotein receptor scavenger receptor class B type I (SR-BI) (29), and two tight junction proteins, claudin-1 (CLDN1) (30) and occludin (OCLN) (31). In the flow cytometric analysis, the expression of CD81, SR-BI, and OCLN in DGAT1-silenced cell lines was well preserved, but that of CLDN1 was markedly diminished (Fig. 3A and B). We observed the downregulation of CLDN1 expression at the mRNA level by real-time quantitative PCR (Fig. 3C). Immunoblotting confirmed the downregulation of CLDN1 expression (Fig. 3D). Notably, CLDN1 expression was not downregulated by the silencing of DGAT2, the other DGAT protein present in human hepatocytes (Fig. 3E). Immunofluorescence staining revealed that CLDN1 expression on the cell membrane was lost in the DGAT1-silenced cells (Fig. 3F). Additionally, a significant proportion of OCLN was localized not only to the cell membrane but also to the cytoplasm in these cells (Fig. 3F). We further confirmed downregulation of CLDN1 at the mRNA and protein levels under DGAT1-silencing conditions in another hepatoma cell line, HepG2, following DGAT1 silencing by lentivirus shRNA transduction (Fig. 3G).
FIG 3.

CLDN1 expression is downregulated in DGAT1-silenced cell lines. (A and B) The expression of CD81, SR-B1, CLDN1, and OCLN in DGAT1-silenced cell lines was analyzed by flow cytometry. Histograms are representative of three independent experiments (A), and the mean fluorescence index (F/F0) of the histogram is presented (B). F, mean fluorescence intensity (MFI) of specific antibody staining; F0, MFI of isotype control antibody staining. Data are representative of three independent experiments. (C) mRNA levels of CD81, SR-B1, CLDN1, and OCLN in DGAT1-silenced cell lines were determined by real-time quantitative PCR and standardized to β-actin mRNA levels (n = 3). (D) CLDN1 protein expression in DGAT1-silenced cell lines was assessed by immunoblotting. (E) Immunoblot analysis was performed to evaluate CLDN1 expression in the control and DGAT2 knockdown Huh-7.5 cell line after 40 days of shRNA lentiviral transduction. Data are representative of two independent experiments. (F) Immunofluorescence staining was performed using anti-OCLN (green) and anti-CLDN1 (red) in Caco-2, shRNA-control, and DGAT1-silenced Huh-7.5 cell lines, and images were analyzed by confocal microscopy. Merged images are also presented. The scale bar represents 20 μm. (G) A stably DGAT1-silenced HepG2 cell line was established by transduction with a lentivirus harboring shRNA-DGAT1-1666. The mRNA levels of DGAT1 and CLDN1 were determined by real-time quantitative PCR (n = 3) (left graph), and the protein expression levels of DGAT1 and CLDN1 were assessed by immunoblotting (right blots). Bar graphs represent means ± SEM. * and **, P < 0.001 and P < 0.01, respectively, compared to the control.
Forced expression of CLDN1 restores HCV entry to DGAT1-silenced cell lines.
To verify whether the downregulation of CLDN1 expression led to HCV entry impairment, we transfected DGAT1-silenced cell lines with the CLDN1 gene and evaluated HCV entry using HCVpp. After 72 h of the transfection, CLDN1 expression was observed by immunoblotting (Fig. 4A) and flow cytometry (Fig. 4B). Importantly, the entry of all of the HCVpp was restored in the CLDN1-expressing DGAT1-silenced cells (Fig. 4C). These results demonstrate that CLDN1 downregulation is responsible for impairment of HCV entry into DGAT1-silenced cells and that the reexpression of CLDN1 is sufficient to restore HCV entry into these cells.
FIG 4.

Forced expression of CLDN1 restores HCV entry to DGAT1-silenced cell lines. (A) CLDN1 protein expression was assessed by immunoblotting after mock or CLDN1 transfection of DGAT1-silenced cell lines. (B) Surface expression of CLDN1 was analyzed by flow cytometry after mock or CLDN1 transfection of DGAT1-silenced cell lines. Data are representative of three independent experiments. (C) HCV entry was assessed after mock or CLDN1 transfection of DGAT1-silenced cell lines using HCVpp of various genotypes. Virus pseudoparticles harboring the VSV-G envelope glycoprotein served as a positive control. HCVpp entry was determined by luciferase activity. Data are expressed as percentages of the shRNA-control cell line (n = 4). The statistical analysis was conducted for differences between the mock-transfected and CLDN1-transfected cells. Bar graphs represent means ± SEM. *, P < 0.001.
Expression of hepatocyte-specific genes is reduced in DGAT1-silenced cell lines.
In DGAT1-silenced cells, we examined the expression of hepatocyte nuclear factor 4α (HNF4α) because this factor has been reported to be associated with CLDN1 expression in mice (32–34). In fact, HNF4α expression was markedly reduced in DGAT1-silenced cell lines at both the mRNA and protein levels (Fig. 5A). HNF4α is a central regulator of hepatocyte differentiation and function (35, 36), and the expression of other hepatocyte-specific genes, such as the albumin and α1-antitrypsin genes, was also decreased in the DGAT1-silenced cells (Fig. 5B), suggesting that their differentiation status had been altered by DGAT1 silencing. However, HNF4α expression was not downregulated by the silencing of DGAT2 (Fig. 5C). We also examined temporal changes in DGAT1, HNF4α, and CLDN1 expression during the establishment of a stable cell line following lentivirus shRNA-DGAT1 transduction into Huh-7.5 cells. DGAT1 expression was silenced at 10 days following transduction, whereas the expression of HNF4α and CLDN1 was preserved (Fig. 5D). HNF4α expression then decreased gradually, followed by a slow decrease in CLDN1 expression. Ultimately, the expression of both HNF4α and CLDN1 was markedly diminished at 45 days following transduction (Fig. 5D).
FIG 5.

Expression of hepatocyte-specific genes is reduced in DGAT1-silenced cell lines. (A) HNF4α mRNA levels in DGAT1-silenced cell lines were determined by real-time quantitative PCR (n = 3) (upper graph), and HNF4α protein expression was assessed by immunoblotting (lower blots). (B) mRNA expression of representative hepatic differentiation markers in control and DGAT1-silenced cell lines. Data are standardized to β-actin mRNA levels (n = 3). Bar graphs represent means ± SEM. *, P < 0.001 compared to the control. (C) Immunoblot analysis was performed to evaluate HNF4α expression in control and DGAT2 knockdown Huh-7.5 cell lines after 40 days of lentivirus shRNA transduction. Data are representative of two independent experiments. (D) Huh-7.5 cells were transduced with lentivirus harboring shRNA-DGAT1-1402, and temporal changes in protein expression were examined during the establishment of a stable cell line. The cell pellets were harvested at days 10, 25, 32, and 45 after transduction and subjected to immunoblot analyses to detect DGAT1, HNF4α, and CLDN1. Data are representative of two independent experiments.
To verify whether DGAT1 silencing directly caused the downregulation of HNF4α and CLDN1, we transfected shRNA-DGAT1-transduced cells with the shRNA-resistant DGAT1 gene 10 days after the transduction. Following the transfection, DGAT1 expression was restored, and expression of HNF4α and CLDN1 was maintained (Fig. 6A). In addition, intracellular lipid droplets were restored by transfection (Fig. 6B). Furthermore, HCVpp entry was recovered by the restoration of DGAT1 expression (Fig. 6C). Next, we examined the requirement for the catalytic activity of DGAT1 for the maintenance of CLDN1 and HNF4α expression. We constructed a catalytically inactive DGAT1 mutant gene (H415A) (11, 37) that was also shRNA resistant. Unlike the catalytically active DGAT1, the catalytically inactive form did not maintain the expression of CLDN1 and HNF4α (Fig. 6D). Collectively, these results indicate that DGAT1 silencing causes HNF4α and CLDN1 downregulation and HCV entry impairment and that the loss of DGAT1 catalytic activity is important in this process.
FIG 6.

Catalytically active DGAT1 is responsible for maintaining CLDN1 expression and the hepatocyte-specific phenotype in Huh-7.5 cells. (A) An shRNA-1402-resistant DGAT1 gene was constructed by introducing 6 point mutations into the sequence that is recognized by the shRNA-1402 oligonucleotide without altering the amino acid sequence. The shRNA-1402-resistant DGAT1 gene was transfected into shRNA-DGAT1-1402-harboring cells 10 days after transduction, and temporal changes in protein expression were examined. The cell pellets were harvested at days 10, 31, and 45 after transduction with lentivirus harboring shRNA-DGAT1-1402 (days 0, 21, and 35 after the shRNA-1402-resistant DGAT1 transfection) and were subjected to immunoblot analysis to detect DGAT1, HNF4α, and CLDN1. Data are representative of two independent experiments. (B) Intracellular lipid droplet staining was performed using the BODIPY lipid probe 493/503 in cells that had been transfected with mock or shRNA-resistant DGAT1. The scale bar represents 20 μm. (C) HCV entry into DGAT1-silenced cell lines was assessed after mock or shRNA-resistant DGAT1 transfection using JFH1 HCVpp. Data are expressed as percentages of the pretransfected cell line (n = 3). Bar graphs represent means ± SEM. *, P < 0.001. (D) Immunoblot analysis of DGAT1-silenced cell lines harboring a catalytically active or inactive DGAT1 gene was performed. A catalytically inactive DGAT1-encoding plasmid was constructed by site-directed mutagenesis, replacing the histidine residue at position 415 (encoded by CAU) with alanine (encoded by GCU), and the catalytically inactive DGAT1 gene was also shRNA resistant. We confirmed the sequence of the mutated construct, which was then used to transfect Huh-7.5 cells on 10 days after DGAT1 silencing. Data are representative of two independent experiments. (E, F) The chromatin immunoprecipitation assay was performed using anti-HNF4α antibody. In the schematic of the CLDN1 promoters, the putative HNF4α binding site is shaded in gray, and the positions of the primers for real-time qPCR are designated with arrows (E). Real-time qPCR analysis was performed using DNA precipitated by rabbit anti-HNF4α (clone number H171) (Santa Cruz Biotechnology, Inc.) or IgG. Transthyretin (TTR) was used as a positive control (F). *, P < 0.01 (n = 3). (G) Real-time qPCR analysis of HNF4α and CLDN1 expression in the Huh-7.5 cell line was conducted after siHNF4α treatment. Data are standardized to β-actin mRNA levels (n = 3). Bar graphs represent means ± SEM. *, P < 0.001. (H) Immunoblot analysis of HNF4α and CLDN1 in the Huh-7.5 cell line was performed after siHNF4α treatment. Data are representative of two independent experiments. (I) Immunoblot analysis of HNF4α and CLDN1 in DGAT1-silenced cell lines was performed at 72 h after transfection with HNF4α1 plasmid. Data are representative of two independent experiments.
As HNF4α was reported to upregulate CLDN1 expression in mice (32, 33), we examined whether HNF4α downregulation was responsible for the CLDN1 downregulation that was observed in the DGAT1-silenced cells. In fact, HNF4α bound to the promoter region of human CLDN1 in the ChIP assay (Fig. 6E and F). However, HNF4α silencing by siRNA (siHNF4α) did not significantly decrease CLDN1 expression (Fig. 6G and H). We also transfected DGAT1-silenced cells with the HNF4α gene. For the transfection, we used the gene encoding HNF4α1, which is a major isoform in adult human hepatocytes (38, 39). However, CLDN1 expression was not restored by this transfection (Fig. 6I). Taking these findings together, we conclude that CLDN1 expression is not regulated solely by HNF4α but also by other factors that are influenced by DGAT1 silencing.
Exogenous palmitic acid treatment prevents downregulation of CLDN1 and HNF4α expression after DGAT1 silencing.
Considering that DGAT1 regulates intracellular lipid homeostasis (10) and our data showing that intracellular lipid droplets are depleted in DGAT1-silenced cells (Fig. 1E), we hypothesized that CLDN1 downregulation might be attributed to a dysregulation of intracellular fatty acid metabolism. Therefore, we examined the effect of exogenous fatty acid treatment following DGAT1 silencing. We applied BSA-conjugated palmitic acid (C16:0) at a low concentration to shRNA-DGAT1-transduced cells 10 days after the transduction and continued the treatment for 17 days. The expression of CLDN1 and HNF4α was preserved by palmitic acid treatment, although DGAT1 expression was silenced (Fig. 7A). Importantly, HCVpp entry was also recovered by palmitic acid treatment (Fig. 7B). These results indicate that altered fatty acid homeostasis in DGAT1-silenced cells is associated with CLDN1 downregulation, which leads to HCV entry impairment.
FIG 7.
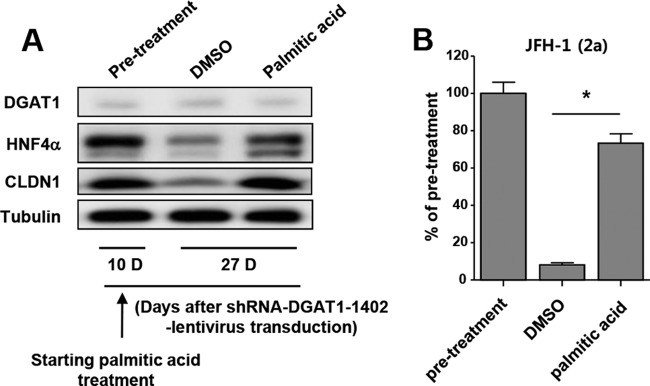
Exogenous palmitic acid treatment prevents downregulation of CLDN1 and HNF4α expression after DGAT1 silencing. (A) From day 10 after transduction with lentiviruses harboring shRNA-DGAT1-1402, the transduced cells were cultured in medium that had been supplemented with 10% charcoal-stripped fetal bovine serum and 1% bovine serum albumin with either 50 μM palmitic acid or DMSO. The cell pellets were harvested at days 10 and 27 after transduction (days 0 and 17 after the palmitic acid treatment) and subjected to immunoblot analysis to detect DGAT1, HNF4α, and CLDN1. Data are representative of two independent experiments. (B) HCV entry (of JFH1 HCVpp) into DGAT1-silenced cell lines was assessed after DMSO or palmitic acid treatment. Data are expressed as percentages of the values for the pretreatment cell lines (n = 3). Bar graphs represent means ± SEM. *, P < 0.001.
DISCUSSION
In the present study, we attempted to identify the effects of complete, long-term silencing of DGAT1 on the whole life cycle of HCV in Huh-7.5 cells. We demonstrated that CLDN1 downregulation impairs HCV entry into DGAT1-silenced cell lines and that this effect is associated with altered fatty acid homeostasis. However, these findings were made using the Huh-7.5 human hepatoma cell line and need to be confirmed in primary human hepatocytes.
One of the main roles of DGAT1 is in catalyzing the final step of triglyceride biosynthesis (5, 6). However, studies of knockout mice have shown that DGAT1 deficiency does not result in elevated levels of diacylglycerol or acyl coenzyme A (acyl-CoA) in the liver, heart, muscle, and adipose tissue following the ingestion of high-fat diets (7, 40). These data suggest that DGAT1 does not simply catalyze the final step of triglyceride biosynthesis but is involved in the regulation of intracellular lipid homeostasis. In fact, DGAT1 recycles the products of triglyceride hydrolysis, which are partial glycerides, for triglyceride synthesis. In addition, DGAT1 inhibition decreases the intracellular lipid pool comprised of triglycerides, partial glycerides, and free fatty acids in human liver-derived cells (10, 41). In the present study, a low-dose exogenous palmitic acid treatment prevented the downregulation of CLDN1 and HNF4α expression in DGAT1-silenced cells. These results show that expression of CLDN1 and HNF4α is regulated by altered intracellular lipid homeostasis in DGAT1-silenced cells and that the restoration of intracellular fatty acid levels by exogenous palmitic acid rescued CLDN1 and HNF4α expression.
We also made a DGAT2-silenced cell line in this study. However, the expression of CLDN1 and HNF4α was not downregulated in the DGAT2-silenced cells (Fig. 3E and 5C). DGAT2 also catalyzes the final step of triglyceride synthesis, but recent reports suggest that DGAT1 and DGAT2 play different roles in human hepatocytes (10). DGAT2 is involved in the de novo synthesis of triglycerides, whereas DGAT1 functions in the reesterification of partial glycerides that are generated by intracellular lipolysis. Moreover, DGAT2 encompasses 30% of the total DGAT activity in human hepatocytes, whereas DGAT1 contributes the other 70% (10).
HNF4α is a central regulator of hepatocyte differentiation and function (33, 34) and is a major transcription factor that regulates the expression of genes that are involved in lipid homeostasis (42). HNF4α is also known to regulate the expression of proteins that are required for hepatic and intestinal tight junction assembly, including CLDN1, in mice (30–32). A recent study demonstrated the binding of HNF4α to the CLDN1 gene by an electrophoretic mobility shift assay and ChIP assay of the mouse fetal liver (30). In accordance with this previous report, our present data showed that HNF4α bound to the promoter region of human CLDN1 in a ChIP assay (Fig. 6E and F). However, HNF4α silencing did not significantly decrease CLDN1 expression (Fig. 6G and H), and HNF4α1 transfection did not restore CLDN1 expression in DGAT1-silenced cells (Fig. 6I). Taking these findings together, we conclude that CLDN1 expression is not regulated solely by HNF4α but also by other factors that are influenced by DGAT1 silencing.
A previous study has shown that DGAT1 is involved in the assembly of HCV by facilitating the trafficking of the HCV core protein to the lipid droplet (11). The authors suppressed DGAT1 activity by using a DGAT1 inhibitor or silenced DGAT1 expression by shRNA, but they observed no impairment of HCV entry to Huh-7.5 cells. The discrepancy between the studies may be explained by the duration of DGAT1 inhibition or silencing. The earlier study involved the use of the DGAT inhibitor for a short duration of approximately 72 h. In contrast, we used stable DGAT1-silenced cell lines to study the long-term effects of DGAT1 silencing. In fact, CLDN1 expression decreased gradually in shRNA-DGAT1-transduced cells and was markedly downregulated only after long-term culturing for over 45 days (Fig. 5D). In the present study, we also knocked out the DGAT1 gene by using the TALEN technique to silence DGAT1 expression completely and found no DGAT1 expression in the DGAT1-TALEN cell line (Fig. 1C and D). Interestingly, DGAT1-TALEN transfection facilitated CLDN1 downregulation compared to shRNA-DGAT1 transduction in the process of stable cell line establishment (data not shown). CLDN1 downregulation may be induced only by the complete silencing of DGAT1, which could be achieved by a long-term culture of shRNA-DGAT1-transduced cells or by highly efficient silencing with DGAT1-TALEN.
In the present study, we found that HCV RNA replication was also slightly impaired in DGAT1-silenced cell lines (Fig. 2E). This impairment can be partially attributed to the loss of lipid droplets in these cells (Fig. 1E). In fact, recent studies have emphasized the association of lipid droplets and HCV RNA replication (43). Further research is required to clarify the manner by which HCV RNA replication is influenced by intracellular fatty acid homeostasis.
In conclusion, our data demonstrate that complete, long-term silencing of DGAT1 causes CLDN1 downregulation and thus inhibits HCV entry and that CLDN1 downregulation is associated with altered fatty acid homeostasis in the absence of DGAT1. Future research is needed to elucidate the exact mechanisms and consequences of CLDN1 downregulation in DGAT1-silenced hepatocytes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a National Agenda Project grant from the Korea Research Council of Fundamental Science and Technology (NTM1311423), the Korea Research Institute for Bioscience and Biotechnology (KRIBB) Initiative Program (KGM3121423), and by the Project of Global Ph.D. Fellowship (NRF-2012H1A2A1012809, to P.S.S.) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning of Korea. A.M. and T.K. were supported by a grant-in-aid from the Japan Society for the Promotion of Science and from the Ministry of Health, Labor, and Welfare of Japan. This work was partly supported by the KAIST (Korea Advanced Institute of Science and Technology) Future Systems Healthcare Project from the Ministry of Science, ICT, and Future Planning of Korea.
Footnotes
Published ahead of print 4 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01428-14.
REFERENCES
- 1.Lavanchy D. 2011. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 17:107–115. 10.1111/j.1469-0691.2010.03432.x [DOI] [PubMed] [Google Scholar]
- 2.Liang TJ, Ghany MG. 2013. Current and future therapies for hepatitis C virus infection. N. Engl. J. Med. 368:1907–1917. 10.1056/NEJMra1213651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aloia AL, Locarnini S, Beard MR. 2012. Antiviral resistance and direct-acting antiviral agents for HCV. Antivir. Ther. 17:1147–1162. 10.3851/IMP2426 [DOI] [PubMed] [Google Scholar]
- 4.Zeisel MB, Lupberger J, Fofana I, Baumert TF. 2013. Host-targeting agents for prevention and treatment of chronic hepatitis C—perspectives and challenges. J. Hepatol. 58:375–384. 10.1016/j.jhep.2012.09.022 [DOI] [PubMed] [Google Scholar]
- 5.Cases S, Smith SJ, Zheng YW, Myers HM, Lear SR, Sande E, Novak S, Collins C, Welch CB, Lusis AJ, Erickson SK, Farese RV., Jr 1998. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. U. S. A. 95:13018–13023. 10.1073/pnas.95.22.13018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yen CL, Stone SJ, Koliwad S, Harris C, Farese RV., Jr 2008. Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 49:2283–2301. 10.1194/jlr.R800018-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith SJ, Cases S, Jensen DR, Chen HC, Sande E, Tow B, Sanan DA, Raber J, Eckel RH, Farese RV., Jr 2000. Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat. Nat. Genet. 25:87–90. 10.1038/75651 [DOI] [PubMed] [Google Scholar]
- 8.Devita RJ, Pinto S. 3 September 2013. Current status of the research and development of diacylglycerol O-acyltransferase 1 (DGAT1) inhibitors. J. Med. Chem. 10.1021/jm4007033 [DOI] [PubMed] [Google Scholar]
- 9.Haas JT, Winter HS, Lim E, Kirby A, Blumenstiel B, DeFelice M, Gabriel S, Jalas C, Branski D, Grueter CA, Toporovski MS, Walther TC, Daly MJ, Farese RV., Jr 2012. DGAT1 mutation is linked to a congenital diarrheal disorder. J. Clin. Invest. 122:4680–4684. 10.1172/JCI64873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wurie HR, Buckett L, Zammit VA. 2012. Diacylglycerol acyltransferase 2 acts upstream of diacylglycerol acyltransferase 1 and utilizes nascent diglycerides and de novo synthesized fatty acids in HepG2 cells. FEBS J. 279:3033–3047. 10.1111/j.1742-4658.2012.08684.x [DOI] [PubMed] [Google Scholar]
- 11.Herker E, Harris C, Hernandez C, Carpentier A, Kaehlcke K, Rosenberg AR, Farese RV, Jr, Ott M. 2010. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 16:1295–1298. 10.1038/nm.2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Camus G, Herker E, Modi AA, Haas JT, Ramage HR, Farese RV, Jr, Ott M. 2013. Diacylglycerol acyltransferase-1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J. Biol. Chem. 288:9915–9923. 10.1074/jbc.M112.434910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014. 10.1128/JVI.76.24.13001-13014.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kato T, Miyamoto M, Furusaka A, Date T, Yasui K, Kato J, Matsushima S, Komatsu T, Wakita T. 2003. Processing of hepatitis C virus core protein is regulated by its C-terminal sequence. J. Med. Virol. 69:357–366. 10.1002/jmv.10297 [DOI] [PubMed] [Google Scholar]
- 15.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. 10.1126/science.1114016 [DOI] [PubMed] [Google Scholar]
- 16.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796. 10.1038/nm1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299. 10.1073/pnas.0503596102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park J, Kang W, Ryu SW, Kim WI, Chang DY, Lee DH, Park DY, Choi Y-H, Choi K, Shin E-C, Choi C. 2012. Hepatitis C virus infection enhances TNFalpha-induced cell death via suppression of NF-kappaB. Hepatology 56:831–840. 10.1002/hep.25726 [DOI] [PubMed] [Google Scholar]
- 19.Kang W, Shin EC. 2012. Colorimetric focus-forming assay with automated focus counting by image analysis for quantification of infectious hepatitis C virions. PLoS One 7:e43960. 10.1371/journal.pone.0043960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197:633–642. 10.1084/jem.20021756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsumura T, Kato T, Sugiyama N, Tasaka-Fujita M, Murayama A, Masaki T, Wakita T, Imawari M. 2012. 25-Hydroxyvitamin D3 suppresses hepatitis C virus production. Hepatology 56:1231–1239. 10.1002/hep.25763 [DOI] [PubMed] [Google Scholar]
- 22.Kim Y, Kweon J, Kim A, Chon JK, Yoo JY, Kim HJ, Kim S, Lee C, Jeong E, Chung E, Kim D, Lee MS, Go EM, Song HJ, Kim H, Cho N, Bang D, Kim S, Kim JS. 2013. A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol. 31:251–258. 10.1038/nbt.2517 [DOI] [PubMed] [Google Scholar]
- 23.Kim H, Kim MS, Wee G, Lee CI, Kim H, Kim JS. 2013. Magnetic separation and antibiotics selection enable enrichment of cells with ZFN/TALEN-induced mutations. PLoS One 8:e56476. 10.1371/journal.pone.0056476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doyon Y, Choi VM, Xia DF, Vo TD, Gregory PD, Holmes MC. 2010. Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat. Methods 7:459–460. 10.1038/nmeth.1456 [DOI] [PubMed] [Google Scholar]
- 25.Takeuchi T, Katsume A, Tanaka T, Abe A, Inoue K, Tsukiyama-Kohara K, Kawaguchi R, Tanaka S, Kohara M. 1999. Real-time detection system for quantification of hepatitis C virus genome. Gastroenterology 116:636–642. 10.1016/S0016-5085(99)70185-X [DOI] [PubMed] [Google Scholar]
- 26.Fang B, Mane-Padros D, Bolotin E, Jiang T, Sladek FM. 2012. Identification of a binding motif specific to HNF4 by comparative analysis of multiple nuclear receptors. Nucleic Acids Res. 40:5343–5356. 10.1093/nar/gks190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Runkle EA, Rice SJ, Qi J, Masser D, Antonetti DA, Winslow MM, Mu D. 2012. Occludin is a direct target of thyroid transcription factor-1 (TTF-1/NKX2–1). J. Biol. Chem. 287:28790–287801. 10.1074/jbc.M112.367987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941. 10.1126/science.282.5390.938 [DOI] [PubMed] [Google Scholar]
- 29.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21:5017–5025. 10.1093/emboj/cdf529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. 10.1038/nature05654 [DOI] [PubMed] [Google Scholar]
- 31.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886. 10.1038/nature07684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Battle MA, Konopka G, Parviz F, Gaggl AL, Yang C, Sladek FM, Duncan SA. 2006. Hepatocyte nuclear factor 4alpha orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proc. Natl. Acad. Sci. U. S. A. 103:8419–8424. 10.1073/pnas.0600246103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walesky C, Gunewardena S, Terwilliger EF, Edwards G, Borude P, Apte U. 2013. Hepatocyte-specific deletion of hepatocyte nuclear factor-4α in adult mice results in increased hepatocyte proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 304:G26–G37. 10.1152/ajpgi.00064.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong W, Zhao Y, McClain CJ, Kang YJ, Zhou Z. 2010. Inactivation of hepatocyte nuclear factor-4α mediates alcohol-induced downregulation of intestinal tight junction proteins. Am. J. Physiol. Gastrointest. Liver Physiol. 299:G643–G651. 10.1152/ajpgi.00515.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolotin E, Liao H, Ta TC, Yang C, Hwang-Verslues W, Evans JR, Jiang T, Sladek FM. 2010. Integrated approach for the identification of human hepatocyte nuclear factor 4alpha target genes using protein binding microarrays. Hepatology 51:642–653. 10.1002/hep.23357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tirona RG, Lee W, Leake BF, Lan LB, Cline CB, Lamba V, Parviz F, Duncan SA, Inoue Y, Gonzalez FJ, Schuetz EG, Kim RB. 2003. The orphan nuclear receptor HNF4alpha determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat. Med. 9:220–224. 10.1038/nm815 [DOI] [PubMed] [Google Scholar]
- 37.Hofmann K. 2000. A superfamily of membrane-bound O-acyltransferases with implications for wnt signaling. Trends Biochem. Sci. 25:111–112. 10.1016/S0968-0004(99)01539-X [DOI] [PubMed] [Google Scholar]
- 38.Nakhei H, Lingott A, Lemm I, Ryffel GU. 1998. An alternative splice variant of the tissue specific transcription factor HNF4alpha predominates in undifferentiated murine cell types. Nucleic Acids Res. 26:497–504. 10.1093/nar/26.2.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Babeu JP, Boudreau F. 2014. Hepatocyte nuclear factor 4-alpha involvement in liver and intestinal inflammatory networks. World J. Gastroenterol. 20:22–30. 10.3748/wjg.v20.i1.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu L, Yu S, Khan RS, Ables GP, Bharadwaj KG, Hu Y, Huggins LA, Eriksson JW, Buckett LK, Turnbull AV, Ginsberg HN, Blaner WS, Huang LS, Goldberg IJ. 2011. DGAT1 deficiency decreases PPAR expression and does not lead to lipotoxicity in cardiac and skeletal muscle. J. Lipid Res. 52:732–744. 10.1194/jlr.M011395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wurie HR, Buckett L, Zammit VA. 2011. Evidence that diacylglycerol acyltransferase 1 (DGAT1) has dual membrane topology in the endoplasmic reticulum of HepG2 cells. J. Biol. Chem. 286:36238–36247. 10.1074/jbc.M111.251900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. 2001. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 21:1393–1403. 10.1128/MCB.21.4.1393-1403.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka T, Kuroda K, Ikeda M, Wakita T, Kato N, Makishima M. 2013. Hepatitis C virus NS4B targets lipid droplets through hydrophobic residues in the amphipathic helices. J. Lipid Res. 54:881–892. 10.1194/jlr.M026443 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.