

ABSTRACT
The ephrin receptor tyrosine kinase A2 (EphA2) is an entry receptor for Kaposi's sarcoma-associated herpesvirus (KSHV) that is engaged by the virus through its gH/gL glycoprotein complex. We describe here that natural ephrin ligands inhibit the gH/gL-EphA2 interaction. The effects of point mutations within EphA2 demonstrated that KSHV gH/gL interacts with EphA2 through a restricted set of the same residues that mediate binding of A-type ephrins. Two previously described inhibitors of the EphA2 interaction with ephrin A5 also inhibited binding of KSHV gH/gL to EphA2. The more potent of the two compounds inhibited KSHV infection of blood vessel and lymphatic endothelial cells in the micromolar concentration range. Our results demonstrate that interaction of KSHV with EphA2 occurs in a fashion similar to that of the natural ephrin ligands. Our data further indicate a new avenue for drug development against KSHV.
IMPORTANCE Our study reports two important findings. First, we show that KSHV engages its receptor, the receptor tyrosine kinase EphA2, at a site that overlaps the binding site of the natural ephrin ligands. Second, we demonstrate that KSHV infection of target cells can be blocked by a small-molecule inhibitor of the viral glycoprotein-EphA2 interaction. These findings represent a novel avenue for the development of strategies to treat KSHV-associated diseases.
INTRODUCTION
Not only is the Kaposi's sarcoma-associated herpesvirus (KSHV) the causative agent of Kaposi's sarcoma (KS) in humans (1–3), it is also found in most forms of primary effusion lymphoma (PEL) and multicentric Castleman's disease (MCD) (4, 5). KS is a relatively rare malignancy in the industrialized world, where it occurs mostly in immunocompromised individuals (AIDS KS and iatrogenic KS) (6); it also occurs in a milder form in elderly men (classic KS) (7). This contrasts with the situation in sub-Saharan Africa, where KS is among the leading types of cancer, even among small children (6, 8). A substantial portion of cases in Africa are not associated with the human immunodeficiency virus (HIV) epidemic (8, 9).
The extent to which continuous rounds of de novo infection of new cells are needed to sustain KS lesions or maintain growth-transformed B cells in PEL is currently not clear. The extent to which KSHV lytic genes are expressed in KS varies considerably with the stage and type of disease (10). Anti-KSHV therapy has been associated with sustained remission of PEL after chemotherapy (11), and ganciclovir therapy was found to reduce the signs and symptoms of MCD in parallel with reduction in KSHV loads in plasma (12). The latter finding most likely reflects the fact that a substantial portion of the cells in MCD support lytic viral replication (13, 14). However, neither cidofovir (15) nor valganciclovir (16) was successful in slowing the progression of KS in clinical trials. Thus, the ability of anti-KSHV agents to reverse, block, or slow disease progression may vary with the stage and nature of the disease.
De novo infection of cells by KSHV seems to be required for the establishment of KS. This can be deduced from the fact that the characteristic spindle cells of KS are mostly polyclonal in origin (17) and the observation that monoclonal KS lesions are only rarely found, mostly at later stages (18). Furthermore, treatment with the anti-herpesviral agent ganciclovir has been found to reduce the rate of new KS development (19, 20). Consequently, therapies that block infection of new cells could logically impact the maintenance or progression of KS and MCD.
EphA2 is a cellular receptor for the glycoprotein complex gH/gL of KSHV (21, 22). EphA2 is a member of the ephrin receptor tyrosine kinase family (Ephs). The 14 Ephs are classified into nine A- and five B-type Ephs according to their ligand specificity for the five A-type ephrins and three B-type ephrins (23). EphA2 was shown to be the principal receptor used for entry of KSHV into various types of cells, including fibroblasts, blood and lymphatic endothelial cells, and epithelial cells. Also, EphA2 is heavily overexpressed in KS lesions, on spindle cells and also in surrounding tissue (21). Thus, blocking EphA2 may prevent KSHV from infecting this potential pool of fresh target cells. KSHV also triggers activation of EphA2, a process that can be observed after stimulation with virus or with antibody-clustered recombinant gH/gL glycoprotein (21). The facts that EphA2 is upregulated in a wide variety of cancers (24) and that the Eph-ephrin signaling axis is deregulated in many malignancies (25) suggest the possibility that this receptor may not only be important for entry of KSHV into target cells but may also be involved in other ways in the complex biology of KS pathogenesis. Recently, it was shown that activation of EphA2 through ephrin A1 induces an inflammatory response in endothelial cells (26). Thus, EphA2 may be a rational target for intervention, not only based on its role in mediating KSHV entry, but also based on its inflammatory and cell-stimulatory properties.
MATERIALS AND METHODS
Cell culture, transfections, and virus.
293T cells and primary rhesus fibroblasts were cultured in Dulbecco's modified Eagle's Glutamax medium with 25 mM HEPES, high glucose (Invitrogen), and penicillin/streptomycin (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Invitrogen). Transfections were performed using JetPrime (Polyplus transfection) following the manufacturer's recommendations. Human umbilical endothelial cells (Lonza) and lymphatic endothelial cells (Promocell) were maintained in EGM-2 (Lonza). KSHV.219 and vesicular stomatitis virus (VSV) G-pseudotyped simian immunodeficiency virus mac239deltaNef (SIVmac239deltaNef)-green fluorescent protein (GFP) was prepared as described previously (22).
DNA constructs and proteins.
Expression plasmids for KSHV gH-Fc (22), gL (27), and gH and EphA2 (22) are described elsewhere. Soluble gH/gL complex was expressed in 293T cells through transfection of pcDNA6-based (Invitrogen) expression plasmids encoding codon-optimized (Genscript) KSHV gH and gL. The gH ectodomain (amino acids 1 to 704) was fused to the polypeptide IEGRTSAWSHPQFEKGGGSGGGSGGGSAWSHPQFEK, which includes a factor Xa site followed by a tandem Strep epitope. Expression plasmids for EphA2 mutants were purchased from Genscript. Recombinant gH-Fc was purified via a C-terminal tandem Strep tag from the supernatant of 293T cells transfected with expression plasmids for gH-Fc and gL at a 1:3 ratio. The supernatant was harvested 2 days after transfection and cleared by centrifugation at 3,000 × g and filtration through a 0.22-μm PES filter (Corning). Two hundred milliliters of cleared cell culture supernatant was then passed manually over 0.5 ml Streptactin (Qiagen) in a 10-ml OmniPrep (Bio-Rad) column, followed by washing with 50 ml phosphate-buffered saline (PBS). Protein was eluted in 3 mM desthiobiotin (Sigma-Aldrich) in PBS, aliquoted, and stored at −80°C. Purity was assessed by colloidal Coomassie staining (Fig. 1) (SafeStain; Invitrogen), and the protein concentration was determined by absorbance at 280 nm. Recombinant murine ephrin A1-Fc, ephrin A4-Fc, and ephrin A5-Fc and recombinant soluble EphA2 (amino acids 25 to 534) were purchased from R&D Systems. Fc fusion proteins become dimeric via dimerization of the Fc portion.
FIG 1.
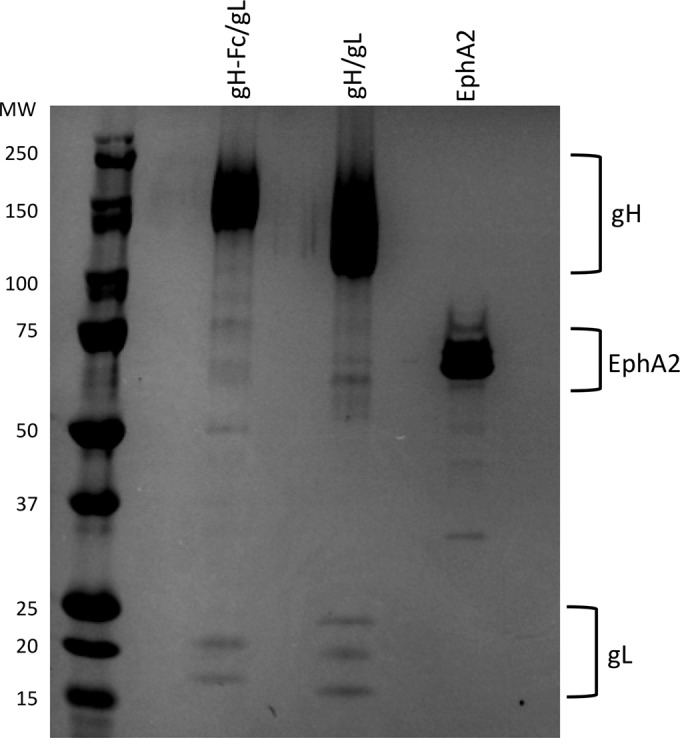
Recombinant proteins. Shown is colloidal Coomassie staining of gH-Fc/gL, gH/gL, and soluble EphA2 (R&D Systems) as used in our studies. gH-Fc/gL (0.93 μg), gH/gL (1.86 μg), and EphA2 (1.33 μg) were separated on an 8 to 16% gradient gel (Invitrogen), followed by staining with colloidal Coomassie (Safestain; Invitrogen). Note that the gL in the gH-Fc/gL complex runs slightly higher because of a C-terminal Flag epitope tag. gL runs in several different glycosylation forms.
ELISA.
Compounds 1 and 2 (Matrix Chemicals) were dissolved to 1 M in dimethyl sulfoxide (DMSO). The stocks were aliquoted and stored at −20°C. For further use, the compounds were prediluted in DMSO to 200 mM. Compounds 1 and 2 were diluted from 200 mM in DMSO to 2 mM in enzyme-linked immunosorbent assay (ELISA) buffer (10% FBS in PBS-T [PBS with 0.02% Tween 20] supplemented with 25 mM HEPES) (see Fig. 3B, C, and D) or to 1 mM (see Fig. 4A). DMSO was diluted accordingly in ELISA buffer for the “0 μM inhibitor”/DMSO control. The DMSO concentration was kept constant over all inhibitor concentrations by diluting further in the 0 μM inhibitor control. Half-well-size white 96-well cell culture-treated plates (Fig. 2B, 3B and C, and 4A) or radioimmunoassay (RIA)/ELISA polystyrene plates (Fig. 2C and 3D) (Becton Dickenson) were coated with recombinant EphA2 ectodomain (R&D Systems) or gH/gL at 2 μg/ml in PBS overnight (for the ELISAs in Fig. 3B and 4A, B, C, and D, only 1 μg/ml was used). No difference in half-maximum binding concentrations between plate types was observed (not shown). After three washes, the wells were blocked with ELISA buffer for 2 h. Incubation with proteins with or without inhibitors was performed for 2 h at room temperature in ELISA buffer. The plates were washed four times with ELISA buffer, followed by incubation with secondary detection reagent, either anti-human-horseradish peroxidase (HRP) (Southern Biotech) or Streptactin-HRP (IBA), 1:5,000 in ELISA buffer, for 2 h at room temperature, followed by four washes with PBS-T. Bound EphA2 ectodomain (R&D) was detected via its C-terminal 6-His epitope using HRP-conjugated THE His tag monoclonal antibody (Genscript) at 67 ng/ml. Finally, 40 μl of SuperSignal West Pico ECL solution (Pierce) was added to each well, and the luminescence was quantified with a Victor 3 plate reader (PerkinElmer).
FIG 3.

Binding of KSHV gH/gL to EphA2 can be competed with small-molecule inhibitors. (A) Chemical structures of compound 1 [4-(2,5-dimethyl-pyrrol-1-yl)-2-hydroxy-benzoic acid] and compound 2 [5-(2,5-dimethyl-pyrrol-1-yl)-2-hydroxy-benzoic acid]. (B) Binding of gH-Fc/gL at 1 μg/ml to immobilized EphA2 ectodomain in the presence of compound 1 and compound 2 at the indicated concentrations was measured by ELISA. (C) Binding of ephrin A4-Fc and ephrin A5-Fc at 1 μg/ml to immobilized EphA2 ectodomain in the presence of compound 1 and compound 2 at the indicated concentrations was measured as for panel B.
FIG 4.

KSHV gH/gL and ephrins bind EphA2 with nanomolar and subnanomolar affinity, resulting in different sensitivities to inhibition. (A) Binding of ephrin A4-Fc, ephrin A5-Fc, and gH-Fc/gL at various concentrations to immobilized EphA2 ectodomain in the presence of compound 1 at 1 mM (squares) or DMSO only (circles) was measured by ELISA. (B) Binding of gH-Fc/gL at increasing concentrations to immobilized EphA2 ectodomain measured in the absence (circles) or presence of compound 1 at 500 μM (squares) in the same fashion as for panel A. (C) Binding of the monoclonal antibody 6F8 to the EphA2 ectodomain at two different dilutions within the dynamic range of the binding reaction was measured as for panel B in the absence (gray bars) and presence of compound 1 at 500 μM (black bars). (D) Binding of soluble gH/gL to immobilized EphA2 or of soluble EphA2 to immobilized gH/gL was measured by ELISA in the presence of compound 1 at 1 mM or 2 mM or DMSO only. The error bars represent the standard errors of the mean.
FIG 2.

KSHV gH/gL binds to the ligand binding domain of EphA2, and the interaction can be competed with ephrins. (A) KSHV gH/gL interacts with the ligand binding domain of EphA2. 293T cells were transfected with expression constructs for the complete soluble EphA2 ectodomain (amino acids [aa] 1 to 534) or the ligand binding domain (aa 1 to 208), both fused C terminally to a hemagglutinin (HA) tag. Equal amounts of cell culture supernatants from cells expressing the complete EphA2 ectodomain or the ligand binding domain were mixed with equal amounts of supernatants from cells transfected with expression plasmids for either soluble gH-Fc alone or gH-Fc and gL, which leads to a soluble gH-Fc/gL complex. After immunoprecipitation (IP) of gH-Fc or gH-Fc/gL with protein G Sepharose beads, samples were analyzed by Western blotting. The input lanes were loaded with the supernatants from cells transfected with constructs encoding the EphA2 ectodomain (aa 1 to 534) or the ligand binding domain (aa 1 to 208). (B) KSHV gH/gL binding to EphA2 can be competed with soluble ephrins. An ELISA plate was coated with recombinant soluble EphA2 ectodomain. The plate was then incubated with gH-Fc/gL at 1 μg/ml in the presence of mephrin A1-Fc, ephrin A4-Fc, EGFR-Fc as a control, and EphA2 (the same protein used for coating) as an assay control, all at 5 μg/ml. Bound gH-Fc/gL was detected with Streptactin-HRP, specifically recognizing a C-terminal tandem Strep tag on gH-Fc that is not present on the two ephrin-Fc proteins. The error bars represent the standard errors of the mean. (C) Dose-dependent inhibition of gH-Fc/gL binding by ephrin A4-Fc and ephrin A5-Fc. Binding of gH-Fc/gL to immobilized EphA2 in the presence of various concentrations of ephrin A4-Fc or ephrin A5-Fc was measured as for panel B. Each data point represents a duplicate measurement.
Infections.
Compounds 1 and 2 were prediluted to a 200 mM solution in DMSO, from which they were diluted to 1 mM in cell culture medium. The DMSO was diluted accordingly for the 0 μM inhibitor control. The DMSO concentration was kept constant over all inhibitor concentrations by diluting further in the 0 μM inhibitor control. Cells were preincubated for 20 min with the inhibitor, followed by addition of virus in one-fifth of the final volume. For some infections (see Fig. 7A to C), the medium was not changed. In some infections (see Fig. 7D), the medium was changed to fresh DMEM with 10% FBS 24 h postinfection. Cells were harvested for analysis by flow cytometry 2 days postinfection.
FIG 7.

Small-molecule inhibitors of the gH/gL-EphA2 interaction inhibit entry of KSHV. (A) Inhibition of KSHV entry by two dimethylpyrrolyl benzoic acid derivatives (compounds 1 and 2). 293T cells were preincubated with compound 1 (diamonds) or compound 2 (×) at the indicated concentrations for 30 min before infection with rKSHV.219 (GFP reporter gene) in the continued presence of the inhibitor. The error bars represent the standard deviations; n = 3. (B) Inhibition of viral entry by compound 1 is specific for KSHV. 293T cells were infected as for panel A with GFP encoding rKSHV.219 (diamonds) or a VSV G-pseudotyped SIV239-GFP (circles) in the presence of compound 1 at the indicated concentrations. The error bars represent the standard deviations; n = 3. (C) 293T cells were infected with rKSHV.219 or a VSV G-pseudotyped SIVmac239-GFP as for panel A in the presence of the indicated concentrations of compound 1. The top row of each pair shows GFP fluorescence, the bottom row phase-contrast. The images were taken at ×100 magnification after 2 days. (D) Inhibition of rKSHV.219 infection by compound 1 on LEC, HUVEC, and rhesus fibroblasts. Infection and preincubation with different concentrations of compound 1 was carried out as for panel B. After 24 h, the medium was changed to medium without inhibitor, and the cells were cultured for an additional 24 h. The percentage of GFP-expressing cells was determined after 2 days (diamonds). Infection with VSV G-pseudotyped SIV239-GFP was performed in parallel to control for specificity (circles). EC50 values as calculated by linear approximation of the respective curve segments are given. The error bars represent the standard deviations; n = 3.
Immunoprecipitation.
293T cells were transfected with the indicated plasmids and harvested after 2 days. The cells were lysed in lysis buffer (1% NP-40, 150 mM NaCl, 1 mM EDTA, 25 mM HEPES, pH 7.4) with protease inhibitor cocktail (Pierce). Mouse monoclonal antibodies to myc and V5 epitope tags were purchased from Serotec. Lysates were incubated for 4 h with protein G beads (GE Healthcare) that had been preadsorbed with 0.5 ml of gH-Fc/gL-containing cell culture supernatant or 100 μg ephrin-Fc fusion proteins (R&D Systems) in 500 μl 10% FBS in DMEM. After three washes in lysis buffer, samples were subjected to Western blot analysis. For Fig. 5B, each mutant was expressed separately by transfection of a single well of a six-well plate. After lysis, each mutant was immobilized to protein G beads by immunoprecipitation with 1 μg of 9E10 anti-myc epitope monoclonal antibody. After aspiration of the cell lysate, 500 μl of a lysate from cells expressing KSHV gH (V5 epitope tagged) and gL was added, followed by 4 h of incubation and three washes with 1 ml of lysis buffer. Samples were analyzed by Western blotting. Band intensities were quantified with ImageGauge (Kodak). For the quantification of binding in Fig. 5B, the ratio of intensities for gH (V5) over each EphA2 mutant (myc) was calculated, and wild-type (wt) EphA2 was set to 1 (hence, there is no error bar for the wt). The experiment was repeated five times under identical conditions for each mutant. Averages and standard errors of the mean were calculated. The significance of reduction in binding was calculated using Student's one-tailed t test versus the control.
FIG 5.

KSHV gH/gL and ephrins interact with an overlapping set of residues on the surface of EphA2. (A) Immunoprecipitation of EphA2 point mutants with KSHV gH-Fc/gL, ephrin A4-Fc, or ephrin A5-Fc. The indicated EphA2 (myc epitope-tagged full-length EphA2) point mutants were recombinantly expressed in 293T cells. Cellular lysates were prepared and subjected to immunoprecipitation with protein G beads that were preadsorbed with either supernatant from gH-Fc/gL-expressing cells (top), 500 ng of recombinant ephrin A4-Fc (2nd from top), or ephrin A5-Fc (3rd from top) protein. Samples were analyzed by Western blotting as indicated. The expression level of each mutant in the lysate is shown in the bottom blots. (B) 293T cells were transfected with expression plasmids for the indicated EphA2 mutants (myc epitope tagged), and lysates were prepared 2 days after transfection. The myc epitope-tagged EphA2 mutants were immobilized to protein G Sepharose through immunoprecipitation with anti-myc monoclonal antibody. After aspiration of the lysate, equal amounts of cell lysate prepared from 293T cells that had been transfected with expression plasmids for KSHV gH (V5 epitope tagged) and gL were added to the immobilized EphA2 mutants. After 4 h of incubation, the samples were washed and analyzed by Western blotting. One representative Western blot is shown. The ratio of the signals for gH (V5) to EphA2 (myc) was calculated (bottom) from five independent experiments. The signal ratio for the wt was set to 1 for each independent experiment. Reductions in binding compared to the wt that reached significance (P < 0.05; Student's one-tailed t test) are marked with asterisks. The error bars represent the standard errors of the mean.
Molecule models.
Images of the EphA2 ectodomain structure were generated with the BALLview free software (www.BALLview.org; 28) using the 3MX0 structure of EphA2 complexed with ephrin A5 (29) downloaded from the RCSB Protein Data Bank. Alignments between EphA4 and EphA2 were generated using the BLAST algorithm (30).
RESULTS
Ephrins and KSHV gH/gL compete for binding to EphA2.
The ability of soluble ephrins to inhibit entry of KSHV into target cells (21, 22) raises the possibility that KSHV interacts with a region of EphA2 that is similar to the target region of the natural ephrin ligands. A construct comprising only the first 208 amino acids corresponding to the ligand binding domain of EphA2 (amino acids 1 to 208) as described by Himanen et al. (31) still bound KSHV gH/gL with efficiency equal to that of the full-length EphA2 ectodomain, as determined by immunoprecipitation and Western blotting (Fig. 2A). We further found that the KSHV gH/gL complex competes with natural ephrin ligands for binding to EphA2 in an ELISA with solid-phase immobilized EphA2 ectodomain (Fig. 2B). Murine ephrin (mephrin) A1-Fc and human ephrin A4-Fc, two ligands for EphA2 that were used previously to block entry of KSHV and that bind EphA2 (22), did specifically block binding of gH-Fc/gL to EphA2; a control protein (EGFR-Fc) did not alter binding of gH-Fc/gL. Competition with soluble EphA2, the same protein used for coating, reduced binding of gH-Fc/gL by 99%, indicating specific binding of gH-Fc/gL under the conditions of the ELISA. We also tested ephrin A4-Fc and ephrin A5-Fc in a dose-response experiment, and both proteins inhibited binding of gH/gL to immobilized EphA2 in a dose-dependent manner (Fig. 2C), with ephrin A4-Fc being more potent than ephrin A5-Fc.
Two small molecules inhibit binding of KSHV gH/gL to EphA2.
Based on these findings, we hypothesized that compounds that inhibit binding of ephrins to EphA2 might also inhibit binding of gH/gL. A number of chemical compounds were described by Noberini et al. that inhibit the interaction of EphA2 with ephrin A5 and of EphA4 with ephrin A5 (32). We tested the two most potent compounds. Compound 1 [4-(2,5-dimethyl-pyrrol-1-yl)-2-hydroxy-benzoic acid] and compound 2 [5-(2,5-dimethyl-pyrrol-1-yl)-2-hydroxy-benzoic acid] were purchased from Matrix Scientific; their structures are shown in Fig. 3A. Both were found to inhibit the interaction of gH-Fc/gL with EphA2 in our receptor binding ELISA (Fig. 3B). The 50% inhibitory concentration (IC50) values for inhibition of the gH-Fc/gL interaction with EphA2 were estimated to be 408 μM for compound 1 and 812 μM for compound 2, as calculated from the data in Fig. 3B; in contrast, the interactions of ephrin A4-Fc with EphA2 and of ephrin A5-Fc with EphA2 were much more resistant to inhibition by both compounds in the same assay. IC50s toward the ephrin A5-Fc–EphA2 interaction in our ELISA were 1,863 μM for compound 1, >2,000 μM for compound 2, and >2,000 μM for both compounds toward the ephrin A4-Fc–EphA2 interaction (calculated from the data shown in Fig. 3C). The greater sensitivity of the gH-Fc/gL–EphA2 interaction to inhibition is consistent with the poorer affinity of this interaction than ephrin A4-Fc–EphA2 or ephrin A5-Fc–EphA2, as described below. The IC50s reported here for inhibition of the ephrin A5-Fc–EphA2 interaction by compounds 1 and 2 are considerably higher than the IC50s of 65 μM and 33 μM reported by Noberini et at al. (32) for the EphA2-ephrin A5 interaction; however, very different assay conditions and reagent concentrations were used, and therefore, the IC50s from their study and ours are not directly comparable. We measured binding of ephrin-Fc to immobilized EphA2 (amino acids 25 to 534), whereas Noberini et al. (32) measured binding of soluble EphA2 (amino acids 1 to 219 fused to alkaline phosphatase) to immobilized ephrin A5-Fc.
KSHV gH/gL binds EphA2 with nanomolar affinity.
We next measured binding of gH-Fc/gL, ephrin A4-Fc, and ephrin A5-Fc to immobilized EphA2 in the presence and absence of compound 1 at various concentrations of the protein ligands (Fig. 4A and B). These measurements allowed us to calculate half-maximal binding concentrations, which we refer to here as K1/2max, for the three interactions. The strongest interaction with EphA2 was for ephrin A4-Fc, with a K1/2max of 0.01 μg/ml (0.1 nM). Next was ephrin A5-Fc, with a K1/2max of 0.06 μg/ml (0.6 nM). The weakest interaction with EphA2 was observed for gH-Fc/gL, with a K1/2max of 0.4 μg/ml (1.6 nM). This rank ordering is consistent with our previous finding that ephrin A4-Fc inhibits entry of KSHV with considerably greater potency than ephrin A5-Fc (22). Although binding constants of ephrins A4 and A5 for EphA2 have not to our knowledge been reported so far, other ephrin-Eph interactions exhibit binding constants in the nanomolar and subnanomolar range (33), affinities compatible with our findings. The results in Fig. 4A also confirmed the inhibitory activity of compound 1 and its greatest potency against the gH-Fc/gL–EphA2 interaction. We used the monoclonal antibody 6F8 to the extracellular domain of EphA2 as a control for specificity (Fig. 4C). Binding of this antibody within the dynamic range of the binding reaction was at most marginally affected by the presence of compound 1 at 500 μM.
Next, we used a soluble monomeric version of gH/gL lacking the Fc portion that leads to dimerization and measured binding to immobilized EphA2 (Fig. 4D, left). We also performed the reverse experiment using soluble EphA2 and measured binding to immobilized gH/gL (Fig. 4D, right). Both measurements were performed in the absence and presence of compound 1. Half-maximal binding of soluble gH/gL occurred at 0.9 μg/ml (9 nM) and half-maximal binding of soluble EphA2 at 0.16 μg/ml (2.9 nM). The value of 9 nM for soluble gH/gL is slightly higher than the value of 2.9 nM for soluble EphA2. It is likely that incomplete formation of complexes between gH and gL in the purified material contributed to the slightly higher value; gH without gL does not bind EphA2. Compound 1 again inhibited both binding reactions in a dose-dependent manner (Fig. 4D).
To test whether our results regarding the differing potencies of compound 1 for inhibition of binding of EphA2 to KSHV gH-Fc/gL and of EphA2 to ephrin A5-Fc are consistent with our affinity measurements and are in line with published data, we calculated Ki values of compound 1 toward EphA2 using the Cheng-Prusoff equation (34), with the half-maximal binding concentrations derived from Fig. 4A and B as a substitute for the dissociation constant. Using the formula Ki = IC50/(concentration/K1/2max + 1), this calculation with gH-Fc/gL results in an apparent Ki of compound 1 for EphA2 of 126 μM. For the same assay with ephrin A5-Fc (Fig. 3C, left), the same calculation yields an apparent Ki of 109 μM. Being derived from the inhibition of binding of two different ligands of EphA2, these values are highly consistent.
KSHV gH/gL binds a subset of the same residues of EphA2 that mediate binding of ephrin A4 and ephrin A5.
In order to examine the extent to which residues within EphA2 that are important for binding A-type ephrins and compound 1 may also be important for binding gH/gL, we generated a set of point mutations within EphA2 targeted to the region of binding of the small-molecule inhibitor as described by Qin et al. (35). In the study by Qin et al., compound 1 was found to interact with residues corresponding to EphA2 residues I58, M59, S68, F156, and E157 (Table 1). We selected 13 sites including and adjacent to these residues for mutagenesis. Either single amino acids or pairs were changed to alanine, D155 was changed to proline to kink the backbone (D155P), and one proline was changed to leucine (P109L) in order to introduce a steric hindrance like that present in EphB4 at this position. Most point mutations did not affect the gH/gL-EphA2 interaction as tested by immunoprecipitation in a first set of experiments. Only replacement of R103 with alanine severely abrogated the interaction; replacement of M55 seemed to slightly decrease binding of gH-Fc/gL to EphA2 (Fig. 5A, top). Binding to ephrin A4-Fc and ephrin A5-Fc was affected by mutations at several positions (Fig. 5A, second and third pairs of blots from top), among them M55, R103, I58 plus M59, D155, R159, and F156. For ephrin A5-Fc, we also observed decreased binding for mutations at positions M73 and P109.
TABLE 1.
Residues on EphA4 that were found to interact with compound 1 in solution by Qin et al
| EphA4 residuea | Corresponding EphA2 residueb | Interaction data for EphA4c |
|---|---|---|
| I31 | I58 | NMR + HADDOCK |
| M32 | M59 | NMR + HADDOCK |
| I39 | I64 | NMR |
| Q43 | S68 | NMR + HADDOCK |
| D123 | D148 | NMR |
| I131 | F156 | NMR + HADDOCK |
| G132 | E157 | NMR + HADDOCK |
As numbered in reference 35, starting at position 28 as the first amino acid.
Corresponding residues on EphA2 were determined by us using the BLAST algorithm and were confirmed visually in the structure to match the positions on EphA4.
From reference 35. Only residues that were identified by NMR and could be fitted into a computational docking model (nuclear magnetic resonance [NMR] plus HADDOCK) were considered real interactions by Qin et al.
The sites corresponding to the residues found by Qin et al. to interact with compound 1 are highlighted in the representation of the EphA2 crystal structure, viewed from the top, in Fig. 6A; the sites that we chose for mutagenesis are highlighted in Fig. 6B. Mutations affecting binding to ephrin A4 and ephrin A5 are highlighted in Fig. 6C and D, respectively. As we observed a clear effect on gH/gL binding only with the R103A substitution, we decided to also perform the reverse experiment in a semiquantitative fashion. We immobilized the EphA2 mutants with anti-myc monoclonal antibody on protein G Sepharose beads in an initial immunoprecipitation and then pulled down KSHV gH/gL from an equal amount of lysate with each mutant (Fig. 5B), followed by Western blotting. Bound gH was quantified densitometrically and normalized to the EphA2 signal. Apart from R103A and the R103A plus R159A double mutant (indicated in Fig. 5B with a black arrow), a modest reduction in the amount of bound gH/gL was detectable for replacement of M55 with alanine and S68 with alanine (highlighted in Fig. 5B with a gray arrow). In addition, the double mutant I58A plus M59A in this region of EphA2 also exhibited a significant reduction in bound gH/gL in this assay (also highlighted with a gray arrow). Mutants that exhibited a significant reduction in binding in our assay are marked with arrows and are highlighted in the structure in Fig. 6E. It must be noted that the effects of those changes were minor compared to the effect achieved by mutating R103. Nonetheless, there was significant overlap with respect to residues critical for gH-Fc/gL binding and residues critical for binding the ephrins and compound 1 (shown in Fig. 6).
FIG 6.

Structure of the extracellular domain of EphA2 viewed from the top. The crystal structure of the EphA2 extracellular domain is represented as a solvent-excluded surface. (A) Residues I58, M59, S68, F156, and E157 on EphA2 are highlighted in yellow. These residues correspond to the binding interface of compound 1 on EphA4 as determined by Qin et al. (B) All residues that were mutated for binding experiments are highlighted in yellow. (C) All residues that exhibited a clearly visible decrease in binding to ephrin A4 upon mutation are highlighted in yellow. (D) All residues that exhibited a clear decrease in binding to ephrin A5 upon mutation are highlighted in yellow. (E) M55, the amino acid pair I58 and M59, S68, and R103, whose mutation affected binding of KSHV gH/gL, are highlighted in yellow. (F) Same representation as in panel D, but with a “stick and ball” model of the structure of the bound receptor binding domain of ephrin A5 superimposed.
Compound 1 inhibits KSHV infection.
We tested the more potent compound 1 for its ability to inhibit entry of KSHV into different cell types, as measured by the number of green cells 48 h after inoculation with recombinant rKSHV.219, which carries a GFP reporter gene. Compound 1 inhibited entry of rKSHV.219 into 293T cells (Fig. 7A to C), primary lymphatic endothelial cells (LEC) (Fig. 7D), and human umbilical vein endothelial cells (HUVEC) (Fig. 7D) with an EC50 between approximately 14 μM (293T cells) and 276 μM (LEC). Cell viability as assayed by forward scatter/side scatter (FSC/SSC) analysis in flow cytometry was not affected by either compound (data not shown), which is in agreement with the publication by Noberini et al. (32), who found both substances to be nontoxic. As an additional control, entry of a GFP-expressing VSV G-pseudotyped SIVmac239deltaNef lentivirus was assayed in parallel. This pseudotyped virus is an appropriate control in that it depends on functional endocytotic machinery and vesicle acidification, as well as the availability of nucleotides for reverse transcription, to achieve infection (36). Entry of the VSV G SIVmac239deltaNef-GFP pseudotype into 293T cells and rhesus fibroblasts was at most only marginally affected by high concentrations of compound 1 (Fig. 7B, C, and D, right), and entry of this GFP-encoding lentivirus into LEC and HUVEC was not affected at all at concentrations up to 1 mM (Fig. 7D).
DISCUSSION
Our study not only demonstrates that the KSHV gH/gL binding site on the EphA2 receptor overlaps with the ephrin binding interface, but also highlights the fact that this site can be targeted with small molecules to inhibit infection.
The measurements that we have described here allow comparison of half-maximal binding concentrations for three different ligands of the EphA2 receptor: ephrin A4, ephrin A5, and KSHV gH/gL. As all three proteins were expressed as Fc fusions, these values may differ slightly from those for non-Fc-fused versions but allow comparison in the same system. The K1/2max values for these three binding partners of the EphA2 receptor were calculated to be 0.1 nM, 0.6 nM, and 1.6 nM, respectively. The values of 1.6 nM (soluble gH-Fc/gL-immobilized EphA2), 2.9 nM (soluble EphA2-immobilized gH/gL), and 9 nM (soluble gH/gL-immobilized EphA2) for the gH/gL interaction with the EphA2 receptor are comparable to those of interactions of other viral envelope glycoprotein with their receptors. For example, interaction of the HIV-1-encoded envelope glycoprotein gp140/gp120 with the soluble form of its receptor CD4 typically occurs with half-maximal binding values in the range of 0.5 nM or higher (37, 38), depending on the strain. The affinity of the natural ephrin A4 ligand for EphA2 (K1/2max = 0.1 nM) is considerably stronger than that of gH/gL. The higher affinity of ephrin A4 than ephrin A5 measured here is consistent with the greater potency of ephrin A4 than ephrin A5 in inhibiting KSHV entry (22).
Our results show for the first time that previously identified chemical inhibitors of the ephrin A5-EphA2 interaction also inhibit the KSHV gH/gL-EphA2 interaction and KSHV infection of target cells. The potency of the chemical inhibition is greater for the gH/gL-EphA2 interaction than for the ephrin A5-EphA2 interaction, consistent with the poorer affinity of gH/gL for EphA2. Independent calculations of the Ki of compound 1 based on our measurements of the ephrin A5-EphA2 interaction and of the gH/gL-EphA2 interaction gave very similar values: 109 μM and 126 μM. Others have reported Ki values for compound 1 of 7 μM (32) and 20 μM (35) toward EphA4 and 13 μM toward EphA2 (32). Different methodologies and our use of a dimeric Fc-containing molecule could potentially contribute to the range of differences in the Ki values obtained.
KSHV has apparently evolved to use the same binding surface on EphA2 as the natural ephrin ligands. R103 of EphA2 was the most important residue of the 13 studied for gH/gL engagement. This residue was also key for ephrin A4 and ephrin A5 engagement. Less substantial contributions to gH/gL engagement were noted for positions 55, 58 and 59, and 68; positions 55 and 58 and 59 also contributed importantly to ephrin binding. Other positions that were important for ephrin engagement were not noted to contribute to gH/gL binding. Thus, although the same general surface of EphA2 appears to be used for binding gH/gL and the natural ephrin ligands, the exact points of critical contact appear to be somewhat different. Crystal structures will be needed to precisely define the exact details of the gH/gL-EphA2 interaction. It is tempting to speculate whether the slightly different binding interface and lower affinity of KSHV gH/gL for EphA2 than cellular ephrins might translate into a different quality of EphA2 signaling.
Although the evidence is not definitive, maintenance of the early stages of KS appears to be dependent on continuous KSHV replication (19, 20). Certainly, anti-KSHV agents appear to contribute importantly to sustained remission of PEL (11). It is also likely that multicentric Castleman's disease is a result of ongoing KSHV replication. Thus, pursuit of these chemical compounds, or derivatives of them, may be a useful direction to follow for the potential treatment of KSHV-associated diseases. Animal model studies, such as those described by Ashlock and coworkers (39), certainly seem warranted.
Targeting of a cellular protein with a small molecule may of course result in unwanted side effects, and this may represent a significant obstacle. However, the gH/gL-EphA2 interaction is considerably more sensitive to inhibition by compound 1 than the natural ephrin-EphA2 interactions, and this could potentially provide selectivity to the effects of such a drug. Also, EphA2 may play a critical role in secondary effects, such as vascularization and inflammation, that are key components of KS (reviewed in reference 40), and thus, their inhibition could possibly provide an additional benefit.
ACKNOWLEDGMENTS
This work was supported by the DFG research fellowship HA 6013/1-1 “Rhadinovirus Entry” to A.S.H. and by grants RO1 AI072004 and AI063928 from the National Institutes of Health (NIH) to R.C.D.
We thank Armin Ensser, William A. Lauer, Effi Wies, and Jose Maria Martinez-Navio for critical readings of the manuscript.
Footnotes
Published ahead of print 4 June 2014
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. 10.1126/science.7997879 [DOI] [PubMed] [Google Scholar]
- 2.Moore PS, Chang Y. 2014. The conundrum of causality in tumor virology: the cases of KSHV and MCV. Semin. Cancer Biol. 26C:4–12. 10.1016/j.semcancer.2013.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boshoff C, Whitby D, Hatziioannou T, Fisher C, van der Walt J, Hatzakis A, Weiss R, Schulz T. 1995. Kaposi's-sarcoma-associated herpesvirus in HIV-negative Kaposi's sarcoma. Lancet 345:1043–1044 [DOI] [PubMed] [Google Scholar]
- 4.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191. 10.1056/NEJM199505043321802 [DOI] [PubMed] [Google Scholar]
- 5.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay M, Clauvel J, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 6.Stiller CA. 2007. International patterns of cancer incidence in adolescents. Cancer Treat. Rev. 33:631–645. 10.1016/j.ctrv.2007.01.001 [DOI] [PubMed] [Google Scholar]
- 7.Schwartz RA, Micali G, Nasca MR, Scuderi L. 2008. Kaposi sarcoma: A continuing conundrum. J. Am. Acad. Dermatol. 59:179–206. 10.1016/j.jaad.2008.05.001 [DOI] [PubMed] [Google Scholar]
- 8.Amir H, Kaaya EE, Manji KP, Kwesigabo G, Biberfeld P. 2001. Kaposi's sarcoma before and during a human immunodeficiency virus epidemic in Tanzanian children. Pediatr. Infect. Dis. J. 20:518–521. 10.1097/00006454-200105000-00009 [DOI] [PubMed] [Google Scholar]
- 9.Wamburu G, Masenga EJ, Moshi EZ, Schmid-Grendelmeier P, Kempf W, Orfanos CE. 2006. HIV-associated and non-HIV associated types of Kaposi's sarcoma in an African population in Tanzania. Status of immune suppression and HHV-8 seroprevalence. Eur. J. Dermatol. 16:677–682 [PubMed] [Google Scholar]
- 10.Dittmer DP. 2003. Transcription profile of Kaposi's sarcoma-associated herpesvirus in primary Kaposi's sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 63:2010–2015 [PubMed] [Google Scholar]
- 11.Crum-Cianflone NF, Wallace MR, Looney D. 2006. Successful secondary prophylaxis for primary effusion lymphoma with human herpesvirus 8 therapy. AIDS 20:1567–1569. 10.1097/01.aids.0000237381.92303.61 [DOI] [PubMed] [Google Scholar]
- 12.Casper C, Nichols WG, Huang M-L, Corey L, Wald A. 2004. Remission of HHV-8 and HIV-associated multicentric Castleman disease with ganciclovir treatment. Blood 103:1632–1634. 10.1182/blood-2003-05-1721 [DOI] [PubMed] [Google Scholar]
- 13.Staskus KA, Sun R, Miller G, Racz P, Jaslowski A, Metroka C, Brett-Smith H, Haase AT. 1999. Cellular tropism and viral interleukin-6 expression distinguish human herpesvirus 8 involvement in Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease. J. Virol. 73:4181–4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katano H, Sato Y, Kurata T, Mori S, Sata T. 2000. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi's sarcoma, and multicentric Castleman's disease. Virology 269:335–344. 10.1006/viro.2000.0196 [DOI] [PubMed] [Google Scholar]
- 15.Little RF, Merced-Galindez F, Staskus K, Whitby D, Aoki Y, Humphrey R, Pluda JM, Marshall V, Walters M, Welles L, Rodriguez-Chavez IR, Pittaluga S, Tosato G, Yarchoan R. 2003. A pilot study of cidofovir in patients with Kaposi sarcoma. J. Infect. Dis. 187:149–153. 10.1086/346159 [DOI] [PubMed] [Google Scholar]
- 16.Krown SE, Dittmer DP, Cesarman E. 2011. Pilot study of oral valganciclovir therapy in patients with classic Kaposi sarcoma. J. Infect. Dis. 203:1082–1086. 10.1093/infdis/jiq177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delabesse E, Oksenhendler E, Lebbé C, Vérola O, Varet B, Turhan AG. 1997. Molecular analysis of clonality in Kaposi's sarcoma. J. Clin. Pathol. 50:664–668. 10.1136/jcp.50.8.664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duprez R, Lacoste V, Brière J, Couppié P, Frances C, Sainte-Marie D, Kassa-Kelembho E, Lando M-J, Essame Oyono J-L, Nkegoum B, Hbid O, Mahé A, Lebbé C, Tortevoye P, Huerre M, Gessain A. 2007. Evidence for a multiclonal origin of multicentric advanced lesions of Kaposi sarcoma. J. Natl. Cancer Inst. 99:1086–1094. 10.1093/jnci/djm045 [DOI] [PubMed] [Google Scholar]
- 19.Mocroft A, Youle M, Gazzard B, Morcinek J, Halai R, Phillips AN. 1996. Anti-herpesvirus treatment and risk of Kaposi's sarcoma in HIV infection. Royal Free/Chelsea and Westminster Hospitals Collaborative Group. AIDS 10:1101–1105 [PubMed] [Google Scholar]
- 20.Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H, Robinson CA. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. N. Engl. J. Med. 340:1063–1070 [DOI] [PubMed] [Google Scholar]
- 21.Hahn AS, Kaufmann JK, Wies E, Naschberger E, Panteleev-Ivlev J, Schmidt K, Holzer A, Schmidt M, Chen J, Konig S, Ensser A, Myoung J, Brockmeyer NH, Sturzl M, Fleckenstein B, Neipel F. 2012. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi's sarcoma-associated herpesvirus. Nat. Med. 18:961–966. 10.1038/nm.2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hahn AS, Desrosiers RC. 2013. Rhesus monkey rhadinovirus uses Eph family receptors for entry into B cells and endothelial cells but not fibroblasts. PLoS Pathog. 9:e1003360. 10.1371/journal.ppat.1003360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eph Nomenclature Committee. 1997. Unified nomenclature for Eph family receptors and their ligands, the ephrins. Cell 90:403–404. 10.1016/S0092-8674(00)80500-0 [DOI] [PubMed] [Google Scholar]
- 24.Wykosky J, Debinski W. 2008. The EphA2 receptor and EphrinA1 ligand in solid tumors: function and therapeutic targeting. Mol. Cancer Res. 6:1795–1806. 10.1158/1541-7786.MCR-08-0244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J. 2012. Regulation of tumor initiation and metastatic progression by Eph receptor tyrosine kinases. Adv. Cancer Res. 114:1–20. 10.1016/B978-0-12-386503-8.00001-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Funk SD, Yurdagul A, Albert P, Traylor JG, Jin L, Chen J, Orr AW. 2012. EphA2 activation promotes the endothelial cell inflammatory response. Arterioscler. Thromb. Vasc. Biol. 32:686–695. 10.1161/ATVBAHA.111.242792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hahn A, Birkmann A, Wies E, Dorer D, Mahr K, Sturzl M, Titgemeyer F, Neipel F. 2009. Kaposi's sarcoma-associated herpesvirus gH/gL: glycoprotein export and interaction with cellular receptors. J. Virol. 83:396–407. 10.1128/JVI.01170-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moll A, Hildebrandt A, Lenhof H-P, Kohlbacher O. 2005. BALLView: an object-oriented molecular visualization and modeling framework. J. Comput. Aided Mol. Des. 19:791–800. 10.1007/s10822-005-9027-x [DOI] [PubMed] [Google Scholar]
- 29.Himanen JP, Yermekbayeva L, Janes PW, Walker JR, Xu K, Atapattu L, Rajashankar KR, Mensinga A, Lackmann M, Nikolov DB, Dhe-Paganon S. 2010. Architecture of Eph receptor clusters. Proc. Natl. Acad. Sci. U. S. A. 107:10860–10865. 10.1073/pnas.1004148107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 31.Himanen JP, Goldgur Y, Miao H, Myshkin E, Guo H, Buck M, Nguyen M, Rajashankar KR, Wang B, Nikolov DB. 2009. Ligand recognition by A-class Eph receptors: crystal structures of the EphA2 ligand-binding domain and the EphA2/ephrin-A1 complex. EMBO Rep. 10:722–728. 10.1038/embor.2009.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noberini R, Koolpe M, Peddibhotla S, Dahl R, Su Y, Cosford NDP, Roth GP, Pasquale EB. 2008. Small molecules can selectively inhibit ephrin binding to the EphA4 and EphA2 receptors. J. Biol. Chem. 283:29461–29472. 10.1074/jbc.M804103200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gale NW, Holland SJ, Valenzuela DM, Flenniken A, Pan L, Ryan TE, Henkemeyer M, Strebhardt K, Hirai H, Wilkinson DG, Pawson T, Davis S, Yancopoulos GD. 1996. Eph receptors and ligands comprise two major specificity subclasses and are reciprocally compartmentalized during embryogenesis. Neuron 17:9–19. 10.1016/S0896-6273(00)80276-7 [DOI] [PubMed] [Google Scholar]
- 34.Cheng Y, Prusoff WH. 1973. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22:3099–3108. 10.1016/0006-2952(73)90196-2 [DOI] [PubMed] [Google Scholar]
- 35.Qin H, Shi J, Noberini R, Pasquale EB, Song J. 2008. Crystal structure and NMR binding reveal that two small molecule antagonists target the high affinity ephrin-binding channel of the EphA4 receptor. J. Biol. Chem. 283:29473–29484. 10.1074/jbc.M804114200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diamond TL, Roshal M, Jamburuthugoda VK, Reynolds HM, Merriam AR, Lee KY, Balakrishnan M, Bambara RA, Planelles V, Dewhurst S, Kim B. 2004. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J. Biol. Chem. 279:51545–51553. 10.1074/jbc.M408573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeffs SA, Goriup S, Kebble B, Crane D, Bolgiano B, Sattentau Q, Jones S, Holmes H. 2004. Expression and characterisation of recombinant oligomeric envelope glycoproteins derived from primary isolates of HIV-1. Vaccine 22:1032–1046. 10.1016/j.vaccine.2003.08.042 [DOI] [PubMed] [Google Scholar]
- 38.Jeffs SA, McKeating J, Lewis S, Craft H, Biram D, Stephens PE, Brady RL. 1996. Antigenicity of truncated forms of the human immunodeficiency virus type 1 envelope glycoprotein. J. Gen. Virol. 77:1403–1410. 10.1099/0022-1317-77-7-1403 [DOI] [PubMed] [Google Scholar]
- 39.Ashlock BM, Ma Q, Issac B, Mesri EA. 2014. Productively infected murine Kaposi's sarcoma-like tumors define new animal models for studying and targeting KSHV oncogenesis and replication. PLoS One 9:e87324. 10.1371/journal.pone.0087324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J. Clin. Invest. 120:939–949. 10.1172/JCI40567 [DOI] [PMC free article] [PubMed] [Google Scholar]