

ABSTRACT
The structure of adenovirus outer capsid was revealed recently at 3- to 4-Å resolution (V. Reddy, S. Natchiar, P. Stewart, and G. Nemerow, Science 329:1071–1075, 2010, http://dx.doi.org/10.1126/science.1187292); however, precise details on the function and biochemical and structural features for the inner core still are lacking. Protein V is one the most important components of the adenovirus core, as it links the outer capsid via association with protein VI with the inner DNA core. Protein V is a highly basic protein that strongly binds to DNA in a nonspecific manner. We report the expression of a soluble protein V that exists in monomer-dimer equilibrium. Using reversible cross-linking affinity purification in combination with mass spectrometry, we found that protein V contains multiple DNA binding sites. The binding sites from protein V mediate heat-stable nucleic acid associations, with some of the binding sites possibly masked in the virus by other core proteins. We also demonstrate direct interaction between soluble proteins V and VI, thereby revealing the bridging of the inner DNA core with the outer capsid proteins. These findings are consistent with a model of nucleosome-like structures proposed for the adenovirus core and encapsidated DNA. They also suggest an additional role for protein V in linking the inner nucleic acid core with protein VI on the inner capsid shell.
IMPORTANCE Scant knowledge exists of how the inner core of adenovirus containing its double-stranded DNA (dsDNA) genome and associated proteins is organized. Here, we report a purification scheme for a recombinant form of protein V that allowed analysis of its interactions with the nucleic acid core region. We demonstrate that protein V exhibits stable associations with dsDNA due to the presence of multiple nucleic acid binding sites identified both in the isolated recombinant protein and in virus particles. As protein V also binds to the membrane lytic protein VI molecules, this core protein may serve as a bridge from the inner dsDNA core to the inner capsid shell.
INTRODUCTION
Adenoviruses (Ad) are nonenveloped, icosahedral viruses containing a linear double-stranded DNA (dsDNA) genome of ∼36 kbp. These viruses infect many different vertebrate species, causing acute diseases of the eye and upper respiratory and gastrointestinal tracts in humans (1, 2). The adenovirus capsid consists of three main proteins: hexon, penton base, and fiber, which have been characterized structurally both in the virus particle and as isolated molecules (3–7). Two-hundred-forty hexon trimers comprise the majority of the capsid structure. Each of the 12 vertices of the capsid contains a penton base noncovalently linked to the trimeric fiber protein. During cell entry, the fiber and penton base (8) mediate attachment and internalization, respectively (9). The outer Ad capsid also contains four cement proteins, IIIa, VI, VIII, and IX, that stabilize the 150-MDa virion (6, 7). The crystal structure of the fiber (4), penton base (5), and hexon (3) as isolated molecules have been previously reported. In contrast, much less information exists on the functional, biochemical, and structural features of the capsid proteins that comprise the inner nucleoprotein core of the virion. This is due in part to the fact that the Ad core is not icosahedrally ordered, hindering modeling of the interior of the capsid by cryo-electron microscopy (cryo-EM) or X-ray diffraction.
The Ad nucleoprotein core consists of genomic dsDNA and six proteins. Two copies of terminal protein (TP) are covalently linked to the 5′ ends of the DNA. The 23K maturation protease is required to process preproteins in the immature virion following assembly (2). Fully mature virions also contain ∼5 copies of IVa2, which is required to package viral DNA via specific contacts with viral DNA-packing sequence (10) as well as with the adenovirus major late promoter (11). The core region also contains three arginine-rich, highly basic proteins, V, VII, and μ (Mu) (12, 13). Protein VII, present at ∼800 copies per virion (14), is the most abundant core protein and is tightly associated with the DNA in a sequence-independent manner. Protein VII is thought to help organize the viral genome in nucleosome-like structures mediated by the basic regions in separate α-helices in protein VII and the phosphate backbone of DNA, producing a condensed nucleic acid structure (15, 16).
Present in ∼157 copies per virion, protein V is one of the most important proteins in the core due to its involvement in core condensation (15, 17). Moreover, protein V bridges the viral DNA core with the outer capsid by interacting with protein VI (18–20). The importance of the latter cementing function was revealed in an adenovirus harboring a protein V deletion (Ad5-dV). This deletion significantly disrupted viral assembly concomitant with alterations in the capsid morphology and a substantial reduction in thermostability and infectivity (21). Overall, these studies indicate that protein V has a critical role in capsid assembly and in the formation of infectious virions (21). Protein V contains multiple nuclear and nucleolar localization signals and is imported into the nucleus very shortly after viral infection, suggesting that it has a role in the delivery of viral DNA during the infection process (20, 22). Protein V is also capable of relocating nucleolin and B23 from the nucleus to the cytoplasm (23).
A previous study employing a cross-linking approach identified a variety of interactions among the core proteins, including formation of V-V and VII-VII dimers, supporting the notion that the core DNA packing unit contains six molecules of protein VII and one molecule of protein V. Moreover, protein μ was found associated both with protein V and in complex with V-VII but never with protein VII alone, suggesting that protein VII and protein μ are in close contact with protein V (19).
Despite the knowledge gained from these early biochemical studies, there are still gaps in our understanding of the interactions between protein V and DNA. Here, we report for the first time the expression of protein V in a soluble form, which is in dimer-monomer equilibrium. We mapped DNA binding motifs by reversible cross-linking affinity purification and present direct evidence for association between protein V and protein VI. These studies pave the way for further structural characterization of the adenovirus core structure.
MATERIALS AND METHODS
Recombinant protein purification.
A cDNA construct, encoding amino acid residues 1 to 263 of protein V and containing a C-terminal 2× Strep-tag (24), was cloned into the bacterial expression plasmid pET28 (EMD Biosciences).
The recombinant proteins were expressed in BL21(DE3)-RIPL (Stratagene). Cells were grown to an optical density at 600 nm (OD600) of 0.6 in LB media containing 100 μg/ml kanamycin, and protein expression was induced by addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 1 h at 37°C. Cells were harvested, pelleted by centrifugation, and subsequently stored at −20°C. Pellets were resuspended in 20 mM Tris, 1 mM EDTA, 100 mM NaCl at pH 8.0 and then lysed by sonication using a half-inch-long probe sonicator (S4000; Misonix). Cell debris was removed by centrifugation (27,000 × g for 20 min at 4°C), and then polyethyleneimine (PEI; Polysciences, Inc.) was added to the supernatant to a final concentration of 0.2% from a 10% stock solution at pH 8.0 in order to precipitate contaminating bacterial DNA. Following 30 min of incubation on ice, the DNA was pelleted at 9,000 × g for 5 min at 4°C. The supernatant was further treated with 0.1 mg of DNase (Roche) overnight at 4°C, followed by a final round of clarifying centrifugation at 27,000 × g for 20 min at 4°C. The recombinant, DNA-free protein V was further purified by affinity chromatography on Tactin resin (IBA) based on the tandem Strep-tag, followed by nickel-nitrilotriacetic acid (Ni-NTA) agarose (Qiagen), and finally subjected to size-exclusion chromatography (SEC) using a HiLoad 16/60 S200 column and an AKTA purifier (GE Healthcare) in 20 mM Tris, 1 mM EDTA, 100 mM NaCl, pH 8.0. The yield and estimation of protein purity at various stages of the purification were determined by SDS-PAGE analysis using equivalent amounts of total protein from each stage and employing the ImageQuant 5.2 software to estimate the yield and purity of protein V for each stage, as indicated in Table 1.
TABLE 1.
Quantification of protein V purity and yield
| Sample sourcea | Total protein concn (mg/liter of culture) | Purity (%) | Protein V (mg) | A260/280 ratiob |
|---|---|---|---|---|
| Total | 1,851.5 | 12.7 | 235.3 | 1.85 |
| Supernatant | 1,382.5 | 15.1 | 209.3 | 1.4 |
| ElutionStrep | 14.1 | 94.4 | 13.3 | 0.8 |
| ElutionNi | 8.8 | 99.0 | 8.7 | 0.7 |
| Protein V | 4.7 | 99.1 | 4.7 | 0.7 |
Supernatant, supernatant after PEI precipitation; ElutionStrep, elution with Strep-tag; ElutionNi, elution with Ni-NTA; Protein V, protein V without tags after SEC.
Ratio of absorbance at 260 and 280 nm.
A construct encoding residues 34 to 114 of protein VI with an N-terminal 6-histidine tag was purified as described by Moyer and Nemerow (25).
CD.
The secondary structures of proteins V and V263 were analyzed by circular dichroism (CD) spectropolarimetry. Purified proteins were concentrated to 1 mg/ml in 10 mM Tris (pH 8.0) and 150 mM NaCl and analyzed using a Jasco J-820 spectropolarimeter with a 0.1-cm quartz cuvette. Spectra were collected at 25°C in triplicate from 190 to 260 nm in 0.2-nm increments. Secondary structure content was evaluated using the DichroWeb software package (26–28). The data are the average values obtained from Selcon3 analyses (29, 30).
Cross-linking with DMP.
Purified protein V (1 mg/ml) was mixed with different amounts of dimethyl pimelidate (DMP) (Sigma-Aldrich) in 20 mM HEPES buffer, pH 7.2, and incubated for 30 min at room temperature. The reactions were stopped by the addition of 0.5 M Tris, pH 6.8, with 1× SDS-PAGE loading buffer, boiled, and subjected to electrophoresis on a 4 to 20% polyacrylamide gel.
SEC-MALS characterization.
To determine the oligomeric state of recombinant protein V, approximately 300 μg of purified protein V was loaded onto a Superdex 75 10/30GL column (GE Healthcare, NJ) using an AKTA fast pressure liquid chromatography (FPLC) system (GE Healthcare, NJ). SEC was coupled in line with the following calibrated detectors: (i) a MiniDawn Treos multiangle light-scattering (MALS) detector (Wyatt Corporation, CA) and (ii) an Optilab T-reX refractive index (RI) detector (Wyatt Corporation, CA). The Astra VI software (Wyatt Corporation, CA) was used to combine these measurements to determine the absolute molar mass of the eluted protein (31, 32).
Electrophoretic mobility shift assay (EMSA).
A constant amount of 10 μM dsDNA oligomers comprised of 15 or 42 bp or linearized bacmid was mixed in binding buffer (10 mM Tris, 50 mM KCl, 1 mM dithiothreitol [DTT], pH 7.5) with 5 to 20 μg of purified recombinant protein V, followed by a 20-min incubation at room temperature. The DNA-protein samples then were electrophoresed on a 0.8% morpholinepropanesulfonic acid (MOPS) agarose gel that was stained with SYBR green (Life Tech, NY) to visualize nucleic acids and, subsequently, Simply Blue (Life Tech) to visualize protein. The following oligomer sequences were used: 15-bp oligomer, 5′CGCAAACTGTACTAC; 42-bp oligomer, 5′TAAATTTGGGCGTAACCGAGTAAGATTTGGCCATTTTCGCGG (see Fig. 4D for the 100-bp oligomer).
FIG 4.

Mapping the DNA binding sites on protein V. (A) Scheme used for the reversible cross-linking affinity purification protocol (see Materials and Methods for details) and for the identification of DNA sequences that contact protein V. (B) Summary of the results from MALDI-TOF mass spectra of peptides that cross-linked to dsDNA of 42 bp or to the genomic DNA within the virion. Peptides that are overlapping are shown in the regions of the figure colored gray. (C) Plot of protein V intrinsic disorder prediction. The prediction was made using PONDR-FIT, version VL-XT (55). The upper gray lines show the peptides identified in the RCAP assay superimposed on the regions predicted to have secondary structure or intrinsic disorder. The heavy black lines represent portions of protein V predicted to be intrinsically disordered. (D) Mapping of the sequences in a 100-bp DNA that contacts protein V. DNA fragments that bind protein V are identified by the gray lines.
Reversible cross-linking affinity purification (RCAP).
Protein V was dialyzed in phosphate-buffered saline to remove residual Tris, which interferes with formaldehyde cross-linking, as previously described (33). Briefly, 10 μg of protein V was incubated with dsDNA at a 2:1 molar ratio and then cross-linked with 0.1% formaldehyde and digested with 0.2 μg of trypsin. The DNA-peptide heteroconjugates next were precipitated with a 0.3 M final concentration of ammonium acetate and two volumes of ethanol, followed by extensive washing to remove un-cross-linked peptides. The DNA-peptide conjugates were reversed by incubation at 70°C for 1 h, and peptides eluted were analyzed by a matrix-assisted laser desorption ionization–time-of-flight (MALDI-TOF) instrument (Bruker Autoflex III; Agilent Technologies) in positive-ion mode. All assigned peaks correspond to theoretical masses of peptides from V protein ±0.5 Da.
For analysis of protein-DNA interaction in adenovirus virions, Ad5 (10 μg) was cross-linked to its genomic contents and digested with trypsin. The genome-peptide heteroconjugates then were purified by silica adsorption using a QIAprep spin column (Qiagen, MD). The samples were washed 3× with PE buffer (10 mM Tris-HCl, pH 7.5, in 80% ethanol) and eluted in 50 mM sodium acetate, pH 5.5. Two-hundred mM sodium chloride then was added to the elutions, and the cross-links were reversed as previously described. Peptides then were identified using an LTQ Velos Pro mass spectrometer (Velos Pro Dual-Pressure Linear Ion Trap; Thermo Scientific). Peptide assignments all were confirmed by tandem mass spectrometry (MS/MS) analysis and identified using MASCOT (Matrix Science, MA) with an ion cutoff score of 30.
Protein V DNA binding specificity.
To examine specificity for protein V binding to DNA, 20 μg of protein V was incubated with the 100-bp DNA at a 2:1 molar ratio and cross-linked with 0.1% formaldehyde. After quenching the reaction with the addition of 200 mM glycine, the 100-bp DNA was digested with 1 U of DNase I (New England BioLabs) for 1 h at 37°C. Protein V then was bound to Ni-NTA resin (Invitrogen) and washed extensively with buffer containing 1 M NaCl. The formaldehyde cross-links then were reversed, and Taq DNA polymerase (New England BioLabs) was used to add adenines to the DNA fragments. The DNA solution then was adjusted to contain 0.5 M ammonium sulfate, 10 μg of glycogen, and 3 volumes of ethanol and stored overnight at −20°C. The DNA then was pelleted by centrifugation for 30 min at 16,000 × g at 4°C. The pellet was washed with 70% ethanol and resuspended in T4 DNA ligase buffer containing pGEM-T Easy vector and T4 DNA ligase. After ligation overnight at 4°C, the reaction mixture was transformed into Top-10 cells (Invitrogen), and the plasmids from white and light-blue transformants were purified and sequenced to identify the DNA inserts.
Protein V-VI binding assay.
Equal amounts of protein V containing a C-terminal 2× Strep-tag were incubated with constant rocking with protein VI (residues 34 to 114) or the Ad5 fiber knob (Fk) containing an N-terminal 6-histidine tag for 1 h at 4°C in binding buffer (20 mM Tris, 1 mM EDTA, 200 mM NaCl, pH 8.0). Each sample then was loaded onto Tactin resin (IBA), washed three times with binding buffer, and eluted with α-desthiobiotin. Flowthrough, wash, and elution fractions were analyzed by SDS-PAGE.
Competition of protein V-DNA interaction with protein VI.
A constant amount of dsDNA was mixed with 2.5 μg of purified recombinant protein V in binding buffer (10 mM Tris, 50 mM KCl, 1 mM DTT, pH 7.5) and increasing amounts (5 to 30 μg) of purified recombinant protein VI, followed by 20 min of incubation at room temperature. The DNA-protein samples then were electrophoresed on a 0.6% MOPS agarose gel that was stained with SYBR green (Life Tech, NY) to visualize nucleic acids.
RESULTS
Expression and purification of recombinant protein V.
Structural predictions indicated that few secondary structural elements are present in residues 259 to 368 of protein V (Fig. 1A) (PSIPRED v3.3). Since this C-terminal region of protein V likely is less structured and is prone to proteolytic degradation, a truncated form of protein V containing residues 1 to 263 and fused to a 6His tag at the N terminus and a double Strep-tag at the C terminus was expressed in bacteria (Fig. 1B). The dual-affinity tags at opposing ends of protein V were essential to the removal of contaminating degradation products during purification (Fig. 1D). We also utilized circular dichroism (CD) to compare the secondary structures of V263 to that of the wild-type V protein (Table 2). As expected from the secondary structure predictions, the V263 protein contains a higher percentage of certain secondary structural elements (i.e., helices) and less disorder than the wild-type molecule.
FIG 1.

Expression and purification of recombinant protein V. (A) Secondary structure prediction of protein V with α-helical elements shown as cylinders and β-strands as arrows. The secondary structure prediction was made using PSIPRED V3.3. AA, amino acid sequence; Pred, H-helix, C-coil, and E-strand. The black arrow indicates the residue (T263) where protein V was truncated for expression. (B) Schematic of recombinant protein V used for bacterial expression. The entire construct contains an N-terminal polyhistidine tag (6His), a thrombin (Th) cleavage site, the V coding region (residues 1 to 263), a tobacco etch virus (TEV) cleavage site, and a double Strep-tag (2×Strep) at the C terminus. (C) Chromatogram from SEC showing absorption at 280 nm. The gray elution profile corresponds to protein V purified without PEI, eluting at 48 ml (void volume), while the black profile indicates migration for protein V purified with 0.2% PEI, eluting at 68 ml. (D) SDS-PAGE analyses of protein V at 50 μg (lanes 2 and 3) or 5 μg (lanes 4 to 7) of protein at various stages of purification on 4 to 20% denaturing gels and stained with Simply Blue. Lanes: 1, molecular mass markers; 2, bacterial lysate; 3, supernatant after PEI treatment; 4, elution from Tactin resin; 5, elution from Ni-NTA agarose; 6, protein after SEC; 7, protein treated with thrombin and TEV to remove the tags after SEC.
TABLE 2.
Secondary structure of recombinant wild-type and 263 protein V measured by CD spectroscopy
| Secondary structure | Protein V structure proportions (%) |
|
|---|---|---|
| WT | 263 | |
| α-Helix | 34.50 | 80.4 |
| β-Strand | 11.00 | 0.1 |
| Turns | 23.10 | 5.2 |
| Unordered | 31.30 | 14.4 |
Protein V binds dsDNA in a sequence-independent manner, causing a substantial problem during our initial attempts to purify the recombinant protein. In particular, protein V binds bacterial DNA, resulting in the aggregation of protein V (Fig. 1C). To remedy this situation, a PEI precipitation step was incorporated into the purification process. PEI is a positively charged polymeric molecule that binds tightly to nucleic acid and negatively charged proteins (34, 35). The PEI precipitation step reduced the heterogeneity of the starting material and removed the contaminating nucleic acids. PEI at 0.2% was found to precipitate contaminating DNA away from protein V, allowing protein V to be purified by affinity chromatography using a Tactin resin combined with a high-salt Tris-EDTA wash buffer prior to elution with desthiobiotin. A second affinity purification step using Ni-NTA agarose removed low-molecular-mass fragments of protein V lacking the amino-terminal 6His tag. Following proteolytic removal of both fusion tags, a final purification and buffer exchange SEC step was used to remove remaining small peptides as well as to determine the aggregation state of the protein. This purification procedure allowed isolation of soluble DNA-free protein V that was ∼99% pure (Fig. 1C and D and Table 1).
Oligomerization state of recombinant protein V.
Cross-linking studies using pentonless virions suggested that protein V forms dimers (19). To determine the oligomeric state of soluble recombinant protein V, we performed a cross-linking experiment under conditions that likely preserve the native conformation and net charge of the protein. Protein V was incubated with an imidoester cross-linker (DMP), and the resulting molecules were resolved by SDS-PAGE (Fig. 2A). At the lowest concentration of DMP cross-linker, protein V migrated at the predicted sizes for monomer and dimer. At the largest amount of DMP, higher-order oligomers of protein V also were observed (Fig. 2A). The oligomeric distribution of protein V in solution was determined by an SEC-MALS analyzer. Protein V eluted as a single symmetrical peak that corresponds to a mass of 35 to 60 kDa, with an average molecular mass of 43.1 kDa and polydispersity of 1.071 (Fig. 2B). These results show that protein V exists in equilibrium between monomers and dimers in solution.
FIG 2.
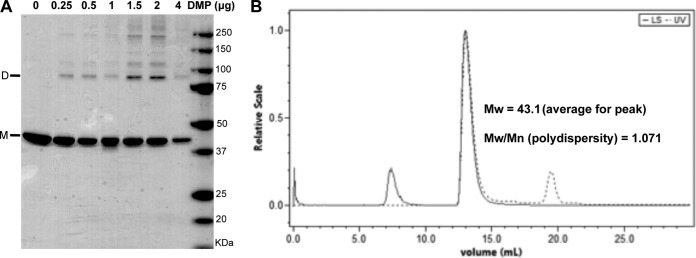
Oligomeric state of recombinant protein V. (A) SDS-PAGE analyses of recombinant protein V (5 μg) after cross-linking with different amounts of DMP. Samples were analyzed on a 4 to 20% SDS-PAGE and stained with Simply Blue dye. The locations of the monomer (M) and dimer (D) are indicated. (B) Chromatogram of purified protein V subjected to SEC-MALS showing the average molecular weight (Mw; in thousands) of the elution peak and the polydispersity values. LS, light scatter.
Protein V-DNA binding analyses.
We next sought to evaluate the DNA binding capacity of recombinant protein V as a function of dsDNA length in an agarose electrophoretic mobility shift assay at 25°C. Protein V formed complexes with dsDNA fragments of 15 or 42 bp, as indicated by its retarded migration in an agarose gel as observed by the comigration of protein V with the dsDNA (Fig. 3B), which does not occur with DNA alone (compare to Fig. 3A). Furthermore, the extent of DNA retardation was proportional to the amount of protein V added. We also observed that a relatively larger amount of protein V is required with longer dsDNAs, such as linearized bacmids (Fig. 3C), likely due to the larger number of negative charges needed to be completely neutralized to achieve complete retardation (36). Overall, these results suggest that the recombinant protein V can bind dsDNA and that binding does not require a specific sequence or DNA length.
FIG 3.
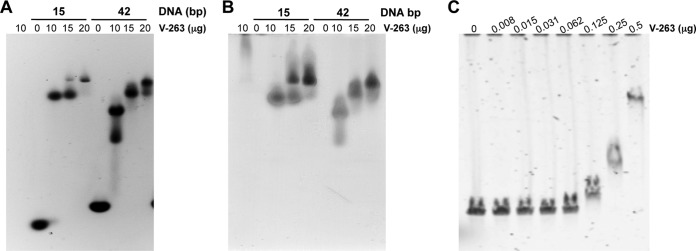
Protein V-DNA binding assays. (A) An electrophoretic mobility shift assay (EMSA) was performed with 2 μg of 15 or 42 bp dsDNA that was incubated with the indicated amounts of recombinant protein V at 25°C for 20 min prior to analysis on an 0.8% agarose gel and then stained with SYBR green to visualize the DNA. (B) The same gel was stained with Simply Blue to visualize protein. (C) EMSA was performed with 150 ng of linearized bacmid (39 kbp) that was incubated with the indicated amounts of protein V at 25°C for 20 min prior to analysis on a 0.8% agarose gel and then stained with SYBR green.
Mapping the DNA binding sites on protein V.
RCAP was used to map the DNA binding sequence in protein V (33). RCAP uses the bifunctional cross-linker formaldehyde to reversibly cross-link DNA to protein V. Following trypsin digestion of the complex and selective recovery of DNA molecules and cross-linked peptides from protein V, the cross-links were reversed and mass spectrometric analysis of the peptides was performed (Fig. 4A). RCAP has been used previously to examine the interaction between viral capsids and viral RNAs (37, 38). The assay was performed with DNA of 42 bp and 100 bp at two different molar ratios to protein V. The control reactions, performed without formaldehyde treatment, resulted in few ions larger than 800 Da, with the smaller ones likely being buffer components. With both the 42-bp and 100-bp (data not shown) DNA oligomers, peptides from similar regions within protein V were identified, with the notable exception of the region containing peptides 24 to 67, which interacted solely with the 42-bp DNA (Fig. 4B). Furthermore, a series of peptides containing residues 68 to 88 and 164 to 181 contained one or more missed cleavages, likely due to the DNA cross-link to arginines and lysines blocking trypsin digestion (Fig. 4B). Lastly, when cross-linking was done with Ad virions, some (but not all) regions of protein V that contacted the 42- and 100-bp DNAs were found to contact the viral DNA. Since formaldehyde cross-linking blocks trypsin cleavage, this result suggests that contacts within the virion are more specific and are constrained by the higher-order structure within the virion. Given that the genome and the 42- and 100-bp duplexes all have distinct DNA sequences, protein V binds DNA in a sequence-independent manner. DNA binding sequences were located in regions of protein V that are predicted to be ordered as well as disordered. A summary of the locations of the peptides within protein V is shown in Fig. 4C.
We modified the RCAP protocol to determine whether protein V has a preferred DNA sequence (Fig. 4A). The 100-bp DNA cross-linked to protein V was digested with DNase I. Protein V and associated DNAs then were purified using a metal affinity column. The cross-linked elements then were reversed and the DNA fragments cloned and sequenced to determine their identities. Thirteen independent DNA sequences of 3 to 8 nucleotides in length were identified. The sequences mapped to locations throughout the 100-bp dsDNA and did not exhibit an obvious bias in nucleotide composition (Fig. 4D). These results are consistent with results from our DNA binding analyses (Fig. 3C) and confirm that protein V has little to no binding specificity.
Protein V-DNA binding is stable.
We sought to examine whether the multiple regions for DNA binding would affect the stability of the protein V-dsDNA complex. Fifteen-bp DNA was incubated with protein V for 20 min at either 25°C or 95°C and then resolved on an agarose gel. Strikingly, the retardation of DNA migration by protein V was observed even upon incubation of the complex at 95°C (Fig. 5). In contrast, other DNA binding proteins, such as the EBV EBNA1 protein or Ad protein IVa2, did not retain dsDNA binding activity at elevated temperatures (Fig. 5).
FIG 5.
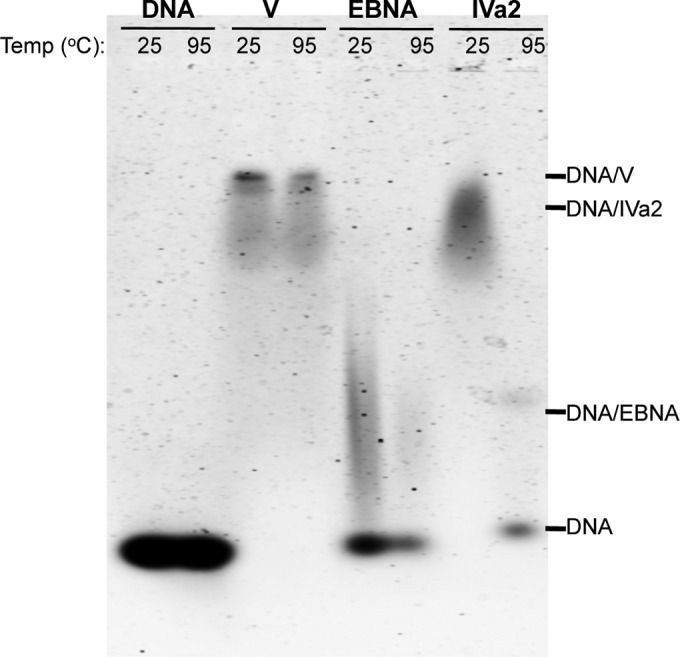
Thermostability of DNA binding proteins. An EMSA was performed with 2 μg of 15 bp DNA after incubation with 10 μg of protein V, protein IVa2, or EBNA at 25 or 95°C for 20 min and analyzed on a 0.8% agarose gel stained with SYBR green.
Protein V interaction with the membrane lytic protein VI.
Although previous studies using Western blot overlay (West-Western) (20) or cross-linking studies (19, 39) suggested that protein V can bind protein VI inside virions, a direct association of these molecules has not been demonstrated using highly purified recombinant proteins. For this purpose, a pulldown assay was performed where protein V and protein VI were mixed for 1 h and then applied to Tactin resin. The pulldown assay was performed with mature protein VI (residues 34 to 239; data not shown) and a truncated version of protein VI114 (residues 39 to 114, containing the membrane lytic domain of VI [25]) (Fig. 6). Due to mature protein VI being susceptible to proteolysis, we obtained more clear-cut results with the bp 39 to 114 domain of VI. All fractions resulting from the pulldown, flowthrough, wash, and elution were resolved by SDS-PAGE (Fig. 6A). Although a small amount of protein VI was in the flowthrough fraction, most of it coeluted with protein V. We repeated the experiment with Ad fiber knob as a negative control and, as expected, did not observe interaction between the two proteins (Fig. 6B). A negative control that was performed in the absence of protein V detected protein VI in the flowthrough fraction and none in the elution fractions (data not shown). A similar pulldown assay was performed using Ni-NTA agarose to bind His-tagged protein VI or the fiber knob with tag-free protein V. We again observed binding between proteins V and VI but not between protein V and the Ad5 fiber knob (data not shown). These results confirm a direct interaction between protein V and protein VI in a system using highly purified recombinant proteins. They also support the observation that protein V serves as a link between the inner DNA core and the outer capsid shell via VI association (19, 20, 39).
FIG 6.

Protein V interacts with the membrane lytic protein VI. A Strep-tag pulldown assay was performed with 100 μg of recombinant protein V following incubation with 100 μg of recombinant protein VI or the Ad fiber knob (control). The proteins were added to Tactin resin, the resin was washed, and bound proteins were eluted with α-desthiobiotin. All of the fractions were analyzed by 4 to 20% SDS-PAGE and stained with Simply Blue. (A) SDS-PAGE of protein V-VI pulldown assay. Lanes: 1, protein V; 2, protein VI; 3, input; 4, flowthrough; 5 to 7, washes; 8 to 10, elutions; 11, molecular mass markers. (B) SDS-PAGE of protein V-fiber knob pulldown assay. Lanes: 1, protein V; 2, fiber knob; 3, input; 4, flowthrough; 5 to 7, washes; 8 to 10, elutions; 11, molecular mass markers. (C) Protein VI competition for protein V-DNA interaction. EMSA was performed with 0.5 μg of DNA and 2.5 μg of recombinant protein V and incubated with increased amounts of recombinant protein VI at 25°C for 20 min prior to analysis on a 0.8% agarose gel and then stained with SYBR green to visualize the DNA.
In order to investigate whether protein V association with VI impacts association with DNA, we performed a competition assay (Fig. 6C). Protein VI also can bind dsDNA (40, 41). As shown in Fig. 6C, the retardation of dsDNA by proteins V and VI is distinct (compare lanes 2 and 3), and protein VI has much less capacity to alter the mobility of dsDNA than protein V. After adding increased amounts of protein VI to a constant amount of protein V in the presence of dsDNA, we observed reduced retardation of nucleic acid relative to V alone (lanes 5 to 7), suggesting that VI association with dsDNA can compete for protein V-DNA binding.
DISCUSSION
Adenovirus particles contain approximately 157 molecules of protein V per capsid (42). A mutant adenovirus that lacks protein V (Ad5-dV) has reduced thermostability and infectivity, demonstrating that protein V plays a critical role in the generation of infectious particles (21). However, relatively little biochemical or structural information has been obtained for protein V. During the infection process, protein V is not released from the viral particles either when the virus penetrates the cells during infection (43) or upon heating purified particles (42). However, upon disruption of virions by a variety of agents (18, 44, 45), protein V has been found in complex with the other core proteins, VII and μ (19, 39). Moreover, protein V appears to bind viral DNA in a sequence-independent manner. Protein V is thought to serve as a bridge between the viral DNA core and the outer capsid by interacting with protein VI and was also reported to be in close proximity to a penton base (19, 20, 39).
Protein V is a highly basic protein with a calculated pI of 10.32 and is enriched for basic residues (Fig. 1A). Protein V contains multiple nuclear and nucleolar localization signals and is imported into the nucleus very shortly after viral infection, suggesting that it has a role in viral DNA delivery (20, 22). Further characterization of protein V in functional and biochemical studies independent of other AdV capsid proteins has proven difficult. While protein V can be isolated from viral particles treated with urea (12), yields are not of sufficient quantity for further biochemical analyses. A bacterial expression system yielded larger amounts of the molecule, but the protein was extensively proteolyzed (20). Additionally, the sequence-independent nature of the interaction between protein V and DNA results in large amounts of contaminating bacterial DNA. Despite these limitations, we developed a modified procedure to express and purify recombinant protein V lacking the unstructured C-terminal region (Table 1) under native conditions. In particular, a PEI precipitation step allowed removal of contaminating bacterial DNA that otherwise causes protein V aggregation (Fig. 1C). Inclusion of dual affinity tags at the N and C termini of protein V (Fig. 1B) allowed isolation of the full-length molecule and efficient removal of unwanted degradation products (Fig. 1D). This purification scheme yields highly purified, DNA-free protein V of sufficient quantity and quality for biochemical and structural analyses. The use of this truncated protein V rather than the full-length molecule was necessary to overcome the poor yield of full-length recombinant protein V. Although the truncated V may not completely duplicate the functional properties of full-length V, we noted that the C-terminal region of full-length protein V in virions did not interact with Ad5 DNA (Fig. 4B).
Cross-linking studies have indicated that protein V forms dimers inside viral particles (19, 39). We found that the recombinant protein V (amino acids 1 to 263) also can form dimers and higher-order oligomers in the absence of DNA (Fig. 2A). Using SEC-MALS, we observed a single protein peak with a molecular mass between 35 and 60 kDa (Fig. 2B), suggesting that protein V exists in solution in an equilibrium of monomers and dimers. An equilibrium of monomer-dimer was previously observed for the DNA binding protein P6 of bacteriophage 29 (Φ29) (46). These results suggest that protein V can have two different protein-protein interaction modes, one as a monomer and one as a dimer. Association with DNA or the core proteins VII and μ could influence the dimeric or monomeric state of protein V.
Protein V is rich in arginine and lysine residues, promoting its interaction with DNA in a sequence-independent manner (17). We confirmed that binding of recombinant protein V to different sizes of dsDNA was also independent of DNA sequence (Fig. 3A and 4D). In the RCAP assays performed with the recombinant V263 protein, several regions of protein V were found to possess DNA binding activity (Fig. 4B). In support of this, the protein V-DNA complex was maintained even after heating to 95°C (Fig. 5). However, the C-terminal region of protein V, which is mostly disordered, is also devoid of DNA binding. However, RCAP analysis of adenovirus virions for DNA binding contained far fewer regions of protein V to contact DNA, likely due to these regions of protein V contacting other viral proteins within the virion. Some of these masked DNA binding sites might become exposed upon transfer of the viral nucleic acid into the nucleus during infection. We also observed that certain V peptides were detected more frequently than others, suggesting that certain regions of protein V, such as residues 68 to 88, are more accessible to DNA than others. Surprisingly, several of the regions of protein V predicted to be intrinsically disordered did not contact DNA when in the context of the virion. This suggests that the intrinsically disordered regions in protein V contact other adenoviral proteins within the virion.
Given the apparent multiple DNA binding sites in protein V, we favor the possibility that protein V has a histone-like role (47) within the virion. It is possible that the DNA binding sites in protein V can be altered upon interaction of protein V with other viral proteins, such as protein VI, VII, or μ. The proteins VII, V, and μ appear to be histone-like proteins that condense and package acetylated DNA. In fact, electron microscopy studies of the Ad core show an organization in nucleosome-like core structures (44, 48–50). Four core histones, H2A, H2b, H3, and H4, are found in cellular nucleosomes. Minimally, H3 and H4 are required to package cellular DNA into a nucleosome array (51). Histone H1 seals off each nucleosome to form tightly compacted structures (52). Furthermore, H1 is the first histone to be displaced from the chromatin, and after cross-linking experiments it was not found to be part of the nucleosome protein core (53). Proteins VII and V have different associations with the viral DNA, as indicated by the selective release of protein V from the cores after a number of treatments (18, 44, 45). Salt-disrupted cores contain only DNA and protein VII, suggesting that protein VII is more tightly associated with viral DNA than protein V (15, 54). Protein V was been suggested to be responsible for the stability and to form more condensed core structures (15, 18).
A large number of interactions among the core proteins have been revealed by cross-linking experiments, including V-V and VII-VII homodimers, as well as heterocomplexes of V-VI, μ-V, and μ-V-VII (19, 39). Furthermore, the effects on morphology and thermostability of Ad5-dV (21) together with our results show that protein V has a strong direct association with DNA through multiple binding sites. These results suggest that protein V not only surrounds the complex of protein VII-DNA but also could be a part of the adenovirus nucleosomes (54). Protein V's ability to facilitate interactions with DNA, protein VII, and μ to maintain condensed core structures suggests that it has functions akin to those of histone H1.
Protein V has other important associations in the virus particle, including interactions with the membrane lytic protein VI (19, 20, 39). We also demonstrated a direct and specific interaction of protein VI with highly purified recombinant protein V (Fig. 6A). One important question is whether individual molecules of protein V can bind DNA and protein VI simultaneously or whether separate populations of protein V mediate these associations. We observed that protein VI can compete with DNA for the binding of protein V (Fig. 6C). These observations raise the possibility that there are two locations for protein V in the virion, one in direct association with DNA in the core and the other serving as a bridge to protein VI association at the vertex region. This interaction between protein V and VI could strengthen the connection between the DNA core and the inner surface of the outer capsid.
A key feature that may contribute to its wide range of interactions is the high degree of intrinsic disorder within protein V. The interconversion of protein V between monomer and dimer, as well as the relatively high rate of proteolysis, are consistent with the presence of intrinsically disordered regions (55). Predictions using computer programs yielded results consistent with this notion (Fig. 4C). Intriguingly, some of the DNA binding regions mapped using RCAP were to the intrinsically disordered regions of protein V. Intrinsically disordered proteins could gain specificity for ligand interaction upon formation of the complex. At this point, we do not have direct evidence for DNA sequence-specific interaction between protein V and DNA.
Although we still lack important structural information on interactions among the Ad core proteins, our studies with purified recombinant protein V and the characterization of its interaction with DNA set the stage for further structural analysis with or without DNA. The structure of protein V will help guide our understanding of the molecular interactions occurring within the AdV core. In particular, it will be interesting to determine how protein V interacts with the other core proteins (VII and μ) and the viral DNA, as well as other capsid proteins, including protein VI.
ACKNOWLEDGMENTS
We thank Thomas Krey for the double Strep vector. We thank Crystal L. Moyer and Sébastien Igonet for critical readings of the manuscript.
This work was supported by NIH grants HL 054352 to G.R.N. and 1R01AI090280 to C.C.K.
Footnotes
Published ahead of print 4 June 2014
This is manuscript 25066 from The Scripps Research Institute.
REFERENCES
- 1.Stewart P, Fuller S, Burnett R. 1993. Difference imaging of adenovirus: bridging the resolution gap between X-ray crystallography and electron microscopy. EMBO J. 12:2589–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Russell W. 2009. Adenoviruses: update on structure and function. J. Gen. Virol. 90:1–20. 10.1099/vir.0.003087-0 [DOI] [PubMed] [Google Scholar]
- 3.Burnett RM. 1985. The structure of the adenovirus capsid. J. Mol. Biol. 185:125–143 [DOI] [PubMed] [Google Scholar]
- 4.van Raaij M, Mitraki A, Lavigne G, Cusack S. 1999. A triple beta-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 401:935–938. 10.1038/44880 [DOI] [PubMed] [Google Scholar]
- 5.Zubieta C, Schoehn G, Chroboczek J, Cusack S. 2005. The structure of the human adenovirus 2 penton. Mol. Cell 17:121–135. 10.1016/j.molcel.2004.11.041 [DOI] [PubMed] [Google Scholar]
- 6.Reddy V, Natchiar S, Stewart P, Nemerow G. 2010. Crystal structure of human adenovirus at 3.5 A resolution. Science 329:1071–1075. 10.1126/science.1187292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu H, Jin L, Koh S, Atanasov I, Schein S, Wu L, Zhou Z. 2010. Atomic structure of human adenovirus by cryo-EM reveals interactions among protein networks. Science 329:1038–1043. 10.1126/science.1187433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu H, Wu L, Zhou Z. 2011. Model of the trimeric fiber and its interactions with the pentameric penton base of human adenovirus by cryo-electron microscopy. J. Mol. Biol. 406:764–774. 10.1016/j.jmb.2010.11.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wickham T, Mathias P, Cheresh D, Nemerow G. 1993. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 73:309–319. 10.1016/0092-8674(93)90231-E [DOI] [PubMed] [Google Scholar]
- 10.Zhang W, Imperiale M. 2000. Interaction of the adenovirus IVa2 protein with viral packaging sequences. J. Virol. 74:2687–2693. 10.1128/JVI.74.6.2687-2693.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tribouley C, Lutz P, Staub A, Kedinger C. 1994. The product of the adenovirus intermediate gene IVa2 is a transcriptional activator of the major late promoter. J. Virol. 68:4450–4557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hosokawa K, Sung M. 1976. Isolation and characterization of an extremely basic protein from adenovirus type 5. J. Virol. 17:924–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson C, Young M, Flint S. 1989. Characterization of the adenovirus 2 virion protein, mu. Virology 172:506–512. 10.1016/0042-6822(89)90193-1 [DOI] [PubMed] [Google Scholar]
- 14.van Oostrum J, Burnett R. 1985. Molecular composition of the adenovirus type 2 virion. J. Virol. 56:439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vayda M, Rogers A, Flint S. 1983. The structure of nucleoprotein cores released from adenovirions. Nucleic Acids Res. 11:441–460. 10.1093/nar/11.2.441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vayda M, Flint S. 1987. Isolation and characterization of adenovirus core nucleoprotein subunits. J. Virol. 61:3335–3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chatterjee P, Vayda M, Flint S. 1986. Identification of proteins and protein domains that contact DNA within adenovirus nucleoprotein cores by ultraviolet light crosslinking of oligonucleotides 32P-labelled in vivo. J. Mol. Biol. 188:23–37. 10.1016/0022-2836(86)90477-8 [DOI] [PubMed] [Google Scholar]
- 18.Brown D, Westphal M, Burlingham B, Winterhoff U, Doerfler W. 1975. Structure and composition of the adenovirus type 2 core. J. Virol. 16:366–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chatterjee P, Vayda M, Flint S. 1985. Interactions among the three adenovirus core proteins. J. Virol. 55:379–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthews D, Russell W. 1998. Adenovirus core protein V interacts with p32–a protein which is associated with both the mitochondria and the nucleus. J. Gen. Virol. 79(Part 7):1677–1685 [DOI] [PubMed] [Google Scholar]
- 21.Ugai H, Borovjagin A, Le L, Wang M, Curiel D. 2007. Thermostability/infectivity defect caused by deletion of the core protein V gene in human adenovirus type 5 is rescued by thermo-selectable mutations in the core protein X precursor. J. Mol. Biol. 366:1142–1160. 10.1016/j.jmb.2006.11.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matthews D, Russell W. 1998. Adenovirus core protein V is delivered by the invading virus to the nucleus of the infected cell and later in infection is associated with nucleoli. J. Gen. Virol. 79(Part 7):1671–1675 [DOI] [PubMed] [Google Scholar]
- 23.Matthews D. 2001. Adenovirus protein V induces redistribution of nucleolin and B23 from nucleolus to cytoplasm. J. Virol. 75:1031–1038. 10.1128/JVI.75.2.1031-1038.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krey T, d'Alayer J, Kikuti C, Saulnier A, Damier-Piolle L, Petitpas I, Johansson D, Tawar R, Baron B, Robert B, England P, Persson MA, Martin A, Rey F. 2010. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 6:e1000762. 10.1371/journal.ppat.1000762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moyer C, Nemerow G. 2012. Disulfide-bond formation by a single cysteine mutation in adenovirus protein VI impairs capsid release and membrane lysis. Virology 428:41–47. 10.1016/j.virol.2012.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lobley A, Whitmore L, Wallace B. 2002. DichroWeb: an interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics 18:211–212. 10.1093/bioinformatics/18.1.211 [DOI] [PubMed] [Google Scholar]
- 27.Whitmore L, Wallace B. 2004. DichroWeb, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32:W668–W673. 10.1093/nar/gkh371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitmore L, Wallace B. 2008. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89:392–400. 10.1002/bip.20853 [DOI] [PubMed] [Google Scholar]
- 29.Sreerama N, Venyaminov SY, Woody RW. 2000. Estimation of the number of helical and strand segments in proteins using CD spectroscopy. Anal. Biochem. 287:243–251. 10.1006/abio.2000.4879 [DOI] [PubMed] [Google Scholar]
- 30.Sreerama NW, Woody RW. 1993. A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem. 209:32–44. 10.1006/abio.1993.1079 [DOI] [PubMed] [Google Scholar]
- 31.Wen J, Arakawa T, Philo J. 1996. Size-exclusion chromatography with on-line light-scattering, absorbance, and refractive index detectors for studying proteins and their interactions. Anal. Biochem. 240:155–166. 10.1006/abio.1996.0345 [DOI] [PubMed] [Google Scholar]
- 32.Folta-Stogniew E. 2006. Oligomeric states of proteins determined by size-exclusion chromatography coupled with light scattering, absorbance, and refractive index detectors. Methods Mol. Biol. 328:97–112. 10.1385/1-59745-026-X:97 [DOI] [PubMed] [Google Scholar]
- 33.Vaughan R, Fan B, You J-S, Kao C. 2012. Identification and functional characterization of the nascent RNA contacting residues of the hepatitis C virus RNA-dependent RNA polymerase. RNA 18:1541–1552. 10.1261/rna.031914.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang M, Szoka F. 1997. The influence of polymer structure on the interactions of cationic polymers with DNA and morphology of the resulting complexes. Gene Ther. 4:823–832. 10.1038/sj.gt.3300454 [DOI] [PubMed] [Google Scholar]
- 35.Duellman S, Burgess R. 2009. Large-scale Epstein-Barr virus EBNA1 protein purification. Protein Expr. Purif. 63:128–133. 10.1016/j.pep.2008.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harbottle R, Cooper R, Hart S, Ladhoff A, McKay T, Knight A, Wagner E, Miller A, Coutelle C. 1998. An RGD-oligolysine peptide: a prototype construct for integrin-mediated gene delivery. Hum. Gene Ther. 9:1037–1047. 10.1089/hum.1998.9.7-1037 [DOI] [PubMed] [Google Scholar]
- 37.Hema M, Murali A, Ni P, Vaughan R, Fujisaki K, Tsvetkova I, Dragnea B, Kao C. 2010. Effects of amino-acid substitutions in the Brome mosaic virus capsid protein on RNA encapsidation. Mol. Plant-Microbe Interact. 23:1433–1447. 10.1094/MPMI-05-10-0118 [DOI] [PubMed] [Google Scholar]
- 38.Yi G, Vaughan R, Yarbrough I, Dharmaiah S, Kao C. 2009. RNA binding by the brome mosaic virus capsid protein and the regulation of viral RNA accumulation. J. Mol. Biol. 391:314–326. 10.1016/j.jmb.2009.05.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Everitt E, Lutter L, Philipson L. 1975. Structural proteins of adenoviruses. XII. Location and neighbor relationship among proteins of adenovirion type 2 as revealed by enzymatic iodination, immunoprecipitation and chemical cross-linking. Virology 67:197–208 [DOI] [PubMed] [Google Scholar]
- 40.Graziano V, McGrath W, Suomalainen M, Greber U, Freimuth P, Blainey P, Luo G, Xie X, Mangel W. 2013. Regulation of a viral proteinase by a peptide and DNA in one-dimensional space. I. Binding to DNA and to hexon of the precursor to protein VI, pVI, of human adenovirus. J. Biol. Chem. 288:2059–2067. 10.1074/jbc.M112.377150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vellinga J, Van der Heijdt S, Hoeben R. 2005. The adenovirus capsid: major progress in minor proteins. J. Gen. Virol. 86:1581–1588. 10.1099/vir.0.80877-0 [DOI] [PubMed] [Google Scholar]
- 42.Benevento M, Di Palma S, Snijder J, Moyer C, Reddy V, Nemerow G, Heck A. 2014. Adenovirus composition, proteolysis and disassembly studied by in-depth qualitative and quantitative proteomics J. Biol. Chem. 289:11421–11430. 10.1074/jbc.M113.537498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ugai H, Wang M, Le L, Matthews D, Yamamoto M, Curiel D. 2010. In vitro dynamic visualization analysis of fluorescently labeled minor capsid protein IX and core protein V by simultaneous detection. J. Mol. Biol. 395:55–78. 10.1016/j.jmb.2009.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Newcomb W, Boring J, Brown J. 1984. Ion etching of human adenovirus 2: structure of the core. J. Virol. 51:52–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prage L, Pettersson U, Philipson L. 1968. Internal basic proteins in adenovirus. Virology 36:508–511. 10.1016/0042-6822(68)90178-5 [DOI] [PubMed] [Google Scholar]
- 46.Abril A, Salas M, Andreu J, Hermoso J, Rivas G. 1997. Phage phi29 protein p6 is in a monomer-dimer equilibrium that shifts to higher association states at the millimolar concentrations found in vivo. Biochemistry 36:11901–11908. 10.1021/bi970994e [DOI] [PubMed] [Google Scholar]
- 47.Alva V, Ammelburg M, Söding J, Lupas A. 2007. On the origin of the histone fold. BMC Struct. Biol. 7:17. 10.1186/1472-6807-7-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lischwe M, Sung M. 1977. A histone-like protein from adenovirus chromatin. Nature 267:552–554. 10.1038/267552a0 [DOI] [PubMed] [Google Scholar]
- 49.Mirza M, Weber J. 1982. Structure of adenovirus chromatin. Biochim. Biophys. Acta 696:76–86. 10.1016/0167-4781(82)90012-4 [DOI] [PubMed] [Google Scholar]
- 50.Wei Q, Jung HJ, Hwang DS, Hwang BH, Gim Y, Cha HJ. 2007. Escherichia coli-based expression of functional novel DNA-binding histone H1 from Carassius auratus. Enzyme Microb. Technol. 40:1484–1490. 10.1016/j.enzmictec.2006.10.028 [DOI] [Google Scholar]
- 51.Luger K, Mäder A, Richmond R, Sargent D, Richmond T. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251–260. 10.1038/38444 [DOI] [PubMed] [Google Scholar]
- 52.Harvey A, Downs J. 2004. What functions do linker histones provide? Mol. Microbiol. 53:771–775. 10.1111/j.1365-2958.2004.04195.x [DOI] [PubMed] [Google Scholar]
- 53.Hardison R, Eichner M, Chalkley R. 1975. An approach to histone nearest neighbours in extended chromatin. Nucleic Acids Res. 2:1751–1770. 10.1093/nar/2.10.1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sung M, Cao T, Coleman R, Budelier K. 1983. Gene and protein sequences of adenovirus protein VII, a hybrid basic chromosomal protein. Proc. Natl. Acad. Sci. U. S. A. 80:2902–2906. 10.1073/pnas.80.10.2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xue B, Dunbrack R, Williams R, Dunker A, Uversky V. 2010. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 1804:996–1010. 10.1016/j.bbapap.2010.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]