

ABSTRACT
There are nine subtypes of influenza A virus neuraminidase (NA), N1 to N9. In addition, influenza B virus also contains NA, and there are two influenza virus NA-like molecules, N10 and N11, which were recently identified from bats. Crystal structures for all of these proteins have been solved, with the exception of N7, and there is no published report of N6, although a structure has been deposited in the Protein Data Bank. Here, we present the N7 and N6 structures at 2.1 Å and 1.8 Å, respectively. Structural comparison of all NA subtypes shows that both N7 and N6 highly resemble typical group 2 NA structures with some special characteristics, including an additional cavity adjacent to their active sites formed by novel 340-loop conformations. Comparative analysis also revealed new structural insights into the N-glycosylation, calcium binding, and second sialic acid binding site of influenza virus NA. This comprehensive study is critical for understanding the complexity of the most successful influenza drug target and for the structure-based design of novel influenza inhibitors.
IMPORTANCE Influenza viruses impose a great burden on society, by the human-adapted seasonal types as well as by variants that occasionally jump from the avian reservoir to infect humans. The surface glycoprotein neuraminidase (NA) is essential for the propagation of the virus and currently the most successfully drug-targeted molecule. Therefore, the structural and functional analysis of NA is critical for the prevention and control of influenza infections. There are nine subtypes of influenza A virus NA (N1 to N9). In addition, influenza B virus also contains NA, and there are two influenza NA-like molecules, N10 and N11, which were recently identified in bats. Crystal structures for all of these proteins have been solved and reported with the exception of N7 and N6. The structural analysis of influenza virus N7 and N6 presented in this study therefore completes the puzzle and adds to a comprehensive understanding of influenza virus NA.
INTRODUCTION
Influenza virus is the causative agent of endemic and occasional pandemic flu infections (1). The outbreak of the 2009 swine origin (S-OIV) H1N1 influenza A virus strain (09-pH1N1) reminded the world about the pandemic potential of influenza (2, 3). The recently emerged strain H7N9 also has caused great concern (4). H7N7 is another potential pandemic pathogen that can be either highly pathogenic or of low pathogenicity (5, 6). H7N7 can infect humans, birds, pigs, seals, and horses and has also infected mice, ferrets, and monkeys in laboratory studies (7, 8). In 2003, there were 89 confirmed human-infection cases of H7N7 in the Netherlands following a poultry outbreak on several farms (9). In 2008, highly pathogenic H7N7 was found in England and caused a large number of deaths in poultry (10). Moreover, highly pathogenic H7N7 was recently found in Chinese markets during a search for H7N9 (11). Therefore, this unusual zoonotic potential represents a pandemic threat that should be monitored and studied carefully. Numerous works have focused on the characterization of H7; however, very few studies have been done on the N7 neuraminidase (NA).
Influenza virus contains two important surface glycoproteins, HA (hemagglutinin) and NA, in addition to the matrix M2 protein. HA is responsible for receptor binding and membrane fusion (12). NA functions as the receptor-destroying element. Specifically, NA cleaves terminally linked sialic acid (SA) from various glycoconjugates on the host cell surface to facilitate the release of progeny virions and prevent their aggregation (13). Excluding influenza B virus NA and the newly identified NA-like molecules N10 and N11, the nine subtypes of influenza A virus NA are classified into two groups according to their primary sequences (14, 15). Group 1 comprises N1, N4, N5, and N8, and group 2 comprises N2, N3, N6, N7, and N9 (16). NA is the most successful influenza drug target (17, 18). Extensive structural and functional investigations of the group 2 NAs N2 and N9 led to the development of the most commonly used influenza drugs, oseltamivir (Tamiflu) and zanamivir (Relenza), which mimic the binding mode of the transition state analogue 2-deoxy-2,3-dehydro-N-acetylneuraminic acid (Neu5Ac2en) (19). Later, structural analysis of N1, N4, N5, and N8 revealed an additional cavity adjacent to the NA active site, found only in group 1 NA (20, 21). This cavity is formed by the 150-loop and was therefore named the 150-cavity. Novel inhibitors, including 3-(p-tolyl)allyl-Neu5Ac2en, have been successfully designed to target this cavity, thereby conferring selectivity toward group 1 NAs (22).
Interestingly, we found that the group 1 N1 from 2009 swine origin (S-OIV) H1N1 influenza A virus strain 09-pH1N1 (09N1) contained no 150-cavity in its crystal structure, thereby resembling group 2 NAs (23). Yet, group 1-specific inhibitors like 3-(p-tolyl)allyl-Neu5Ac2en also inhibit 09N1 with an affinity similar to that of other group 1 NAs (22). Therefore, we speculate that the atypical group 1 09N1 may possess a 150-loop that is more easily opened than those of group 2 NAs. However, the exact role of the 150-cavity in NA catalysis and inhibitor binding is not well understood. Our recent work demonstrated that oseltamivir carboxylate can induce opening of the rigid N2 150-loop, which further illustrates the complexity of influenza virus NA structures (24). Skehel's group reported that the 09N1 150-cavity could be found under a condition of phosphate ion binding with an Ile223Arg mutation (N1 numbering) (25). These discrepancies have raised questions as to whether or not the 150-cavity is conserved only for group 1 members and whether group 2 NAs have their own specificities.
Although structures for 8 of the 9 influenza A virus NA subtypes as well as NA-like N10, N11, and influenza B virus NA have all been solved, the structure of one final NA subtype, N7, remains unreported. Moreover, the structural analysis of N6 remains unpublished, although the structure data have been deposited in the Protein Data Bank (PDB ID 1V0Z). Therefore, we expressed purified and crystallized N7 and N6 proteins, solving their structures at 2.1 Å and 1.8 Å, respectively. The structural comparison of all NA subtypes included in this study offers a comprehensive understanding of the NA evolution and useful data for the design of next-generation NA inhibitors (26).
MATERIALS AND METHODS
Recombinant NA production.
NA proteins were prepared based upon an original method by Xu et al. with some modifications (21, 27). cDNAs encoding residues 83 to 468 (N2 numbering is used throughout the text, except for 09N1-Ile223Arg in order to be consistent with the original article) of strain A/Netherlands/219/03 (H7N7) N7, strain A/mallard/ALB/196/1996 (H10N7) N7, and strain A/Chicken/Nanchang/7-010/2000 (H3N6) N6 were cloned into pFastBac1 (Invitrogen), separately, with a GP67 signal peptide, a 6×His tag, a tetramerization sequence, and a thrombin cleavage site at the N terminus. Recombinant baculovirus was prepared following the manufacturer's protocol (Invitrogen). Hi5 suspension cultures were grown in X-press serum-free medium (Lonza) and shaken at a speed of 120 rpm at 301 K. Cells were infected with high-titer recombinant baculovirus when the density reached 2 million cells per ml. After 48 h, the cultures were collected and centrifuged, and then the supernatant was filtered and loaded onto a 5-ml HisTrap FF column (GE Health). The HisTrap column was then washed with 20 mM imidazole, and NA protein was eluted using 300 mM imidazole. After dialysis, protein was treated with thrombin (3 U/mg of NA; Sigma) overnight at 277 K, and the digested NA was purified using gel filtration chromatography with a Superdex-200 10/300 GL column (GE Healthcare). NA fractions were analyzed by SDS-PAGE. High-purity NA fractions were pooled and concentrated using a membrane concentrator with a molecular mass cutoff of 10 kDa (Millipore). A buffer of 20 mM Tris–50 mM NaCl (pH 8.0) was used for gel filtration and protein crystallization.
Crystallization and soaking experiments.
Crystallization conditions were screened using the sitting drop vapor diffusion method with commercial krorits (Hampton Research). Initial N7 crystals from the H7N7 strain were obtained by mixing 1 μl of the concentrated protein at 7 mg/ml in 20 mM Tris, pH 8.0, and 50 mM NaCl with 1 μl buffer of 0.1 M sodium citrate tribasic dihydrate (pH 5.0)–30% (vol/vol) Jeffamine ED-2001 (pH 7.0) at 291 K. However, all the crystals of N7 NA from H7N7 did not diffract well. After rounds of optimization, the best crystals diffracted at only 3.7 Å. We then tried to crystallize N7 from H10N7 and obtained 2.1-Å resolution data with a buffer of 0.1 M calcium acetate, 0.1 M sodium acetate (pH 4.5), and 10% (wt/vol) polyethylene glycol (PEG) 4000. Quality N6 crystals were obtained with 0.1 M sodium chloride, 0.1 M Tris (pH 8.0), and 8% (wt/vol) PEG 20000. N2-Tyr406Asp and N5 crystals were prepared using hanging drops as previously reported (28, 29), and these crystals were soaked with 3′sialyllactose and LSTa (Neu5Acα2-3Galβ1-4GlcNAcβ1-3Galβ1-4Glc), respectively, for 3 h at 291 K and then flash cooled at 100 K. For inhibitor soaking, NA crystals were soaked in 10 mM inhibitor for 2 h at 291 K and then flash cooled at 100 K. Diffraction data were collected at KEK beamline NE3A and SSRF beamline BL17U.
Data collection, processing, and structure solution.
Diffraction data were processed and scaled using HKL2000 (30). Data collection and processing statistics are summarized in Table 1. The structures of N7 and N6 were solved by molecular replacement using Phaser from the CCP4 program suite with the structures of N2 (PDB ID 1NN2) and N6 (PDB ID 1V0Z) as the search models, respectively (31). Initial restrained rigid-body refinement and manual model building were performed using Refmac5 (32) and Coot (33), respectively. Further rounds of refinement were performed using the phenix.refine program implemented in the Phenix package with coordinate refinement, isotropic ADP refinement, and bulk solvent modeling (34). The stereochemical quality of the final model was assessed with the program Procheck (35). The surface area and volume of the NA active site and 150-cavity were calculated using the program Computed Atlas of Surface Topography of proteins (CASTp; http://sts-fw.bioengr.uic.edu/castp/calculation.php).
TABLE 1.
Crystallographic X-ray diffraction and refinement statisticsa
| Characteristic (unit) | N7 | N6 | N5-LSTa | N6-laninamivir | N7-oseltamivir |
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | P4212 | P4212 | P4 | P4212 | P4212 |
| Cell dimensions | |||||
| a (Å) | 110.50 | 138.10 | 112.30 | 137.90 | 115.50 |
| b (Å) | 110.50 | 138.10 | 112.30 | 137.90 | 115.50 |
| c (Å) | 121.40 | 150.00 | 66.80 | 148.80 | 121.70 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50–2.10 (2.18–2.10) | 50–1.80 (1.86–1.80) | 50–1.70 (1.76–1.70) | 50–1.95 (2.02–1.95) | 50–2.30 (2.38–2.30) |
| Rmerge | 0.125 (0.714) | 0.115 (0.540) | 0.146 (0.586) | 0.082 (0.320) | 0.113 (0.365) |
| I/σI | 19.29 (2.85) | 14.9 (3.1) | 16.1 (5.8) | 25.66 (7.12) | 22.59 (8.966) |
| Completeness (%) | 98.5 (91.1) | 98.1 (96.3) | 100.0 (100.0) | 99.9 (100.0) | 100.0 (100.0) |
| Redundancy | 9.4 (8.3) | 5.1 (4.6) | 8.1 (7.9) | 7.7 (7.6) | 12.8 (12.9) |
| Refinement | |||||
| Resolution (Å) | 33.59–2.10 | 47.69–1.80 | 42.96–1.70 | 47.48–1.95 | 41.89–2.30 |
| No. of reflections | 44,404 | 131,314 | 88,296 | 105,174 | 37,039 |
| Rwork/Rfree | 0.1746/0.2108 | 0.1408/0.1562 | 0.1172/0.1759 | 0.1451/0.1611 | 0.1669/0.2066 |
| No. of atoms | |||||
| Protein | 6,092 | 6,268 | 6,172 | 6,144 | 6,112 |
| Ligand/ion | 82 | 30 | 96 | 136 | 102 |
| Water | 520 | 1,056 | 743 | 846 | 463 |
| B-factors | |||||
| Protein | 26.2 | 10.9 | 14.5 | 20.2 | 24.9 |
| Water | 33.8 | 28.5 | 28.0 | 35.7 | 31.3 |
| RMSD | |||||
| Bond lengths (Å) | 0.005 | 0.006 | 0.003 | 0.008 | 0.005 |
| Bond angles (°) | 1.007 | 1.173 | 1.043 | 1.183 | 1.028 |
| Ramachandran plot | |||||
| Most favored (%) | 83.8 | 86.9 | 86.5 | 86.6 | 83.8 |
| Additionally favored (%) | 15.6 | 13.0 | 13.2 | 13.4 | 15.1 |
| Generally allowed (%) | 0.6 | 0.2 | 0.3 | 0 | 1.0 |
| Disallowed (%) | 0 | 0 | 0 | 0 | 0 |
Values in parentheses are for the highest-resolution shell. Rmerge = ∑|I(k) − <I(k)>|/∑I(k), where I(k) is the value of the kth measurement of the intensity of reflection and <Ik> is the mean intensity of that reflection; Rwork = ∑|Fo − Fc|/∑Fo for the 95% of the reflection data used in the refinement, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively; Rfree is the equivalent of Rwork, except that it was calculated for a randomly chosen 5% test set excluded from the refinement. I, intensity of a reflection; σI, standard deviation of intensity.
NA enzymatic inhibition assay.
NA activity was assayed using the fluorogenic substrate 2′-(4-methylumbelliferyl)-N-acetylneuraminic acid (MUNANA) as described by Potier et al. with some modifications (36). To quantify the potency of NA inhibitors, 10 μl purified N7 protein (10 nM) was mixed with 10 μl inhibitor and incubated for 30 min at room temperature. NA and inhibitors were carefully diluted in fresh phosphate-buffered saline (PBS). At least five concentrations of each inhibitor were used for each repeat. After incubation, 30 μl of 166 μM MUNANA in 33 mM morpholineethanesulfonic acid (MES) buffer and 4 mM CaCl2 (pH 6.0) was added to the solution to start the reaction using a multichannel pipette (Eppendorf). A positive control and a negative control were included in each lane. After starting the reaction for each condition on the plate, the reaction mixture was immediately loaded onto a SpectraMax M5 instrument (Molecular Devices). Fluorescence was quantified over the course of 30 min at an excitation wavelength of 355 nm and an emission wavelength of 460 nm. Single time points at which the positive control produced a relative fluorescence signal of approximately 1,000 were chosen. At least two replicates were used for each condition, and the 50% inhibitory concentration (IC50) for each inhibitor was calculated by sigmoidal fitting of the log[inhibitor] versus percent inhibition data using GraphPad Prism.
Protein structure accession numbers.
The structures determined in this study have been deposited in the Protein Data Bank (PDB) with the following codes: 4QN3 (N7), 4QN4 (N6), 4QN5 (N5-LSTa), 4QN6 (N6-laninamivir), and 4QN7 (N7-oseltamivir).
RESULTS
Overall structures of N7 and N6.
The crystal structures of N7 and N6 were solved at 2.1 Å and 1.8 Å, respectively, and exhibited overall structures similar to that of canonical influenza virus NA. N7 and N6 subunits are assembled into box-shaped tetramers, with each monomer containing a six-bladed propeller-like arrangement of antiparallel β-sheets (Fig. 1). Most NA structures contain 4 β-strands in each blade; however, N7 was found to contain only three β-strands in blade 6, similar to the NA-like molecule N10 (Fig. 1, left) (37). N6 was also found to contain only three β-strands in both blade 4 and blade 6 (Fig. 1, right). Our A/Chicken/Nanchang/7-010/2000 (H3N6) N6 structure is highly similar to that of A/duck/England/1/1956 (H11N6) N6 (PDB ID 1V0Z), with an RMSD (root mean square deviation) of 0.164 (38). There are just 12 amino acids that are different, and none are located near the active site.
FIG 1.
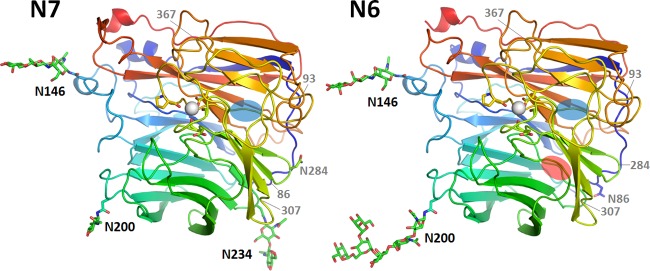
Overall structures of N7 and N6. N7 (left) adopts a propeller-like structure whereby each monomer has six β-sheets (named blades 1 to 6), with blade 6 containing only three β-strands. Each N6 monomer (right) has six blades, with blades 4 and 6 containing only three β-strands. The missing β-sheets in blade 6 and blade 4 are indicated by blue ovals and a red oval, respectively. Three N-glycosylation sites, Asn146, Asn200, and Asn234, are observed in N7, and two N-glycosylation sites, Asn146 and Asn200, are observed in N6. These occupied N-glycosylation sites are indicated by black labels, the putative N-glycosylation sites with no observed glycans are labeled with an N followed by the position number, and N-glycosylation sites that are present in other influenza virus NA structures are indicated by gray labels with only the position number. There is a single calcium ion (white sphere) binding site in each N7 and N6 monomer. In both structures, the calcium ion is coordinated by Asp293, Gly297, Asp324, and Pro347.
Comprehensive structural alignment among N7, N6, and all available influenza NA subtypes reveals RMSDs ranging from 0.433 to 1.952 for Cα atoms of single NA monomers (Table 2). N7 and N6 are most structurally similar to each other and least similar to influenza virus B NA, N10, and N11. As expected, N7 and N6 are also more structurally similar to group 2 influenza A NAs than to group 1 NAs. Therefore, the structural alignment is consistent with the phylogenetic analysis, confirming that typical N1 to N9 can be accurately divided into two distinct groups.
TABLE 2.
Comprehensive structural alignment among N1 to N9, Flu B, N10, and N11
| NA | PDB ID | RMSD (Å) of the Cα atoms of one NA monomer |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N7a | N6b | N2 | N3 | N9 | VN04N1 | 09N1 | N4 | N5 | N8 | Flu B NAc | N10 | ||
| N6 | 0.433 | ||||||||||||
| N2 | 1NN2 | 0.683 | 0.706 | ||||||||||
| N3 | 4HZV | 0.598 | 0.731 | 0.566 | |||||||||
| N9 | 7NN9 | 0.448 | 0.337 | 0.726 | 0.722 | ||||||||
| VN04N1 | 2HTY | 0.823 | 0.787 | 0.910 | 0.765 | 0.536 | |||||||
| 09N1 | 3NSS | 0.761 | 0.738 | 0.734 | 0.713 | 0.714 | 0.311 | ||||||
| N4 | 2HTV | 0.887 | 0.806 | 0.865 | 0.817 | 0.561 | 0.371 | 0.434 | |||||
| N5 | 3SAL | 0.920 | 0.862 | 0.815 | 0.791 | 0.434 | 0.540 | 0.536 | 0.544 | ||||
| N8 | 2HT5 | 0.916 | 0.818 | 0.850 | 0.815 | 0.536 | 0.536 | 0.561 | 0.547 | 0.364 | |||
| Flu B NA | 1NSB | 1.654 | 1.330 | 1.636 | 1.542 | 1.752 | 1.384 | 1.269 | 1.522 | 1.306 | 1.490 | ||
| N10 | 4FVK | 1.687 | 1.952 | 1.925 | 1.741 | 1.737 | 1.529 | 1.463 | 1.753 | 1.591 | 1.691 | 2.557 | |
| N11 | 4K3Y | 1.686 | 1.572 | 1.544 | 1.479 | 1.408 | 1.586 | 1.471 | 1.359 | 1.602 | 1.390 | 2.161 | 0.903 |
N7 from strain A/mallard/ALB/196/1996 (H10N7).
N6 from strain A/Chicken/Nanchang/7-010/2000 (H3N6).
Flu B NA from strain B/Beijing/1/87.
We also solved the structure of N7 in complex with oseltamivir carboxylate at 2.3 Å and that of N6 in complex with laninamivir at 2.0 Å (Fig. 2). The binding modes of these classical inhibitors are virtually the same as in previously reported complex structures (39, 40). Oseltamivir carboxylate, zanamivir, peramivir, and laninamivir inhibited N7 at the nanomolar level, with IC50s of 0.85, 0.87, 3.0, and 0.54 nM, respectively. The novel inhibitor MS-257 (26) also inhibits N7 with an IC50 of 0.63 nM (Table 3). These values are highly consistent with our data for inhibition of other group 2 NAs (27, 40).
FIG 2.

Analysis of the N7 and N6 NA active sites compared to group 2 N2 and group 1 N5 NA. N7 (A) and N6 (B) both contain a 340-cavity adjacent to their active site. This 340-cavity has not been found in any other group 1 or group 2 NA structures, including group 2 N2 (PDB ID 1NN2) (C) or group 1 N5 (PDB ID 3SAL) (D). Similar to the bottom of the NA active site cavity, the 340-cavity is highly negatively charged. N7 oseltamivir carboxylate (A) binding and N6 laninamivir (B) binding are virtually identical to those of other group 2 NA structures.
TABLE 3.
IC50s and 95% confidence intervals (CI) for the inhibition of N7
| IC50 (nM), 95% CI | ||||
|---|---|---|---|---|
| N7-oseltamivir | N7-zanamivir | N7-laninamivir | N7-peramivir | N7-MS-257 |
| 0.84, 0.69–1.02 | 0.72, 0.60–0.87 | 1.71, 1.23–2.38 | 0.57, 0.51–0.63 | 0.61, 0.49–0.74 |
Structural comparison of influenza NA N-glycosylation sites.
Now that the structures of all subtypes of influenza virus NA are available, it is possible to compare their N-glycosylation (Table 4). We have omitted N10 and N11 from this overview, as their glycosylation has been extensively compared with that of influenza A viruses recently (37, 41). It must be noted that in several crystal structures no glycan was detected at some of the predicted N-glycosylation sites (indicated as an open circle in Table 4). It is hard to state whether this is due to technical issues related to the crystal structure or is a reflection of the in vivo expression system.
TABLE 4.
Comprehensive N-glycosylation analysis of reported NA crystal structuresa
| NA (PDB ID) | Asn86 | Asn93 | Asn146 | Asn200 | Asn234 | Asn284 | Asn307 | Asn367 |
|---|---|---|---|---|---|---|---|---|
| N1 | ||||||||
| VN04N1 (2HTY) | ○ | × | ◆ | × | ○ | × | × | × |
| 18N1 (3BEQ) | ○ | × | ◆ | × | ○ | × | × | × |
| 09N1 (3NSS) | ◆ | × | ◆ | × | ○ | × | × | × |
| 09N1-I223R (4B7M) | ◆ | × | ◆ | × | ◆ | × | × | × |
| N2 | ||||||||
| N2 (1NN2, 2AEQ) | ◆ | × | ◆ | ◆ | ◆ | × | × | × |
| N2 (4GZO) | ◆ | × | ◆ | ○ | ○ | × | × | ◆ |
| N2 (4KIH) | ○ | × | ◆ | ◆ | ○ | × | × | × |
| N3 (4HZV) | × | × | ◆ | × | × | × | ◆ | × |
| N4 (2HTV) | × | × | ◆ | × | × | × | × | × |
| N5 (3SAL) | ○ | ◆ | ◆ | × | × | × | × | × |
| N6 | ||||||||
| N6 (1V0Z) | ◆ | × | ◆ | ◆ | × | × | × | × |
| N6 | ○ | × | ◆ | ◆ | × | × | × | × |
| N7 | ○ | × | ◆ | ◆ | ◆ | × | × | × |
| N8 (2HT5) | ○ | × | ◆ | × | × | × | × | × |
| N9 (7NN9)b | ◆ | × | ◆ | ◆ | × | × | × | × |
| Flu B (1INF, 1NSB) | × | × | ○ | × | × | ◆ | × | × |
Symbols: ◆, a glycan is found in the crystal structure to occupy the N-glycosylation site; ○, no glycan is observed to occupy the putative N-glycosylation site in the crystal structure; ×, no N-glycosylation sequon is present in the sequence.
All available N9 structures are included (PDB IDs 7NN9, 1A14, 1NMC, 2B8H, 1NCD, 4MWJ, and 4MWL).
In total, eight different putative N-glycosylation sites are predicted in the complete set of NAs. Of these, the N-glycosylation site of Asn146 contained N-glycans in all structures elucidated (indicated by a black diamond in Table 4), with the exception of influenza virus B structures (PDB ID 1INF and 1NSB). The influenza virus B NA structures (PDB ID 1INF and 1NSB) have only one occupied N-glycosylation site at Asn284, which is not found in any of the influenza A virus structures, while the putative N-glycosylation site Asn146 contains no glycan electron density in the crystal structures. The remaining six N-glycosylation sites can be divided into unique sites and the ones that are shared by several different NA subtypes. We discuss them in order of appearance in the sequence, starting with Asn86. This site is predicted or shown in all influenza A virus NAs, except for N3 (PDB ID 4HZV) and N4 (PDB ID 2HTV). For subtype N1, N-glycans of the Asn86 site are observed in A/Vietnam/3028/2004 (H5N1) N1 (PDB ID 2HTY) (VN04N1) and A/Brevia/Mission/1/18 (H1N1) N1 (PDB ID 3BEQ) (18N1), and for subtype N2, it is not observed in A/RI/5+/57 (H2N2) N2 (PDB ID 4K1H) only. Asn93 is uniquely found in N5 (PDB ID 3SAL). Asn200 is found in four subtypes: N2 (PDB IDs 1NN2, 2AEQ, and 4K1H), N6 (PDB ID 1V0Z), and N7 and N9 (PDB IDs 7NN9, 1A14, 1NMC, 2B8H, 1NCD, 4MWJ, and 4MWL). For Asn234, it is predicted in all the N1 and N2 solved structures but shown only in 09N1-Ile223Arg (PDB ID 4B7M) (N1 numbering), A/Tokyo/3/67 (H2N2) N2 (PDB ID 1NN2), and A/Memphis/31/98 (H3N2) N2 (PDB ID 2AEQ). Asn234 is also observed in our N7 structure. Asn307 and Asn367 are unique sites, found only in N3 (PDB ID 4HZV) and A/Tanzania/205/2010 (H3N2) N2 (PDB ID 4GZO), respectively. The latter site is interesting, as it is shown only in A/Tanzania/205/2010 (H3N2) N2 (PDB ID 4GZO) and not predicted in any of the other solved N2 structures, including A/RI/5+/57(H2N2) N2 (PDB ID 4K1H), A/Tokyo/3/67 (H2N2) N2 (PDB ID 1NN2), and A/Memphis/31/98 (H3N2) N2 (PDB ID 2AEQ). Also, for the Asn86, Asn200, and Asn234 sites, intrasubtype glycosylation variation is observed in the structures. This suggests that N-glycosylation may be one of the factors to differentiate individual NA proteins within each subtype, as there is currently very little known intrasubtype variation in terms of tertiary structure.
Comprehensive analysis of influenza virus NA loop variations.
A close comparison of the loop regions of N7 and N6 with all other available influenza virus A NA structures was carried out to search for novel features. The 150-loops (residues 147 to 152), 270-loops (residues 267 to 276), 380-loops (residues 380 to 390), and 430-loops (residues 429 to 433) of N7 and N6 adopt the typical group 2 conformation described in our previous analysis of N3 (Fig. 3A) (39). Interestingly, one loop, the 340-loop (residues 342 to 347), was found to adopt novel conformations in both N7 and N6 (Fig. 3A and B).
FIG 3.

Comprehensive analysis of influenza virus NA loops. (A) Superimposition of NA monomers with an emphasis on the 150-loop, 270-loop, 380-loop, and 430-loop. The colors of different NAs are as follows: N3 (PDB ID 4HZV), cyan; N6 (PDB ID 1V0Z), pink; A/Chicken/Nanchang/7–010/2000 (H3N6) N6, orange; A/mallard/ALB/196/1996 (H10N7) N7, yellow-orange; VN04N1 (PDB ID 2HTY), 09N1 (PDB ID 3NSS), N2 (PDB ID 1NN2), N4 (PDB ID 2HTV), N5 (PDB ID 3SAL), N8 (PDB ID 2HT5), and N9 (PDB ID 7NN9) are all in gray. (B) Cartoon view of the 340-loop. The 340-loops of N7 and N6 adopt novel conformations. A calcium ion is shown as a sphere. (C) Sequence alignment of the 320-loop (residues 324 to 328) and the 340-loop (residues 342 to 347) in N1 to N9 subtypes. All the N1 to N9 NA sequences in the NCBI database are included. The image was created with the WebLogo program (http://weblogo.berkeley.edu/). The overall height of the stack indicates the sequence conservation at that position, while the height of the symbols within the stack indicates the relative frequency of each amino acid at that position. Amino acids hypothetically responsible for the loop orientation are boxed in red.
The 340-loops of N7 and N6 are oriented further away from the conserved calcium ion than in all other known influenza virus NA structures (Fig. 3B). N7 and N6 both contain a conserved Ser326 residue, which forms hydrogen bonds with the 340-loop. All other influenza virus A NA subtypes contain a conserved Pro326, with no side chain to hydrogen bond with the 340-loop (Fig. 3C). Therefore, Ser326 probably is a key factor underlying the unique 340-loop conformation of N7 and N6. In addition, unlike other NA subtypes, N3, N6, and N7 all contain a conserved Pro347 residue, yet N7 and N6 each contain a second, additional conserved proline in their 340-loop. Specifically, N7 contains Pro344 and N6 contains Pro345 (Fig. 3C). We suspect that the additional proline residue may be another important factor in determining the unique 340-loop conformation. In N7, the distance between Pro344 and Trp295 is 12.66 Å, and in N6 the distance between Pro345 and Trp295 is 12.7 Å. This is much further than the corresponding distance in N2 (Gly345-Trp295) and N5 (Asn345-Trp295), with distances of 5.66 Å and 5.59 Å, respectively.
Most importantly, the unique 340-loop conformation results in the formation of a novel cavity adjacent to the active site in both N7 and N6, which we refer to as the 340-cavity (Fig. 2). No distinct 340-cavity is present in any other NA structures, including N2 and N5, which were chosen as representatives of group 2 and group 1 NAs, respectively. The surface of the 340-cavity is about 100 Å2 and is highly electronegative (Fig. 2). Similar to the 150-cavity, the 340-cavity is also adjacent to the active site and therefore may exert some effects on NA ligand binding or catalysis.
This is seen in the case of group 1 NAs that contain a conserved Tyr347 residue, which can hydrogen bond with the carboxylate of sialic acid and its analogues (20). Residue 347 of the 340-loop is also directly involved in calcium ion binding, which we discuss next.
Comprehensive analysis of influenza virus NA calcium ion binding.
The 340-loop is also important for the conserved NA calcium binding site. Most NA structures are able to maintain two interactions between the 340-loop and the conserved calcium ion via the backbone carbonyl groups of the 345 and 347 residues. Position 347 has previously been demonstrated to be especially important for calcium binding in N1 (42). With the unique conformation of the 340-loop in N7 and N6, only Pro347 is able to form a calcium interaction (Fig. 3B), which may have some effect on the stability of N7 and N6.
Some influenza virus NA structures, including those of B/Lee/40 NA, B/Beijing/1/87, N3, and 09N1, contain an additional calcium ion in the center of their tetramer. Interestingly, 09N1 also contains an additional calcium ion in each monomer, and A/NWS/whale/Maine/1/84 (H1N9) N9 structures sometimes contain no conserved calcium ion (PDB IDs 2B8H and 1NMA) (43). In this regard, N7 and N6 resemble typical influenza virus NA structures with a single calcium ion.
The second sialic acid binding site of influenza virus NA.
In addition to the NA active site, which binds sialic acid and its analogues, a second sialic acid binding site has been discovered (38, 44, 45) in some avian influenza virus NAs, including N6 (PDB ID 1W20) and N9 (PDB ID 1MWE) (Fig. 4B). When bound to the NA active site, αNeu5Ac adopts a twisted boat conformation, which appears to be a critical event in the NA enzymatic mechanism. However, in both N6 and N9, the αNeu5Ac in the second binding site adopts a chair conformation, similar to Neu5Ac in α2,3 and α2,6 receptors bound to influenza virus HA. It is postulated that a terminal α-sialic acid oligosaccharide should be a better ligand than sialic acid monosaccharide, which is present mostly as the β-anomer (15). However, there have been no reports of any α2,3 or α2,6 receptor bound to the NA second sialic acid binding site.
FIG 4.

Influenza virus NA second sialic acid binding site. (A) Sequence alignment of the key residues of the influenza virus A NA second sialic acid binding site. All the N1 to N9 NA sequences in the NCBI database are included. The image was created with the WebLogo program (http://weblogo.berkeley.edu/). The overall height of the stack indicates the sequence conservation at that position, while the height of the symbols within the stack indicates the relative frequency of each amino acid at that position. Amino acids that interact with sialic acid are marked with a red star. (B) Structural analysis of the second binding site in N6 (PDB ID 1W20), N9 (PDB ID 1MWE), N2-Tyr406Asp (PDB ID 4H53), and N5. Ser367, Ser370, Ser372, and Asn400 hydrogen bond with Neu5Ac, and Trp403 forms hydrophobic interactions with the Neu5Ac N-acetyl group.
Here, we soaked N5 with the α2,3-linked glycan LSTa, which was identified in the second binding site with Neu5Ac in the active site (Fig. 4B; Table 5). This is the first direct evidence of a second SA-binding site in N5. Moreover, as far as we know, this is the first report of soaking an intact influenza virus receptor into the NA second sialic acid binding site. Detailed analysis of the N5 SA binding site shows that the Neu5Ac carboxylate hydrogen bonds with the Ser367 hydroxyl oxygen and the Asn400 amide side chain. The main-chain carbonyl oxygen of Asn400 interacts with both the 4-hydroxyl oxygen and the 5-acetamido nitrogen of Neu5Ac. The Neu5Ac 5-acetamido nitrogen hydrogen bonds with Ser372 and also forms a hydrophobic interaction with Trp403, as it rests 3.8 Å above the indole side chain. The Neu5Ac O8 hydroxyl interacts with the Ser370 hydroxyl, whereas the neighboring hydroxyl groups (O7 and O9) are exposed to the solvent. Details of the hydrogen bond interactions are listed in Table 5. There is no direct interaction between the galactose residue and the second binding site, and there is no electron density for the rest of the glycan. This is quite different from HA receptor binding, where the galactose and glucose residues also form important interactions with the HA receptor binding site. The Neu5Ac in the active site or second binding site adopts the boat or chair conformation, respectively.
TABLE 5.
Interactions between the ligands in the second sialic acid binding site and the NA molecules
| Ligands in the second sialic acid binding site and hydrogen bond distancea (Å) | |||
|---|---|---|---|
| N5 | N2Tyr406Asp | N6 | N9 |
| Ser367, 2.55; Ser370, 2.78; Ser372, 2.91; Asn400, 3.16, 2.76, 3.11. | Ser367, 2.53; Ser370, 2.67; Ser372, 2.81; Asn400, 3.18, 3.04, 2.71. | Ser367, 2.85; Ser370, 2.58, 3.29; Asn400, 3.25, 2.79, 2.94. | Ser367, 2.55; Ser370, 2.66; Ser372, 2.75; Asn400, 2.96, 2.90, 2.61; Lys432, 2.74. |
All hydrogen bond distances refer to molecule A of each structure.
Our previous attempt to gain an NA active site receptor complex by soaking 3′sialyllactose into N2-Tyr406Asp (PDB ID 4H53) (28), which has impaired activity, resulted in a complex structure with 3′sialyllactose in the second binding site as well (Fig. 4B). As far as we know, this is the first direct evidence of a second SA binding site in N2 seen in the crystal structure.
Comparison of all the structures with a second SA binding site illustrates that this site is comprised of the 370-loop (residues 366 to 373), 400-loop (residues 399 to 404), and 430-loop (residues 430 to 433), as previously reported (45). Through the amino acid alignment of the three loops, we can see that many more NAs are predicted to contain a second binding site (Fig. 4A). The second binding site is near the active site and may facilitate the binding of SA-containing glycoconjugates or provide supplemental receptor binding activity (44). In addition, the NA inhibitor also appears in the second sialic acid binding site in the N6-zanamivir complex (PDB ID 2CML), while the second binding site in the PDB file is listed with only 50% occupancy (38). However, the exact role of the second SA binding site in influenza virus infection remains to be clearly established.
DISCUSSION
Influenza virus NA is currently the most successful anti-influenza drug target, and therefore the structural and functional analysis of NA is critical for the prevention and control of influenza infections. The structural analysis of influenza virus N7 and N6 is therefore necessary for a comprehensive understanding of influenza virus NA. In this study, the crystal structures of A/mallard/ALB/196/1996 (H10N7) N7 and A/Chicken/Nanchang/7-010/2000 (H3N6) N6 were solved in order to address this problem. From the overall structural comparison of N7 and N6 with all other NA subtypes (Table 2), it is clear that they both fall into the typical group 2 NA classification.
The 150-cavity has been found to be the most distinct group-specific feature of influenza virus NA and an important target for drug design. Therefore, the comprehensive comparison of 150-loops (residues 147 to 152) and the active sites of all NA subtypes is of particular interest (Fig. 5). N7 and N6 both possess no 150-cavity and have the conserved 150-loop sequence of GTIHDR, which is also conserved in N9 and sometimes found in N2. This further illustrates that the structures of N7 and N6 fit into the group-specific model originally reported by Russell et al. (20).
FIG 5.

Comparison of the active sites in all NA subtypes. Structures of the active sites in 09N1 (PDB ID 3NSS), VN04N1 (PDB ID 2HTY), N2 (PDB ID 1NN2), N3 (PDB ID 4HZV), N4 (PDB ID 2HTV), N5 (PDB ID 3SAL), A/chicken/Nanchang/7-010/2000 (H3N6) N6, A/mallard/ALB/196/1996 (H10N7) N7, N8 (PDB ID 2HT5), N9 (PDB ID 7NN9), N10 (PDB ID 4FVK), N11 (PDB ID 4K3Y), Flu B NA (B/Beijing/1/87; PDB ID 1NSB), 09N1-Ile223Arg (PDB ID 4B7M) (N1 numbering), N2-oseltamivir carboxylate (PDB ID 4K1K), and VN04N1-oseltamivir carboxylate (PDB ID 2HU4) are shown in surface presentation. The 09N1-Ile223Arg structure is shown in a slightly different size in order to show the phosphate ions.
With the exception of our previously solved wild-type 09N1 structure, all solved group 1 uncomplexed native structures (N1, N4, N5, and N8) have a 150-cavity, and all solved group 2 uncomplexed structures (N2, N3, N4, N6, and N9) have no 150-cavity (23). The conserved 150-loop sequence of typical group 1 NAs is GTVKDR. However, 09N1 has the conserved sequence GTIKDR, which is also conserved in group 2 N3. Interestingly, the 150-cavity of 09N1-Ile223Arg (PDB ID 4B7M), a drug-resistant mutant, turns out to be open, with a phosphate ion in the crystallization buffer, and the phosphate ion can interact with the Lys150 residue, which helps to stabilize the 150-loop open conformation (40). N5 and N2 are also unique in terms of their 150-loop properties. N5 contains a more extended 150-loop conformation, leading to an unusually large 150-cavity (29). The conserved 150-loop sequence of N5 is NTVKDR, in which Asn147 contributes to the formation of the extended 150-cavity. N2 is also unique in that it often contains an Asp147 residue, which can form a salt bridge with His150 and influence the flexibility of the 150-loop. Interestingly, we previously found that the N2 150-loop can adopt a half-open conformation after soaking with oseltamivir carboxylate (PDB ID 4K1K) (24). This is in contrast to the situation for typical group 1 NAs, in which the 150-loop closes upon inhibitor binding (20). Influenza virus NA active site and 150-cavity are shown as surface presentation in Fig. 5, and the sizes are outlined in Table 6, calculated using the program Computed Atlas of Surface Topography of proteins (CASTp). These comparisons illustrate the complexity of the NA active site and provide some basis for drug design. The influenza virus B NA active site resembles that of group 2 NAs, and the more-divergent NA-like N10 and N11 also show no distinct 150-cavity in their structures (Fig. 5).
TABLE 6.
Surface area and volume of NA active sitea
| NA (PDB ID) | Surface area (Å2) | Vol (Å3) |
|---|---|---|
| VN04N1 (2HTY) | 941.3 | 574.8 |
| 09N1 (3NSS) | 517.4 | 642.6 |
| N2 (1NN2) | 439.6 | 564.8 |
| N3 (4HZV) | 354.2 | 437.9 |
| N4 (2HTV) | 453 | 600.1 |
| N5 (3SAL) | 886.1 | 1,336.1 |
| N6b | 415.8 | 570.8 |
| N7c | 391.6 | 556.2 |
| N8 (2HT5) | 1,052.9 | 1,556.8 |
| N9 (7NN9) | 421.5 | 574.8 |
The surface area and volume of the enzyme activity site were calculated using the program Computed Atlas of Surface Topography of proteins (CASTp).
N6 from strain A/Chicken/Nanchang/7-010/2000 (H3N6).
N7 from strain A/mallard/ALB/196/1996 (H10N7).
Of particular interest, through the structural comparison of all the NA subtypes, we found unique 340-loop (residues 342 to 347) conformations in both N7 and N6. These novel orientations create a novel small cavity adjacent to the active site, named the 340-cavity. Similar to the previously identified 150-cavity, the 340-cavity may present a novel target for structure-based drug design. The close proximity of the 340-loop to the calcium binding site and Gly348, which interacts with Arg292 and Arg371 of the active site triarginyl cluster, suggests that the 340-loop and 340-cavity may have some functional implications.
Although the comprehensive structural analysis of N7, the last remaining influenza virus NA subtype, has now been completed, there still might be some structural novelties hidden within each subtype. N1 and N2 subtypes are especially diverse, as they are known to recombine into avian-, swine-, and human-infecting viruses. However, three distinct N1 structures, VN04N1 (PDB ID 2HTY), 18N1 (PDB ID 3BEQ), and 09N1 (PDB ID 3NSS), as well as four N2 structures, from A/Tokyo/3/67 (H2N2) (PDB ID 1NN2), A/RI/5+/57 (H2N2) (PDB ID 4K1H), A/Memphis/31/98 (H3N2) (PDB ID 2AEQ), and A/Tanzania/205/2010 (H3N2) (PDB ID 4GZO), are available. Besides the previously characterized 150-loop variations, there is little structural variation within the N1 and N2 subtypes, indicating that this analysis is truly comprehensive. In the future, structural studies of NA will still be of value in order to understand the mechanisms underlying drug resistance and binding of ligands, including inhibitors, antibodies, and influenza virus receptors.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (NSFC grants 81301465 and 81341002) and the Intramural Special Grant for Influenza Virus Research from the Chinese Academy of Sciences (KJZD-EW-L09). G.F.G. is a leading principal investigator of the NSFC Innovative Research Group (grant no. 81321063).
Assistance by the staff at SSRF of China is acknowledged. We thank the Genewiz Corporation for the continued synthesis of NA genes. We particularly thank Boris Tefsen in the Institute of Microbiology for the manuscript edit. We highly appreciate the help of Di Liu and Wei Li in the Institute of Microbiology for the sequence alignment.
Author contributions: X.S., Q.L., and Y.L. performed the experiments. X.S. wrote the manuscript with the help of C.J.V.; M.W. and Y.L. assisted with protein expression, purification, and crystallization experiments and edited the manuscript; Y.W. and C.J.V. discussed the experiments and edited the manuscript; J.Q. performed the crystal data collection, solved the crystal structures, and edited the manuscript; G.F.G. conceived and supervised the research and edited the manuscript.
We declare that we have no competing financial interests.
Footnotes
Published ahead of print 4 June 2014
REFERENCES
- 1.Medina RA, Garcia-Sastre A. 2011. Influenza A viruses: new research developments. Nat. Rev. Microbiol. 9:590–603. 10.1038/nrmicro2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dawood FS, Jain S, Finelli L, Shaw MW, Lindstrom S, Garten RJ, Gubareva LV, Xu X, Bridges CB, Uyeki TM. 2009. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N. Engl. J. Med. 360:2605–2615. 10.1056/NEJMoa0903810 [DOI] [PubMed] [Google Scholar]
- 3.Neumann G, Noda T, Kawaoka Y. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931–939. 10.1038/nature08157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu D, Shi W, Shi Y, Wang D, Xiao H, Li W, Bi Y, Wu Y, Li X, Yan J, Liu W, Zhao G, Yang W, Wang Y, Ma J, Shu Y, Lei F, Gao GF. 2013. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: phylogenetic, structural, and coalescent analyses. Lancet 381:1926–1932. 10.1016/S0140-6736(13)60938-1 [DOI] [PubMed] [Google Scholar]
- 5.Lang G, Gagnon A, Geraci JR. 1981. Isolation of an influenza A virus from seals. Arch. Virol. 68:189–195. 10.1007/BF01314571 [DOI] [PubMed] [Google Scholar]
- 6.Banks J, Speidel E, Alexander DJ. 1998. Characterisation of an avian influenza A virus isolated from a human—is an intermediate host necessary for the emergence of pandemic influenza viruses? Arch. Virol. 143:781–787. 10.1007/s007050050329 [DOI] [PubMed] [Google Scholar]
- 7.Shinya K, Watanabe S, Ito T, Kasai N, Kawaoka Y. 2007. Adaptation of an H7N7 equine influenza A virus in mice. J. Gen. Virol. 88:547–553. 10.1099/vir.0.82411-0 [DOI] [PubMed] [Google Scholar]
- 8.Min JY, Vogel L, Matsuoka Y, Lu B, Swayne D, Jin H, Kemble G, Subbarao K. 2010. A live attenuated H7N7 candidate vaccine virus induces neutralizing antibody that confers protection from challenge in mice, ferrets, and monkeys. J. Virol. 84:11950–11960. 10.1128/JVI.01305-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du Ry van Beest Holle M, Meijer A, Koopmans M, de Jager CM. 2005. Human-to-human transmission of avian influenza A/H7N7, The Netherlands, 2003. Euro Surveill. 10:264–268 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=584 [PubMed] [Google Scholar]
- 10.Gibbens N. 2008. Avian influenza outbreak in Oxfordshire. Vet. Rec. 162:795. 10.1136/vr.162.24.795 [DOI] [PubMed] [Google Scholar]
- 11.Lam TT, Wang J, Shen Y, Zhou B, Duan L, Cheung CL, Ma C, Lycett SJ, Leung CY, Chen X, Li L, Hong W, Chai Y, Zhou L, Liang H, Ou Z, Liu Y, Farooqui A, Kelvin DJ, Poon LL, Smith DK, Pybus OG, Leung GM, Shu Y, Webster RG, Webby RJ, Peiris JS, Rambaut A, Zhu H, Guan Y. 2013. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature 502:241–244. 10.1038/nature12515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569. 10.1146/annurev.biochem.69.1.531 [DOI] [PubMed] [Google Scholar]
- 13.Colman PM. 1994. Influenza virus neuraminidase: structure, antibodies, and inhibitors. Protein Sci. 3:1687–1696. 10.1002/pro.5560031007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Y, Wu Y, Tefsen B, Shi Y, Gao GF. 2014. Bat-derived influenza-like viruses H17N10 and H18N11. Trends Microbiol. 22:183–191. 10.1016/j.tim.2014.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Air GM. 2012. Influenza neuraminidase. Influenza Other Respir. Viruses 6:245–256. 10.1111/j.1750-2659.2011.00304.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Air GM, Laver WG. 1989. The neuraminidase of influenza virus. Proteins 6:341–356. 10.1002/prot.340060402 [DOI] [PubMed] [Google Scholar]
- 17.von Itzstein M. 2007. The war against influenza: discovery and development of sialidase inhibitors. Nat. Rev. Drug Discov. 6:967–974. 10.1038/nrd2400 [DOI] [PubMed] [Google Scholar]
- 18.von Itzstein M. 2012. Influenza virus sialidase: a drug discovery target. Springer, Basel, Switzerland [Google Scholar]
- 19.Kim CU, Lew W, Williams MA, Liu H, Zhang L, Swaminathan S, Bischofberger N, Chen MS, Mendel DB, Tai CY, Laver WG, Stevens RC. 1997. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 119:681–690. 10.1021/ja963036t [DOI] [PubMed] [Google Scholar]
- 20.Russell RJ, Haire LF, Stevens DJ, Collins PJ, Lin YP, Blackburn GM, Hay AJ, Gamblin SJ, Skehel JJ. 2006. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 443:45–49. 10.1038/nature05114 [DOI] [PubMed] [Google Scholar]
- 21.Xu X, Zhu X, Dwek RA, Stevens J, Wilson IA. 2008. Structural characterization of the 1918 influenza virus H1N1 neuraminidase. J. Virol. 82:10493–10501. 10.1128/JVI.00959-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudrawar S, Dyason JC, Rameix-Welti MA, Rose FJ, Kerry PS, Russell RJ, van der Werf S, Thomson RJ, Naffakh N, von Itzstein M. 2010. Novel sialic acid derivatives lock open the 150-loop of an influenza A virus group-1 sialidase. Nat. Commun. 1:113. 10.1038/ncomms1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Q, Qi J, Zhang W, Vavricka CJ, Shi Y, Wei J, Feng E, Shen J, Chen J, Liu D, He J, Yan J, Liu H, Jiang H, Teng M, Li X, Gao GF. 2010. The 2009 pandemic H1N1 neuraminidase N1 lacks the 150-cavity in its active site. Nat. Struct. Mol. Biol. 17:1266–1268. 10.1038/nsmb.1909 [DOI] [PubMed] [Google Scholar]
- 24.Wu Y, Qin G, Gao F, Liu Y, Vavricka CJ, Qi J, Jiang H, Yu K, Gao GF. 2013. Induced opening of influenza virus neuraminidase N2 150-loop suggests an important role in inhibitor binding. Sci. Rep. 3:1551. 10.1038/srep01551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Vries E, Collins PJ, Vachieri SG, Xiong X, Liu J, Walker PA, Haire LF, Hay AJ, Schutten M, Osterhaus AD, Martin SR, Boucher CA, Skehel JJ, Gamblin SJ. 2012. H1N1 2009 pandemic influenza virus: resistance of the I223R neuraminidase mutant explained by kinetic and structural analysis. PLoS Pathog. 8:e1002914. 10.1371/journal.ppat.1002914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohan S, McAtamney S, Haselhorst T, von Itzstein M, Pinto BM. 2010. Carbocycles related to oseltamivir as influenza virus group-1-specific neuraminidase inhibitors. Binding to N1 enzymes in the context of virus-like particles. J. Med. Chem. 53:7377–7391. 10.1021/jm100822f [DOI] [PubMed] [Google Scholar]
- 27.Wu Y, Bi Y, Vavricka CJ, Sun X, Zhang Y, Gao F, Zhao M, Xiao H, Qin C, He J, Liu W, Yan J, Qi J, Gao GF. 2013. Characterization of two distinct neuraminidases from avian-origin human-infecting H7N9 influenza viruses. Cell Res. 23:1347–1355. 10.1038/cr.2013.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vavricka CJ, Liu Y, Kiyota H, Sriwilaijaroen N, Qi J, Tanaka K, Wu Y, Li Q, Li Y, Yan J, Suzuki Y, Gao GF. 2013. Influenza neuraminidase operates via a nucleophilic mechanism and can be targeted by covalent inhibitors. Nat. Commun. 4:1491. 10.1038/ncomms2487 [DOI] [PubMed] [Google Scholar]
- 29.Wang M, Qi J, Liu Y, Vavricka CJ, Wu Y, Li Q, Gao GF. 2011. Influenza A virus N5 neuraminidase has an extended 150-cavity. J. Virol. 85:8431–8435. 10.1128/JVI.00638-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326. 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 31.Read RJ. 2001. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr. D Biol. Crystallogr. 57:1373–1382. 10.1107/S0907444901012471 [DOI] [PubMed] [Google Scholar]
- 32.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255. 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 33.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 34.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221. 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. 1993. Procheck: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291. 10.1107/S0021889892009944 [DOI] [Google Scholar]
- 36.Potier M, Mameli L, Belisle M, Dallaire L, Melancon SB. 1979. Fluorometric assay of neuraminidase with a sodium (4-methylumbelliferyl-alpha-D-N-acetylneuraminate) substrate. Anal. Biochem. 94:287–296. 10.1016/0003-2697(79)90362-2 [DOI] [PubMed] [Google Scholar]
- 37.Li Q, Sun X, Li Z, Liu Y, Vavricka CJ, Qi J, Gao GF. 2012. Structural and functional characterization of neuraminidase-like molecule N10 derived from bat influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 109:18897–18902. 10.1073/pnas.1211037109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rudino PE, Tunnah P, Crennell SJ, Webster RG, Laver WG, Garman EF. 2006. The crystal structure of type A influenza virus neuraminidase subtype N6 reveals the existence of two separate Neu5Ac binding sites. PDB IDs: 1W1X, 1V0Z,1W20, 1W21, 2CML. http://www.rcsb.org/pdb/explore/explore.do?structureId=1W1X
- 39.Li Q, Qi J, Wu Y, Kiyota H, Tanaka K, Suhara Y, Ohrui H, Suzuki Y, Vavricka CJ, Gao GF. 2013. Functional and structural analysis of influenza virus neuraminidase N3 offers further insight into the mechanisms of oseltamivir resistance. J. Virol. 87:10016–10024. 10.1128/JVI.01129-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vavricka CJ, Li Q, Wu Y, Qi J, Wang M, Liu Y, Gao F, Liu J, Feng E, He J, Wang J, Liu H, Jiang H, Gao GF. 2011. Structural and functional analysis of laninamivir and its octanoate prodrug reveals group specific mechanisms for influenza NA inhibition. PLoS Pathog. 7:e1002249. 10.1371/journal.ppat.1002249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, Yang H, Chen X, Recuenco S, Gomez J, Chen L-M, Johnson A, Tao Y, Dreyfus C, Yu W, McBride R, Carney PJ, Gilbert AT, Chang J, Guo Z, Davis CT, Paulson JC, Stevens J, Rupprecht CE, Holmes EC, Wilson IA, Donis RO. 2013. New World bats harbor diverse influenza A viruses. PLoS Pathog. 9:e1003657. 10.1371/journal.ppat.1003657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lawrenz M, Wereszczynski J, Amaro R, Walker R, Roitberg A, McCammon JA. 2010. Impact of calcium on N1 influenza neuraminidase dynamics and binding free energy. Proteins 78:2523–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith BJ, Huyton T, Joosten RP, McKimm-Breschkin JL, Zhang JG, Luo CS, Lou MZ, Labrou NE, Garrett TP. 2006. Structure of a calcium-deficient form of influenza virus neuraminidase: implications for substrate binding. Acta Crystallogr. D Biol. Crystallogr. 62:947–952. 10.1107/S0907444906020063 [DOI] [PubMed] [Google Scholar]
- 44.Sung JC, Van Wynsberghe AW, Amaro RE, Li WW, McCammon JA. 2010. Role of secondary sialic acid binding sites in influenza N1 neuraminidase. J. Am. Chem. Soc. 132:2883–2885. 10.1021/ja9073672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Varghese JN, Colman PM, van Donkelaar A, Blick TJ, Sahasrabudhe A, McKimm-Breschkin JL. 1997. Structural evidence for a second sialic acid binding site in avian influenza virus neuraminidases. Proc. Natl. Acad. Sci. U. S. A. 94:11808–11812. 10.1073/pnas.94.22.11808 [DOI] [PMC free article] [PubMed] [Google Scholar]