

Abstract
Viruses efficiently block the host antiviral response in order to replicate and spread before host intervention. The mechanism initiating antiviral immunity during stealth viral replication is unknown, but recent data demonstrate that defective viral genomes generated at peak virus replication are critical for this process in vivo. This article summarizes the supporting evidence and highlights gaps in our understanding of the mechanisms and impact of immunostimulatory defective viral genomes generated during natural infections.
BATTLE FOR VIRUS AND HOST COEXISTENCE
To ensure survival, living organisms must recognize and counteract harmful invaders. In higher species, an army of proteins and cells has evolved to quickly and effectively eliminate viruses and other dangerous microbes. To prevent unnecessary damage to the host, this defense system is tightly controlled and only responds following recognition of pathogen-associated molecular patterns (PAMPs). Viruses capable of escaping or counteracting the antiviral host response have an evolutionary advantage, as they can replicate to high titers and spread before being eradicated. Many examples of viruses that effectively counteract host defenses are found among seasonally circulating viruses. For example, influenza virus and respiratory syncytial virus encode proteins that antagonize the host response, allowing these viruses to replicate for 1.5 to 3 days in the absence of host antiviral activity (1). This so called “incubation period” is characterized by rapid virus growth in the absence of symptoms, such as fever and malaise, and is followed by an effective antiviral response that controls the infection and clears the virus.
Synthetic viral mimics and viruses unable to counteract the host immune response elicit rapid and strong antiviral immunity. These systems have allowed for the identification of both viral PAMPs and cellular proteins involved in the initiation of the antiviral response. However, while it is clear that additional factors are required to trigger a potent host response in the presence of viral antagonists, the specific signals that are utilized remain to be identified. In the following sections, I summarize evidence indicating that for many viruses bearing a negative-sense RNA genome, those signals are provided by defective viral genomes (DVGs) that are generated as replication by-products when the virus reaches high titers. The relatively late generation of DVGs in the virus infection cycle, coupled with the effective detection of DVGs by host cellular proteins and the induction of a potent antiviral response, is a testament to the evolutionary compromise to sustain virus-host coexistence.
ORIGIN AND ACTIVITY OF DVGs
DVGs are truncated forms of the viral genome that are generated during virus replication at high titers. DVGs lack essential genes and cannot propagate in the absence of helper virus. Two major types of DVGs have been described for RNA viruses. Deletion DVGs are truncated versions of the original virus genome that normally share the 3′ and 5′ ends with the parental virus. They are generated when the viral polymerase falls off the original template and reattaches further downstream, resulting in a genomic deletion. Copy-back DVGs, and the related snap-back DVGs, consist of a segment of the viral genome flanked by reverse complementary versions of its 5′ end. Copy-back DVGs occur when the viral polymerase detaches from the template and reattaches to the newly synthesizing strand, copying back the 5′ end of the genome (reviewed in references 2 and 3). In the absence of viral nucleoproteins, copy-back genomes can form a hairpin structure through interactions between their 5′ and 3′ ends. It remains to be demonstrated whether these structures are formed in the context of virus infection, as nascent RNA folds during synthesis and is quickly bound by virus nucleoproteins.
DVGs of RNA viruses were first described for influenza virus in the late 1940s (4). Since then, they have been identified for a variety of viruses passaged at high multiplicities of infection in vitro (5). DVGs were initially characterized as the genomes of defective viral particles able to interfere with standard virus replication, hence earning the name of defective interfering (DI) particles (5). In the late 1970s and early 1980s, a series of reports demonstrated that density-purified defective viral particles from vesicular stomatitis virus, Sindbis virus, Sendai virus (SeV), and other viruses strongly induced type I interferon (IFN) expression during infection of cells in vitro (6, 7). The ability of DI particles to promote strong type I IFN induction was subsequently confirmed using recombinant viruses (8). The interfering activity and the type I IFN-inducing potential led to the study of DI particles as vaccine adjuvants and as broad-spectrum antivirals (reviewed in reference 2). Although DI particles have not yet been tried as immunostimulatory molecules in the clinic setting, large amounts of DI particles were found in vaccine strains of poliovirus and measles virus (9, 10).
CELLULAR RESPONSE TO DVGs
Using the mouse Paramyxovirus pathogen SeV as a model, my group showed that in addition to promoting type I IFN expression, DI particles stimulate the rapid and strong expression of a large panel of proinflammatory cytokines, chemokines, antiviral genes, and cell surface molecules on mouse and human dendritic cells. DI particles rendered these cells fully capable of stimulating T cells, thereby promoting the transition from the innate to the adaptive immune response (11–14). Stocks of SeV lacking DI particles failed to induce a potent response even at a high multiplicity of infection, but potent activation of dendritic cells was restored following supplementation with purified DI particles. Remarkably, the stimulatory activity of DVGs in vitro was independent of protein secretion and was maintained in the absence of type I IFN signaling and in the presence of functional virus antagonists of the antiviral response (13, 14), supporting their potential ability to induce a host response during periods of stealth replication (Fig. 1).
FIG 1.
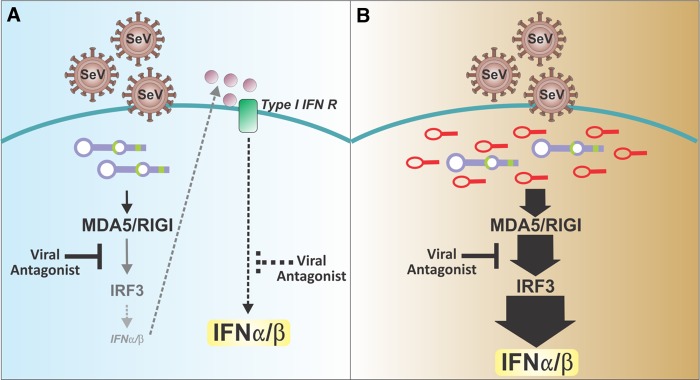
Current and proposed models of the events required for the development of effective antiviral responses to infections with paramyxoviruses. (A) According to current paradigms, viral recognition is maximized by type I IFN signaling that promotes the expression of essential viral sensors and signaling molecules. This paradigm assumes a low level of expression of type I IFN. In the depicted example of SeV infection, this scenario is possible if the virus-encoded V and C antagonistic proteins are only partially active, for example, during a zoonotic infection. However, this scenario cannot explain the induction of the antiviral response in the natural host of this virus, which is effectively blocked by the viral antagonists. (B) The model developed by my laboratory proposes that defective viral genomes that arise at high levels of virus replication provide potent danger signals that stimulate the host antiviral response to overcome viral antagonism, independently of type I IFN feedback.
RNA viruses are primarily recognized in infected cells by two intracellular helicases, the retinoic acid-inducible gene 1-like receptors (RLRs) retinoic acid-inducible gene 1 (RIG-I), and melanoma differentiation-associated protein 5 (MDA5). The binding of viral PAMPs to the RNA-binding domain of RLRs triggers their phosphorylation, oligomerization, and signaling through the adaptor protein mitochondrial antiviral signaling protein (MAVS) (reviewed in reference 15). RLR signaling activates transcription factors that drive the expression of the major antiviral cytokine type I IFN, as well as of proinflammatory and antiviral proteins. A type I IFN signaling loop that amplifies the expression of many key molecules in these pathways, including RLRs themselves, maximizes the RLR response (15). Notably, many of the viral proteins that antagonize the host immune response directly target the RLR pathway. For example, the parainfluenza virus V protein directly blocks MDA5 signaling, while its C protein blocks type I IFN signaling and amplification of the RLR response; respiratory syncytial virus NS1 and NS2 interfere with the expression and signaling of type I IFNs; and the influenza virus NS1 protein interferes with the RLR pathway at multiple levels (reviewed in reference 16). Recombinant viruses lacking these antagonistic proteins induce a fast and potent immune response that results in rapid viral clearance (17), demonstrating that virus-encoded proteins inhibit early recognition of viruses by RLRs in vivo.
Although the mechanism for the efficient and potent response to DVGs in the presence of virus antagonists has not been fully elucidated, data show that DVGs are recognized by RLRs in infected cells. In support of this idea, MAVS deficiency abrogates the generation of the antiviral response to DVGs (12). In addition, RIG-I binds DVGs preferentially over standard virus genomes in cells infected with SeV or influenza virus (18) and RIG-I overexpression enhances the response to SeV DVGs (14). MDA5 also participates in the response to DVGs and is essential for a fast response in vitro (13). Notably, the expression of MDA5 is amplified by DVGs independently of type I IFNs (19). Toll-like receptors, another major family of virus-sensing molecules, are dispensable for the recognition of DVGs in infected cells and it remains to be determined if they participate in DVG recognition during more complex in vivo infections.
IMMUNOSTIMULATORY MOLECULAR MOTIFS OF DVGs
Studies using synthetic viral RNA analogs or purified viral RNA revealed that RIG-I binds to uncapped 5′-di- or -triphosphates (5′ppp) coupled to short double-stranded RNA (dsRNA) stretches or single-stranded RNA, while MDA5 binds to long stretches of dsRNA and complex RNA secondary structures (reviewed in reference 15). Although both deletion and copy-back DVGs preserve the RIG-I stimulatory uncapped 5′ppp motif, copy-back DVGs have significantly more potent immunostimulatory activity than that of other forms of the genome (8, 14). While the potent activity of DVGs may be partially explained by a faster accumulation in infected cells (12) due to their strong flanking antigenomic promoters, infection with large amounts of standard virus lacking DVGs does not compensate for their poor immunostimulatory activity (12, 14), suggesting that the relative level of PAMPs does not fully account for the more potent recognition of DVGs. Interestingly, treatment of SeV DVGs with phosphatase to eliminate the 5′ppp motif significantly diminishes but does not eliminate their immunostimulatory ability (11, 12), suggesting that other RNA motifs play a role in DVG recognition. Using in vitro-transcribed RNA from a highly immunostimulatory SeV DVG, a critical role for secondary structures in the immunostimulatory activity of DVG RNA is beginning to emerge (11, 20).
DVGs IN NATURAL INFECTIONS
Although the accumulation of immunostimulatory DVGs in cells infected in vitro has been demonstrated for many viruses (3, 5–7), it remained unclear if DVGs have a fundamental biological role during natural infections. Early observations of infected cell lines suggested that the interplay between the interfering capacity of DI particles and the requirement for standard virus for DI expansion would lead to alternating cycles of standard virus genome and DI particle replication, thereby promoting virus persistence. However, evidence for a role for DI particles in promoting virus persistence in vivo is lacking. Historically, technical difficulties in differentiating standard and defective viral genomes, together with a widespread belief that DVGs were an epiphenomenon of in vitro virus replication discouraged further investigation of DVGs in natural infections. However, as DVGs that have a strong immunostimulatory activity have been reported to be present in sera from patients infected with various viruses, including hepatitis B virus, dengue virus, and influenza virus (reviewed in reference 2), my laboratory set out to investigate potential roles for DVGs in the virus-host interaction during natural infections. Using SeV and mouse-adapted influenza virus, we demonstrated that DVGs are first detected in the lung at the time of peak virus replication (12), coincident with the first signs of expression of type I IFNs and other host antiviral effector molecules. Importantly, DVGs were detected in mice lacking the type I IFN receptor, indicating that their generation is not driven by type I IFNs. Strikingly, lung cells containing high levels of standard virus genomes, but not DVGs, failed to express type I IFNs, while those containing both standard virus genomes and DVGs expressed type I IFN (12). Altogether, these data are the first demonstration of a pivotal role for DVGs in determining the onset of the antiviral immune response in natural infections.
CONTRIBUTIONS TO THE FIELD AND FUTURE DIRECTIONS
Technological advances and a new understanding of the molecular mechanisms for virus recognition in infected cells have revived an interest in studying the biology and immunostimulatory function of DVGs. A recent demonstration that DVG accumulation during infections in vivo stimulates a potent antiviral response (12) proves that these ubiquitous by-products of virus replication play a pivotal role in natural virus-host interactions.
A number of knowledge gaps in our understanding of immunostimulatory DVGs still exist. It is unclear whether DVG-mediated interference with standard virus replication is required for potent induction of antiviral activity or if interfering and immunostimulatory DVGs can be distinct. The molecular mechanisms driving the generation of DVGs are yet unknown and it remains to be determined if they arise as a consequence of the error-prone viral RNA polymerase that allows for the generation of quasispecies essential to maintain virus fitness or if distinct mechanisms drive DVG generation. The cellular pathways and secondary RNA motifs involved in the efficient recognition and potent response to DVGs in the presence of functional virus antagonists remain poorly understood, and it is likely that novel circuits that modulate the function of RLRs are involved. Importantly, the role of DVGs in determining virus pathogenesis remains to be studied. Vigorous research on the mechanisms and impact of immunostimulatory DVGs in various viral infections will fill these gaps of knowledge and open novel opportunities for harnessing the DVG-host interactions for therapeutic intervention.
ACKNOWLEDGMENTS
I thank all past and present members of my laboratory for their hard work and valuable contributions to the study of defective viral genomes. I thank Debora Argento for help with the figure and Leslie King and Susan Weiss for critically reading the manuscript. I apologize to all colleagues whose work could not be referenced because of length restrictions.
Work in my laboratory is currently supported by grants from the NIH AI083284 and The University of Pennsylvania Research Foundation.
Footnotes
Published ahead of print 28 May 2014
REFERENCES
- 1.Lessler J, Reich NG, Brookmeyer R, Perl TM, Nelson KE, Cummings DA. 2009. Incubation periods of acute respiratory viral infections: a systematic review. Lancet Infect. Dis. 9:291–300. 10.1016/S1473-3099(09)70069-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dimmock NJ, Easton AJ. 2014. Defective interfering influenza virus RNAs: time to reevaluate their clinical potential as broad-spectrum antivirals? J. Virol. 88:5217–5227. 10.1128/JVI.03193-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lazzarini RA, Keene JD, Schubert M. 1981. The origins of defective interfering particles of the negative-strand RNA viruses. Cell 26:145–154. 10.1016/0092-8674(81)90298-1 [DOI] [PubMed] [Google Scholar]
- 4.von Magnus P. 1947. Studies on interference in experimental influenza. Arkiv för Kemi, Mineralogi och Geologi, vol 24 Almqvist & Wiksell, Stockholm, Sweden [Google Scholar]
- 5.Pathak KB, Nagy PD. 2009. Defective interfering RNAs: foes of viruses and friends of virologists. Viruses 1:895–919. 10.3390/v1030895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuller FJ, Marcus PI. 1980. Interferon induction by viruses. IV. Sindbis virus: early passage defective-interfering particles induce interferon. J. Gen. Virol. 48:63–73 [DOI] [PubMed] [Google Scholar]
- 7.Johnston MD. 1981. The characteristics required for a Sendai virus preparation to induce high levels of interferon in human lymphoblastoid cells. J. Gen. Virol. 56:175–184. 10.1099/0022-1317-56-1-175 [DOI] [PubMed] [Google Scholar]
- 8.Strahle L, Garcin D, Kolakofsky D. 2006. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 351:101–111. 10.1016/j.virol.2006.03.022 [DOI] [PubMed] [Google Scholar]
- 9.McLaren LC, Holland JJ. 1974. Defective interfering particles from poliovirus vaccine and vaccine reference strains. Virology 60:579–583. 10.1016/0042-6822(74)90352-3 [DOI] [PubMed] [Google Scholar]
- 10.Shingai M, Ebihara T, Begum NA, Kato A, Honma T, Matsumoto K, Saito H, Ogura H, Matsumoto M, Seya T. 2007. Differential type I IFN-inducing abilities of wild-type versus vaccine strains of measles virus. J. Immunol. 179:6123–6133. 10.4049/jimmunol.179.9.6123 [DOI] [PubMed] [Google Scholar]
- 11.Mercado-Lopez X, Cotter CR, Kim WK, Sun Y, Munoz L, Tapia K, Lopez CB. 2013. Highly immunostimulatory RNA derived from a Sendai virus defective viral genome. Vaccine 31:5713–5721. 10.1016/j.vaccine.2013.09.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tapia K, Kim WK, Sun Y, Mercado-Lopez X, Dunay E, Wise M, Adu M, Lopez CB. 2013. Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. PLoS Pathog. 9:e1003703. 10.1371/journal.ppat.1003703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yount JS, Gitlin L, Moran TM, Lopez CB. 2008. MDA5 participates in the detection of paramyxovirus infection and is essential for the early activation of dendritic cells in response to Sendai virus defective interfering particles. J. Immunol. 180:4910–4918. 10.4049/jimmunol.180.7.4910 [DOI] [PubMed] [Google Scholar]
- 14.Yount JS, Kraus TA, Horvath CM, Moran TM, Lopez CB. 2006. A novel role for viral-defective interfering particles in enhancing dendritic cell maturation. J. Immunol. 177:4503–4513. 10.4049/jimmunol.177.7.4503 [DOI] [PubMed] [Google Scholar]
- 15.Dixit E, Kagan JC. 2013. Intracellular pathogen detection by RIG-I-like receptors. Adv. Immunol. 117:99–125. 10.1016/B978-0-12-410524-9.00004-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Versteeg GA, Garcia-Sastre A. 2010. Viral tricks to grid-lock the type I interferon system. Curr. Opin. Microbiol. 13:508–516. 10.1016/j.mib.2010.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moltedo B, Lopez CB, Pazos M, Becker MI, Hermesh T, Moran TM. 2009. Cutting edge: stealth influenza virus replication precedes the initiation of adaptive immunity. J. Immunol. 183:3569–3573. 10.4049/jimmunol.0900091 [DOI] [PubMed] [Google Scholar]
- 18.Baum A, Sachidanandam R, Garcia-Sastre A. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. U. S. A. 107:16303–16308. 10.1073/pnas.1005077107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yount JS, Moran TM, Lopez CB. 2007. Cytokine-independent upregulation of MDA5 in viral infection. J. Virol. 81:7316–7319. 10.1128/JVI.00545-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel JR, Jain A, Chou YY, Baum A, Ha T, Garcia-Sastre A. 2013. ATPase-driven oligomerization of RIG-I on RNA allows optimal activation of type-I interferon. EMBO Rep. 14:780–787. 10.1038/embor.2013.102 [DOI] [PMC free article] [PubMed] [Google Scholar]