

ABSTRACT
The tegument layer of herpesviruses comprises a collection of proteins that is unique to each viral species. In rhesus monkey rhadinovirus (RRV), a close relative of the human oncogenic pathogen Kaposi's sarcoma-associated herpesvirus, ORF52 is a highly abundant tegument protein tightly associated with the capsid. We now report that ORF52 knockdown during RRV infection of rhesus fibroblasts led to a greater than 300-fold reduction in the viral titer by 48 h but had little effect on the number of released particles and caused only modest reductions in the levels of intracellular viral genomic DNA and no appreciable change in viral DNA packaging into capsids. These data suggested that the lack of ORF52 resulted in the production and release of defective particles. In support of this interpretation, transmission electron microscopy (TEM) revealed that without ORF52, capsid-like particles accumulated in the cytoplasm and were unable to enter egress vesicles, where final tegumentation and envelopment normally occur. TEM also demonstrated defective particles in the medium that closely resembled the accumulating intracellular particles, having neither a full tegument nor an envelope. The disruption in tegument formation from ORF52 suppression, therefore, prevented the incorporation of ORF45, restricting its subcellular localization to the nucleus and appearing, by confocal microscopy, to inhibit particle transport toward the periphery. Ectopic expression of small interfering RNA (siRNA)-resistant ORF52 was able to partially rescue all of these phenotypic changes. In sum, our results indicate that efficient egress of maturing virions and, in agreement with studies on murine gammaherpesvirus 68 (MHV-68), complete tegumentation and secondary envelopment are dependent on intact ORF52.
IMPORTANCE The tegument, or middle layer, of herpesviruses comprises both viral and cellular proteins that play key roles in the viral life cycle. A subset of these proteins is present only within members of one of the three subfamilies (alphaherpesviruses, betaherpesviruses, or gammaherpesviruses) of Herpesviridae. In this report, we show that the gammaherpesvirus-specific tegument protein ORF52 is critical for maturation of RRV, the closest relative of Kaposi's sarcoma-associated herpesvirus (KSHV) (a human cancer-causing pathogen) that has undergone this type of analysis. Without ORF52, the nascent subviral particles are essentially stuck in maturation limbo, unable to acquire the tegument or outer (envelope) layers. This greatly attenuates infectivity. Our data, together with earlier work on a murine homolog, as well as a more distantly related human homolog, provide a more complete understanding of how early protein interactions involving virus-encoded tegument proteins are critical for virus assembly and are also, therefore, potentially attractive therapeutic targets.
INTRODUCTION
Rhesus monkey rhadinovirus (RRV), a gamma-2 herpesvirus, is a close homolog of Kaposi's sarcoma associated herpesvirus (KSHV), or human herpesvirus 8 (HHV-8), the causative agent of three human tumors: Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease (1–3). Like all herpesviruses, KSHV and RRV have both latent and lytic phases of infection (reviewed in references 4 to 6). KSHV produces a primarily latent infection, and even with reactivation by phorbol esters or histone deacetylase (HDAC) inhibitors, titers in culture are low (7, 8). In contrast, RRV efficiently infects cultured primary or immortalized rhesus monkey fibroblasts (RhFs), enters the lytic phase, and replicates to a relatively high titer (9). These qualities, along with the high levels of conservation in the genomic sequence and organization (10, 11), make RRV a useful model to study the structure and lytic (productive) replication of KSHV and other gammaherpesviruses, as well as the roles of individual lytic viral genes in these processes.
RRV also shares with all members of the family Herpesviridae the same overall structural architecture: a linear double-stranded DNA genome surrounded by an icosahedral capsid; then a proteinaceous layer, or tegument, comprised of a limited subset of viral and cellular proteins; and finally an outermost layer derived from the host cell envelope studded with viral glycoproteins (reviewed in reference 5). During host cell entry, the herpesvirus envelope fuses with either the plasma membrane or an endocytic vesicle membrane, releasing the majority of tegument proteins (12, 13; reviewed in reference 14). Investigators have suggested that these tegument proteins likely play important roles in preparing the host cell for viral replication (15–21). Though the process of tegumentation remains poorly defined, previous studies have found both cellular and viral proteins within the layer (22–27). This gives rise to a proteomic tegument profile that is distinct for each viral species and possibly even within the same species grown in different cell types or under different conditions (28).
Until recently, structural data for the tegument were limited to images from electron microscopy (EM) that suggested that the layer was comprised of an amorphous collection of proteins; however, more recent studies with human cytomegalovirus (HCMV) (29), herpes simplex virus 1 (HSV-1) (30, 31), murine gammaherpesvirus 68 (MHV-68) (32), RRV (33), and KSHV (34) indicate the presence of ordered tegument structures (30). Investigators have further divided the tegument into inner and outer layers, which are evident by cryo-electron tomography of MHV-68 (32), with inner tegument proteins more tightly associated with the capsid and more resistant to Triton X-100 detergent treatment (26, 30, 35). The majority of functional data regarding the herpesvirus tegument comes primarily from research on alpha- and betaherpesviruses, which has shown that tegument proteins function in crucial roles in viral replication, including transcytosis of the herpesvirus capsid toward the nucleus during initial infection and egress from the nucleus toward the periphery during lytic replication (12, 36–39). Additional functions of tegument proteins modulate the host cell environment during the immediate-early phase of infection (40), including shutoff of host gene expression (21, 41, 42), antagonism of the innate antiviral host response (43–47), and assembly and egress of herpesvirus virions (38, 48–50; reviewed in reference 51).
Previous biochemical and mass spectrometry analyses of RRV from our laboratory indicate the presence of at least 33 virus-encoded proteins comprising the viral particles (26). Among these proteins in RRV, we identified ORF52 as a gammaherpesvirus-specific, highly abundant tegument protein that tightly associates with the capsid (26) but with a function that remains uncharacterized for this primate gammaherpesvirus. ORF52 carried by RRV is a late gene (52), encoding a protein of 139 amino acids with a molecular mass of 15 kDa. Homologs are present within other gammaherpesvirus virions and share varying degrees of identity/similarity, including MHV-68 (41%/63%) (38, 53), KSHV (47%/67%) (19, 25), and Epstein-Barr virus (EBV) (38%/55%) (23). MHV-68 ORF52 plays a key role in tegumentation and secondary envelopment of viral particles (38, 54).
In the present study, we examined the function of ORF52 during lytic infection with wild-type (WT) RRV in culture using small interfering RNA (siRNA) knockdown (kd) and rescue approaches. We assessed the role of ORF52 in various steps during the viral life cycle, including DNA replication and packaging, capsid production and assembly, nuclear egress, tegumentation and envelopment, and finally egress with the production and release of infectious virions. Our data argue that, much like its murine homolog from MHV-68, RRV ORF52 is necessary for late stages in the viral life cycle and that when ORF52 is absent or limited, particles fail to undergo tegumentation and secondary envelopment. We additionally found that in the absence (or severe reduction) of ORF52, another tegument protein, ORF45, remained restricted to the nucleus. Furthermore, and in contrast to transfections with an MHV-68 bacterial artificial chromosome (BAC) containing a stop codon in its ORF52 homolog, RRV ORF52 knockdown still resulted in the release of subviral particles that lacked tegument and envelope and were also unable to remain cell surface associated, unlike mature and fully infectious virions.
MATERIALS AND METHODS
Cell culture.
Telomerase-immortalized RhFs were grown in complete medium (Dulbecco's modified Eagle's medium [Gibco] supplemented with 1 nM puromycin, 1 mM sodium pyruvate, and 10% fetal bovine serum [Gibco]), as described previously (26).
RRV stocks.
RhFs were grown to confluence, approximately 2 × 107 cells in a T182 flask, and infected with RRV strain H26-95 at a multiplicity of infection (MOI) of 0.05 in 5 ml complete medium for 1 h. The cells were then supplemented with an additional 100 ml of complete medium per flask. The media were collected 5 to 7 days postinfection (p.i.) and cleared of cellular debris by low-speed centrifugation at 350 × g. Cleared media containing virus were passed through a 0.45-μm-pore-size filter. Virus was concentrated by centrifugation for 3 h at 12,855 × g in a Sorvall SL250T rotor. The resulting viral pellets were resuspended in 1.0 ml TNE (20 mM Tris [pH 7.5], 100 mM NaCl, 1 mM EDTA).
Antibodies.
Mouse monoclonal anti-RRV ORF52 and anti-RRV ORF65 (small capsomer-interacting protein [SCIP]) were generated in the Lymphocyte Culture Center at the University of Virginia. Following PCR amplification and cloning of RRV orf52 and orf65 from RRV-derived DNA, full-length ORF52 and ORF65 (SCIP) glutathione S-transferase (GST) fusion proteins were made in Escherichia coli. Following purification and cleavage of GST, the proteins were used by the University of Virginia Lymphocyte Culture Center to produce mouse monoclonal antibodies to ORF52 and ORF65/SCIP. Rabbit polyclonal anti-RRV ORF45 was generated by Open Biosystems, Inc. (28). Mouse monoclonal anti-RRV major capsid protein (MCP) and anti-glycoprotein B (gB) were kindly provided by Scott Wong at Oregon Health and Science University. Rabbit polyclonal anti-actin (sc-1616-R) was purchased from Santa Cruz Biotechnology. Conjugated secondary antibodies anti-infrared dye (IRDye) 800 anti-mouse and IRDye 800 anti-rabbit were purchased from LiCor Biosciences and Rockland Immunochemicals, respectively.
siRNA.
Silencer Select custom siRNA specific to the RRV ORF52 coding sequence 5′-AACCCGTAAGATTGAAGCTAA-3′ and siControl 1 were purchased from Life Technologies.
siRNA transfection followed by RRV infection.
ORF52 siRNA or control siRNA (siCNL) (20 nM) was transfected into 4 × 106 RhFs in 10-cm plates using 25 μl Lipofectamine RNAiMax (Invitrogen) following the manufacturer's reverse transfection protocol. Twenty-four hours later, the cells were infected with RRV at an MOI of 5 for 1 h at 37°C with rocking every 15 min to ensure uniform distribution of the virus. One hour later, the virus was removed and the cells were washed with 1× phosphate-buffered saline (PBS) 3 times and then replaced into complete medium. The cells were incubated for an additional 48 h.
Expression of viral lytic proteins. (i) RRV-infected RhFs.
Following removal of the medium, the plated cells were washed 3 times in 1× PBS at room temperature (RT). The cells were trypsinized off the plates and pelleted. The pellets were washed once in 1× PBS prior to being resuspended in lysis buffer as described above. For each sample, approximately 45 μg of total protein was loaded onto a precast 12% Bis-Tris gel (NuPage; Life Technologies).
(ii) Viral supernatants.
Forty-eight hours p.i., medium was collected and cell debris was cleared by centrifugation at 350 × g. To concentrate and isolate viral particles, 3 ml of the cleared medium was layered over a 20% sucrose cushion in TNE. The medium was centrifuged at 65,204 × g for 30 min at 4°C in a SW-41Ti rotor. Following centrifugation, the medium was decanted, and pellets containing viral particles were resuspended in 60 μl sample buffer. The samples were boiled for 10 min, and equal volumes were loaded onto a 12% Bis-Tris gel. Statistical significance between conditions was determined using an unpaired Student t test. Calculations were performed using GraphPad Prism online software.
Protein electrophoresis and immunoblotting.
Cells were trypsinized off the plates, pelleted, and washed once in 1× PBS. The pelleted cells were lysed for 15 min at 4°C with whole-cell lysis buffer (50 mM Tris [pH 7.3], 150 mM NaCl, 1% Igepal, 5 mM EDTA, 10% glycerol) supplemented with 1× protease inhibitor cocktail (Roche Applied Science) immediately prior to use. The lysed cells were centrifuged for 30 min at 4°C, and the media were removed for protein analysis.
Cell lysates and concentrated supernatants containing viral particles from each experiment were resuspended in lithium dodecyl sulfate (LDS) sample buffer (NuPage; Life Technologies) with NuPage sample reducing agent (50 mM dithiothreitol [DTT]). Following denaturation by boiling for 10 min, proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 12% Bis-Tris gels (NuPage; Life Technologies).
For immunoblot analyses, proteins separated by SDS-PAGE were transferred to nitrocellulose membranes for 60 min at 300 mA at 4°C. The membranes were blocked in 5% nonfat milk–Tris-buffered saline (TBS) (20 mM Tris base, 150 mM NaCl, 3 mM Tris-HCl) for 90 min at RT and then incubated with primary antibodies overnight at 4°C. Primary antibodies were used at the following dilutions: anti-RRV ORF52 (1:1,000), anti-RRV SCIP (1:2,500), anti-RRV ORF45 (1:10,000), anti-RRV MCP (1:1,000), anti-RRV gB (1:5,000), and anti-mouse actin (1:15,000). After three washes with TBS-Tween (0.05%) at RT, the membranes were incubated with secondary antibodies for 45 min at RT. For quantitative immunoblotting, membranes were incubated with Infrared Dye 800-conjugated anti-mouse (Rockland Immunochemicals) or Infrared Dye 800-conjugated anti-rabbit (LiCor Biosciences) diluted 1:10,000 in 5% nonfat milk in TBS-Tween (0.05%). Images were scanned and analyzed using an Odyssey infrared imaging system and Odyssey 3.0 software (LiCor Biosciences).
Quantitative PCR (qPCR). (i) RRV-infected RhFs.
Cells were washed 3 times in 1× PBS prior to collection and, after pelleting, were resuspended in proteinase K (PK) digestion buffer (100 mM NaCl, 10 mM Tris-Cl [pH 8], 25 mM EDTA [pH 8], 0.5% SDS, and 0.1 mg/ml PK [Sigma-Aldrich]) and incubated at 55°C overnight. DNA was extracted with phenol-chloroform and precipitated with ethanol. A standard DNA concentration curve was based on serial dilutions of an ORF45 plasmid, pCMV-Tag2A-R45. Primers for human glyceraldehyde-3-phosphate dehydrogenase (GAPDH), GAPDH-F (5′-GAAGATGGTGATGGGATTTCCA-3′) and GAPDH-R (5′-GATTCCACCCATGGCAAATT3′), were used to normalize the samples. Biological triplicates were performed for each condition and time point, and each triplicate was analyzed by PCR in triplicate. Quantitative data are presented as means and standard deviations (SD) (the error bars show only the positive component of the SD).
(ii) Viral supernatants.
Precleared medium was collected under each condition at various time points p.i., and particles were pelleted by centrifugation at 65,204 × g for 30 min at 4°C in an SW-41Ti rotor through a 20% sucrose cushion. The pellet containing viral particles was resuspended in DNase buffer (100 mM Tris-Cl, 25 mM MgCl2, 1 mM CaCl2) in PBS overnight at 4°C. Viral particles were then dissociated by bath sonication in an ice bath slurry 3 times for 5 s each time, and samples were treated with 5 U RNase-free DNase I (Stratagene) for 30 min at 37°C to leave only encapsidated DNA. DNase I was inactivated with 50 mM EDTA and an additional 10 min of incubation at 65°C to inactivate the DNase. Samples were then treated with 0.1 mg/ml PK (Sigma-Aldrich) at 55°C overnight. The DNA was extracted with phenol-chloroform and precipitated with ethanol, along with 1 μg of glycogen carrier (Glycoblue; Ambion). RRV genomic copy numbers were assayed in triplicate by real-time PCR (SYBR green PCR master mix; Applied Biosystems), using primers specific to the ORF45 coding sequence (ORF45F, 5′-TGATTCGTCCCATGTCTCAA-3′; ORF45R, 5′-CCTGTTGTTGCTGGATCAAA-3′), and amplified and detected with an ABI Prism 7900 HT detection system instrument at the University of Virginia Biomolecular Research Center. Quantification was based on serial dilution of a plasmid bearing the ORF45 coding sequence, pCMV-Tag2A-R45.
Plaque assay.
Determination of viral titers was performed essentially as described previously (55). In brief, 2 × 105 RhFs were plated in 12-well plates; 48 h later, confluent monolayers were infected with 5-fold serial dilutions of precleared medium containing an unknown concentration of virus. Each dilution was done in triplicate. The plates were incubated for 1 h at 37°C with rocking every 15 min to ensure uniform distribution of the virus. After 1 h, overlay medium containing 0.6% methylcellulose was added. The plates were incubated for 5 days at 37°C. The overlay medium was removed, and the cells were stained with crystal violet for 10 min at RT. Plaques were counted using an inverted microscope (Nikon Eclipse TE-2000-E) at ×100 magnification. Absolute titers were determined based on the dilution used and the number of plaques counted. Data are presented as means and SD. Statistical significance between conditions was determined using an unpaired Student t test.
Immunofluorescence microscopy (IF) assay.
A total of 2 × 104 cells per well were transfected with RNAiMax (Invitrogen) with either ORF52 siRNA or control siRNA and plated on 48-well plates containing a Cell-Tak (BD Biosciences)-coated 8-mm round coverslip (Electron Microscopy Sciences) and incubated for 24 h. The cells were then infected with RRV at an MOI of 5 or mock infected.
Forty-eight hours p.i., cells were fixed with 4% formaldehyde in PHEM buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2, pH 6.9) solution for 15 min at RT. The cells were washed 3 times with PHEM buffer and then permeabilized in 0.25% Triton in PHEM buffer for 10 min and washed 3 times with PHEM buffer. The samples were blocked overnight at 4°C in 10% normal goat serum (in PHEM).
Samples were stained at RT with antibodies diluted in 5% normal goat serum (in PHEM). Primary antibodies were incubated for 1 h, and secondary antibodies were incubated for 30 min. The staining was sequential (i.e., primary antibody and then the corresponding secondary antibody, followed by the next primary antibody and its corresponding secondary antibody, and so forth), with 3 washes of PHEM buffer between each pair of antibodies. Primary antibodies to ORF52 (1:500), ORF45 (1:250), and SCIP (1:250), each conjugated to fluorophore 488 with a Mix-n-Stain CF488A kit (Biotium), were used. The secondary antibodies Alexa Fluor 647 goat anti-mouse (ORF52) and Alexa Fluor 555 goat anti-rabbit (ORF45) (Life Technologies; both 1:500), respectively, were used. After the last secondary antibody, the cells were counterstained with DAPI (4,6-diamidino-2-phenylindole) (Sigma; 1.0 μg/ml in double-distilled water) for 5 min and washed once with double-distilled water. Coverslips were mounted onto microscope slides (Fisher) with Fluro-Gel (Electron Microscopy Sciences) and imaged using the Zeiss 710 confocal microscope.
Transmission electron microscopy (TEM). (i) RRV-infected RhFs.
Following removal of medium, the cells were washed 3 times with 1× PBS at RT; 2.5% electron microscopy grade gluteraldehyde (Electron Microscopy Sciences) in 1× PBS was added for 10 min at RT to fix the cells. The cells were then scraped into a tube and pelleted by centrifugation at 350 × g for 5 min at RT. The fixed cell pellet was stored at 4°C until it was processed at the University of Virginia Advanced Microscopy (AVM) Core.
(ii) Supernatants.
Forty-eight hours p.i., medium was collected and concentrated by centrifugation over a sucrose cushion as described above. Following centrifugation, the medium was decanted, and the remaining pellet was fixed in 2.5% gluteraldehyde in 1× PBS. Samples were stored at 4°C until processing.
The samples were postfixed in 2% osmium tetroxide, dehydrated in graded ethanol, and embedded in epoxy resin. Ultrathin sections, approximately 60 to 70 nm in thickness, were collected on 200-mesh copper grids and contrast stained with lead citrate and uranyl acetate. The sections were examined at 80 kV on a JEOL 1230 transmission electron microscope at the University of Virginia AVM Core.
Complementation of RRV ORF52 with exogenous ORF52. (i) siORF52-resistant plasmid.
N-terminally myc-tagged siORF52-resistant RRV ORF52 was generated from a pK-myc WT RRV ORF52 plasmid. The pK-myc plasmid was derived from the pKH3 backbone and was a gift from Deborah Lannigan and Ian Macara. The full-length ORF52 sequence was amplified by PCR using purified RRV DNA as a template and primers that added a NotI and an EcoRI sequence to ORF52 for cloning (NotI-ORF52-F, 5′-ATAAGAATGCGGCCGCTTATGTCTTCCACGCGT-3′, and EcoRI-ORF52-R, 5′-GGAATTCCTAGTCCGCGTCGTTATT-3′). RRV ORF52 was cloned into the pK-myc vector at the NotI and EcoRI sites. The sequence of the WT pK-myc-RRV52 plasmid was confirmed prior to further use. The WT pK-myc RRV ORF52 plasmid was used as a template to generate an RRV ORF52 siRNA (siORF52)-resistant plasmid. Primers were designed that would alter 2 nucleotides (225 and 228) in the wobble base position of two adjacent amino acid coding sequences of ORF52 that are complementary to the ORF52 siRNA (the changed nucleotides are underlined): 5′-AACCCGTAAAATCGAAGCTAA-3′. The siORF52-resistant plasmid was generated using the primers containing the desired mutations, the WT pK-myc ORF52 plasmid, and the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies). The mutations were verified by sequencing of the pK-myc-siORF52-resistant plasmid.
(ii) Complementation assay.
RhFs (6 × 106) were transfected via Amaxa nucleofector program T-016 with 6 μg of pK-myc-empty vector or pK-myc-siORF52 resistant plasmid DNA in primary fibroblast nucleofection reagent and plated in a 10-cm plate. Twenty-four hours later, the cells were reverse transfected with siRNA by lifting cells off the plate with 0.05% trypsin and replating them in 8 ml of complete medium on 10-cm plates containing 2 ml Opti-MEM, 20 nM control siRNA (siCNL) or siORF52, and RNAiMax (Invitrogen). Twenty-four hours later, the cells were infected in 2 ml complete medium for 1 h with RRV at an MOI of 5 at 37°C and rocked every 15 min to ensure even distribution of virus. Following infection, the virus was removed, the cells were washed 3 times in 1× PBS, and complete medium was added. Medium and cells were collected 48 h p.i., and the protein composition was determined by gel electrophoresis and Western blotting (the methods for determining the cell pellet and protein compositions in the medium are identical to those described in “Protein electrophoresis and immunoblotting” and “Expression of viral lytic proteins” above).
(iii) IF assay for complementation.
RhFs (6 × 106) were transfected via the Amaxa nucleofector program T-016 with 6 μg of pK-myc-empty vector or pK-myc-siORF52 resistant plasmid DNA in primary fibroblast nucleofection reagent and incubated in a 10-cm plate for 24 h. Twenty-five thousand cells/well were subsequently reverse transfected with RNAiMax (Invitrogen) with either ORF52 siRNA or control siRNA, plated onto 48-well plates containing a Cell-Tak (BD Biosciences)-coated 8-mm round coverslip (Electron Microscopy Sciences), and incubated for 24 h. The cells were then infected with RRV at an MOI of 5 or mock infected. The remainder of the staining and imaging methods were the same as those described in “Immunofluorescence microscopy (IF) assay” above, except that the secondary antibodies used were Alexa Fluor 488 goat anti-mouse and Alexa Fluor 555 goat anti-rabbit (Life Technologies; both 1:500).
Particle purification by velocity sedimentation.
RhFs in 10-cm plates were treated with ORF52 or control siRNA and then infected at an MOI of 5 as described above; 48 h p.i., the medium was collected and cell debris was cleared. To concentrate and isolate viral or subviral particles, 85 ml of the cleared medium was layered over a 20% sucrose cushion in TNE and centrifuged at 65,204 × g for 30 min at 4°C in a SW-41Ti rotor. Following centrifugation, the medium was decanted, and pellets containing viral particles were resuspended in 60 μl (total) of 1× PBS supplemented with 1× protease inhibitor cocktail (Roche Applied Science) and rocked overnight at 4°C. The 60 μl of concentrated supernatant was then layered above a 20 to 60% sucrose step gradient prepared in TNE and centrifuged at 75,746 × g for 30 min at 4°C in a SW-55Ti rotor. One-drop fractions (approximately 30 to 40 μl each) were collected by bottom puncture with a 21-gauge needle, and 10 μl of each fraction was loaded onto a precast 12% Bis-Tris gel (NuPage; Life Technologies).
Cell-to-cell spread assay.
RhFs (1.85 × 105 per well) were reverse transfected with either ORF52 siRNA or control siRNA as described above and then plated onto 12-well plates. Twenty-four hours later, the cells were infected with RRV as described above, but at an MOI of 0.1. One hour later, the virus was removed and the cells were washed with 1× PBS 3 times, and then the medium was replaced with overlay medium containing 0.6% methylcellulose. The plates were incubated for 7 days at 37°C. The overlay medium was removed, and images of RhFs were obtained using an inverted microscope (Nikon Eclipse TE-2000-E) at ×100 magnification.
RESULTS
Efficient knockdown of ORF52 has little effect on other viral structural proteins.
To begin investigating the function of ORF52 during lytic RRV infection, we first knocked down its expression by transfecting RhFs with either control (siCNL) or ORF52-specific (siORF52) siRNA 24 h prior to the addition of the virus. Using quantitative Western blotting, we then measured the relative levels of ORF52, as well as several other lytic structural proteins, in the cell lysates (Fig. 1A and B). In cells targeted with ORF52 siRNA, the knockdown of ORF52 was nearly complete at 97% compared to the robust expression in controls (siCNL). In contrast, another tegument protein, ORF45, and capsid proteins, small capsomer-interacting protein (SCIP/ORF65) and major capsid protein (MCP/ORF25), showed no statistically significant change (Fig. 1B). We noted similar results with the use of a second ORF52-targeting siRNA (data not shown). These results suggested that expression of the structural proteins we examined was independent of the synthesis of ORF52 and that the knockdown was specific.
FIG 1.
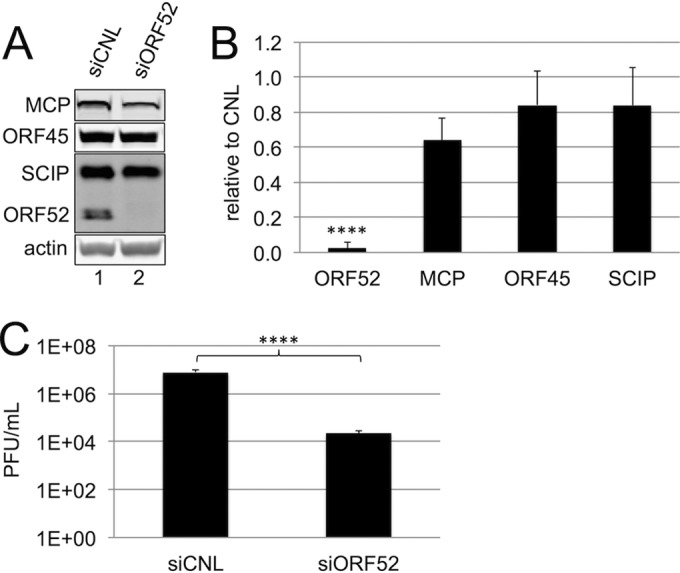
Efficient knockdown of ORF52 significantly decreased the RRV titer with only minimal effects on intracellular levels of other structural viral proteins. (A) Immunoblot analysis of cell lysates from RhFs transfected with siCNL (lane 1) or siORF52 (lane 2) and then infected with RRV 24 h later at an MOI of 5. Cells were harvested 48 h p.i., and immunoblotting was performed, probing for the viral tegument proteins ORF52 and ORF45, the capsid proteins MCP and SCIP, and actin to normalize for loading differences. (B) Intracellular levels of the indicated viral structural proteins after siORF52 treatment relative to siCNL in RRV-infected RhFs. The data represent the means and SD of 6 individual experiments. (C) Viral titers in the medium 48 h p.i. from RhFs treated with siCNL or siORF52 were determined in 6 different experiments by viral plaque assay. The values are means and SD. ****, P < 0.0001.
Loss of ORF52 markedly decreases the viral titer.
We next determined if the depletion of ORF52 had an effect on productive RRV infection by assessing the release of infectious virions. In earlier work with MHV-68, cells transfected with a BAC construct containing a stop codon in its ORF52 (ORF52Stop-BAC) did not result in the release of measurable infectious virus (38). Therefore, we hypothesized that if ORF52 were likewise required for RRV production, then the titers that we measured from siORF52-treated cells would be significantly lower than those from controls. In six separate experiments, we found that the titers from siORF52-treated cells were over 300-fold lower than those from controls (Fig. 1C), indicating that ORF52 was indeed critical for efficient production and/or release of infectious virions.
We subsequently asked whether depletion of ORF52 affected intracellular levels of viral DNA, thereby leading to the decrease in viral titer we observed following ORF52 knockdown. Since viral production of lytic proteins seemed grossly unaffected despite the near absence of ORF52 (Fig. 1A and B) and since ORF52 is a late gene expressed after viral DNA replication (52), we hypothesized that viral DNA copies would not be markedly affected in siORF52-treated cells. We instead predicted that the block in virus production would be at a stage after viral DNA replication. qPCR analysis showed that viral DNA increased by approximately 3 orders of magnitude between 12 and 24 h following infection for siCNL-treated cultures and only modestly less with ORF52 knockdown (Fig. 2A). While DNA in cell lysates with ORF52 knocked down had 25% and 50% fewer copies of viral DNA at 24 and 48 h postinfection, respectively, the overall kinetics and expression profile closely paralleled those of siCNL-treated cells, suggesting that knockdown of ORF52 did not grossly inhibit viral DNA replication (Fig. 2A). Combined, these data indicated that neither the production of structural viral proteins (Fig. 1A and B) nor the lytic replication of viral DNA (Fig. 2A) could account for the profound reduction (>300-fold) in viral titers that resulted from ORF52 knockdown. Rather, the findings suggested a block at a subsequent stage of particle maturation.
FIG 2.
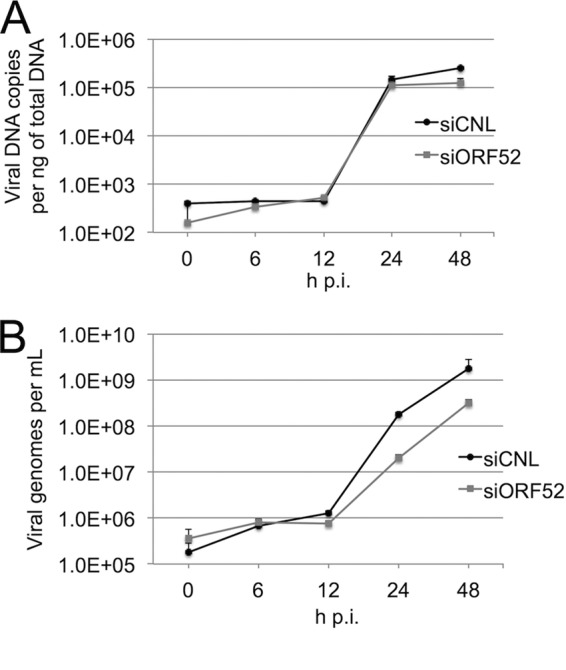
Intracellular viral DNA production and packaging within particles appeared to be independent of ORF52. (A) RhFs were transfected with siCNL or siORF52 and infected 24 h later with RRV at an MOI of 5. At the time points indicated, cells were collected and total intracellular DNA was isolated and purified. Viral DNA was quantified by SYBR green qPCR with primers to the RRV ORF45 coding region. (B) RhFs were transfected with siCNL or siORF52 and infected 24 h later with RRV at an MOI of 5. At the time points indicated, particles were treated with DNase, followed by PK, and viral DNA was isolated, purified, and quantified by SYBR green qPCR with primers for RRV ORF45. The values are the means and SD of 2 or 3 qPCR replicates from 3 separate experiments, with only the positive component of the errors shown for increased graphical clarity.
Release of an increased proportion of empty particles.
Although intracellular viral DNA and lytic structural proteins were present at close to control levels despite the near absence of ORF52, we could not rule out the possibility that ORF52 might be necessary for DNA packaging in capsids within the nuclei. Therefore, we next asked whether ORF52 knockdown led to a decrease in the DNA content of released particles, measuring encapsidated viral DNA in medium from cells treated with siCNL or siORF52 at multiple time points following infection with RRV. DNase-resistant (encapsidated) viral DNA in the media of both siCNL- and siORF52-treated RRV-infected cells began to appreciably accumulate 12 h p.i. and continued to increase through 48 h p.i. (Fig. 2B). However, the levels of encapsidated viral DNA released from siORF52-treated cells were approximately 9-fold and 6-fold lower at 24 and 48 h p.i., respectively, than control levels at these times (Fig. 2B). These results suggested that ORF52 kd led to the release of either fewer overall particles or a lower proportion of particles containing viral DNA, or both. In either case, however, the modest difference in encapsidated viral DNA at 48 h p.i. in the medium from the ORF52 kd condition was also insufficient to explain the large drop in titers (>2.5 orders of magnitude) (Fig. 1C). Therefore, it followed that the major contributor to the loss in titer likely stemmed from a defect in particle infectivity other than DNA packaging or a drop in particle numbers.
ORF52 knockdown led to the release of subviral particles lacking tegument.
To test whether the decrease in encapsidated viral DNA from ORF52 knockdown resulted from a concomitant decrease in released particles, we first collected medium from siCNL- or siORF52-treated RhFs 48 h p.i., isolated potential viral particles from the medium by centrifugation through a sucrose cushion, and then used quantitative immunoblotting to measure the levels of particle-associated structural proteins from equal volumes of concentrated medium. Since ORF52 knockdown led to such a dramatic decrease in the viral titer, nearly 2 orders of magnitude greater than the relatively modest reduction in encapsidated viral DNA, we predicted that we would observe qualitative, as well as quantitative, differences in the protein compositions of the released particles. As we anticipated, siCNL treatment of RhFs led to the production of particles containing the capsid proteins MCP and SCIP, as well as tegument proteins ORF45 and ORF52 (Fig. 3, lane 1). The particles from siORF52-treated cells also contained the capsid proteins MCP and SCIP, and not surprisingly, they lacked ORF52. We noted, however, that the difference in particle-associated capsid proteins (MCP and SCIP) released from siCNL- and siORF52-treated cells was minimal (Fig. 3B). Since the stoichiometry of both SCIP and MCP is fixed in the icosahedral capsid of all herpesviruses (56–59), these data suggested that the overall abundances of released virion- or subvirion-like particles were similar under the two conditions following RRV infection.
FIG 3.
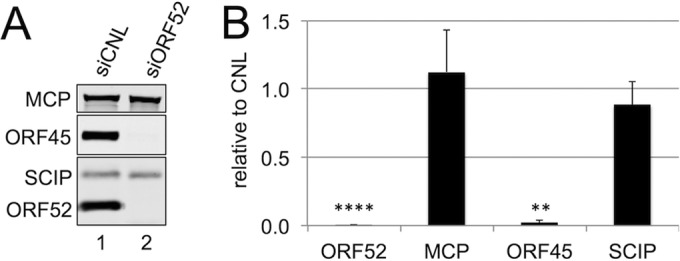
In the absence of ORF52, RRV infection led to release of immature particles lacking tegument. (A) RhFs were transfected with siCNL (lane 1) or siORF52 (lane 2) and then infected 24 h later with RRV at an MOI of 5. Forty-eight hours p.i., the medium was collected and concentrated over a 20% sucrose cushion to isolate particles, and equal volumes of medium were separated by SDS-PAGE and immunoblotted for MCP, ORF45, SCIP, and ORF52. (B) Effect of siORF52 relative to siCNL on the levels of the indicated viral proteins within the released viral particles. The data represent the means and SD of 6 individual experiments. **, P < 0.01; ****, P < 0.0001.
In contrast, we noted that the released particles from the ORF52 kd conditions also lacked the tegument protein ORF45, despite its presence within the siORF52-treated cell lysates (Fig. 1A and B). Figure 3B graphically depicts the protein levels from six separate experiments, quantifying the ratio of each protein associated with particles released from siORF52- compared to siCNL-treated cells. These results demonstrated that both ORF52 and ORF45 were virtually absent in particles present in the medium of siORF52-treated cells. Since the decrease in viral titers between siORF52- and siCNL-treated cells was 300-fold (Fig. 1C), despite the suggestion of similar particle numbers (Fig. 3B), we reasoned that the much lower infectivity of particles in the medium from ORF52 knockdown cells likely resulted from the production of incomplete particles lacking, at a minimum, a full complement of tegument proteins.
Loss of ORF52 prevents ORF45 incorporation into the particle.
As we have mentioned above, though the intracellular abundance of the tegument protein ORF45 remained essentially unaffected following ORF52 knockdown (Fig. 1A and B), the protein was absent from released particles (Fig. 3A and B). Earlier studies with KSHV documented that K-ORF45 and K-ORF52 interact by both coimmunoprecipitation (co-IP) and yeast two-hybrid studies, though the latter was evident only when ORF52 served as bait but not as prey (34), and similar interactions are less clear for the murine gammaherpesvirus MHV-68 (54, 60). To gain further insight into the interactions between ORF52 and ORF45 during RRV infection, we assessed their subcellular localization using IF in the absence or presence of ORF52 knockdown (Fig. 4). In siCNL cells, IF revealed that ORF45 localization was diffuse in the nucleus, with nucleolar sparing, but highly punctate in the cytoplasm, while ORF52, in agreement with MHV-68 ORF52 staining (38), displayed a discrete punctate pattern in the cytoplasm and was absent from the nucleus. Additionally, a merge of the two staining patterns demonstrated that the punctate patterns of ORF52 and ORF45 in the cytoplasm overlapped, consistent with their representing individual or collections of viral or subviral tegumented particles (Fig. 4A, top row, and B, left). To help assess whether the punctate cytoplasmic staining of the two tegument proteins did reflect maturing virions, we also costained for SCIP, reasoning that it would localize to trafficking capsids and would colocalize with both ORF52 and ORF45. However, we found that cytoplasmic SCIP staining was weak compared to nuclear SCIP staining, possibly reflecting a partial masking of the SCIP epitope due to the overlying tegument proteins. In contrast, following ORF52 knockdown, ORF45 remained strictly nuclear, and SCIP staining in the cytoplasm was now prominent, though it mainly adopted an aggregated perinuclear distribution (Fig. 4A, bottom row, and B, right). Together, these images suggested a role for ORF52, directly or indirectly, in the export of ORF45 into the cytoplasm. Of note, co-IP experiments failed to demonstrate an interaction between ORF45 and ORF52, either from RRV-infected RhFs or from HEK293 cells, following ectopic expression of the two tegument proteins (data not shown). Thus, during unperturbed assembly of the virions, ORF45 appeared to exit the nucleus by its incorporation within maturing particles as they acquired tegument. The exact mechanism underlying the ORF52 dependence of this incorporation, however, remains unclear (see Discussion).
FIG 4.

ORF52 knockdown restricted ORF45 to the nucleus. (A) RhFs were reverse transfected with siCNL (i to v) or siORF52 (vi to x) and plated on coverslips. Twenty-four hours later, the cells were infected with RRV at an MOI of 5, and then at 48 h p.i., the cells were fixed and stained with conjugated anti-SCIP, anti-ORF52, and anti-ORF45 antibodies, as indicated, followed by the secondary antibody Alexa Fluor 647 goat anti-mouse or Alexa Fluor 555 goat anti-rabbit. The cells were stained with DAPI. (B) Boxed areas in panel A, v and x, magnified (3×). The arrows in siCNL (i) point to punctate orange staining (due to ORF45-ORF52 colocalization) in the cytoplasm, consistent with tegumented viral particles during maturation/egress. The arrowheads in siORF52 (ii) point to yellow cytoplasmic staining consistent with SCIP antibody reactivity in the absence of ORF45 and ORF52 reactivity and suggesting untegumented cytoplasmic capsids.
Secondary envelopment depends on ORF52.
Since the immunofluorescence images of infected cells with ORF52 knockdown showed little to no evidence of either ORF52 or ORF45 in the cytoplasm and immunoblots of the released particles from these cells likewise demonstrated an absence of these two tegument proteins, we hypothesized that the particles would more closely resemble capsids than mature trilaminar virions. Further, since we also detected lower levels of encapsidated DNA in the medium from ORF52 kd cells, we anticipated observing a greater proportion of empty particles lacking electron-dense genomic viral DNA otherwise typical of electron micrographs of mature herpesviruses (27, 30, 31, 33; reviewed in references 5, 51, and 61).
After centrifuging the medium over a sucrose cushion, we fixed the pelleted samples and visualized any particles from both ORF52 knockdown and control conditions using thin-section TEM. The majority of particles from siCNL-treated cells appeared to be mature virions containing DNA, an icosahedral capsid, tegument, and envelope (Fig. 5A). In contrast, the particles from siORF52-treated cells had the shape and diameter of capsids and lacked evidence of either tegument or envelope (Fig. 5B). A number of these subviral particles contained the electron-dense DNA that is typical of TEM images of herpesviruses; however, compared to siCNL, there was an increase in the proportion of empty capsids (lacking DNA) present (Fig. 5B). This result suggested that ORF52 was necessary for the production and release of fully formed virions at a stage subsequent to capsid protein production, assembly, and DNA replication and packaging but before tegumentation and envelopment.
FIG 5.
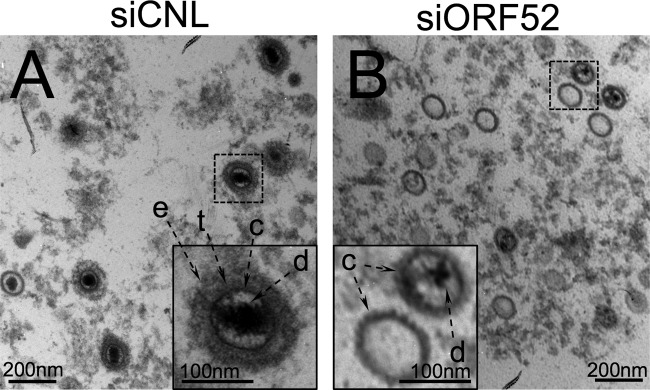
TEM images of the concentrated medium from RRV-infected RhFs after ORF52 knockdown demonstrated release of untegumented and unenveloped capsid-like particles, many of which lacked DNA. RhFs were transfected with siCNL or siORF52 and infected 24 h later with RRV at an MOI of 5. Forty-eight hours p.i., the medium was removed and layered over a sucrose cushion prior to centrifugation. Pelleted particles from the medium were fixed and examined by TEM at ×30,000 magnification. In contrast to the siCNL sample (A), which contained mature virions with envelope (e) and tegument (t) surrounding a capsid (c), particles from siORF52-treated RhFs (B) appeared to lack both envelope and tegument. d, encapsidated viral DNA. The inset images are expanded (3×) from the original images (dashed boxes).
To ascertain that ORF52 kd led to the release of particles lacking envelope, as well as tegument, we purified particles by velocity sedimentation through a sucrose gradient collected from the medium of infected cells pretreated with either control or ORF52 siRNA. We then blotted the fractions for capsid, tegument, and the envelope gB. Consistent with TEM images (Fig. 5), the control condition demonstrated the presence of gB in peak fractions (maximal MCP signal), consistent with the presence of envelope (Fig. 6A, top). In contrast, under the kd condition, peak fractions showed a clear lack of gB, in addition to a lack of the two tegument proteins, ORF45 and ORF52 (Fig. 6A, bottom). The capsid protein MCP is present under both control and kd conditions (Fig. 6A and B), indicating, as we showed previously (Fig. 3A and 5B), a release of untegumented and unenveloped capsids following ORF52 knockdown (Fig. 6B). As we predicted, the majority of gB remained trapped in the cellular fraction under the kd conditions (Fig. 6C), providing further evidence that the lack of ORF52 abrogated glycoprotein egress due to the block in tegumentation and secondary envelopment rather than disruption of viral glycoprotein synthesis.
FIG 6.

Knockdown of ORF52 leads to release of capsids lacking envelope and tegument. RhFs were transfected with either siCNL or siORF52. After 24 h, the cells were infected with RRV at an MOI of 5 and then harvested 48 h p.i. (A) Medium was collected and concentrated over a 20% sucrose cushion to isolate particles and subsequently layered over a 20 to 60% sucrose gradient. Fractions were collected by bottom puncture, and equal volumes of medium were separated by SDS-PAGE and immunoblotted for gB, MCP, ORF45, and ORF52. (B) Levels of the viral capsid protein MCP present in each fraction. (C) Immunoblot analysis of cell lysates from infected RhFs transfected with siCNL (lane 1) or siORF52 (lane 2). The blots were probed for the viral tegument proteins ORF52 and ORF45, the capsid proteins MCP and SCIP, the glycoprotein gB, and actin to normalize for loading differences.
ORF52 is necessary for tegumentation and envelopment.
Although we had anticipated that knockdown of a conserved tegument protein such as ORF52 might interfere with virion maturation, we also expected that this block would inhibit particle release. Thus, we were struck by the apparent efficiency with which infected cells knocked down for ORF52 still released subviral (defective) particles in quantities that approximated the numbers of presumably mature virions from control cells (Fig. 3B). To better characterize this unexpected phenomenon and to more precisely elucidate the stage that requires ORF52 for virion maturation, we used TEM to examine the morphology and distribution of particles within infected cells with or without ORF52 knockdown.
The fact that ORF52 appeared to play a critical structural role in tegumentation and secondary envelopment, similar to that of its homolog in MHV-68 (38), was consistent with its cytoplasmic localization during lytic infection (Fig. 4) and our earlier finding that it belongs to the inner tegument of RRV (26). TEM revealed that cells receiving either siCNL or siORF52 produced all three major types of capsid species within their nuclei, which is typical for infection with RRV and other herpesviruses (56, 58, 59, 62, 63; reviewed in reference 57): A, empty capsids; B, capsids containing scaffold protein; and C, capsids containing viral DNA (Fig. 7A and B). These results demonstrated that capsid assembly and DNA packaging did not require ORF52 expression and provided strong support for our earlier data showing that released particles still had DNase-resistant, packaged viral DNA, even after kd of ORF52 (Fig. 2B).
FIG 7.

TEM of RRV-infected cells following ORF52 knockdown showed a block in tegumentation and secondary envelopment, leading to perivesicular accumulation of capsids, as well as a lack of cell surface-associated virions. RhFs were transfected with siCNL or siORF52 and infected as described in the legend to Fig. 5. Forty-eight hours p.i., the cells were fixed and examined by TEM at ×40,000 magnification. (A and B) The nuclei of siCNL-treated (A) and siORF52-treated (B) cells contained A (empty) capsids (arrowheads), B capsids containing scaffold (chevrons), and C capsids with DNA (arrows). (C) The cytoplasm of siCNL cells contained large vesicles (ves) filled with multiple tegumented and enveloped mature virions. (D) In contrast, the cytoplasm of siORF52-treated cells contained untegumented, unenveloped capsids that were juxtaposed to, but not within, vesicles (the arrowheads indicate capsids surrounding a vesicle). (E and F) Multiple extracellular plasma membrane (pm)-associated virions were present in siCNL cells (E), but not in siORF52 samples (F). The inset images are expanded (3×) from the original images. Cyto, cytoplasm; nuc, nucleus.
Likewise, we noted that capsid egress from the nucleus into the cytoplasm also appeared to be independent of ORF52 expression. TEM revealed cytoplasmic particles under both knockdown and control conditions (Fig. 7C and D). In the cytoplasm of siCNL-treated cells, multiple trilaminar, mature virions were evident within large vesicles (Fig. 7C). However, in cells treated with siORF52, the cytoplasm contained large numbers of untegumented and unenveloped subviral particles that were indistinguishable from nuclear capsids and no evidence of mature virions (Fig. 7D).
Since invagination into vesicles appears to be coupled with tegumentation and secondary envelopment (reviewed in reference 5), it seemed that ORF52 was necessary for these maturation steps (Fig. 7D). We also noted that in ORF52 knockdown cells there appeared to be a larger number of total capsids present in the cytoplasm than the combined number of subviral particles and virions in siCNL cells (quantified below), providing further evidence that inhibition of tegumentation in cells lacking ORF52 created a block in the efficient release of particles via the canonical vesicle-based pathway. Comparison of particles from the nuclei of siCNL- or siORF52-treated cells and the cytoplasm of siORF52-treated cells showed that the species appeared to be similar in size and morphology (compare insets in Fig. 7A and B to that in D). This finding suggested the particles in the cytoplasm of cells knocked down for ORF52 likely lacked most, if not all, of their tegument, as well as their envelope.
Efficient release and cell surface association of viral particles are ORF52 dependent.
The presence of cell surface-associated virions postegress is typical during productive infection with herpesviruses. Although the mechanism of particle release into the medium from ORF52 knockdown cells remains unclear, we predicted that these subviral particles, lacking tegument and envelope, would also be defective in adhering to the cell surface, since in other herpesviruses (and enveloped viruses in general), this depends on the interaction between cellular BST2/tetherin and viral envelope glycoproteins (64–69; reviewed in reference 70). To address this question, we examined the surfaces of both siORF52- and siCNL-treated cells and found that knockdown of ORF52 led to a marked absence of cell surface-associated particles (Fig. 7F). In comparison, control cells showed mature virions populating large areas of the plasma membrane (Fig. 7E). Cell-to-cell spread of infection for herpesviruses, including gammaherpesviruses, is an efficient process (71–76) and is likely dependent, in large part, on the presence of plasma membrane-associated virus. Knockdown of ORF52 led to the release of untegumented and unenveloped subviral particles into the medium (Fig. 5B and 6A and B) that do not adhere to the plasma membrane (Fig. 7F), in contrast to infected control cells with abundant virions decorating their surfaces (Fig. 7E). We predicted that infected cells lacking ORF52 would have greatly diminished ability to support cell-to-cell transmission. To examine this prediction, we treated RhFs with siCNL or siORF52 and infected them with WT RRV at a low MOI (0.1). Following infection, we added medium containing 0.6% methylcellulose on top of the cells to inhibit the dissemination of released virions and particles and to favor, instead, cell-to-cell spread, waiting for a sufficient time (7 days) to allow multiple rounds of infection. The inhibition of cell-to-cell spread in ORF52 kd was striking. The cells treated with siORF52 remained confluent even after 7 days p.i., whereas the cells under the siCNL condition were nearly all lysed (Fig. 8).
FIG 8.
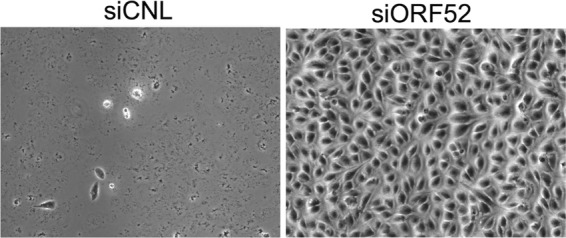
Knockdown of ORF52 prevents cell-to-cell particle spread. RhFs were transfected with either siCNL or siORF52. Twenty-four hours later, the cells were infected with RRV at an MOI of 0.1, and overlay medium containing methylcellulose was added to inhibit dissemination of released virions while allowing cell-to-cell spread. The plates were examined by phase microscopy 7 days postinfection at ×100 magnification.
To assess more precisely the effect of ORF52 on viral maturation, we quantified the different phenotypes of particles within the nuclear and cytoplasmic compartments, as well as those released particles collected from the medium (Table 1). While the total numbers of particles in siCNL and siORF52 were similar overall, the relative proportions of the different viral and subviral species and their locations differed markedly between control and knockdown conditions. In siCNL cells, the nuclei contained approximately twice as many capsids as in the ORF52 knockdown cells; however, the proportions of A, B, and C capsids were similar. Conversely, in the cytoplasm, cells lacking ORF52 contained over four times as many particles as were present in siCNL cells, suggesting that egress from the cell was significantly less efficient. We reasoned that this relative intracellular accumulation of particles might have led to fewer released particles in the medium of the kd than the control conditions; however, we found little difference (Fig. 3B). One possible explanation for this is that a large portion of the particles following egress in the controls remained cell surface associated (Fig. 7E), thereby lowering the number released into the medium.
TABLE 1.
Distribution of intracellular and released viral and subviral particles following RRV infection with and without ORF52 knockdown
| Compartment | No. (%) of particlesa after transfection with: |
|
|---|---|---|
| siCNL | siORF52 | |
| Nucleus | ||
| A | 44 (11) | 23 (11) |
| B | 218 (56) | 86 (43) |
| C | 125 (32) | 93 (46) |
| Total | 387 | 202 |
| Cytoplasm | ||
| A | 20 (27) | 27 (8) |
| B | 0 (0) | 4 (1) |
| C | 9 (12) | 294 (90) |
| Virions in vesicles | 45 (61) | 2 (0.6) |
| Total | 74 | 327 |
| Cell surface virions | 145 (24) | 1 (0.2) |
| Total no. of cell-associated particles | 606 | 530 |
| Media | ||
| A | 43 (19) | 176 (58) |
| B | 9 (4) | 32 (10) |
| C | 32 (14) | 95 (31) |
| Virions | 147 (63) | 2 (0.7) |
| Total no. of particles counted | 231 | 305 |
Total particles in TEM images from 10 RhFs transfected with siCNL or siORF52 for 24 h and then infected at an MOI of 5 for 48 h. The percentages indicate the proportion of a particle species for the specific compartment, except for cell surface, which reflects the proportion of total particles in all compartments.
In siCNL cells, virions within vesicles constituted the majority (61%) of total cytoplasmic particles, while in siORF52 cells, virions within vesicles represented only 2 of the 327 (<1%) viral particles that we detected. Unenveloped and untegumented capsids comprised the remaining cytoplasmic subviral particles in the siORF52-treated cells and were often present surrounding vesicles, as is evident in Fig. 7D (arrowheads). Examining 10 randomly selected infected cells from siCNL and siORF52 treatments, we enumerated the cell surface-associated virions near the cellular plasma membrane and detected 145 cell surface-associated virions in siCNL cells in contrast to 1 subviral particle evident under siORF52 conditions. Combined with TEM (Fig. 5 and 7), these results demonstrated that ORF52 played little if any role in capsid formation, DNA packaging, or nuclear egress but instead was critical for later steps of maturation, including tegumentation, secondary envelopment, release, and virion adherence to the cell surface.
Expression of siRNA-resistant ORF52 in trans partially rescued maturation and release of infectious virions in ORF52 knockdown cells.
To confirm that the block in tegumentation and secondary envelopment was specific to ORF52 knockdown and not to off-target effects, we sought to rescue the defect by providing siRNA-resistant myc-tagged ORF52 in trans. We first transfected RhFs with either empty myc-tagged vector (pK-myc) or myc-tagged ORF52 that we had engineered to have two wobble base changes, making it resistant to siORF52. Twenty-four hours later, we treated the cultures with either siCNL or siORF52 and then infected them at an MOI of 5, as we had in our earlier experiments. After infection, RhFs transfected first with an empty myc-tagged vector (pK-myc) and siCNL expressed MCP and SCIP, as well as ORF45 and ORF52 (Fig. 9A, lane 1), whereas cells transfected with pK-myc and siORF52 showed a significant reduction in ORF52 (Fig. 9A, lane 2), similar to the pattern we discerned in infected cultures pretreated with only siORF52 (Fig. 1A and B). Aside from the near absence of ORF52 in cell lysates, production of the other viral protein that we examined was not significantly different from that in infected cells transfected with empty vector and siCNL (Fig. 9B, black bars). In cell lysates from the complementing RhFs (i.e., transfected with pK-myc-Res52, abbreviated as Res52 here), we detected Res52 that, as we expected, migrated more slowly than wild-type ORF52 due to the myc tag (Fig. 9A, lane 3). With the exception of SCIP, which is modestly depressed (by approximately 25%) under the kd/Res52 conditions compared to the control (kd/vector), the intracellular levels of the nontargeted structural proteins are similar under the two conditions (Fig. 9B). Expression of the transfected Res52 in siORF52-treated, RRV-infected cells is notably lower than that of WT ORF52 encoded by the virus in cells treated with siCNL plus vector, yet it is significantly higher (11-fold) than the WT ORF52 levels following knockdown alone (siORF52 plus pK-myc vector) (Fig. 9B).
FIG 9.

Complementation of ORF52 knockdown with exogenous siRNA-resistant ORF52 partially rescued the wild-type phenotype. RhFs transfected with either empty myc-tagged vector (Vec) or siORF52-resistant myc-tagged ORF52 (Res52) were reverse transfected 24 h later with siCNL or siORF52. After an additional 24 h, the cells were infected with RRV at an MOI of 5 and then harvested 48 h p.i. (A) Immunoblot analysis of cell lysates from infected RhFs first transfected with siCNL plus Vec (lane 1), siORF52 plus Vec (lane 2), or siORF52 plus Res52 (lane 3). The protein blots were incubated with antibodies specific for the viral tegument proteins ORF45 and ORF52, the capsid proteins MCP and SCIP, and cellular actin to normalize for loading differences. (B) Levels of the indicated viral proteins after siORF52 treatment with Vec or Res52 relative to siCNL in infected RhFs. The data represent the means and SD of 4 individual experiments. (C) Immunoblot analysis of concentrated medium collected from RhFs transfected with siCNL plus Vec (lane 1), siORF52 plus Vec (lane 2), or siORF52 plus Res52 (lane 3). Protein blots from the medium were probed for ORF45, ORF52, MCP, and SCIP. (D) Levels of the indicated viral proteins in released particles after siORF52 treatment following transfection with Vec or Res52 relative to those in particles released from siCNL-treated cells. The data represent the means and SD of 4 individual experiments. (B and D) P values were calculated by comparing knockdown (black bars) to rescue (gray bars). (E) Viral titers in the medium from each condition were determined by viral plaque assays of 4 individual experiments. The values are means and SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Since Res52 was present in the rescue cell lysate (Fig. 9A, lane 3), we hypothesized that if it were expressed at the correct time in the viral life cycle and in a useable form, we would observe some rescue of the knockdown phenotype. We first examined particle composition from media collected under the control, knockdown, and rescue conditions. As we had observed previously with infected cells pretreated with siCNL or siORF52 (Fig. 3), the levels of particle-associated capsid proteins (MCP and SCIP) were similar in the presence or absence of complementing Res52 following ORF52 kd compared to controls (Fig. 9C), and the two tegument proteins (ORF45 and ORF52) were absent following kd without ORF52 complementation (Fig. 9C, lane 2). However, in the particles released into the medium under the rescue condition, we now observed Res52 at a significantly increased level (40-fold) over the amount in kd without rescue (Fig. 9C, lane 3 versus lane 2, and D). The presence of Res52 in these released particles indicated that the Res52 that we provided in trans was packaged within the virion. Of note, we also observed partial rescue (albeit proportionally less than for Res52) of ORF45 incorporation into particles (Fig. 9C, lane 3). The average of five experiments demonstrated an approximately 7-fold-greater amount of ORF45 per particle (using MCP as a way to normalize for particle number) than the particles released from infected cells pretreated with siORF52 without rescue (plus pK-myc vector). This suggested not only that ORF45 required the presence of ORF52 on particles for its stable incorporation within the virion (Fig. 9C and D), but also that exogenous expression of Res52 may not be optimal in allowing efficient production of fully WT virions.
We next tested whether the released particles that contained Res52 and ORF45 represented infectious virions that would, in turn, partially restore the suppressed viral titers. To address this, we assessed the viral titers of the media under the rescue versus control conditions (i.e., ORF52 kd with or without Res52 expression in trans, respectively). siORF52 plus pK-myc vector showed a decrease in titer of >400-fold compared to siCNL plus pK-myc vector. However, the presence of Res52 with siORF52 was able to partially reverse this suppression, increasing the titer approximately 24-fold over siORF52 plus pK-myc vector (Fig. 9E). Combined, these data argued that reduction of ORF52 was responsible for the significant decrease in the viral titer and that this decrease was likely due to a lack of formation and release of mature virions. However, to demonstrate the latter more definitively, we next examined whether Res52 could also rescue the siORF52 block in particle maturation that was evident in our earlier TEM (Fig. 7).
ORF52 in trans partially rescued virion maturation.
To address more directly whether the increase in titer after Res52 expression in trans reflected an increase in the production of mature particles and, specifically, tegumentation and envelopment, we next asked whether we could discern an increase in the production and release of trilaminar virions. We again used thin-section TEM to examine the various stages of RRV maturation in cells, this time comparing those transfected with pK-myc or rescued with Res52, both in the presence of siORF52. Since our data indicated that particles packaged exogenously expressed Res52 that resulted in an increase in the production of infectious virions (Fig. 9E), we also expected to observe a partial rescue in normal virion morphogenesis. Not surprisingly, A, B, and C capsids were present in the nuclei of infected cells under all three conditions: control, knockdown, and rescue (Fig. 10A, B, and C, respectively). Likewise, examination of the cytoplasm in siCNL plus pK-myc vector-treated cells revealed multiple mature virions with tegument and envelope within large vesicles (Fig. 10D), as well as cell-associated virions on or near the plasma membrane (Fig. 10G). In cells transfected with siORF52 plus pK-myc vector, however, we once again found untegumented and nonenveloped particles that often surrounded but did not appear to enter vesicles (Fig. 10E). Furthermore, these cells lacking ORF52 and transfected only with vector displayed a lack of cell surface-associated virions (Fig. 10H). In marked contrast, cultures under rescue conditions (siORF52 plus Res52) displayed low but consistent levels of tegumented and enveloped virions located within cytoplasmic vesicles (Fig. 10F), as well as cell surface-associated virions along the outer surface of the cellular plasma membrane (Fig. 10I), reminiscent of the control phenotype. Consistent with the somewhat weak restoration of total intracellular ORF52, this rescued phenotype was less frequent than in the control cells.
FIG 10.

Exogenous ORF52 rescues formation and release of tegumented and enveloped virions. RhFs transfected with either empty myc-tagged vector (Vec) or siORF52-resistant myc-tagged ORF52 (Res52) were transfected 24 h later with siCNL or siORF52 and then, after an additional 24 h, were infected at an MOI of 5. Forty-eight hours later, the cells were fixed and examined by TEM at ×40,000 magnification. The nuclei of cells treated with (A) siCNL plus vec, (B) siORF52 plus vec, (C) or siORF52 plus Res52 all contained A (empty) (arrowheads), B scaffold-containing (chevrons), and C (arrows) capsids. (D and E) The cytoplasm of siCNL plus Vec cells (D) contained large vesicles (ves) filled with multiple tegumented and enveloped virions; the cytoplasm of siORF52 plus Vec cells (E) contained untegumented particles, many of which surrounded but were not within vesicles. (F) siORF52 plus Res52 cells showed virions within cytoplasmic vesicles. (G to I) In cells treated with siORF52 plus Vec (H), cell-associated virions are absent, in contrast to cells treated with siCNL plus vec (G) or siORF52 plus Res52 (I). (J to L) Cells treated with siORF52 plus Vec release particles that appear to be capsids, lacking tegument and envelope (K), in contrast to cells treated with siCNL plus Vec (J) and siORF52 plus Res52 (L), which release intact, mature virions. The inset images are expanded (2×) from the original images.
Likewise, with levels of Res52 and ORF45 in released particles under the rescue conditions lower than those of ORF52 and ORF45, respectively, under control conditions (Fig. 9D), we predicted that we would continue to find immature capsid-like particles in the medium, along with the recovery of at least some mature trilaminar virions. TEMs from the concentrated media of the rescue and control cells bore out these predictions. Cells treated with siORF52 but expressing Res52 still produced naked capsids, but we also observed mature-appearing virions with evidence of tegument, envelope, and glycoproteins that resembled WT virions from infected cells treated with siCNL and pK-myc (compare Fig. 10L with J). Combined, these TEM images provided additional evidence that ORF52 is necessary for RRV tegumentation and envelopment, as well as subsequent release of free and cell surface-associated mature viral particles.
Res52 partially restored nuclear egress of ORF45 in siORF52-treated cells.
Since providing Res52 in trans in the presence of siORF52 partially rescued ORF45 incorporation into viral particles and led to an increase in the production and release of infectious virions, we also predicted that IF images would reveal a partial return to wild-type subcellular distribution of ORF45. To verify this, we again transfected RhFs with vector or Res52 prior to ORF52 knockdown and RRV infection. Forty-eight hours later, pK-myc vector plus siCNL, as we had predicted, resulted in normal cytoplasmic and nuclear distribution of ORF45 and only cytoplasmic localization for ORF52 (Fig. 11, top row). In siORF52-treated cells, ORF45 expression again was only nuclear (Fig. 11, middle row). However, with Res52, some ORF45 signal was again evident in the cytoplasm and appeared as punctate dots that colocalized with ORF52, suggesting that ORF45 and ORF52 were again packaged within virions (Fig. 11, bottom row, arrows).
FIG 11.

Complementation with siRNA-resistant ORF52 partially restored cytoplasmic subcellular localization of ORF45. RhFs transfected with either empty myc-tagged vector (Vec) or siORF52-resistant myc-tagged ORF52 (Res52) were reverse transfected 24 h later on coverslips with siCNL (top row) or siORF52 (middle and bottom rows) and then, after 24 h, infected with RRV at an MOI of 5 and fixed 48 h later. The cells were stained with anti-ORF52 and anti-ORF45 antibodies, followed by the secondary antibodies Alexa Fluor 488 goat anti-mouse and Alexa Fluor 555 goat anti-rabbit, respectively. The cells were then stained with DAPI. The arrows highlight overlapping signals and colocalization of ORF52 and ORF45 in the cytoplasm.
DISCUSSION
While there are a number of proteins (or their homologs) present in the tegument of all herpesvirus species (reviewed in reference 77), others are restricted to a specific subfamily. The latter proteins likely have specialized roles critical in aspects of the life cycle or intracellular environment that are specific to those subfamilies. Most models of herpesvirus maturation suggest that final tegumentation begins once a capsid enters the cytoplasm following nuclear egress (reviewed in references 51 and 77). Capsids in the cytoplasm may then obtain a number of tegument proteins while moving toward vesicles, which may have an additional reservoir of tegument proteins (reviewed in references 5 and 77). These partially tegumented capsids reach the vesicles and then bud into them, acquiring additional tegument proteins, as well as the envelope studded with glycoproteins already embedded in the vesicle membrane (reviewed in references 77 and 78). If tegumentation depends on the coordinated interaction of proteins to assemble, it follows that a lack of, or a defect in, one of these proteins might disturb this process. Similarly, disruption of tegument assembly could also lead to particles lacking the proteins necessary to initiate and ensure receptor-mediated invagination, thereby preventing the acquisition of an envelope and inhibiting vesicle-mediated egress.
ORF52 is a tegument protein found only within the gammaherpesvirus subfamily. While previous studies have investigated both the structure and function of the murine homolog, MHV-68 ORF52 (38, 54, 79), and most recently the importance of phosphorylation to the ability of BLRF2, the EBV homolog, to complement a null mutant of MHV-68 ORF52 (60), this is the first study investigating the role of ORF52 in RRV, a close homolog of the human pathogen KSHV. RRV ORF52 is a tegument protein that is highly abundant within virions and tightly associated with the capsid (26). In this report, we show that ORF52 is necessary for tegumentation and secondary envelopment but is not essential for the production of structural proteins, assembly of capsids, viral DNA replication or packaging, or the ability of capsids to egress from the nucleus. Without ORF52, untegumented particles, which are morphologically indistinguishable from intranuclear capsids, accumulate within the cytoplasm (Fig. 7D), unable to enter but often surrounding cytoplasmic vesicles, where they would otherwise undergo secondary envelopment (reviewed in references 51 and 78). These results are consistent with the observations from disruption of MHV-68 ORF52, which also leads to a defect in tegumentation and secondary envelopment (38, 54).
Increased proportion of immature particles in the absence of ORF52.
Despite the inability of the untegumented and unenveloped particles to enter vesicles, we detected their release into the medium at levels that were similar to those from control cells (Fig. 3). At first, this seemed contradictory, in light of the approximately 4-fold-greater cytoplasmic accumulation of particles in the ORF52 kd than in controls, which we suggest reflects the inability of the untegumented particles to enter the vesicle-mediated egress pathway. However, we also noted that essentially no virions were associated with the cell surface under the kd conditions, whereas the controls displayed large numbers of virions that remained attached to the plasma membrane following egress (Fig. 7E and Table 1), a typical phenomenon for herpesviruses, including KSHV (80, 81). Thus, although the numbers of released particles in the media are similar with or without ORF52 knockdown (Fig. 3), the contribution of the cell surface-associated fraction argues that the number of virions exiting the cells, in total, is greater under the control than under the knockdown conditions. Although the mechanism of release of the immature particles from the siORF52-treated cells remains unclear, we did investigate whether it was simply due to cell lysis. However, we found that even if we harvested medium at 24 h p.i., a time at which infection-induced lysis is negligible (M. S. Anderson and D. H. Kedes, unpublished observations), the numbers of particles in the medium, though lower than at 48 h p.i., were similar under kd and control conditions (data not shown), arguing against this explanation. Furthermore, it is not immediately evident whether our data reflect a bona fide alternative egress pathway or arise as an artifact of the ORF52 kd conditions. Arguing against the last interpretation, however, is the fact that even under control conditions, A, B, and C capsids together comprised over 1/3 of the total released particles. Nevertheless, the mechanism of vesicle-independent particle release, other than through cell lysis, remains unclear and is of continued interest to our laboratory.
ORF52 is essential for virion maturation following egress from the nucleus.
Although ORF52 kd conditions led to an approximately 6-fold reduction in the proportion of DNA-containing particles in the medium, we conclude that their lack of tegument and envelope accounted for most of the drastic loss in titer (>300-fold) that we observed (Fig. 1C) despite similar total concentrations of particles under kd and control conditions (Fig. 3). The released particles were morphologically indistinguishable from the accumulating cytoplasmic particles (and nuclear capsids), lacking any discernible evidence of tegument or envelope (Fig. 5 and 6). Consistent with these observations, immunoblots demonstrated that these particles lacked tegument proteins ORF52 and ORF45 (Fig. 3). We also showed that these phenotypes are specific to ORF52, since myc-tagged ORF52 in trans partially rescued maturation with particle entry into vesicles and release of mature virions that was accompanied by the reappearance of cell-associated mature virions (Fig. 9 and 10).
ORF52 acts as a functional linchpin for tegument formation.
Tegumentation is a complex process involving the layering of multiple interacting cellular and viral proteins, which occurs throughout the cytoplasm and at sites of secondary envelopment (reviewed in references 5, 77, and 82). Earlier studies divided the tegument into inner and outer portions, referring to biochemical or structural evidence describing the relative affinity and proximity of specific sets of proteins to the surface of the capsid (26, 30, 32, 35, 83). When one of these tegument proteins is missing, it affects the abilities of other proteins to be added or perhaps to remain stably attached to the particle. We postulate that this might explain the loss of tegumentation that we observed by TEM and, specifically, the defect in incorporation of tegument protein ORF45 during RRV maturation in the absence of ORF52.
In our work, we found that diminution of ORF52 critically affected the ability of particles to acquire this tegument layer post-nuclear egress, and these particles localized around but were unable to enter vesicles (Fig. 7D). This result is consistent with data published on MHV-68 ORF52, where cells transfected with an ORF52-StopBAC, which expresses the entire viral genome except ORF52, were also unable to undergo tegumentation and envelopment, as mentioned above (38). Recent work from the Deng laboratory demonstrated, by co-IP of transiently transfected HEK-293 cells, that MHV-68 ORF52 interacts with another gammaherpesvirus-specific tegument protein, ORF42, and that an arginine-to-alanine change at amino acid 95 (R95A) in ORF52 inhibited this interaction (54). The specific function of ORF42 is currently unknown; however, R95, which is conserved among ORF52 homologs in gammaherpesviruses, is important in the viral life cycle. The WT MHV-68 ORF52, but not the R95A mutant, complements the defect in replication of the ORF52-StopBAC (79). Other evidence from KSHV studies demonstrated that its ORF52 protein interacts with ORF45 (34).
In our experiments with RRV, we were unable to detect a direct interaction between ORF52 and ORF45 by IP either from HEK-293 cells expressing tagged versions of the two proteins or from RRV-infected RhFs (data not shown). Since there are multiple proteins within the tegument layer interacting in yet unknown configurations, it is possible that ORF52 and ORF45 might indirectly interact through a third protein. A possible candidate is ORF33, a tegument protein present in alpha-, beta-, and gammaherpesviruses. In MHV-68, ORF33 plays a role in virion maturation affecting both egress of capsids from the nucleus and tegumentation and envelopment of capsids reaching the cytoplasm (48). Particles released via freeze-thaw of cells transfected with an MHV-68 ORF33-StopBAC have ORF52, but they lack ORF45 (48), suggesting that ORF33 might be a bridge between inner (e.g., ORF52) and outer (e.g., ORF45) tegument proteins.
Nuclear egress of ORF45 is ORF52 dependent.
ORF45, a multifunctional protein with roles that include antagonizing the host antiviral response (84), is also a gammaherpesvirus-specific tegument protein with homologs in RRV, KSHV, EBV, and MHV-68 (19, 23, 25, 26, 53). KSHV ORF45 also interacts with the kinesin-2 motor protein subunit, KIF3A, and is responsible for loading viral particles onto microtubules for plus-end-directed transport toward the plasma membrane (49). In our experiments, knockdown of ORF52 had no discernible effect on the overall ORF45 expression levels (Fig. 1A and B), yet it was not incorporated into released particles (Fig. 3). This suggests that ORF52 is necessary for ORF45 incorporation (or at least its stable incorporation) into maturing virions and likely reflects the role ORF52 might have in establishing a tegument foundation that allows the stable addition of other tegument proteins rather than a direct interaction with ORF45 itself. In light of these findings, we hypothesized that if ORF45 were responsible for efficient particle transport, then knocking down ORF52 would affect tegumentation of particles in the cytoplasm and result in reduced movement through the cytoplasm and viral egress.
Consistent with a potential defect in anterograde transport, suppression of RRV ORF52 expression also appears to affect the subcellular localization of ORF45. During unperturbed RRV infection, we found that in addition to a diffuse nuclear pattern, ORF45 also colocalized with ORF52 in the cytoplasm in a discrete punctate pattern. Following ORF52 kd, however, we detected ORF45 only in the nucleus. While we have no evidence that ORF52 can enter the nucleus (e.g., no nuclear localization signal [NLS] and no nuclear staining by IF) or interact directly with ORF45, these data argue that an intact tegument, dependent on ORF52 expression, is necessary for ORF45 incorporation into viral particles undergoing transcytosis. Capsid (SCIP-containing) particles from ORF52 kd lacked ORF45 and ORF52 in the cytoplasm (Fig. 3 and 4) and demonstrated a more restricted and aggregated perinuclear distribution. This might reflect an inability to attach to microtubule-based motors (49).
Inability of particles to invaginate into cytoplasmic vesicles.
During secondary envelopment in the cytoplasm, partially tegumented particles enter vesicles, acquiring their final complement of tegument proteins and their glycoprotein-studded envelope. In cells lacking ORF52, there was an accumulation of particles with no detectable tegument within the cytoplasm (Fig. 7D). We found that vesicles in siCNL cells were larger than in siORF52 cells, and many contained multiple trilaminar virions (Fig. 7C and D). Interestingly, cells that we treated with siORF52 still formed vesicles, but the particles failed to enter and were often clustered around them (Fig. 7D), consistent with studies of ORF52-null MHV-68 (38, 54). We speculate that the last step of invagination is dependent on proper tegumentation that provides key protein interactions between the surface of the particle and the surface of the vesicle.
ORF52-dependent tegumentation and envelopment are necessary for cell surface association of viral particles.
Virions undergo release from the cell in a process similar to exocytosis once the vesicle containing virions merges with the cellular plasma membrane (reviewed in references 51, 77, and 85). This leads to opening of the vesicle, where particles are released into the surrounding environment or remain associated with the cell surface. Part of the host immune function is to prevent the spread of infectious virions to other cells. Some virus-infected cells, in fact, trap newly released enveloped virions, including retroviral and filoviral particles (64) and KSHV (66), onto the outer surface of the plasma membrane using bone marrow stromal cell antigen 2 (BST2)/tetherin. BST2, a heavily glycosylated type II transmembrane cellular protein (86), likely acts by forming homodimers that link the viral envelope and plasma membrane (reviewed in reference 87). KSHV partially circumvents this inhibition by encoding K5/MIR2, which ubiquinates BST2, leading to its degradation (66). RRV, interestingly, has no K5 homolog. We observed mature RRV particles lining up along the cell surface during RRV infection under control but not under ORF52 kd conditions (Fig. 7E and F), suggesting that without the envelope and its glycoproteins, no tethering of particles occurs. These released unenveloped subviral particles in ORF52 kd are unable to remain cell surface associated, thereby also leading to near abrogation of cell-to-cell viral spread normally evident under control conditions (Fig. 8).
ORF52 in trans partially rescues the block in particle maturation.
To ensure that ORF52 was responsible for the phenotypes that we observed, we complemented the system by providing siRNA-resistant ORF52 (Res52) in trans. Expression of Res52 (Fig. 9A) during siORF52 treatment of infected cells led to the incorporation of Res52 and, to a lesser extent, ORF45 into virions (Fig. 9C and D), suggesting that at least tegumentation was partially restored. This complementation also had functional consequences, increasing the viral titer compared to ORF52 knockdown alone (Fig. 9E) and suggesting partial restoration of fully infectious trilaminar virions. TEM images from Res52-complemented infections corroborated this hypothesis, demonstrating evidence of mature virions within vesicles (Fig. 10F); cell-associated virions that appeared identical to those without knockdown (Fig. 10G and I); and released virions, also with visible tegument, envelope, and glycoproteins (Fig. 10L). The efficiency of the rescue was likely suboptimal due to the low plasmid transfection efficiency (∼30%) of Res52 in the culture and the possibility that ectopic overexpression likely fails to recapitulate the coordinated expression of interacting tegument proteins.
With MHV-68, the expression in trans of its own ORF52, or the homolog from KSHV or EBV (BLRF2), is able to complement a similar defect in virion maturation and total block in virion release that arises with transfection of cultures with an ORF52-StopBAC (54). We also attempted to complement the ORF52 kd-induced defect in tegumentation in the RRV system with KSHV ORF52 but found that despite its expression being comparable to that of RRV Res52, its incorporation into the virions was poor, and as a consequence, the viral titer increased by only 1.5-fold (compared to 26-fold increase with RRV Res52) over ORF52 knockdown controls (data not shown). The reasons behind the differences between the MHV-68 results and our RRV results are unclear.
In sum, we have shown that the inner tegument protein RRV ORF52 is critical for postnuclear stages of virion maturation, including tegumentation and secondary envelopment, necessary for production and egress of fully mature trilaminar virions. Without ORF52, the necessary layering of tegument proteins—key to the architecture of the virion—is unable to occur, resulting in the cytoplasmic accumulation of immature viral particles surrounding but unable to enter awaiting vesicles.
ACKNOWLEDGMENTS
We thank Nancy Verville for cloning RRV ORF52 and ORF65 and generation of purified protein used to generate monoclonal antibodies. We also thank Scott Wong (Oregon Health and Science University) for providing us with the RRV MCP and gB antibodies and for helpful discussions. Additionally, we thank Barbee Herrmann and Stacey Guillot at the University of Virginia Advanced Microscopy Core for training and technical assistance on the JEOL 1230 electron microscope and sample preparation. Finally, we thank Evonne N. Woodson and Thomas J. Ellison for helpful discussions and critical readings of the manuscript.
This research was supported by the NIH (grants R01CA088768 and R01DE022291 [to D.H.K.] and NRSA T32 AI007046 and F31 F31CA159541 [to M.S.A.]).
Footnotes
Published ahead of print 4 June 2014
REFERENCES
- 1.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191. 10.1056/NEJM199505043321802 [DOI] [PubMed] [Google Scholar]
- 2.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. 10.1126/science.7997879 [DOI] [PubMed] [Google Scholar]
- 3.Cesarman E, Moore PS, Rao PH, Inghirami G, Knowles DM, Chang Y. 1995. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood 86:2708–2714 [PubMed] [Google Scholar]
- 4.Boshoff C, Chang Y. 2001. Kaposi's sarcoma-associated herpesvirus: a new DNA tumor virus. Annu. Rev. Med. 52:453–470. 10.1146/annurev.med.52.1.453 [DOI] [PubMed] [Google Scholar]
- 5.Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234. 10.1016/j.virusres.2009.03.018 [DOI] [PubMed] [Google Scholar]
- 6.Grinde B. 2013. Herpesviruses: latency and reactivation—viral strategies and host response. J. Oral Microbiol. 10.3402/jom.v5i0.22766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foreman KE, Friborg J, Jr, Kong WP, Woffendin C, Polverini PJ, Nickoloff BJ, Nabel GJ. 1997. Propagation of a human herpesvirus from AIDS-associated Kaposi's sarcoma. N. Engl. J. Med. 336:163–171. 10.1056/NEJM199701163360302 [DOI] [PubMed] [Google Scholar]
- 8.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342–346. 10.1038/nm0396-342 [DOI] [PubMed] [Google Scholar]
- 9.Desrosiers RC, Sasseville VG, Czajak SC, Zhang X, Mansfield KG, Kaur A, Johnson RP, Lackner AA, Jung JU. 1997. A herpesvirus of rhesus monkeys related to the human Kaposi's sarcoma-associated herpesvirus. J. Virol. 71:9764–9769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alexander L, Denekamp L, Knapp A, Auerbach MR, Damania B, Desrosiers RC. 2000. The primary sequence of rhesus monkey rhadinovirus isolate 26-95: sequence similarities to Kaposi's sarcoma-associated herpesvirus and rhesus monkey rhadinovirus isolate 17577. J. Virol. 74:3388–3398. 10.1128/JVI.74.7.3388-3398.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Searles RP, Bergquam EP, Axthelm MK, Wong SW. 1999. Sequence and genomic analysis of a Rhesus macaque rhadinovirus with similarity to Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 73:3040–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Granzow H, Klupp BG, Mettenleiter TC. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J. Virol. 79:3200–3205. 10.1128/JVI.79.5.3200-3205.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maurer UE, Sodeik B, Grunewald K. 2008. Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proc. Natl. Acad. Sci. U. S. A. 105:10559–10564. 10.1073/pnas.0801674105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandran B. 2010. Early events in Kaposi's sarcoma-associated herpesvirus infection of target cells. J. Virol. 84:2188–2199. 10.1128/JVI.01334-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKnight JL, Kristie TM, Roizman B. 1987. Binding of the virion protein mediating alpha gene induction in herpes simplex virus 1-infected cells to its cis site requires cellular proteins. Proc. Natl. Acad. Sci. U. S. A. 84:7061–7065. 10.1073/pnas.84.20.7061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He B, Gross M, Roizman B. 1997. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 94:843–848. 10.1073/pnas.94.3.843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato K, Daikoku T, Goshima F, Kume H, Yamaki K, Nishiyama Y. 2000. Synthesis, subcellular localization and VP16 interaction of the herpes simplex virus type 2 UL46 gene product. Arch. Virol. 145:2149–2162. 10.1007/s007050070045 [DOI] [PubMed] [Google Scholar]
- 18.Schlieker C, Korbel GA, Kattenhorn LM, Ploegh HL. 2005. A deubiquitinating activity is conserved in the large tegument protein of the herpesviridae. J. Virol. 79:15582–15585. 10.1128/JVI.79.24.15582-15585.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bechtel JT, Winant RC, Ganem D. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:4952–4964. 10.1128/JVI.79.8.4952-4964.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato A, Yamamoto M, Ohno T, Tanaka M, Sata T, Nishiyama Y, Kawaguchi Y. 2006. Herpes simplex virus 1-encoded protein kinase UL13 phosphorylates viral Us3 protein kinase and regulates nuclear localization of viral envelopment factors UL34 and UL31. J. Virol. 80:1476–1486. 10.1128/JVI.80.3.1476-1486.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taddeo B, Zhang W, Roizman B. 2006. The U(L)41 protein of herpes simplex virus 1 degrades RNA by endonucleolytic cleavage in absence of other cellular or viral proteins. Proc. Natl. Acad. Sci. U. S. A. 103:2827–2832. 10.1073/pnas.0510712103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spear PG, Roizman B. 1972. Proteins specified by herpes simplex virus. V. Purification and structural proteins of the herpesvirion. J. Virol. 9:143–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E. 2004. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. U. S. A. 101:16286–16291. 10.1073/pnas.0407320101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG, II, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960–10966. 10.1128/JVI.78.20.10960-10966.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu FX, Chong JM, Wu L, Yuan Y. 2005. Virion proteins of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:800–811. 10.1128/JVI.79.2.800-811.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Connor CM, Kedes DH. 2006. Mass spectrometric analyses of purified rhesus monkey rhadinovirus reveal 33 virion-associated proteins. J. Virol. 80:1574–1583. 10.1128/JVI.80.3.1574-1583.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 82:8605–8618. 10.1128/JVI.00904-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woodson EN, Kedes DH. 2012. Distinct roles for extracellular signal-regulated kinase 1 (ERK1) and ERK2 in the structure and production of a primate gammaherpesvirus. J. Virol. 86:9721–9736. 10.1128/JVI.00695-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen DH, Jiang H, Lee M, Liu F, Zhou ZH. 1999. Three-dimensional visualization of tegument/capsid interactions in the intact human cytomegalovirus. Virology 260:10–16. 10.1006/viro.1999.9791 [DOI] [PubMed] [Google Scholar]
- 30.Zhou ZH, Chen DH, Jakana J, Rixon FJ, Chiu W. 1999. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J. Virol. 73:3210–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown JC, Newcomb WW. 2011. Herpesvirus capsid assembly: insights from structural analysis. Curr. Opin. Virol. 1:142–149. 10.1016/j.coviro.2011.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dai W, Jia Q, Bortz E, Shah S, Liu J, Atanasov I, Li X, Taylor KA, Sun R, Zhou ZH. 2008. Unique structures in a tumor herpesvirus revealed by cryo-electron tomography and microscopy. J. Struct. Biol. 161:428–438. 10.1016/j.jsb.2007.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu XK, O'Connor CM, Atanasov I, Damania B, Kedes DH, Zhou ZH. 2003. Three-dimensional structures of the A, B, and C capsids of rhesus monkey rhadinovirus: insights into gammaherpesvirus capsid assembly, maturation, and DNA packaging. J. Virol. 77:13182–13193. 10.1128/JVI.77.24.13182-13193.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rozen R, Sathish N, Li Y, Yuan Y. 2008. Virion-wide protein interactions of Kaposi's sarcoma-associated herpesvirus. J. Virol. 82:4742–4750. 10.1128/JVI.02745-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolfstein A, Nagel CH, Radtke K, Dohner K, Allan VJ, Sodeik B. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7:227–237. 10.1111/j.1600-0854.2005.00379.x [DOI] [PubMed] [Google Scholar]
- 36.Sodeik B, Ebersold MW, Helenius A. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007–1021. 10.1083/jcb.136.5.1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luxton GW, Haverlock S, Coller KE, Antinone SE, Pincetic A, Smith GA. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. U. S. A. 102:5832–5837. 10.1073/pnas.0500803102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bortz E, Wang L, Jia Q, Wu TT, Whitelegge JP, Deng H, Zhou ZH, Sun R. 2007. Murine gammaherpesvirus 68 ORF52 encodes a tegument protein required for virion morphogenesis in the cytoplasm. J. Virol. 81:10137–10150. 10.1128/JVI.01233-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203. 10.1016/j.chom.2013.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smiley JR. 2004. Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J. Virol. 78:1063–1068. 10.1128/JVI.78.3.1063-1068.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taddeo B, Roizman B. 2006. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J. Virol. 80:9341–9345. 10.1128/JVI.01008-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shu M, Taddeo B, Zhang W, Roizman B. 2013. Selective degradation of mRNAs by the HSV host shutoff RNase is regulated by the UL47 tegument protein. Proc. Natl. Acad. Sci. U. S. A. 110:E1669–E1675. 10.1073/pnas.1305475110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Full F, Jungnickl D, Reuter N, Bogner E, Brulois K, Scholz B, Sturzl M, Myoung J, Jung JU, Stamminger T, Ensser A. 2014. Kaposi's sarcoma associated herpesvirus tegument protein ORF75 is essential for viral lytic replication and plays a critical role in the antagonization of ND10-instituted intrinsic immunity. PLoS Pathog. 10:e1003863. 10.1371/journal.ppat.1003863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jurak I, Silverstein LB, Sharma M, Coen DM. 2012. Herpes simplex virus is equipped with RNA- and protein-based mechanisms to repress expression of ATRX, an effector of intrinsic immunity. J. Virol. 86:10093–10102. 10.1128/JVI.00930-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ling PD, Tan J, Sewatanon J, Peng R. 2008. Murine gammaherpesvirus 68 open reading frame 75c tegument protein induces the degradation of PML and is essential for production of infectious virus. J. Virol. 82:8000–8012. 10.1128/JVI.02752-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu FX, Sathish N, Yuan Y. 2010. Antagonism of host antiviral responses by Kaposi's sarcoma-associated herpesvirus tegument protein ORF45. PLoS One 5:e10573. 10.1371/journal.pone.0010573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sathish N, Zhu FX, Golub EE, Liang Q, Yuan Y. 2011. Mechanisms of autoinhibition of IRF-7 and a probable model for inactivation of IRF-7 by Kaposi's sarcoma-associated herpesvirus protein ORF45. J. Biol. Chem. 286:746–756. 10.1074/jbc.M110.150920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo H, Wang L, Peng L, Zhou ZH, Deng H. 2009. Open reading frame 33 of a gammaherpesvirus encodes a tegument protein essential for virion morphogenesis and egress. J. Virol. 83:10582–10595. 10.1128/JVI.00497-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sathish N, Zhu FX, Yuan Y. 2009. Kaposi's sarcoma-associated herpesvirus ORF45 interacts with kinesin-2 transporting viral capsid-tegument complexes along microtubules. PLoS Pathog. 5:e1000332. 10.1371/journal.ppat.1000332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chiu YF, Sugden B, Chang PJ, Chen LW, Lin YJ, Lan YC, Lai CH, Liou JY, Liu ST, Hung CH. 2012. Characterization and intracellular trafficking of Epstein-Barr virus BBLF1, a protein involved in virion maturation. J. Virol. 86:9647–9655. 10.1128/JVI.01126-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mettenleiter TC, Muller F, Granzow H, Klupp BG. 2013. The way out: what we know and do not know about herpesvirus nuclear egress. Cell. Microbiol. 15:170–178. 10.1111/cmi.12044 [DOI] [PubMed] [Google Scholar]
- 52.Dittmer DP, Gonzalez CM, Vahrson W, DeWire SM, Hines-Boykin R, Damania B. 2005. Whole-genome transcription profiling of rhesus monkey rhadinovirus. J. Virol. 79:8637–8650. 10.1128/JVI.79.13.8637-8650.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bortz E, Whitelegge JP, Jia Q, Zhou ZH, Stewart JP, Wu TT, Sun R. 2003. Identification of proteins associated with murine gammaherpesvirus 68 virions. J. Virol. 77:13425–13432. 10.1128/JVI.77.24.13425-13432.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang L, Guo H, Reyes N, Lee S, Bortz E, Guo F, Sun R, Tong L, Deng H. 2012. Distinct domains in ORF52 tegument protein mediate essential functions in murine gammaherpesvirus 68 virion tegumentation and secondary envelopment. J. Virol. 86:1348–1357. 10.1128/JVI.05497-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeWire SM, Money ES, Krall SP, Damania B. 2003. Rhesus monkey rhadinovirus (RRV): construction of a RRV-GFP recombinant virus and development of assays to assess viral replication. Virology 312:122–134. 10.1016/S0042-6822(03)00195-8 [DOI] [PubMed] [Google Scholar]
- 56.Haarr L, Skulstad S. 1994. The herpes simplex virus type 1 particle: structure and molecular functions. APMIS 102:321–346. 10.1111/j.1699-0463.1994.tb04882.x [DOI] [PubMed] [Google Scholar]
- 57.Homa FL, Brown JC. 1997. Capsid assembly and DNA packaging in herpes simplex virus. Rev. Med. Virol. 7:107–122. 10.1002/(SICI)1099-1654(199707)7:2<107::AID-RMV191>3.0.CO;2-M [DOI] [PubMed] [Google Scholar]
- 58.Steven AC, Trus BL, Booy FP, Cheng N, Zlotnick A, Caston JR, Conway JF. 1997. The making and breaking of symmetry in virus capsid assembly: glimpses of capsid biology from cryoelectron microscopy. FASEB J. 11:733–742 [DOI] [PubMed] [Google Scholar]
- 59.Nealon K, Newcomb WW, Pray TR, Craik CS, Brown JC, Kedes DH. 2001. Lytic replication of Kaposi's sarcoma-associated herpesvirus results in the formation of multiple capsid species: isolation and molecular characterization of A, B, and C capsids from a gammaherpesvirus. J. Virol. 75:2866–2878. 10.1128/JVI.75.6.2866-2878.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duarte M, Wang L, Calderwood MA, Adelmant G, Ohashi M, Roecklein-Canfield J, Marto JA, Hill DE, Deng H, Johannsen E. 2013. An RS motif within the Epstein-Barr virus BLRF2 tegument protein is phosphorylated by SRPK2 and is important for viral replication. PLoS One 8:e53512. 10.1371/journal.pone.0053512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mettenleiter TC. 2004. Budding events in herpesvirus morphogenesis. Virus Res. 106:167–180. 10.1016/j.virusres.2004.08.013 [DOI] [PubMed] [Google Scholar]
- 62.Gibson W, Roizman B. 1972. Proteins specified by herpes simplex virus. 8. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J. Virol. 10:1044–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O'Connor CM, Damania B, Kedes DH. 2003. De novo infection with rhesus monkey rhadinovirus leads to the accumulation of multiple intranuclear capsid species during lytic replication but favors the release of genome-containing virions. J. Virol. 77:13439–13447. 10.1128/JVI.77.24.13439-13447.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jouvenet N, Neil SJ, Zhadina M, Zang T, Kratovac Z, Lee Y, McNatt M, Hatziioannou T, Bieniasz PD. 2009. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 83:1837–1844. 10.1128/JVI.02211-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. 2009. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 106:2886–2891. 10.1073/pnas.0811014106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mansouri M, Viswanathan K, Douglas JL, Hines J, Gustin J, Moses AV, Fruh K. 2009. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi's sarcoma-associated herpesvirus. J. Virol. 83:9672–9681. 10.1128/JVI.00597-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sakuma T, Noda T, Urata S, Kawaoka Y, Yasuda J. 2009. Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 83:2382–2385. 10.1128/JVI.01607-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pardieu C, Vigan R, Wilson SJ, Calvi A, Zang T, Bieniasz P, Kellam P, Towers GJ, Neil SJ. 2010. The RING-CH ligase K5 antagonizes restriction of KSHV and HIV-1 particle release by mediating ubiquitin-dependent endosomal degradation of tetherin. PLoS Pathog. 6:e1000843. 10.1371/journal.ppat.1000843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zenner HL, Mauricio R, Banting G, Crump CM. 2013. Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J. Virol. 87:13115–13123. 10.1128/JVI.02167-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Ooij C. 2010. Virology: cleaving the tether. Nat. Rev. Microbiol. 8:385. 10.1038/nrmicro2377 [DOI] [PubMed] [Google Scholar]
- 71.Black FL, Melnick JL. 1955. Microepidemiology of poliomyelitis and herpes-B infections: spread of the viruses within tissue cultures. J. Immunol. 74:236–242 [PubMed] [Google Scholar]
- 72.Campadelli-Fiume G, Roizman B. 2006. The egress of herpesviruses from cells: the unanswered questions. J. Virol. 80:6716–6719. 10.1128/JVI.00386-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Christian RT, Ludovici PP, Jeter WS. 1971. Cell-to-cell transmission of herpes simplex virus in primary human amnion cells. Proc. Soc. Exp. Biol. Med. 138:1109–1115. 10.3181/00379727-138-36061 [DOI] [PubMed] [Google Scholar]
- 74.Mothes W, Sherer NM, Jin J, Zhong P. 2010. Virus cell-to-cell transmission. J. Virol. 84:8360–8368. 10.1128/JVI.00443-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sattentau Q. 2008. Avoiding the void: cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 6:815–826. 10.1038/nrmicro1972 [DOI] [PubMed] [Google Scholar]
- 76.Tomescu C, Law WK, Kedes DH. 2003. Surface downregulation of major histocompatibility complex class I, PE-CAM, and ICAM-1 following de novo infection of endothelial cells with Kaposi's sarcoma-associated herpesvirus. J. Virol. 77:9669–9684. 10.1128/JVI.77.17.9669-9684.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo H, Shen S, Wang L, Deng H. 2010. Role of tegument proteins in herpesvirus assembly and egress. Protein Cell 1:987–998. 10.1007/s13238-010-0120-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Henaff D, Radtke K, Lippe R. 2012. Herpesviruses exploit several host compartments for envelopment. Traffic 13:1443–1449. 10.1111/j.1600-0854.2012.01399.x [DOI] [PubMed] [Google Scholar]
- 79.Benach J, Wang L, Chen Y, Ho CK, Lee S, Seetharaman J, Xiao R, Acton TB, Montelione GT, Deng H, Sun R, Tong L. 2007. Structural and functional studies of the abundant tegument protein ORF52 from murine gammaherpesvirus 68. J. Biol. Chem. 282:31534–31541. 10.1074/jbc.M705637200 [DOI] [PubMed] [Google Scholar]
- 80.Granzow H, Weiland F, Jons A, Klupp BG, Karger A, Mettenleiter TC. 1997. Ultrastructural analysis of the replication cycle of pseudorabies virus in cell culture: a reassessment. J. Virol. 71:2072–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Granzow H, Klupp BG, Fuchs W, Veits J, Osterrieder N, Mettenleiter TC. 2001. Egress of alphaherpesviruses: comparative ultrastructural study. J. Virol. 75:3675–3684. 10.1128/JVI.75.8.3675-3684.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sathish N, Wang X, Yuan Y. 2012. Tegument proteins of Kaposi's Sarcoma-associated herpesvirus and related gamma-herpesviruses. Front. Microbiol. 3:98. 10.3389/fmicb.2012.00098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou ZH, Dougherty M, Jakana J, He J, Rixon FJ, Chiu W. 2000. Seeing the herpesvirus capsid at 8.5 A. Science 288:877–880. 10.1126/science.288.5467.877 [DOI] [PubMed] [Google Scholar]
- 84.Zhu FX, King SM, Smith EJ, Levy DE, Yuan Y. 2002. A Kaposi's sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl. Acad. Sci. U. S. A. 99:5573–5578. 10.1073/pnas.082420599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mettenleiter TC, Minson T. 2006. Egress of alphaherpesviruses. J. Virol. 80:1610–1612. 10.1128/JVI.80.3.1610-1612.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, Banting G. 2003. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 4:694–709. 10.1034/j.1600-0854.2003.00129.x [DOI] [PubMed] [Google Scholar]
- 87.Evans DT, Serra-Moreno R, Singh RK, Guatelli JC. 2010. BST-2/tetherin: a new component of the innate immune response to enveloped viruses. Trends Microbiol. 18:388–396. 10.1016/j.tim.2010.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]