

ABSTRACT
Viruses take advantage of host posttranslational modifications for their own benefit. It was recently reported that influenza A virus proteins interact extensively with the host sumoylation system. Thereby, several viral proteins, including NS1 and M1, are sumoylated to facilitate viral replication. However, to what extent sumoylation is exploited by influenza A virus is not fully understood. In this study, we found that influenza A virus nucleoprotein (NP) is a bona fide target of sumoylation in both NP-transfected cells and virus-infected cells. We further found that NP is sumoylated at the two most N-terminal residues, lysines 4 and 7, and that sumoylation at lysine 7 of NP is highly conserved across different influenza A virus subtypes and strains, including the recently emerged human H7N9 virus. While NP stability and polymerase activity are little affected by sumoylation, the NP sumoylation-defective WSN-NPK4,7R virus exhibited early cytoplasmic localization of NP. The growth of the WSN-NPK4,7R virus was highly attenuated compared to that of the wild-type WSN virus, and the lysine residue at position 7 is indispensable for the virus's survival, as illustrated by the rapid emergence of revertant viruses. Thus, sumoylation of influenza A virus NP is essential for intracellular trafficking of NP and for virus growth, illustrating sumoylation as a crucial strategy extensively exploited by influenza A virus for survival in its host.
IMPORTANCE Host posttranslational modifications are heavily targeted by viruses for their own benefit. We and others previously reported that influenza A virus interacts extensively with the host sumoylation system. However, the functional outcomes of viral sumoylation are not fully understood. Here we found that influenza A virus nucleoprotein (NP), an essential component for virus replication, is a new target of SUMO. This is the first study to find that NP from different influenza A viruses, including recently emerged H7N9, is sumoylated at conserved lysine 7. Our data further illustrated that sumoylation of influenza A virus NP is essential for intracellular trafficking of NP and virus growth, indicating that influenza A virus relies deeply on sumoylation to survive in host cells. Strategies to downregulate viral sumoylation could thus be a potential antiviral treatment.
INTRODUCTION
Influenza A viruses are among the most common pathogens and pose a continuous threat to animal and human health. Although aquatic birds are the primary reservoir of these viruses, they can cross species barriers to infect other animals, such as poultry, pigs, and humans. The latest pandemic, in 2009, was caused by the swine origin H1N1 virus and resulted in >18,449 deaths by August 2010 (1). The recent outbreak of a novel reassortant avian H7N9 virus in China has also raised considerable concern both domestically and globally (2, 3). Therefore, continued efforts to elucidate the mechanisms and factors underlying the pathogenesis of these viruses are critical.
On the basis of the antigenic properties of the viral surface glycoproteins hemagglutinin (HA) and neuraminidase (NA), influenza A viruses can be classified into 18 HA subtypes and 11 NA subtypes (4, 5). Within a certain subtype, virus isolates are further clustered into different clades according to their genetic diversity. Given this variability, the identification of general strategies exploited by all influenza A viruses in their infection cycle will facilitate the development of universal antiviral drugs.
Several recent studies have indicated that influenza A virus interacts extensively with host posttranslational modification systems, including those responsible for phosphorylation, ubiquitination, and sumoylation. For instance, a mass spectrometry study of the phosphoproteome of influenza A and B viruses revealed that almost all viral proteins, namely, PB2, PB1, PA, HA, NA, NP, M1, M2, NS1, and NEP, are phosphorylated (6). In addition, NP undergoes ubiquitination and deubiquitination, which may play an important role in regulating viral transcription and replication (7). Another recently identified posttranslational modifier, SUMO (small ubiquitin-related modifier), is covalently conjugated to a target protein via a three-step enzymatic cascade similar to the ubiquitination process and has also been suggested to play a role in viral replication. We previously reported that the NS1 protein of different influenza A virus strains is sumoylated under physiological conditions in infected cells, resulting in accelerated virus growth (8). Others have reported that the M1 protein is sumoylated to facilitate viral ribonucleoprotein (vRNP) export and assembly of virus particles (9). A recent systematic screening of influenza A virus proteins with the PR8 strain suggested that, in addition to NS1 and M1, PB1 and NP might be SUMO targets in vitro and in transfected cells (10). However, so far, no clear evidence has been provided that PB1 and NP are sumoylated in virus-infected cells. Moreover, the actual modification sites of PB1 and NP, as well as the role of PB1 and NP sumoylation in virus infection, remain unknown. Because many viruses take advantage of sumoylation (11, 12) and because influenza A virus intensively hijacks the sumoylation system to assist viral replication (10), a thorough investigation of the sumoylation of influenza A virus proteins, particularly the components of the polymerase, is warranted.
There are four SUMO isoforms in humans, SUMO1, SUMO2, SUMO3, and SUMO4 (13, 14). The conjugation of these SUMO molecules to a target protein occurs in a three-step cascade. After proteolytic processing mediated by sentrin-specific proteases (SENPs), the resulting mature form of SUMO with two glycine residues exposed at the carboxy terminus is first activated by the E1 heterodimer of Aos-Uba2. Subsequently, the SUMO molecule is transferred to the E2-conjugating enzyme Ubc9, the only E2 sumoylation enzyme discovered to date. Finally, the carboxyl group of the glycine residues at the SUMO carboxy terminus forms an isopeptide linkage with the amino group of a lysine residue on the substrate molecule, a step usually facilitated by E3 ligases in vivo. As a reversible posttranslational modification, the SUMO moiety is dynamically removed by SENPs and recovered for a new sumoylation cycle (13). Although SUMO and ubiquitin belong to the same family of ubiquitin-like modifiers, the functional outcome of sumoylation and ubiquitination can be completely different (15). The outcome of sumoylation is largely dependent on the function of its target protein, and many aspects of the target protein can be affected by sumoylation, including stability, structure, interaction partners, cellular localization, and enzymatic activity (16).
The genome of influenza A virus consists of eight segments of RNA, each of which is tightly associated with one copy of the polymerase complex (consisting of the PB1, PB2, and PA proteins) and multiple copies of NP to form vRNPs (17). The vRNP complex, of which NP is the major structural component, is responsible for the replication and transcription of the viral genome. To initiate the replication and transcription of the viral genome, vRNPs must be transported into the nucleus of the host cell by the cellular importin-α/β nuclear import pathway (18). Considering that most of the components of the sumoylation machinery are also localized in the nucleus, we investigated whether the proteins comprising the vRNP could be bona fide SUMO targets.
Here, we report that influenza A virus NP is a sumoylation substrate and the N-terminal lysines of NP at positions 4 and 7 are the SUMO acceptor sites. Lysine 7 in NP is highly conserved across subtypes and strains, indicating that sumoylation of NP is a common strategy of influenza A viruses. We also rescued the WSN-NPK4,7R virus, which carries an NP that cannot be sumoylated, and found that the mutant virus was highly attenuated and had aberrant nucleus and cytoplasm trafficking dynamics. These data, in all, emphasize the crucial role of NP sumoylation in influenza A virus infection.
MATERIALS AND METHODS
Cells and transfection.
Human embryonic kidney 293T cells, Madin-Darby canine kidney (MDCK) cells, human non-small-cell lung carcinoma cells (NCI-H1299), and human lung adenocarcinoma epithelial A549 cells were purchased from the American Type Culture Collection and cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS; Gibco) at 37°C in a CO2 incubator. All transfections were performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's procedure.
Plasmids.
A/WSN/33 (H1N1) virus (WSN) cDNA in pHW2000 and the luciferase reporter plasmid pPolI-NP-luc were provided by Hans-Dieter Klenk (Marburg University, Marburg, Germany). The A/PR/8/34 (H1N1) NP-encoding gene in pHW2000 was kindly provided by Robert G. Webster (St. Jude Children's Research Hospital, University of Tennessee, Memphis, TN). The NP-encoding genes from 2009 pH1N1 A/Sichuan/1/2009 (H1N1) (Sichuan), A/Donghu/312/2006 (H3N2), A/chicken/Henan/12/2004 (H5N1), and A/chicken/Jiangsu/7/2002 (H9N2) were cloned from the viral genomes. The NP-encoding gene from A/Anhui/1/2013 (H7N9) was commercially synthesized by using a sequence from the Epiflu database (isolate ID, EPI_ISL_138739; submitting lab, WHO Chinese National Influenza Center; submitter, Lei Yang; all data submitters may be contacted directly via the Global Initiative on Sharing All Influenza Data [GISAID] website, www.gisaid.org). The NP-encoding genes from the above strains were further cloned into the pCAGGS vector, which was kindly provided by Jun-ichi Miyazaki (Osaka University, Japan) and encodes an HA tag at the 3′ end. WSN NP was further cloned into pCAGGS without any tag (pCAGGS-NP) or with a FLAG tag at the 3′ end (FLAG-tagged NP).
Lysine (K)-to-arginine (R) mutations were introduced into the NP-encoding genes with the QuikChange site-directed mutagenesis kit (Stratagene) by using primers designed in accordance with the manufacturer's protocol. A plasmid expressing an NP in which all of the lysines were mutated to arginines (KO) was also generated with the QuikChange site-directed mutagenesis kit (Stratagene). In addition, the N-terminal 16 amino acids (aa) of NP were eliminated (construct named NPdel16) with primers corresponding to the NP-encoding gene from the beginning of residue 17. All NP mutation genes were cloned into pCAGGS with a 3′ HA tag.
GFP was cloned into the pCAGGS vector. The N-terminal sequence of WSN NP encoding residues 1 to 13 was fused to the green fluorescent protein (GFP) coding sequence to express a GFP fusion protein with aa 1 to 13 of the NP (NP[1-13aa]-GFP). Similarly, the K4R and K7R mutations were introduced into NP[1-13aa]-GFP to generate the sumoylation-defective mutant NPK4,7R[1-13aa]-GFP.
Human SUMO1, SUMO2, and SUMO3 were subcloned into pcDNA3 harboring Cerulean (a variant of enhanced cyan fluorescent protein with a higher quantum yield and an improved fluorescence lifetime [19]) to obtain Cer-SUMO1, Cer-SUMO2, and Cer-SUMO3, as described previously (8). Cer-SUMO1AA was obtained with the QuikChange site-directed mutagenesis kit (Stratagene) and primers containing GG-to-AA mutations at the C terminus of SUMO1. SUMO1 (or SUMO1AA) was further subcloned into pCAGGS directly to obtain SUMO1 (or SUMO1AA) with a 5′ FLAG-encoding sequence to obtain FLAG-SUMO1 (or FLAG-SUMO1AA). The expression plasmids pcDNA3-SENP1 (SENP1), pcDNA3-SENP1mut (SENP1mut), and pcDNA3-Ubc9 were described previously (8). HA-tagged human PIAS family plasmids were kindly provided by Jinke Cheng (Shanghai Jiao-Tong University), and HA-tagged, E3 ligase-deficient PIASxamut (W383A) (20) was generated with the QuikChange site-directed mutagenesis kit (Stratagene). All plasmids were confirmed by commercial sequencing.
Viruses.
The wild-type WSN virus (WSN-WT) was rescued by plasmid-based reverse genetics with eight plasmids, followed by two rounds of plaque purification and propagation in MDCK cells. The NP sumoylation-deficient virus WSN-NPK4,7R was rescued by transfecting 293T cells with the WSN NP-encoding gene harboring the K4R and K7R mutations and another seven WT WSN viral genes in the pHW2000 plasmid. Because plaque purification and propagation in MDCK cells resulted in reverse mutations, the WSN-NPK4,7R virus was collected directly from 293T cell supernatants without further plaque purification. Viral RNAs were extracted from the clarified 293T supernatant with an RNeasy RNA extraction kit (Qiagen, Chatsworth, CA). This was followed by reverse transcription-PCR with Moloney murine leukemia virus reverse transcriptase (Invitrogen) and Uni12 primers to obtain viral cDNAs (21). Sequencing of NP cDNA confirmed a uniform population of WSN-NPK4,7R viruses.
Plaque assays.
Plaque assays were performed by immunostaining as previously described (22). Monolayers of MDCK cells were infected with A/WSN/33 (WSN-WT) virus or its sumoylation-defective mutant WSN-NPK4,7R. At 48 h postinfection, virus-infected MDCK cells were fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 20 min and permeabilized with 0.1% Triton X-100 for 30 min. The cells were then incubated with horseradish peroxidase (HRP)-conjugated anti-NP polyclonal antibodies (Antibodies Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences) for 60 min. True Blue substrate (KPL) was then added to visualize the plaques.
Immunoprecipitation and Western blotting.
To precipitate HA-tagged NP, cells were washed with cold PBS and then lysed in radioimmunoprecipitation assay (RIPA) lysis buffer (50 mM Tris-HCl [pH 7.4],150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA) supplemented with a complete protease inhibitor (Roche) and 10 mM N-ethylmaleimide (Sigma) at 4°C for 20 min. The lysate was centrifuged at maximum speed in an Eppendorf microcentrifuge to remove cellular debris, and the supernatant was subjected to immunoprecipitation with anti-HA–agarose (Sigma) at 4°C for 4 h. The beads were then washed five times with RIPA buffer and eluted with SDS loading buffer, and the resulting eluates were separated by SDS-PAGE. The proteins were transferred to nitrocellulose membranes (Bio-Rad), and the NPs were detected with anti-HA polyclonal antibodies (Sigma) and HRP-conjugated goat anti-rabbit IgG secondary antibodies (Southern Biotech). SUMO1 was detected with an anti-SUMO1 monoclonal antibody (Zymed) and an HRP-conjugated goat anti-mouse IgG secondary antibody (R&D).
To block protein synthesis, 293T cells were treated with 100 μg/ml cycloheximide (CHX; Sigma) at 24 or 36 h posttransfection. Cells were lysed at 0, 2, 4, 6, or 8 h after CHX treatment. The amount of NP or phosducin (phd) was analyzed by Western blotting with an anti-HA antibody.
Anti-FLAG–agarose (Sigma) was used for immunoprecipitation of FLAG-tagged SUMO1, and anti-FLAG polyclonal antibodies (Sigma) were used to detect FLAG-tagged SUMO1 by Western blotting. Cer-SUMO1 was detected with an anti-SUMO1 monoclonal antibody (Zymed) and a goat anti-mouse IgG HRP-conjugated secondary antibody (R&D).
GFP was detected with anti-GFP polyclonal antibodies (Antibodies Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences) and a goat anti-rabbit IgG HRP-conjugated secondary antibody (Southern Biotech). Ubc9 was detected with anti-Ubc9 polyclonal antibodies (Santa Cruz).
Luciferase reporter assay.
To determine the polymerase activities of the replication complexes containing the sumoylation-defective mutant NPs, 293T cells were cotransfected with plasmids expressing the WSN polymerase subunits PB1, PB2, PA, and pPolI-NP-luc, the Renilla luciferase-expressing plasmid pRL-SV40 (Promega), and either WT NP or NPK4,7R. The cells were lysed at 24 h posttransfection, and the luciferase activity was measured with the dual-luciferase assay system (Promega) according to the manufacturer's protocol.
Immunocytochemistry and confocal microscopy.
To achieve equal infectivity for examining NP localization, the WSN-WT and WSN-NPK4,7R viruses were both titrated on A549 cells and then used to infect A549 cells at a multiplicity of infection (MOI) of 0.1 for the time indicated during a single infection cycle. Infected A549 cells cultured on glass slides were fixed with 4% PFA and permeabilized with 0.1% Triton X-100 in PBS. The cells were then immunolabeled with NP monoclonal antibodies (kindly provided by Ningshao Xia, Xiamen University, China) for 2 h, washed five times with PBS, and incubated with goat anti-mouse–Alexa 488 (Molecular Probes) for 1 h. DAPI (4,6′-diamidino-2-phenylindole; Sigma) was used for nuclear staining. All images were acquired with a Leica TCS SP2 confocal microscope (Leica Microsystems).
To block the nuclear export of NP in infected cells, the cells were treated with 10 nM leptomycin B (LMB) beginning at 3 h postinfection and continuing for the duration of the experiment.
Multiple growth curves.
To determine the growth curves of the WSN-WT and WSN-NPK4,7R viruses on A549 cells, the viruses were first titrated on MDCK cells and then used to infect A549 cells at an MOI of 0.001. After 1 h of virus adsorption, the cells were culture in infection medium (DMEM with 2% FBS) and the cell supernatant was separately collected at 24, 36, 48, 72, and 96 h postinfection. The virus amounts in the supernatant were determined by a plaque assay with MDCK cells.
Statistical analyses.
The data were analyzed with Student's t test and are presented as means ± standard errors. A P value of <0.05 was considered significant.
RESULTS
Influenza A virus NP is mainly sumoylated by SUMO1.
Here we investigated whether vRNP component proteins (PB1, PB2, PA, and NP) are targets of SUMO and examined the conjugation of SUMO isoforms SUMO1 and SUMO2/3 to these proteins. Initially, Ubc9-overexpressing 293T cells were cotransfected with HA-tagged vRNPs together with either Cer-SUMO1 or Cer-SUMO1AA (a SUMO1 variant that is conjugation deficient) as previously described (8). The cells were lysed at 36 h posttransfection, and the lysates were immunoprecipitated with anti-HA–agarose. The precipitated proteins were further analyzed by SDS-PAGE and Western blotting with anti-HA polyclonal antibodies or anti-SUMO1 antibody. Of the four vRNPs, we detected a clear additional band for NP at ∼95 kDa, which reflected the modification of NP (∼55 kDa) by Cer-SUMO1 (∼40 kDa). In contrast, this ∼95-kDa band was absent from cells transfected with Cer-SUMO1AA, suggesting that the NP is sumoylated (Fig. 1A). Because only a faint modification band was detected for PB2 and no sumoylated bands were visible for the PB1 and PA proteins under the same experimental conditions (Fig. 1B), we focused on the sumoylation of NP in the subsequent analyses. To determine whether SENP1, which is mainly responsible for the removal of SUMO1 from modified proteins, is involved in the regulation of NP sumoylation, we coexpressed NP, Cer-SUMO1, and Ubc9 with SENP1 or with functionally defective SENP1 (SENP1mut) in 293T cells. As shown in Fig. 1C, the band of sumoylated NP at ∼95 kDa was strongly reduced in the SENP1-expressing cells, whereas it was hardly affected in the SENP1mut-expressing cells. When Cer-SUMO2 and Cer-SUMO3 were overexpressed with Ubc9 and NP, only weak levels of sumoylated NP were observed compared to SUMO1-modified NP (Fig. 1D). Taken together, these results indicate that NP is predominantly modified with SUMO1 and that this modification is removed by SENP1.
FIG 1.

Sumoylation of NP in vivo. (A) 293T cells were transfected with plasmids expressing WSN NP and Ubc9 together with plasmids expressing either Cer-SUMO1 or Cer-SUMO1AA, as indicated. The cells were lysed at 36 h posttransfection, and NP was immunoprecipitated (IP) with anti-HA–agarose. The NPs were immunoblotted (IB) with anti-HA polyclonal antibodies. Cer-SUMO1 was detected in separate blots with an anti-SUMO1 monoclonal antibody. (B) 293T cells were transfected with plasmids expressing an HA-tagged WSN polymerase protein (PB1, PA, or PB2) and Ubc9 together with plasmids expressing either Cer-SUMO1 or Cer-SUMO1AA. The cells were lysed at 36 h posttransfection, and each polymerase protein was immunoprecipitated with anti-HA–agarose and further analyzed by SDS-PAGE and immunoblotting with anti-HA polyclonal antibodies. (C) 293T cells were transfected with WSN NP and Ubc9 together with Cer-SUMO1, SENP1, or SENP1mut, as indicated. NP was immunoprecipitated with anti-HA–agarose, and the precipitated proteins were further analyzed by SDS-PAGE and immunoblotting with anti-HA polyclonal antibodies. (D) WSN NP and Ubc9 were cotransfected with GFP (as a control), Cer-SUMO1, Cer-SUMO1AA, Cer-SUMO2, and Cer-SUMO3, as indicated; NP was immunoprecipitated with anti-HA–agarose, and the precipitated proteins were further analyzed by SDS-PAGE and immunoblotting with anti-HA polyclonal antibodies. Input of the whole-cell lysates was detected by immunoblotting with anti-GFP polyclonal antibodies to show the expression of GFP, Cer-SUMO1, Cer-SUMO2, or Cer-SUMO3; with anti-Ubc9 antibody to show the expression of Ubc9; and with anti-actin antibodies to show the loading controls. The values to the left of the blots are molecular sizes in kilodaltons.
The lysines at positions 4 and 7 of the N terminus of NP are sumoylated.
To identify the sumoylation sites of NP, all 19 lysines of the NP were individually replaced with arginines. We also included double mutations to prevent complementary sumoylation caused by nearby lysine residues. These HA-tagged K-to-R mutant forms of NP were coexpressed with Ubc9 and Cer-SUMO1 and then immunoprecipitated with anti-HA–agarose and subjected to Western blot analysis with anti-HA antibodies. As expected, NP was modified to form an ∼95-kDa sumoylated band, whereas an NP lacking all 19 lysines (KO) was not sumoylated by SUMO1, indicating that the lysine residues of NP are SUMO acceptor sites (Fig. 2A). Of the 19 single K-to-R mutations and the 3 double mutations, only the double mutation of lysines at positions 4 and 7 to arginines (NPK4,7R) completely abolished the sumoylation of NP; in contrast, the individual K4R or K7R mutation did not affect the sumoylation of NP (Fig. 2A). These data suggest that K4 and K7 may compensate for each other in NP sumoylation. To confirm that K4 and K7 are indeed SUMO acceptor sites, we fused the N terminus of NP encompassing the K4 and K7 motif with a second sumoylation-defective protein to determine whether the presence of the NP N terminus would enable sumoylation of the second protein. Thus, the sequence encoding residues 1 to 13 of NP was cloned into the 5′ end of the GFP gene to express a GFP protein fused to the N-terminal 13 aa of NP (NP[1-13aa]-GFP). The data in Fig. 2B show that the GFP protein was not sumoylated when coexpressed with SUMO1 (untagged) and Ubc9, whereas NP[1-13aa]-GFP was clearly sumoylated, with an ∼40-kDa sumoylation band. In contrast, sumoylation did not occur in cells expressing NPK4,7R[1-13aa]-GFP, in which arginines were present at positions 4 and 7. These results further confirmed that the N-terminal sequence of NP contains SUMO acceptor sites and that K4 and K7 are targeted by the SUMO molecules.
FIG 2.

K4 and K7 are SUMO acceptor sites for different influenza A virus subtypes and strains. (A) Expression plasmids encoding WSN-WT NP or NP with K-to-R mutations at the indicated positions and Ubc9 were transfected together with Cer-SUMO1 or Cer-SUMO1AA. The cells were lysed, and NP was immunoprecipitated (IP) with anti-HA–agarose and then detected by Western immunoblotting (IB) with anti-HA polyclonal antibodies. (B) The expression plasmid pCAGGS-GFP, NP[1-13aa]-GFP, or NPK4,7R[1-13aa]-GFP was cotransfected with SUMO1 or SUMO1AA; Ubc9 was also cotransfected with each pCAGGS plasmid. The cells were lysed and directly subjected to Western blotting with anti-GFP or anti-SUMO1 antibodies. (C) Expression plasmids encoding WSN-WT NP or the NPdel16 mutant form and Ubc9 were transfected together with Cer-SUMO1 or Cer-SUMO1AA. The cells were lysed, and NP was immunoprecipitated with anti-HA–agarose and then detected by Western immunoblotting with anti-HA polyclonal antibodies. (D) Schematic representation of the N-terminal amino acid sequences of NPs from different influenza A virus strains. The lysine residues (framed) indicate the SUMO1 modification sites for each NP. (E) 293T cells were cotransfected with plasmids encoding Ubc9 and HA-tagged NPs or the NPK7R mutant form from different influenza A virus subtypes and strains together with Cer-SUMO1 or Cer-SUMO1AA. The cells were lysed, and NP was immunoprecipitated with anti-HA–agarose. The precipitated proteins were further analyzed by SDS-PAGE and Western blotting with anti-HA polyclonal antibodies. The values to the left of the blots are molecular sizes in kilodaltons.
Previous studies have shown that NP of human influenza A virus is cleaved by a caspase-dependent mechanism at the late stage of infection associated with the development of apoptosis and that the cleavage occurs at D16 (23); thus, the sumoylation sites of NP are lost after caspase cleavage. We generated NPdel16, which harbors a deletion of the N-terminal 16 aa to mimic caspase-cleaved NP. In agreement with our findings described above, this truncated form of NP was not modified by Cer-SUMO1 (Fig. 2C). This result confirms that the N-terminal K4 and K7 residues are authentic sumoylation sites of NP and also indicates that sumoylation of NP likely plays a role at early stages of the virus replication cycle, before the onset of an apoptotic response by the host cell.
SUMO1 modification of NP is conserved among influenza A virus subtypes and strains.
Because the genetics of influenza A viruses are highly variable, we next assessed the sumoylation of NP in several avian and human isolates. HA-tagged NPs from the A/Sichuan/1/2009 (H1N1, Sichuan), A/PR/8/34 (H1N1, PR8), A/Donghu/312/2006 (H3N2), A/Chicken/Henan/12/2004 (H5N1), A/Chicken/Jiangsu/7/2002 (H9N2), and recently emerged A/Anhui/1/2013 (H7N9) strains were coexpressed with Cer-SUMO1 or Cer-SUMO1AA in 293T cells. The cells were then lysed, and the NPs were immunoprecipitated and detected by Western blotting with anti-HA antibodies. Sequence analysis revealed that K7 in NP is highly conserved across all of the strains (Fig. 2D). Indeed, all of the NPs tested were sumoylated, suggesting that sumoylation is a common modification of influenza A virus NPs (Fig. 2E). The data in Fig. 2E also confirm that the sumoylation levels of all of the strains tested were greatly reduced when K7 was mutated to R7, indicating that K7 is the major SUMO acceptor site, although K4 could compensate for K7 in the WSN strain.
NP is sumoylated at N-terminal lysines in infected cells.
Because it is unclear whether NP is sumoylated in infected cells, we performed a sumoylation analysis of WSN-infected cells with WSN-WT and a recombinant virus containing NP with mutated sumoylation sites (WSN-NPK4,7R) generated by reverse genetics. However, we observed that the WSN-NPK4,7R virus underwent a reverse mutation at position 7 during multiplication in MDCK cells, resulting in a mixture of viruses producing NPs with either K4R or K4,7R. We therefore directly collected virus from the 293T supernatants with the desired NPK4,7R mutation. To analyze NP sumoylation in infected cells, 293T cells overexpressing Ubc9 and FLAG-SUMO1 or FLAG-SUMO1AA were infected with either the WSN-WT or the WSN-NPK4,7R virus at an MOI of 1. The cells were harvested 16 h later and subjected to immunoprecipitation with anti-FLAG–agarose, followed by Western blot analysis with anti-NP antibodies. As expected, a band at ∼70 kDa corresponding to sumoylated NP was detectable in WSN-WT-infected cells overexpressing SUMO1 but not in cells overexpressing SUMO1AA or those infected with the WSN-NPK4,7R virus (Fig. 3, left panel). Correspondingly, overexpression of SENP1 but not SENP1mut abolished the sumoylation of NP in WSN-WT-infected cells (Fig. 3, right panel). These data confirm that NP is sumoylated at K4 and K7 during infection.
FIG 3.
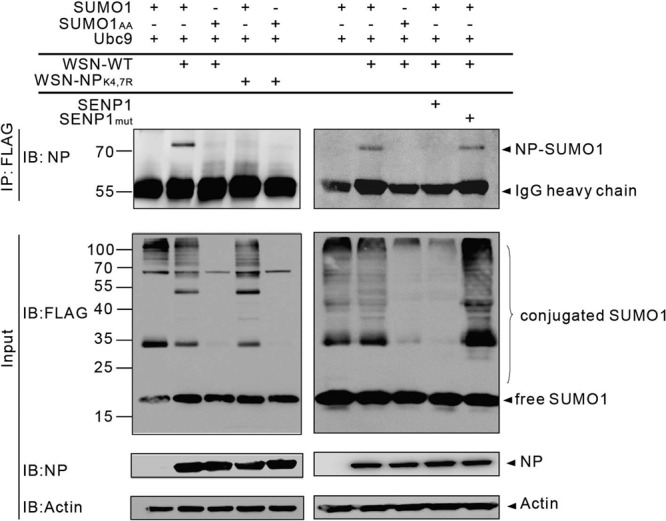
NP is sumoylated in infected cells. 293T cells were cotransfected with FLAG-tagged SUMO1 or SUMO1AA and Ubc9 together with SENP1 or SENP1mut as indicated. At 24 h posttransfection, the cells were infected with the WSN-WT or WSN-NPK4,7R virus at an MOI of 1. The cells were lysed 16 h later, and SUMO1 was immunoprecipitated (IP) with anti-FLAG–agarose. The precipitated proteins were further analyzed by SDS-PAGE and immunoblotting (IB) with anti-NP monoclonal antibody. Immunoblot assays with anti-FLAG, anti-NP, or anti-actin polyclonal antibodies and whole-cell lysates are shown as SUMO1 expression, infectivity, and loading controls (Input), respectively. The values to the left of the blots are molecular sizes in kilodaltons.
NP stability and polymerase activity are not influenced by sumoylation.
After identifying the sumoylation sites of the NP, we investigated the functional significance of NP sumoylation. We first examined whether the stability of NP is affected by sumoylation. For this, cells were transfected with an expression plasmid encoding either HA-tagged NP or HA-tagged sumoylation-defective NPK4,7R. As controls, HA-tagged phd and HA-tagged phdK33R were transfected into the cells in parallel. At 24 h posttransfection, protein synthesis was blocked with CHX and the steady-state levels of NP or phd protein expressed after CHX treatment were monitored. Samples were collected at 0, 4, and 8 h after CHX treatment and subjected to immunoprecipitation with anti-HA antibodies. As previously reported, sumoylation-competent phd remained stable over time while the sumoylation-deficient phdK33R protein underwent degradation (Fig. 4A, top) (24). In contrast, both NP and NPK4,7R protein levels remained stable under CHX treatment (Fig. 4A, bottom). To further prove that sumoylation of NP does not alter protein stability, HA-tagged NP or HA-tagged NPK4,7R was cotransfected with Ubc9 and Cer-SUMO1 to visualize the sumoylated form of NP. The data in Fig. 4B showed that both native NP and sumoylated NP were present in the NP-expressing cells, whereas only unmodified NP was detected in the NPK4,7R-expressing cells. Again, within 8 h of CHX treatment, the levels of both NP and NPK4,7R stayed stable, suggesting that sumoylation does not affect the stability of NP (Fig. 4B).
FIG 4.
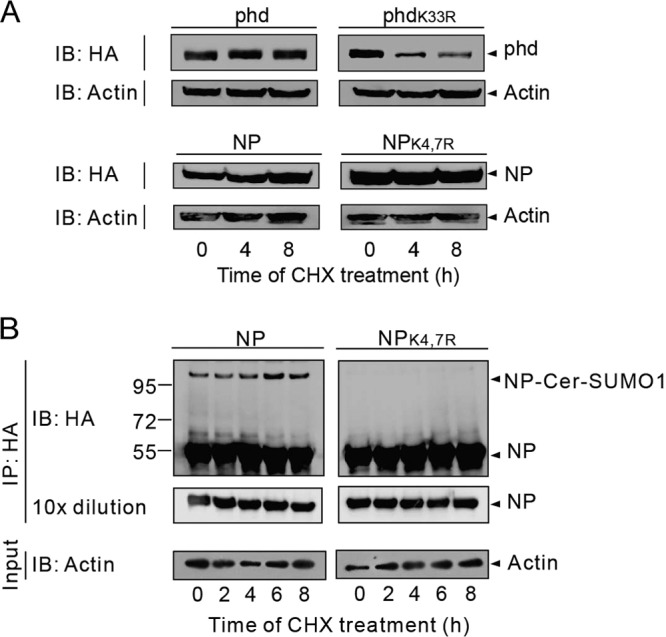
The stability of NP is not affected by sumoylation. (A) 293T cells were transfected with phd or phdK33R and NP or NPK4,7R. At 24 h posttransfection, the cells were treated with 100 μg/ml CHX for the times indicated. The phd or NP was immunoblotted (IB) with anti-HA polyclonal antibodies. Immunoblot assay of whole-cell lysates with anti-actin antibodies are shown as loading controls (Input). (B) 293T cells were transfected with Ubc9 and Cer-SUMO1 together with NP or the NPK4,7R mutant form. At 36 h after transfection, the cells were treated with 100 μg/ml CHX for the times indicated. The NPs were immunoprecipitated (IP) with anti-HA–agarose, and the precipitated proteins were further analyzed by SDS-PAGE and immunoblotting with anti-HA polyclonal antibodies to detect NP. Western blot assays of whole-cell lysates with anti-actin antibodies are shown as loading controls (Input). The values to the left of the blots are molecular sizes in kilodaltons.
Because NP is a major component of vRNP and contributes to RNA-dependent RNA polymerase (RdRp) activity, we next tested whether the RdRp activity of influenza A virus is affected by NP sumoylation. We performed a minireplicon assay with two different cell lines, 293T and NCI-H1299. Different amounts of genes for polymerases (PB1, PB2, and PA) and NP (or NPK4,7R) were transfected into 293T cells (Fig. 5A) or NCI-H1299 cells (Fig. 5B) together with pPolI-NP-luc (a luciferase reporter gene mimicking vRNA) and pRL-SV40 (transfection control) to evaluate RdRp activity. No significant difference in RdRp activity between NP and sumoylation-defective NPK4,7R was observed. To further confirm that sumoylation has little effect on RdRp activity, we cotransfected SUMO1 and Ubc9 to upregulate the overall sumoylation level or cotransfected SENP1 to downregulate the overall SUMO1 modification level (with SENP1mut as a control). No statistically significant change in RdRp activity was observed under either condition (Fig. 5C and D). On the basis of these results, we conclude that the sumoylation of NP does not necessarily contribute to viral RdRp activity.
FIG 5.

Polymerase activity is not affected by NP sumoylation. 293T (A) or NCI-H1299 (B) cells cultured in 24-well plates were transfected with PolI-NP-Luc (100 ng/well) and pRLSV40 (10 ng/well) together with different amounts (100, 50, and 30 ng/well, as indicated) of four polymerase complex genes from the WSN-WT virus (NP) or WSN-NPK4,7R virus (NPK4,7R). A luciferase assay was performed at 24 h posttransfection. All of the data were normalized to the activity of the 100-ng NP-transfected sample at 24 h. 293T (C) or NCI-H1299 (D) cells in the wells of 12-well plates were transfected with polI-NP-Luc (200 ng/well), pRLSV40 (20 ng/well), and the genes for polymerases (PB1, PB2, and PA) (3P) and NP of the WSN-WT virus (200 ng/well) together with either SUMO1 and Ubc9 or SENP1 (or SENP1mut). A luciferase assay was performed at 24 h posttransfection. All data were normalized to the activity of the 200-ng NP-transfected sample at 24 h. The data are from three independent experiments and are presented as means ± standard errors. A P value of <0.05 was considered significant; NS, not significant. Immunoblot assays with anti-NP and anti-actin antibodies using whole-cell lysates are shown as the NP expression and loading controls, respectively.
Sumoylation regulates the intracellular trafficking of NP.
NP is a shuttle protein whose localization is tightly regulated. In infected cells, NP from incoming vRNPs is actively transported into the nucleus and localized in the nucleus at early stages postinfection; conversely, NP is exported to the cytoplasm at late stages postinfection (25). Because sumoylation regulates the nuclear localization of several substrates (26, 27), we investigated whether sumoylation influences the nuclear localization of NP during a single infection cycle. A549 cells were infected with the WSN-WT or WSN-NPK4,7R virus, and the localization of NP was analyzed by immunostaining with anti-NP antibodies. Figure 6A shows that NP from both the WSN-WT and WSN-NPK4,7R viruses was localized mostly in the nucleus at 4 h postinfection, indicating that both NPs could be imported into the nucleus. However, at 6 h postinfection, when cytoplasmic NP localization was already observed in WSN-NPK4,7R-infected cells, NP was still retained in the nuclei of WSN-WT-infected cells. At 8 and 10 h postinfection, NP from both viruses had accumulated largely in the cytoplasm. Quantification of the subcellular distribution of NP by automatic image analysis demonstrates a more pronounced localization of NP in the nuclei of WSN-WT-infected cells than in those of WSN-NPK4,7R-infected cells later than 4 h postinfection (Fig. 6B). Simultaneously, a stronger increase in cytoplasmic NP in WSN-NPK4,7R-infected cells was observed (Fig. 6C), indicating an abnormal nucleus-cytoplasm transition of sumoylation-deficient NP.
FIG 6.

Sumoylation alters the nuclear transport dynamics of NP. (A) A549 cells were infected with the WSN-WT or WSN-NPK4,7R virus at an MOI of 0.1; the cells were collected and fixed at the times indicated and stained with an anti-NP monoclonal antibody and a goat anti-mouse–Alexa 488 secondary antibody. The nuclei were stained with DAPI. At the very bottom are amplified images taken at 6 h postinfection. The subcellular distribution of NP was quantified by automatic image analysis with the CellProfiler software (35). For each time point, 5 to 10 confocal images were analyzed. The means and 95% confidence intervals of nuclear (B) or cytoplasmic (C) NP fluorescence intensity over the total NP fluorescent intensity from 30 to 100 infected cells are shown. Statistical significance was determined by unpaired Student t tests (*, P < 0.05; ***, P < 0.001). (D) A549 cells were infected with the WSN-WT or WSN-NPK4,7R virus at an MOI of 0.1. At 3 h after infection, the cells were treated with 10 nM LMB and fixed at the times indicated. Staining, imaging, and quantification of nuclear (E) or cytoplasmic (F) NP were performed as described above.
To distinguish whether nuclear import or export of NP was affected by sumoylation, we further treated the infected cells at 3 h postinfection with LMB, a drug known to block the chromosome region maintenance 1 protein-dependent vRNP export pathway (28). After LMB treatment, NP was found exclusively in the nuclei of cells infected with either the WSN-WT or the WSN-NPK4,7R virus (Fig. 6D). Quantitative analysis of NP localization in Fig. 6E and F also supported the idea that NP from both viruses was fully retained in the nucleus after LMB treatment. This result indicates that sumoylation regulates the intracellular trafficking of NP without directly affecting the nuclear import of NP.
Viral replication is highly suppressed in the sumoylation-defective WSN-NPK4,7R virus.
We then examined whether the sufficient nuclear localization of sumoylation-competent NP observed contributes to viral growth by analyzing the growth properties of the WSN-WT and WSN-NPK4,7R viruses. We first compared the plaque morphologies of these two viruses in A549 cells and found that the WSN-WT virus formed large plaques, whereas the WSN-NPK4,7R virus formed only pinpoint-size plaques, indicating that the growth of WSN-NPK4,7R was heavily impaired (Fig. 7A). The growth curves shown in Fig. 7B further demonstrate that the growth of the WSN-NPK4,7R virus was severely attenuated, with no progeny virus detectable until 24 h postinfection. Although the growth of the WSN-NPK4,7R virus recovered partially at later time points, the viral titer was 5 to 20 times lower than that of the WSN-WT virus. Consistent with our previous observations, the arginine at position 7 was mutated back to lysine in the WSN-NPK4,7R virus starting at 72 h postinfection and the percentage of reverse mutants of the progeny virus population gradually increased with time (Fig. 7C). These data indicate that replication of the sumoylation-defective WSN-NPK4,7R virus is highly suppressed and that sumoylation at K7 is essential for viral survival, as a virus harboring the K7R mutation consistently generated the reverse mutation during the growth cycle.
FIG 7.

Sumoylation of NP enhances viral growth. (A) Plaque phenotypes of the WSN-WT and WSN-NPK4,7R viruses in A549 cells. (B) Growth curves of the WSN-WT and WSN-NPK4,7R viruses. A549 cells were infected with either the WSN-WT or the WSN-NPK4,7R virus at an MOI of 0.001, and viral production at the postinfection times indicated was determined by a plaque assay with MDCK cells. The means and standard deviations of three independent experiments are shown. (C) Sequencing results of the NP-encoding gene from the WSN-NPK4,7R virus obtained from the cell supernatants in panel B at the times indicated. The arrows indicate the reverse mutation of arginine at position 7 of NP. (D) PIASxa facilitates NP sumoylation. HA-tagged pcDNA3-PIASxa, PIASxb, PIAS1, PIAS3, or PIAS4 was cotransfected with PCAGGS-NP, SUMO1, or SUMO1AA in the absence of Ubc9. The cells were lysed at 36 h after transfection and directly subjected to Western immunoblotting (IB) with anti-NP monoclonal antibodies. The values to the left are molecular sizes in kilodaltons. (E) HA-tagged pcDNA3-PIASxa and FLAG-tagged NP was either individually transfected or cotransfected into 293T cells. The cells were lysed at 36 h after transfection, and NP was immunoprecipitated (IP) with anti-FLAG–agarose. The expression of HA-PIASxa, NP, and actin is shown (Input). (F) HA-tagged pcDNA3-PIASxa or E3 ligase-deficient HA-PIASxamut was cotransfected with PCAGGS-NP, SUMO1, or SUMO1AA in the absence of Ubc9. The cells were lysed at 36 h after transfection and directly subjected to Western immunoblotting with anti-NP monoclonal antibodies. Input of the whole-cell lysates were detected by immunoblot assays with anti-HA polyclonal antibodies to show the expression of PIASxa and with anti-actin antibodies to show the loading controls. (G) PIASxa promotes viral growth. 293T cells were individually transfected with pcDNA3 (vector) or PIASxa. At 24 h posttransfection, the cells were infected with the WSN-WT virus at an MOI of 0.001 and the viral titers in the culture supernatant at the postinfection times indicated were determined by plaque assay with MDCK cells. The means and standard deviations of three independent experiments are shown. A P value of <0.05 was considered significant (*, P < 0.05). (H) 293T cells were individually transfected with pcDNA3 (vector), PIASxa, or PIASxamut. At 24 h posttransfection, the cells were infected with the WSN-WT or WSN-NPK4,7R virus at an MOI of 0.001 and the viral titers in the culture supernatant at 48 h postinfection were determined by plaque assay with MDCK cells. The fold increase in WSN-WT or WSN-NPK4,7R virus was determined by determining the viral titer of cells transfected with PIASxa or PIASxamut divided by that of cells transfected with the vector. The means and standard deviations of three independent experiments are shown. A P value of <0.05 was considered significant (NS, not significant; *, P < 0.05).
To further confirm the role of NP sumoylation in viral growth, we sought to identify the specific E3 ligase for NP. We therefore evaluated whether members of the PIAS family, the best-characterized E3 ligases, could function as an E3 ligase for NP. Several human PIAS isoforms, PIAS1, PIASxa, PIASxb, PIAS3, and PIAS4, were tested for the ability to facilitate NP sumoylation. As shown in Fig. 7D, the sumoylated form of NP was barely detectable without Ubc9 overexpression (lane 1). Among all of the PIAS homologs, overexpression of PIASxa had the strongest effect on NP sumoylation, suggesting that PIASxa is the predominant E3 ligase for NP (Fig. 7D). Moreover, we could detect an interaction between NP and PIASxa in transfected cells (Fig. 7E), supporting the accessibility of NP to PIASxa. In contrast, when E3 ligase-deficient PIASxamut (W383A) was cotransfected, the enhancement of NP sumoylation was totally abolished in PIASxamut-expressing cells, indicating that E3 ligase activity is required to facilitate the sumoylation of NP (Fig. 7F). When PIASxa was overexpressed in WSN-WT-infected cells, viral production was significantly elevated at 48 h postinfection, with a viral titer 6.5-fold higher than that in vector-transfected cells (Fig. 7G). When we repeated this experiment with overexpression of PIASxamut, the increase in the virus titer mediated by PIASxa was significantly reduced in PIASxamut-overexpressing cells (Fig. 7H). Notably, PIASxa promoted the growth of only sumoylation-competent WSN-WT virus. No obvious effect of either PIASxa or PIASxamut on the titer of the WSN-NPK4,7R virus was observed (Fig. 7H). These data illustrate that PIASxa is a potential E3 ligase for NP and that PIASxa-induced NP sumoylation enhancement facilitates viral growth.
DISCUSSION
Sumoylation is an important reversible and highly dynamic posttranslational modification that is involved in many cellular processes, including the control of gene transcription, cell signaling, and even embryonic development (13). Like many other viruses, influenza A viruses have developed distinct strategies to interfere with the host sumoylation process (11). However, the full picture of the interaction between influenza A virus and the host cell sumoylation system is not clear. We and others have demonstrated that several viral proteins are sumoylated in transfected and infected cells; we previously demonstrated that NS1 is sumoylated at C-terminal residues in infected cells and that sumoylation stabilizes NS1 for efficient viral replication (8). Another group confirmed our finding that the C-terminal lysine is the major sumoylation site and that sumoylation ensures NS1 function (29). Sumoylation of M1 has also been reported and found to be involved in virus assembly (9). Evidence of the sumoylation of other viral proteins has been obtained in transfection studies, but these interactions have not been confirmed in infected cells, and the biological significance of the sumoylation for these proteins remains largely unknown. In this study, we clearly demonstrated that NP is a target of SUMO in transfected and infected cells and that sumoylation of NP is essential for efficient virus production.
We demonstrated that the SUMO acceptor site on NP is located at N-terminal residues K4 and K7. The genetic analysis of the N-terminal amino acid sequence of NP indicated that K7 is highly conserved among viral strains of different subtypes, whereas K4 is found only in some strains, such as A/WSN/1933 (H1N1), A/Wisconsin/67/2005 (H3N2), and A/Brisbane/97/2007 (H1N1). We also showed that the single mutation of either K4 or K7 to arginine cannot abolish the sumoylation of NP in the WSN strain, suggesting that both of these sites serve as SUMO acceptors. In fact, the presence of an adjacent lysine residue as a complementary SUMO target site is commonly observed and only the double mutation of these proximal lysines abolishes sumoylation (8). However, K7 is the major acceptor site in viruses possessing a single N-terminal lysine, as exemplified by the A/Sichuan/1/2009 (H1N1), A/PR/8/34 (H1N1), A/Donghu/312/2006 (H3N2), A/chicken/henan/12/2004 (H5N1), A/chicken/Jiangsu/7/2002 (H9N2), and A/Anhui/1/2013 (H7N9) viruses. The presence of K4 might ensure NP sumoylation when K7 is occupied by other interaction partners, but this hypothesis has not been confirmed experimentally. Interestingly, a reverse mutation to reconstitute K7 consistently occurred when we attempted to rescue the recombinant virus harboring the NPK7R mutation in the different strains. These results and the conservation of K7 among influenza A viruses suggest that K7 is the major SUMO acceptor site and that sumoylation at this site is critical for viral survival.
When we screened all of the PIAS family members to identify the possible E3 ligases for NP, we found that PIASxa most effectively enhanced the sumoylation of NP. It is reasonable that NP sumoylation is preferentially mediated by a specific E3 ligase and that this may play a specific role in the process of viral replication. However, the interaction between substrates and endogenous E3 ligases is mostly transient and hard to detect. We therefore evaluated the interaction between NP and PIASxa in transfected cells. Here, we found a strong interaction between NP and PIASxa. Using an E3 ligase-negative mutant form of PIASxa, we showed that PIASxa activity greatly contributed to viral replication. One has to keep in mind that changes in the activity of E3 ligases may also alter the sumoylation of other host proteins (30, 31), which may in turn influence the infection cycle. However, we could show that the virus titer enhancement mediated by PIASxa was pronounced only for the WSN virus with sumoylation-competent NP and not for the WSN-NPK4,7R virus, indicating that facilitation of NP sumoylation is a predominant event for PIASxa during infection.
The functional outcome of SUMO modification is highly diverse and depends largely on the individual target protein (13). The crystal structure of purified vRNP demonstrates that NP homo-oligomerizes to form a nonameric ring with a high affinity for RNA (32). NP also directly interacts with the PB1 and PB2 proteins for viral RNA replication (33). In addition to forming the vRNP scaffold, NP is also involved in many virus-host interactions (17). To investigate the role of sumoylation in NP functions, we examined whether the sumoylation of NP affects viral RdRp activity with a minireplicon system. However, there was no significant difference in RdRp activity between the NP and NPK4,7R minireplicons. Up- or downregulation of NP sumoylation by overexpression of Ubc9 or SENP1, respectively, also had no effect on luciferase expression, further confirming that the sumoylation of NP had no or only a limited effect on viral RdRp activity. We then determined whether sumoylation affects NP stability, as has been demonstrated for NS1. However, in contrast to NS1, sumoylation-defective NP and WT NP were equally stable. When we compared the cellular localization of the WSN-WT and WSN-NPK4,7R viruses in infected cells, we found more cytoplasmic localization and less nuclear localization of NPK4,7R than WT NP, indicating that sumoylation regulates the intracellular trafficking of NP in infected cells. However, the nuclear import of NP was little affected by sumoylation, as NP from both sumoylation-competent and -defective viruses localized in the nucleus after LMB treatment. SUMO may, for instance, create new interfaces for NP to interact with nuclear proteins which will act as nuclear scaffolds to retain NP in the nucleus. It is also possible that the nuclear export of NP was slowed down because of the increased molecular weight of NP after sumoylation, so that sumoylation-defective NP is exported into the cytoplasm more quickly and earlier than WT NP. Further studies are required to understand the details of the mechanisms involved.
The dynamic localization of influenza A virus NP is very important for viral replication. NP is detected in the nucleus at early times postinfection but is distributed in the cytoplasm at late stages postinfection (25). Sufficient nuclear retention of NP at the early stage postinfection will guarantee adequate vRNP assembly to form mature vRNPs. Interestingly, we observed that the sumoylation of NP was lost when the sumoylation motif-containing N terminus of NP was cleaved by caspase. As this process occurs in the late stage of the infection cycle, when cell apoptosis takes place (34), it appears that NP sumoylation is important within the virus replication cycle and that caspase-dependent cleavage may serve as an antiviral strategy to counteract NP sumoylation.
In summary, we identified the influenza A virus NP as a bona fide target of sumoylation, which regulates the intracellular trafficking of NP and efficient virus production. Our data strengthen the concept that influenza A virus relies deeply on sumoylation to survive in host cells. Strategies to downregulate viral sumoylation could thus be a potential antiviral treatment.
ACKNOWLEDGMENTS
This work was supported by grants from the National Key Project of 973 (2013CB530504), the National 863 Project (2012AA02A404, 2012AA020103), the National Natural Science Foundation of China (81201280, 31030029, and 31230024), the Science and Technology Commission of Shanghai Municipality (12ZR1435000), the SA-SIBS Scholarship Program, the CAS Youth Innovation Association, the CAS-SIBS Frontier Research Field Foundation for Young Scientists, the Albert-Ludwigs-Universität Freiburg Foundation for Foreign Visiting Scientists, the National Science and Technology Major Project (2013ZX10004-101-005, 2013ZX10004-003-003, and 2012ZX10002-007-003), CAS Key Project (KJZD-EW-L09-3), the National Ministry of Science and Technology (2007DFC31700), and the Li Kha Shing Foundation.
We thank Otto Haller (Freiburg University), Hans-Dieter Klenk (Marburg University), and Sabrina Schreiner (Leibniz Institute for Experimental Virology, Hamburg) for reviewing the manuscript. We gratefully acknowledge the authors and submitting laboratories for the sequences from the GISAID EpiFlu database.
Footnotes
Published ahead of print 11 June 2014
REFERENCES
- 1.World Health Organization. 2010. Pandemic (H1N1) 2009: update 112. World Health Organization, Geneva, Switzerland: http://www.who.int/csr/don/2010_08_06/en/index.html [Google Scholar]
- 2.Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, Chen J, Jie Z, Qiu H, Xu K, Xu X, Lu H, Zhu W, Gao Z, Xiang N, Shen Y, He Z, Gu Y, Zhang Z, Yang Y, Zhao X, Zhou L, Li X, Zou S, Zhang Y, Li X, Yang L, Guo J, Dong J, Li Q, Dong L, Zhu Y, Bai T, Wang S, Hao P, Yang W, Zhang Y, Han J, Yu H, Li D, Gao GF, Wu G, Wang Y, Yuan Z, Shu Y. 2013. Human infection with a novel avian-origin influenza A (H7N9) virus. N. Engl. J. Med. 368:1888–1897. 10.1056/NEJMoa1304459 [DOI] [PubMed] [Google Scholar]
- 3.Nicoll A, Danielsson N. 2013. A novel reassortant avian influenza A(H7N9) virus in China—what are the implications for Europe. Euro Surveill. 18(15):20452 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20452 [PubMed] [Google Scholar]
- 4.Medina RA, García-Sastre A. 2011. Influenza A viruses: new research developments. Nat. Rev. Microbiol. 9:590–603. 10.1038/nrmicro2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, Yang H, Chen X, Recuenco S, Gomez J, Chen LM, Johnson A, Tao Y, Dreyfus C, Yu W, McBride R, Carney PJ, Gilbert AT, Chang J, Guo Z, Davis CT, Paulson JC, Stevens J, Rupprecht CE, Holmes EC, Wilson IA, Donis RO. 2013. New World bats harbor diverse influenza A viruses. PLoS Pathog. 9(10):e1003657. 10.1371/journal.ppat.1003657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hutchinson EC, Denham EM, Thomas B, Trudgian DC, Hester SS, Ridlova G, York A, Turrell L, Fodor E. 2012. Mapping the phosphoproteome of influenza A and B viruses by mass spectrometry. PLoS Pathog. 8(11):e1002993. 10.1371/journal.ppat.1002993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao TL, Wu CY, Su WC, Jeng KS, Lai MM. 2010. Ubiquitination and deubiquitination of NP protein regulates influenza A virus RNA replication. EMBO J. 29:3879–3890. 10.1038/emboj.2010.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu K, Klenk C, Liu B, Keiner B, Cheng J, Zheng BJ, Li L, Han Q, Wang C, Li T, Chen Z, Shu Y, Liu J, Klenk HD, Sun B. 2011. Modification of nonstructural protein 1 of influenza A virus by SUMO1. J. Virol. 85:1086–1098. 10.1128/JVI.00877-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu CY, Jeng KS, Lai MM. 2011. The SUMOylation of matrix protein M1 modulates the assembly and morphogenesis of influenza A virus. J. Virol. 85:6618–6628. 10.1128/JVI.02401-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pal S, Santos A, Rosas J, Ortiz-Guzman J, Rosas-Acosta G. 2011. Influenza A virus interacts extensively with the cellular SUMOylation system during infection. Virus Res. 158:12–27. 10.1016/j.virusres.2011.02.017 [DOI] [PubMed] [Google Scholar]
- 11.Boggio R, Chiocca S. 2006. Viruses and sumoylation: recent highlights. Curr. Opin. Microbiol. 9:430–436. 10.1016/j.mib.2006.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wimmer P, Schreiner S, Dobner T. 2012. Human pathogens and the host cell SUMOylation system. J. Virol. 86:642–654. 10.1128/JVI.06227-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geiss-Friedlander R, Melchior F. 2007. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 8:947–956. 10.1038/nrm2293 [DOI] [PubMed] [Google Scholar]
- 14.Gill G. 2005. Something about SUMO inhibits transcription. Curr. Opin. Genet. Dev. 15:536–541. 10.1016/j.gde.2005.07.004 [DOI] [PubMed] [Google Scholar]
- 15.Gill G. 2004. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 18:2046–2059. 10.1101/gad.1214604 [DOI] [PubMed] [Google Scholar]
- 16.Hay RT. 2005. SUMO: a history of modification. Mol. Cell 18:1–12. 10.1016/j.molcel.2005.03.012 [DOI] [PubMed] [Google Scholar]
- 17.Portela A, Digard P. 2002. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol. 83:723–734 http://vir.sgmjournals.org/content/83/4/723.long [DOI] [PubMed] [Google Scholar]
- 18.Resa-Infante P, Gabriel G. 2013. The nuclear import machinery is a determinant of influenza virus host adaptation. Bioessays 35:23–27. 10.1002/bies.201200138 [DOI] [PubMed] [Google Scholar]
- 19.Rizzo MA, Springer GH, Granada B, Piston DW. 2004. An improved cyan fluorescent protein variant useful for FRET. Nat. Biotechnol. 22:445–449. 10.1038/nbt945 [DOI] [PubMed] [Google Scholar]
- 20.Kotaja N, Karvonen U, Janne OA, Palvimo JJ. 2002. PIAS proteins modulate transcription factors by functioning as SUMO-1 ligases. Mol. Cell. Biol. 22:5222–5234. 10.1128/MCB.22.14.5222-5234.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. 2001. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 146:2275–2289. 10.1007/s007050170002 [DOI] [PubMed] [Google Scholar]
- 22.Matrosovich M, Matrosovich T, Garten W, Klenk HD. 2006. New low-viscosity overlay medium for viral plaque assays. Virol. J. 3:63. 10.1186/1743-422X-3-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhirnov OP, Konakova TE, Garten W, Klenk H. 1999. Caspase-dependent N-terminal cleavage of influenza virus nucleocapsid protein in infected cells. J. Virol. 73:10158–10163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klenk C, Humrich J, Quitterer U, Lohse MJ. 2006. SUMO-1 controls the protein stability and the biological function of phosducin. J. Biol. Chem. 281:8357–8364. 10.1074/jbc.M513703200 [DOI] [PubMed] [Google Scholar]
- 25.Momose F, Kikuchi Y, Komase K, Morikawa Y. 2007. Visualization of microtubule-mediated transport of influenza viral progeny ribonucleoprotein. Microbes Infect. 9:1422–1433. 10.1016/j.micinf.2007.07.007 [DOI] [PubMed] [Google Scholar]
- 26.Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. 1997. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 88:97–107. 10.1016/S0092-8674(00)81862-0 [DOI] [PubMed] [Google Scholar]
- 27.Müller S, Matunis MJ, Dejean A. 1998. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J. 17:61–70. 10.1093/emboj/17.1.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chase GP, Rameix-Welti MA, Zvirbliene A, Zvirblis G, Gotz V, Wolff T, Naffakh N, Schwemmle M. 2011. Influenza virus ribonucleoprotein complexes gain preferential access to cellular export machinery through chromatin targeting. PLoS Pathog. 7(9):e1002187. 10.1371/journal.ppat.1002187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santos A, Pal S, Chacon J, Meraz K, Gonzalez J, Prieto K, Rosas-Acosta G. 2013. SUMOylation affects the interferon blocking activity of the influenza A nonstructural protein NS1 without affecting its stability or cellular localization. J. Virol. 87:5602–5620. 10.1128/JVI.02063-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang W, Chen Y, Wang S, Hu N, Cao Z, Wang W, Tong T, Zhang X. 2014. PIASxalpha ligase enhances SUMO1 modification of PTEN protein as a SUMO E3 ligase. J. Biol. Chem. 289:3217–3230. 10.1074/jbc.M113.508515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang SH, Sharrocks AD. 2005. PIASx acts as an Elk-1 coactivator by facilitating derepression. EMBO J. 24:2161–2171. 10.1038/sj.emboj.7600690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coloma R, Valpuesta JM, Arranz R, Carrascosa JL, Ortin J, Martin-Benito J. 2009. The structure of a biologically active influenza virus ribonucleoprotein complex. PLoS Pathog. 5(6):e1000491. 10.1371/journal.ppat.1000491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biswas SK, Boutz PL, Nayak DP. 1998. Influenza virus nucleoprotein interacts with influenza virus polymerase proteins. J. Virol. 72:5493–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ludwig S, Pleschka S, Planz O, Wolff T. 2006. Ringing the alarm bells: signalling and apoptosis in influenza virus infected cells. Cell. Microbiol. 8:375–386. 10.1111/j.1462-5822.2005.00678.x [DOI] [PubMed] [Google Scholar]
- 35.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. 2006. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7(10):R100. 10.1186/gb-2006-7-10-r100 [DOI] [PMC free article] [PubMed] [Google Scholar]