

ABSTRACT
The human pathogen Kaposi's sarcoma-associated herpesvirus (KSHV), the etiological agent of Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease, establishes lifelong latency upon infection. Murine gammaherpesvirus 68 (MHV68) is a well-established model for KSHV. Toll-like receptors (TLRs) play a crucial role for the innate immune response to pathogens. Although KSHV and MHV68 are detected by TLRs, studies suggest they modulate TLR4 and TLR9 signaling, respectively. In this study, we show that in bone marrow-derived macrophages (BMDMs), MHV68 did not induce a detectable proinflammatory cytokine response. Furthermore, MHV68 abrogated the response to TLR2, -4, -7, and -9 agonists in BMDMs. Similarly to observations with MHV68, infection with KSHV efficiently inhibited TLR2 signaling in THP-1 monocytes. Using a KSHV open reading frame (ORF) library, we found that K4.2, ORF21, ORF31, and the replication and transcription activator protein (RTA)/ORF50 inhibited TLR2-dependent nuclear factor kappa B (NF-κB) activation in HEK293 TLR2-yellow fluorescent protein (YFP)- and Flag-TLR2-transfected HEK293T cells. Of the identified ORFs, RTA/ORF50 strongly downregulated TLR2 and TLR4 signaling by reducing TLR2 and TLR4 protein expression. Confocal microscopy revealed that TLR2 and TLR4 were no longer localized to the plasma membrane in cells expressing RTA/ORF50. In this study, we have shown that the gammaherpesviruses MHV68 and KSHV efficiently downmodulate TLR signaling in macrophages and have identified a novel function of RTA/ORF50 in modulation of the innate immune response.
IMPORTANCE The Toll-like receptors (TLRs) are an important class of pattern recognition receptors of the innate immune system. They induce a potent proinflammatory cytokine response upon detection of a variety of pathogens. In this study, we found that the gammaherpesviruses murine gammaherpesvirus 68 (MHV68) and Kaposi's sarcoma-associated herpesvirus (KSHV) efficiently inhibit the TLR-mediated innate immune response. We further identified the KSHV-encoded replication and transcription activator protein (RTA) as a novel modulator of TLR signaling. Our data suggest that the gammaherpesviruses MHV68 and KSHV prevent activation of the innate immune response by targeting TLR signaling.
INTRODUCTION
Human herpesvirus 8, also known as Kaposi's sarcoma-associated herpesvirus (KSHV), was first identified in 1994 from Kaposi's sarcoma tissues from AIDS patients (1). It is now known to be the etiological agent of Kaposi's sarcoma and the B-cell disorders primary effusion lymphoma (PEL) and multicentric Castleman's disease (reviewed in reference 2). Murid herpesvirus 4, also known as murine gammaherpesvirus 68 (MHV68), which was first isolated in 1980 (3), has emerged as a model for the human gammaherpesviruses KSHV and Epstein-Barr virus (4). The gammaherpesviruses are double-stranded DNA viruses with an icosahedral nucleocapsid, a tegument protein layer, and an envelope layer containing viral glycoproteins (reviewed in reference 5). KSHV includes at least 90 open reading frames (ORFs) in its central unique genomic region of about 145 kb (5). MHV68 is somewhat smaller, with a unique region of about 120 kb encoding at least 80 ORFs (6).
Like all members of the herpesvirus family, KSHV and MHV68 have both lytic and latent phases of their life cycles. Upon primary infection with KSHV, latent and lytic genes are expressed, but lytic gene expression quickly decreases (5). The latent phase seems to be the default pathway; however, lytic replication is required for viral dissemination and transmission (7). Recently, a recombinant KSHV has been described in which expression of the replication and transcription activator, replication and transcription activator protein (RTA)/ORF50, is under the control of the constitutively active cellular phosphoglycerate kinase promoter (8). This virus enters by default the lytic replication cycle, allowing study of lytic replication without exogenous chemical treatments.
An early mechanism of defense in host-pathogen interactions occurs upon recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs). The Toll-like receptors (TLRs) are type I transmembrane proteins and were the first PRRs to be characterized. Via their ectodomains, TLRs recognize PAMPs and then activate signal transduction through their intracellular Toll-interleukin 1 (IL-1) receptor (TIR) domains (9). To date, 10 human and 12 murine TLRs have been identified, with distinct PAMP recognition functions. TLRs localize to either the plasma membrane or intracellularly in endolysosomal compartments. The cell surface TLRs detect microbial surface molecules, for example bacterial lipopeptides (TLR2), viral components (TLR2 and TLR4), and bacterial lipopolysaccharide (LPS) (TLR4). The endosomal TLRs recognize nucleic acids from pathogens, like double-stranded RNA (dsRNA) (TLR3), single-stranded RNA (ssRNA) (TLR7, TLR8), and dsDNA (TLR9) (9, 10). The membrane protein UNC93B1 interacts with endosomal TLRs and escorts them from the endoplasmic reticulum to the endolysosomal compartment (11, 12), where TLR9 is cleaved yielding a functional receptor (13, 14). Recently, the introduction of transgenic TLR9-green fluorescent protein (GFP) mice has enhanced analysis of TLR9 trafficking and function in antigen-presenting cells (15).
Both surface and endosomal TLRs have been implicated for a role in gammaherpesvirus detection. Of the surface TLRs, TLR2 has been found to detect MHV68 and TLR4 to detect KSHV. An NF-κB reporter in transfected human embryonic kidney 293 cells was activated by MHV68 in a TLR2-dependent manner, and compared to murine embryonic fibroblasts (MEFs) from wild-type (WT) mice, the proinflammatory cytokine IL-6 and alpha interferon (IFN-α) levels in response to MHV68 were reduced in MEFs derived from TLR2-knockout (KO) mice (16). Macrophages derived from TLR4-KO mice showed higher levels of KSHV gene expression than WT macrophages, and similar results were obtained with small interfering RNA (siRNA)-mediated TLR4 knockdown in human lymphatic endothelial cells (LEC) (17).
Studies have linked MHV68 and KSHV to the endosomal TLRs 3, 7, and 9. TLR3 is involved in detection of KSHV, as primary infection of human monocytes with KSHV led to upregulation of TLR3 expression and activation of TLR3 signaling, including production of TLR3-specific chemokines and cytokines (18). Signaling through TLR7, either through TLR7/8 agonists or induced by vesicular stomatitis virus infection, reactivated KSHV from latency (19). TLR9 is involved in detection of both KSHV and MHV68 in plasmacytoid dendritic cells (20, 21). Intraperitoneal infection of TLR9-KO mice resulted in markedly increased viral loads during lytic and latent infection with MHV68, further confirming that TLR9 is involved in detection of MHV68 (20).
Studies show that in addition to detection by TLRs, the gammaherpesviruses can also modulate TLR signaling. For example, KSHV infection of endothelial cells rapidly suppresses TLR4 expression on the mRNA and protein levels, and the ORFs vIRF1 and vGPCR can downregulate TLR4 from the cell surface (17). The vIRFs 1, 2, and 3 have been found to inhibit TLR3-induced IFN transcription factor activation (22). MHV68 may have modulated its recognition by TLR9. Compared to murine cytomegalovirus, the MHV68 genome contains a much lower quantity of immunostimulatory CpG motifs, and MHV68 may have selectively suppressed these motifs through cytosine-to-thymine conversion (23).
In this study, we analyzed TLR sensing of MHV68 in primary bone marrow-derived macrophages (BMDMs) and detected only an extremely weak proinflammatory cytokine response. We furthermore found that MHV68 abolished the production of tumor necrosis factor alpha (TNF-α) upon TLR stimulation of BMDMs. A similar observation was made for KSHV and TLR2 in human monocytic THP-1 cells. We utilized an NF-κB luciferase reporter assay and a KSHV ORF library (24) to identify gene products involved in KSHV inhibition of TLR2 signaling. We then determined whether identified ORFs also affected TLR4 signaling. ORF21, ORF31, and RTA/ORF50 strongly inhibited TLR2 and TLR4 signaling in transfected cells. RTA encoded by ORF50 reduced levels of coexpressed TLR2 and TLR4 and disrupted the plasma membrane localization of TLR2 and TLR4, but not CD95, in transiently transfected cells. This study reveals a new function for RTA/ORF50 in modulation of the innate immune response.
MATERIALS AND METHODS
Cell lines.
Cell lines were maintained at 37°C in a humidified, 7.5% CO2 environment. Standard medium, used for human embryonic kidney (HEK) 293T (ATCC CRL-11268) and murine bone marrow fibroblast M2-10B4 (ATCC CRL-1972) cells and modified as noted, was Dulbecco's modified Eagle's medium (DMEM; high glucose) supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, and 1% penicillin-streptomycin (P-S). hTERT-RPE1 (ATCC CRL-4000) cells were grown with 5% FCS. The immortalized murine bone marrow-derived macrophage cell line (BEI Resources no. NR-9456) was derived from primary bone marrow cells from C57BI6/J wild-type mice immortalized by infection with the ecotropic-transforming replication-deficient retrovirus J2 [carrying v-myc and v-raf(mil) oncogenes] (25) using described techniques (26). Immortalized BMDM medium was supplemented with the addition of 50 μM 2-mercaptoethanol. Immortalized BMDMs expressing mTLR9-GFP or mTLR2-GFP were generated by retroviral transduction as described previously (15) and were selected with 5 μg/ml puromycin. HEK293 hTLR2-yellow fluorescent protein (YFP) (BEI Resources no. NR-9316) medium was supplemented with 1 mM sodium pyruvate and 1 mg/ml G418. HEK293 hTLR4A-MD2-CD14 cells were purchased from Invivogen, and medium was supplemented with 10 μg/ml blasticidin and 50 μg/ml HygroGold. The human acute monocytic leukemia cell line THP-1 (DMSZ no. ACC 16) was grown in RPMI supplemented with 10% FCS, 2 mM glutamine, and 1% P-S.
Antibodies and reagents.
Polyclonal anti-MHV68 antiserum was prepared in rabbits (27). Murine monoclonal anti-β-actin (A5441) and rabbit-anti-c-Myc (C3956) were purchased from Sigma-Aldrich. Rabbit anti-GFP (Ab290) was from Abcam. Rabbit antibodies against IKKα (sc-7184), IKKγ/NEMO (sc-8330), and IκBα (sc-371) were from Santa Cruz Biotechnology. The murine Myc tag antibody (no. 2276) and rabbit antibodies against GFP (D5.1, no. 2956), IKKβ (no. 2370), IRAK1 (no. 4504), NF-κB p65 (no. 4764), RIP1 (no. 3493), Tab2 (no. 3745), and Tak1 (no. 5206) were purchased from Cell Signaling Technology. Anti-MyD88 (AB16527) was from Millipore and anti-TRAF6 (GTX61290) was from GeneTex. BD Pharmingen Alexa Fluor 647-conjugated rat anti-mouse TNF (no. 557730) and allophycocyanin (APC)-conjugated mouse anti-human TNF (no. 554514) were purchased from BD Biosciences. CpG A 2336 and CpG B 1826 were from MWG. FSL-1, Pam3CSK4, and R848 were purchased from Invivogen. Brefeldin A and lipopolysaccharide (LPS) were purchased from Sigma-Aldrich. Lipofectamine 2000 was purchased from Life Technologies, and Fugene HD was purchased from Promega.
Plasmids.
The 85-plasmid KSHV ORF library has been previously described (24). All plasmids were at least partially resequenced for verification. Most express a C-terminal Myc tag; of the ones mentioned in multiple figures, the ORF50 construct expresses an N-terminal Myc tag. For clarity, ORF-Myc is used to describe library ORFs. C-terminally GFP-tagged TLR9 in pMSCVpuro (Clontech) was previously described (12). The plasmids pNF-κB Luc (Agilent Technologies) and pTK-RL (Promega) are commercially available. pCMV1 Flag-hTLR2 (Addgene plasmid 13082) was originally provided by Ruslan Medzhitov. pMSCVpuro mTLR2-GFP expresses full-length murine TLR2 C-terminally fused with enhanced GFP (EGFP), and primer sequences for this construct are available upon request. pcDNA3 hTLR2-YFP and pcDNA3 hTLR4-YFP are available from Addgene and originally provided by Douglas Golenbock (Addgene plasmids 13016 and 13018). pEF Flag-MAVS expresses N-terminally Flag-tagged MAVS and was kindly provided by Zhijian Chen (UT Southwestern Medical Center). pCMV-TAG2B NEMO expresses N-terminally Flag-tagged NEMO and was deposited to Addgene (plasmid 11970) by Jon Ashwell (28). The endoplasmic reticulum (ER) marker mCherry-KDEL pMSCVneo (cherry-KDEL) construct has been previously described (12). pEYFP-N1 CD95 is human CD95 C-terminally fused to YFP and was previously described (29).
Viral preparation and infections.
BAC-derived MHV68-GFP has been described (30). The MHV68 WUMS strain was purchased from ATCC (VR-1465). For simplicity, MHV68 is used in figures. In figures where both MHV68 WUMS and MHV68-GFP are used, MHV68 refers to WUMS. MHV68 strains were amplified in M2-10B4 cells. The supernatant from heavily infected cells was cleared of cell debris, and then virus was pelleted (2 h at 26,000 × g at 4°C). The pellet was soaked overnight in virus standard buffer (VSB; 50 mM Tris-HCl, 12 mM KCl, 5 mM NA-EDTA, pH 7.8) and then purified by pelleting through a 10% Nycodenz cushion in VSB for 3 h at 26,500 × g at 4°C. The virus-containing pellet was soaked in VSB, Dounce homogenized, and flash frozen in liquid nitrogen. Titers of purified virus were determined using the median tissue culture infective dose (TCID50) method on M2-10B4 cells. The JSC1 cell-derived KSHV strain was previously cloned as a bacterial artificial chromosome and termed BAC16 (31). BAC16 was modified to enforce lytic replication by inserting a pgk promoter in front of ORF50 in the same way as previously described for another KSHV clone, BAC36 (8). The resulting virus, KSHVLYT, expresses GFP and was propagated on hTERT-RPE1 cells. For stock production, virus in the supernatant was pelleted (4 h at 15,000 × g at 4°C) and resuspended in 1/100 of the original volume in complete medium. KSHVLYT titers were determined by centrifugal enhancement of infection using TCID50 on hTERT-RPE1 cells. Immortalized BMDMs were infected with MHV68-GFP in serum-free medium, utilizing centrifugal enhancement (30 min at 800 × g at 4°C), followed by further incubation for 1.5 h at 37°C. Medium was changed and cells were incubated for the indicated times. THP-1 cells were infected with KSHVLYT (multiplicity of infection [MOI] of 0.5) in serum-containing medium utilizing centrifugal enhancement (90 min at 450 × g at room temperature [RT]) in the presence of 4 μg/ml Polybrene. Murine cytomegalovirus (MCMV)-GFP (32) was purified as described previously (33), and the titer was determined by TCID50.
Mice.
C57BL/6J mice were bred at the HZI and maintained under specific-pathogen-free conditions in accordance with institutional, state, and federal guidelines.
Preparation, stimulation, and infection of primary cells.
Bone marrow was isolated from the femur and tibia of wild-type (C57BL/6J) mice. For BMDMs, cells were cultured in DMEM, 10% FCS, 2 mM glutamine, 1% P-S, 50 μM 2-mercaptoethanol, and 5% macrophage colony-stimulating factor (MCSF). Cells were seeded at day 7 or day 8 of MCSF culture. For enzyme-linked immunosorbent assays (ELISAs), cells were stimulated with CpG B 1826 or infected with MCMV-GFP or MHV68 WUMS for 16 h. For intracellular cytokine staining, cells were infected with MHV68 WUMS or MHV68-GFP utilizing centrifugal enhancement (30 min at 800 × g at RT), followed by further incubation for 1.5 h at 37°C. Medium was changed and cells were incubated for 20 h prior to stimulation.
ELISA.
TNF-α production was detected by ELISA using a hamster anti-mouse/rat TNF capture antibody (BD Biosciences Pharmingen no. 557516) and a rabbit anti-mouse/rat TNF detection antibody (BD Biosciences no. 557432) as described previously (11). Interleukin 6 (IL-6) and IL-12 (p40) levels were detected using OptEIA kits (BD Biosciences) according to the manufacturer's instructions.
Intracellular cytokine staining and flow cytometry.
For 4 h prior to harvesting, cells were incubated with TLR agonists (primary BMDMs: 100 ng/ml LPS, 0.5 μM CpG B 1826, 500 ng/ml FSL-1; immortalized BMDMs: 100 ng/ml LPS, 1,000 ng/ml Pam3CSK4, 1,000 ng/ml FSL-1, 1 μM CpG B 1826, 0.5 μM R848; THP-1: 100 ng/ml FSL-1). During TLR stimulation, cells were treated with 10 μg/ml brefeldin A to inhibit secretion of TNF-α. Cells were collected, fixed for 15 min in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS), and then washed with PBS. Cells were permeabilized with 0.5% saponin in PBS-2% FCS (FACS buffer) and then stained with Alexa Fluor 647-conjugated anti-mouse TNF-α or APC-conjugated anti-hTNF-α. For BMDM experiments, cells were costained with rabbit polyclonal anti-MHV68 and anti-rabbit Alexa Fluor 594. Samples were analyzed on an LSRII or Fortessa flow cytometer (BD), followed by data analysis using FlowJo. Mean fluorescence intensity (MFI) for TNF-α is shown.
Immunoblotting.
For analysis of steady-state levels of TLR pathway components, 2 × 106 immortalized BMDMs (WT, expressing mTLR9-GFP, or expressing mTLR2-GFP) were uninfected or infected with MHV68 WUMS at an MOI of 2 or 5 and then incubated for the indicated times prior to lysing in radioimmunoprecipitation (RIPA) buffer (20 mM Tris [pH 7.5], 1 mM EDTA, 100 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) with protease and phosphatase inhibitors (Roche). For analysis of protein levels in cotransfected cells, 200,000 HEK293T cells were transfected with 600 ng KSHV ORFs or empty vector and 200 ng of Flag-hTLR2, mTLR2-GFP, hTLR4-YFP, hTLR2-YFP, hCD95-YFP, Flag-MAVS, or Flag-NEMO expression plasmids, using 2 μl Lipofectamine 2000 according to the manufacturer's instructions and then lysed 24 h posttransfection in RIPA buffer. Whole-cell lysates were cleared by centrifugation and separated by SDS-PAGE or Bis-Tris PAGE, transferred to nitrocellulose, and probed with specific antibodies as indicated. Anti-mouse horseradish peroxidase (HRP) and anti-rabbit HRP antibodies (Dianova) were used, followed by development with Lumi-Light (Roche Applied Science) or SuperSignal West Femto (Thermo Scientific) chemiluminescence substrates and film exposure. Films were scanned and images prepared using Photoshop and Illustrator (Adobe). Bands were quantified using ImageJ software analysis. Intensities were measured, normalized to actin levels, and scaled to empty vector or uninfected cells.
Screen for KSHV ORFs which inhibit TLR2 signaling.
For the KSHV plasmid library screen, HEK293 hTLR2-YFP cells (40,000/well in 96-well plates) were transiently transfected with 8 μl of Fugene HD/DNA complexes using a ratio of 7 μl Fugene HD to 2 μg DNA (approximately 150 ng total DNA/well). The three-plasmid cotransfection mixture was 50 ng pNF-κB Luc, 50 ng pTK-RL, and 1,900 ng ORF expression plasmids or empty vector. Twenty-four hours posttransfection, cells were stimulated with 100 ng/ml FSL-1 for 18 h and then lysed in 50 μl/well 1× passive lysis buffer (Promega). Luciferase production from 10 μl lysate was measured using the dual-luciferase reporter assay system (Promega) and a GloMax 96-microplate luminometer (Promega) with autoinjection of 50 μl each substrate, 0.4-s delay, and 2-s read. Values shown were calculated by dividing renilla-normalized stimulated sample values by renilla-normalized unstimulated values and then scaling to empty vector (set to 100% induction).
Titration analysis of TLR2-dependent inhibition by selected KSHV ORFs.
HEK293T cells (25,000/well in 96-well plates) were cotransfected with 8 μl of Fugene HD/DNA complexes using a ratio of 7 μl Fugene HD to 2 μg DNA. The four-plasmid cotransfection mixture was 50 ng pNF-κB Luc, 50 ng pTK-RL, 500 ng Flag-TLR2, and 1,400 ng pcDNA3.1(−) or 1,400, 700, or 350 ng ORF. DNA quantity was brought to 2 μg using pcDNA3.1(−). Eighteen hours posttransfection, cells were stimulated with 50 ng/ml FSL-1 for 6 h and then lysed in 50 μl/well 1× passive lysis buffer (Promega). Values were measured and calculated as described above. Results shown are means plus standard deviations from 3 to 4 replicates total from 2 independent experiments.
Titration analysis of TLR4-dependent inhibition by selected KSHV ORFs.
Two days prior to transfection, HEK293 hTLR4A-MD2-CD14 cells (20,000/well in 96-well plates) were plated on poly-d-lysine-coated plates. Cells were transfected as for HEK293 hTLR2-YFP cells, with DNA mixes of 50 ng pNF-κB Luc, 50 ng pTK-RL, and 1,900 ng pcDNA3.1(−) or 1,900, 950, or 475 ng ORF. DNA quantity was brought to 2 μg using pcDNA3.1(−). Approximately 18 h posttransfection, cells were stimulated with 25 ng/ml LPS for 9 h and then lysed and processed as described above. Results shown are means plus standard deviations from duplicates from 3 independent experiments.
Confocal microscopy.
One day prior to transfection, HEK293T cells were seeded in phenol red-free DMEM supplemented with 10% FCS, 2 mM glutamine, and 1% P-S into poly-d-lysine-coated coverglass cell culture chambers (Sarstedt). Cells were transfected with 280 ng KSHV ORFs or empty vector, 95 ng mTLR2-GFP, hTLR2-YFP, hTLR4-YFP, or CD95-YFP, and 25 ng cherry-KDEL per well using Lipofectamine 2000. Twenty-four hours posttransfection, images were taken using a Nikon ECLIPSE Ti-E inverted microscope equipped with a spinning-disc confocal device (UltraView VOX; PerkinElmer), solid state diode lasers (PerkinElmer), an OrcaR2 charge-coupled-device (CCD) camera (Hamamatsu), and a Nikon ×60-magnification 1.4-numerical-aperture planapochromat objective. During imaging, cells were maintained in a climate chamber (EMBLEM, Germany) at 37°C and 5% CO2. Image processing was performed using Volocity and Photoshop.
Statistical analysis.
Statistical analysis was performed using Prism version 5 software (GraphPad). P values for pairwise comparisons were determined using a two-tailed unpaired t test. The KSHV library screen was analyzed by one-way analysis of variance (ANOVA), and pairwise comparisons were analyzed by a two-tailed unpaired t test. ORF titrations were analyzed by one-way ANOVA followed by Dunnett's multiple-comparison test. P values of <0.05 are indicated with asterisks in the figures; P values of >0.05 are not otherwise indicated.
RESULTS
MHV68 induces a minimal proinflammatory cytokine response in BMDMs.
First, we analyzed the proinflammatory cytokine response to MHV68 in primary and immortalized bone marrow-derived macrophages (BMDMs) (Fig. 1). As measured by ELISA, primary BMDMs produced TNF-α (Fig. 1A) and IL-6 (Fig. 1B) in response to the TLR9 agonist CpG B and the betaherpesvirus murine cytomegalovirus (MCMV), respectively. In contrast, very little TNF-α and IL-6 were detected upon infection with MHV68. The amount of TNF-α was less than one-tenth of that induced by CpG or MCMV, and IL-6 levels induced by MHV68 were barely above the ELISA detection limit (Fig. 1A and B).
FIG 1.

The proinflammatory cytokine response of BMDMs to MHV68 is very weak. Primary BMDMs were stimulated with CpG B or infected with MCMV-GFP or MHV68 for 16 h. TNF-α (A) or IL-6 (B) production was detected by ELISA. Results are shown as means + standard deviations (SD) from duplicates from 2 (CpG B, A) or 3 independent experiments. Immortalized BMDMs were infected for 4 to 24 h with MHV68-GFP (MOI of 5) (C). Four hours prior to harvesting, cells were incubated with brefeldin A to prevent TNF-α secretion and then fixed and processed for intracellular TNF-α staining. Samples were analyzed by flow cytometry. The means ± SD of mean fluorescence intensity (MFI) for the anti-TNF-α signal from 2 independent experiments are shown. *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
Next, we used a more sensitive assay to determine TNF-α production at various time points after MHV68-GFP infection in immortalized BMDMs. Intracellular cytokine staining for TNF-α followed by flow cytometry also allows better determination of kinetics than ELISA, as recent rather than total accumulation is measured. To prevent cytokine secretion, cells were treated for the last 4 h prior to harvesting with brefeldin A. Using a rabbit anti-MHV68 polyclonal antibody, we found that approximately 85 to 90% of infected cells at 24 h postinfection were MHV68 positive. For this reason, we determined the mean fluorescent intensity (MFI) of the anti-TNF-α signal of the total cell population at 4, 8, 12, and 24 h postinfection for MHV68-infected cells as well as uninfected controls (Fig. 1C). The MFIs of uninfected and infected samples at all time points were similar, indicating that MHV68 did not induce a TNF-α response at any time. However, uninfected cells still responded appropriately to stimulation with various TLR ligands at all time points (see Fig. 5), validating the assay. The results in both primary and immortalized BMDMs were in agreement with each other and indicated that MHV68 does not strongly induce TNF-α or IL-6 in macrophages.
FIG 5.
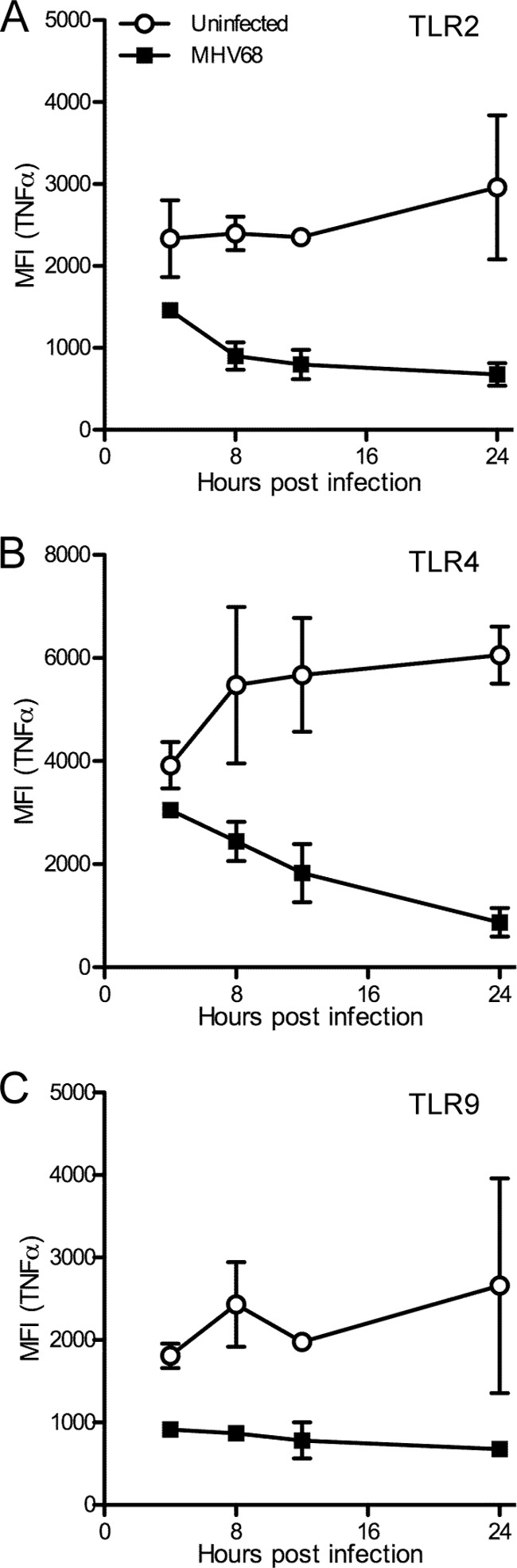
MHV68 inhibits TLR-induced TNF-α production in BMDMs early after infection. Immortalized BMDMs were infected for 4 to 24 h with MHV68-GFP (MOI of 5). Four hours prior to harvesting, cells were incubated with TLR agonists for TLR2 (FSL-1; A), TLR4 (LPS; B), or TLR9 (CpG B; C) in the presence of brefeldin A to prevent TNF-α secretion and then fixed and processed for intracellular TNF-α staining. Samples were analyzed by flow cytometry. The means ± SD of mean fluorescence intensity (MFI) for the anti-TNF-α signal from 2 independent experiments are shown. For reference, the MFI for unstimulated samples was at all times less than 1,000 (see Fig. 1C). For each time course, P < 0.0001.
MHV68 inhibits TLR-induced TNF-α signaling in primary macrophages.
Several studies have suggested manipulation of TLR signaling pathways by gammaherpesviruses (16–19). Next, we analyzed the ability of MHV68-infected primary BMDMs to respond to stimulation of plasma membrane and endosomal TLRs, using TLR2 and TLR9 as representative members of the respective categories. We utilized intracellular cytokine staining as described for Fig. 1C, which allows gating on infected cells. At 24 h postinfection with MHV68 WUMS or MHV68-GFP, we stimulated primary BMDMs for 4 h with the TLR2 agonist FSL-1 or the TLR9 agonist CpG or left cells unstimulated. All cells were treated with brefeldin A. Uninfected cells produced TNF-α in response to FSL-1 and CpG, as shown in representative contour plots (Fig. 2A, top row). MHV68 WUMS- or MHV68-GFP-infected cells did not respond to TLR agonists (Fig. 2A, middle and bottom rows). We quantified the TNF-α MFI of uninfected or infected populations and found that while the TNF-α MFI was similar in both uninfected unstimulated and infected unstimulated cells (P > 0.05), the MFI in FSL-1- or CpG-treated cells was much lower in infected populations than in uninfected cells (P < 0.05 for MHV68) (Fig. 2B).
FIG 2.
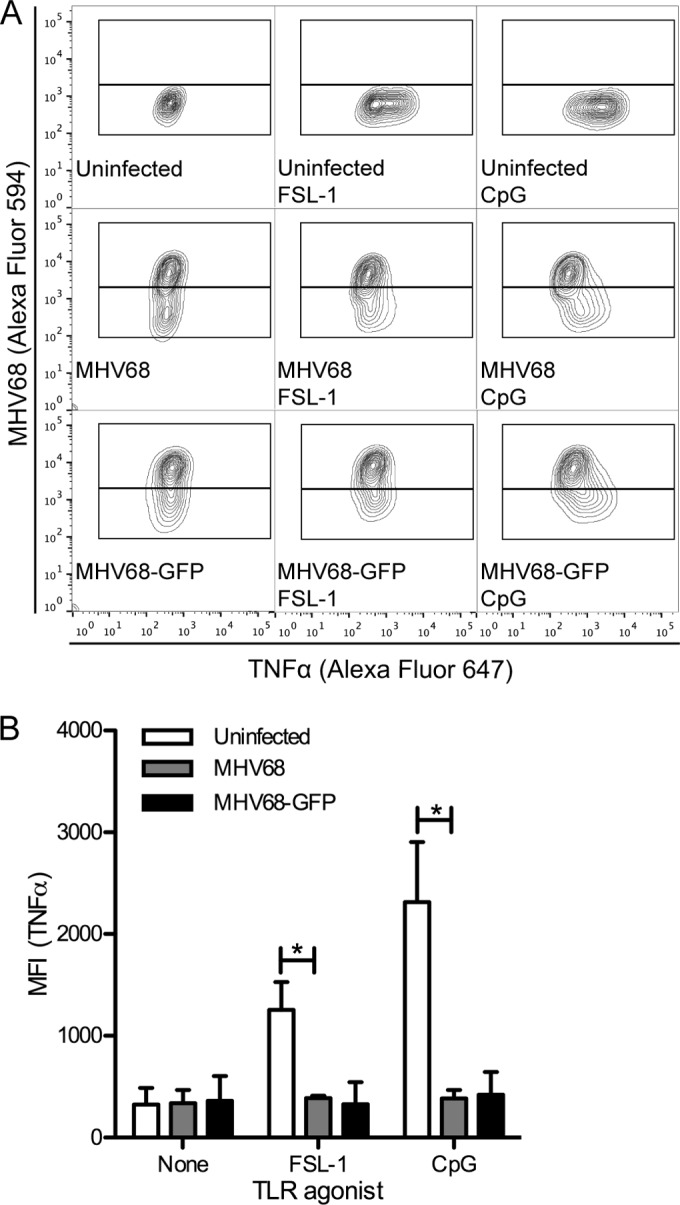
MHV68-infected primary BMDMs do not respond to TLR stimulation. At 24 h postinfection, MHV68- or MHV68-GFP-infected (MOI of 2) or uninfected primary BMDMs were stimulated for 4 h with TLR agonists (TLR2, FSL-1; TLR9, CpG B) in the presence of brefeldin A to prevent TNF-α secretion and then processed for intracellular MHV68 and TNF-α staining and analyzed by flow cytometry. (A) Representative contour plots. (B) The averages + SD of the mean fluorescence intensity of the anti-TNF-α channel gated on MHV68-negative (uninfected) or MHV68-positive (MHV68 or MHV68-GFP) populations from 2 independent experiments are shown. *, P < 0.05.
MHV68 inhibits TLR-induced TNF-α signaling in immortalized BMDMs.
We next wanted to examine the effect of MHV68 on additional TLRs. For these studies, we used immortalized BMDMs, which divide rapidly and are easily infected. We analyzed the TNF-α response to a panel of TLR agonists in MHV68-GFP-infected immortalized BMDMs stimulated with TLR agonists at 24 h postinfection for 4 h in the presence of brefeldin A. TLR3 is omitted, because in our assay, 4 h of poly(I·C) stimulation was insufficient to induce a TNF-α response (data not shown). First, as in primary BMDMs, TNF-α production was similar between unstimulated uninfected and MHV68-infected cells (P > 0.05) (Fig. 3A and B). However, we found that the TNF-α response to TLR2 (Pam3CSK4, FSL-1), TLR4 (LPS), TLR7 (R848), and TLR9 (CpG B) agonists was completely inhibited in MHV68-infected cells (Fig. 3A and B). Not only does MHV68 prevent its own detection, MHV68 also renders macrophages unresponsive to TLR agonist stimulation of the proinflammatory cytokine response.
FIG 3.

MHV68-infected immortalized BMDMs do not respond to TLR stimulation. (A, B) At 24 h postinfection, MHV68-GFP-infected (MOI of 5) or uninfected immortalized BMDMs were stimulated for 4 h with TLR agonists (TLR2, Pam3CSK4, FSL-1; TLR4, LPS; TLR7, R848; TLR9, CpG B) in the presence of brefeldin A to prevent TNF-α secretion and then processed for intracellular TNF-α staining and analyzed by flow cytometry. (A) Representative contour plots (uninfected, upper row; MHV68-GFP infected, lower row). (B) The averages + SD of the mean fluorescence intensity of the anti-TNF-α channel gated on MHV68-negative (uninfected) or MHV68-positive (MHV68 or MHV68-GFP) populations from 3 independent experiments are shown. *, P < 0.05; ***, P < 0.001. (C) Immortalized BMDMs expressing TLR9-GFP were infected with MHV68 at the indicated MOIs or left uninfected, and then TLR9 levels at 4 h and 24 h postinfection were determined by immunoblotting with an anti-GFP antibody (FL, full-length TLR9-GFP; Ct, C-terminal cleavage fragment of TLR9-GFP). Immunoblots for MHV68 and actin are included as controls.
One possible explanation for the inhibition of TLR signaling is that MHV68 downregulates TLR expression. For this reason, we used an anti-GFP antibody to determine the levels of the endosomally located TLR9 in MHV68 WUMS-infected TLR9-GFP-expressing immortalized BMDMs (Fig. 3C). We observed similar levels of full-length TLR9-GFP in uninfected and MOI 2 and MOI 5 infected samples at both 4 and 24 h postinfection. Since we observed the C-terminal cleavage fragment of TLR9-GFP, TLR9 trafficking to the endolysosomal compartment, where cleavage occurs, is functional in MHV68-infected BMDMs. To confirm infection, we used a polyclonal anti-MHV68 antibody (Fig. 3C). We also examined levels of TLR2-GFP upon MHV68 infection of TLR2-GFP-expressing immortalized BMDMs and observed very similar TLR2-GFP protein levels in uninfected and MHV68-infected cells (data not shown). We also analyzed TLR2-GFP localization in MHV68-infected cells by confocal microscopy but did not detect obvious differences in TLR2-GFP expression levels or localization (data not shown). These results indicate that MHV68 does not negatively modulate TLR2-GFP or TLR9-GFP protein expression levels in stably transduced immortalized BMDMs in steady-state lysates at the time points analyzed.
Next, we determined if levels of other TLR-induced cytokines were reduced by MHV68 infection. At 8 h postinfection, uninfected or MHV68 WUMS- or MHV68-GFP-infected immortalized BMDMs were stimulated for 16 h with the TLR2 agonist FSL-1 or the TLR9 agonist CpG. The levels of TNF-α, IL-6, and IL-12 (p40) in culture supernatants were then determined by ELISA. As observed for intracellular cytokine staining, the TNF-α response by ELISA was strongly reduced in FSL-1 or CpG-treated MHV68-infected cells, compared to uninfected cells (Fig. 4A). Similar results were observed for IL-6 (Fig. 4B) and IL-12 (p40) (Fig. 4C). Infection was confirmed by staining with an anti-MHV68 antibody and flow cytometric analysis (data not shown).
FIG 4.
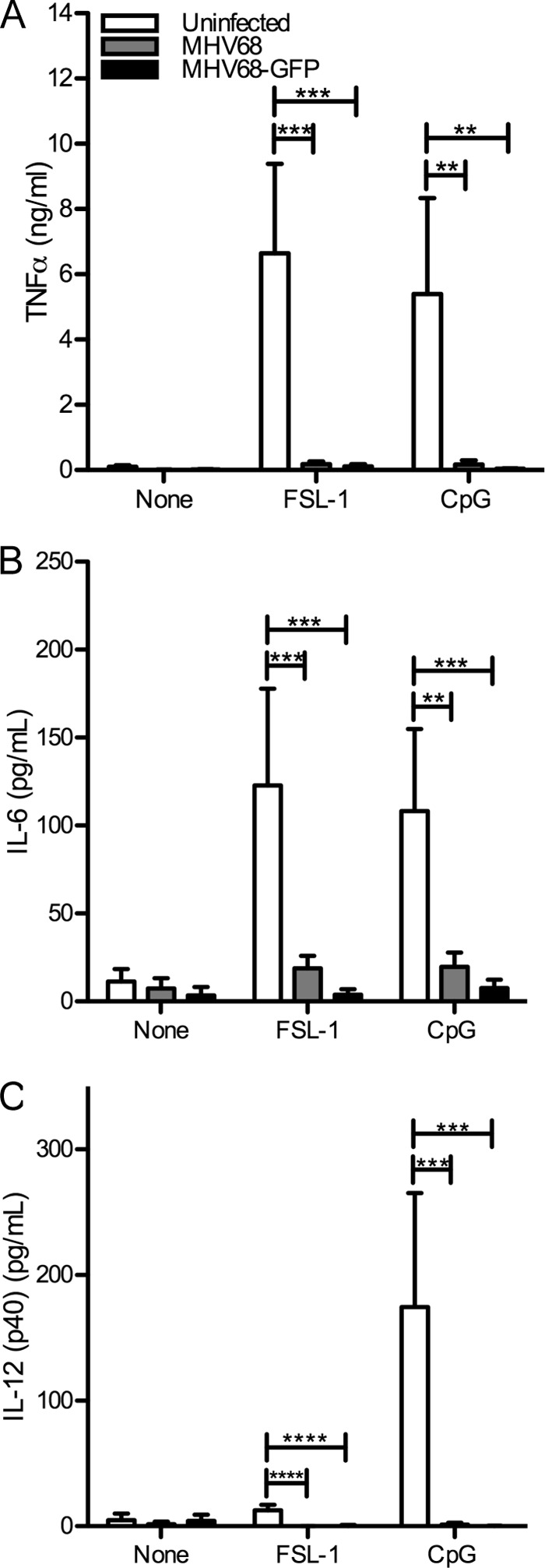
MHV68-infected BMDMs do not produce proinflammatory cytokines in response to TLR stimulation. At 8 h postinfection, MHV68- or MHV68-GFP-infected (MOI of 5) or uninfected immortalized BMDMs were stimulated with TLR agonists (TLR2, FSL-1; TLR9, CpG B) for 16 h, and then TNF-α (A), IL-6 (B), and IL-12 (p40) (C) levels were determined by ELISA. The averages + SD from triplicate samples from 2 (A, C) or 4 (B) independent infection experiments are shown. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
We then determined the time dependency of MHV68 inhibition of TLR signaling. Immortalized BMDMs were infected for 4, 8, 12, or 24 h with MHV68-GFP. During the last 4 h of infection for each time point, cells were stimulated with various TLR agonists, or left unstimulated, in the presence of brefeldin A. We then determined the TNF-α response using intracellular cytokine staining. Compared to uninfected cells, which responded well to TLR agonist stimulation, we found that MHV68 inhibits the response to TLR2, TLR4, and TLR9 agonists (Fig. 5A, B, and C, respectively) to a limited extent at 4 h postinfection and more strongly as early as 8 h postinfection.
Because the response to all TLR agonists tested was inhibited, it is possible that MHV68 targets a downstream effector of TLR signaling, rather than individual TLRs themselves. To identify potential cellular protein targets of MHV68, which may be downregulated to disrupt TLR signaling, we examined the protein levels of a variety of TLR pathway components at 4 and 24 h after MHV68 infection in immortalized BMDMs (Fig. 6). As expected, actin quantities were increased at 24 h compared to at 4 h. MHV68 is known to induce an NF-κB response independently of TLR signaling. In agreement, we found that levels of the NF-κB inhibitor IκBα were reduced by 67% in MOI 5 infected cells 24 h postinfection (Fig. 6), although the reduction we observed in macrophages was not as complete as that reported for MEFs (34). We did not observe great differences in levels of the TLR adapter MyD88 or the further-downstream signaling components IRAK1, RIP1, TRAF6, Tab2, IKKα, IKKβ, IKKγ, or p65. With the exception of IκBα as noted, protein levels in uninfected or infected samples varied by less than 2-fold. These results indicate that either MHV68 targets other pathway components for TLR signaling modulation or that the mechanism is not through degradation of pathway components.
FIG 6.

Steady-state expression levels of proteins involved in TLR signaling are not affected in MHV68-infected cells. Immortalized BMDMs were infected with MHV68 at an MOI of 2 or 5 or left uninfected and then lysed at 4 and 24 h with RIPA buffer. Equal volumes of lysates were separated by SDS-PAGE or Bis-Tris PAGE and transferred to nitrocellulose membranes, and then protein levels for the indicated proteins were determined by immunoblotting with the indicated antibodies. Separate gels were used for each antibody, and actin blots are included as controls. Results are representative of 3 independent experiments.
KSHV-infected monocytes show reduced TNF-α production upon TLR stimulation.
To determine whether the human gammaherpesvirus KSHV blocks TLR signaling in human macrophage precursors similarly to MHV68, we utilized the permissive human monocytic THP-1 cell line. Nondifferentiated THP-1 cells have been previously found to respond poorly to LPS (35), and we also observed a poor response to LPS. However, using intracellular cytokine staining, we detected distinguishable TNF-α responses in nondifferentiated THP-1 cells to the TLR2/6 agonist FSL-1 and the TLR1/2 agonist Pam3CSK4 (data not shown).
We infected nondifferentiated THP-1 cells at an MOI of 0.5 with KSHVLYT in the presence of Polybrene or treated cells with Polybrene only. Twenty-four hours postinfection, cells were stimulated for 4 h with FSL-1 in the presence of brefeldin A. We then analyzed TNF-α levels by intracellular cytokine staining (Fig. 7). The use of GFP-expressing KSHVLYT allowed gating on GFP-positive infected cells. Compared to uninfected cells, we observed a slight increase in TNF-α levels in KSHVLYT-infected cells (Fig. 7B). The uninfected cell population responded to FSL-1, as indicated by increased MFI. However, in KSHV-infected cells, we observed a reduced response to FSL-1 (Fig. 7A and B). The MFI for GFP-positive cells is shown (Fig. 7B). Similar results were obtained using Pam3CSK4 (data not shown). We conclude from these data that KSHV, like MHV68, is able to downmodulate TLR2 signaling in infected macrophages.
FIG 7.

KSHV inhibits the TLR2-induced TNF-α response in THP-1 monocytes. THP-1 cells were treated with Polybrene or infected with GFP-expressing KSHVLyt (MOI of 0.5) in the presence of Polybrene. Twenty-four hours postinfection, cells were stimulated or not with FSL-1 in the presence of brefeldin A and then processed for TNF-α intracellular cytokine staining. (A) Representative contour plots. (B) The averages + SD of mean fluorescence intensity (MFI) of duplicates from 2 independent experiments are shown. For KSHVLyt-infected samples, MFI was determined for the GFP-positive population. **, P < 0.01; ***, P < 0.001.
Multiple KSHV ORFs inhibit TLR2 signaling in HEK293 TLR2-YFP cells.
Based on these observations, we wished to identify viral proteins that modulate TLR signaling. As TNF-α production downstream of TLRs is activated by NF-κB (36, 37), we chose to use an NF-κB-dependent luciferase reporter assay. In combination with a pcDNA plasmid-based KSHV ORF-Myc library (24, 38), we analyzed the NF-κB-driven luciferase response in HEK293-derived cells constitutively expressing human TLR2 fused to YFP. The TLR2-YFP fusion protein is functional and restores TLR2 signaling in HEK293 cells (39). We cotransfected empty vector or KSHV ORFs of interest with an NF-κB-luciferase reporter plasmid, along with a renilla luciferase plasmid for normalization. Transfection of empty vector alone did not induce an NF-κB response. ORF71/K13 has been previously shown to induce a strong NF-κB response in HEK293T cells (38), and ORF71/K13 reproducibly induced a strong NF-κB response in HEK293 TLR2-YFP cells as expected. In some experiments, ORF74 induced a strong NF-κB response, although it was not as consistent as with ORF71/K13 (data not shown).
As expected, upon stimulation with the TLR2 agonist FSL-1, we detected a very strong NF-κB response for empty vector-transfected cells (Fig. 8). Most ORFs did not affect the cellular response to FSL-1, and luciferase values were similar to those observed for empty vector-transfected cells. However, several ORFs, including K4.2, ORF21, ORF31, ORF50, and K10.5, strongly inhibited the cellular response to FSL-1 stimulation (Fig. 8). Because ORF71/K13 and ORF74 induced a strong NF-κB response upon transfection, the apparent fold change upon stimulation is very low. However, this does not necessarily indicate they actively block TLR2 signaling.
FIG 8.

Screen for KSHV ORFs that may inhibit the TLR2-induced NF-κB response. HEK293 TLR2-YFP cells were transfected with pNF-κB-Luc (firefly), pTK-RL (renilla), and the indicated KSHV ORF-Myc or empty pcDNA vector. At 24 h posttransfection, cells were unstimulated or stimulated with the TLR2 agonist FSL-1 overnight and then lysed in passive lysis buffer for measurement of luciferase activity. Fold induction of NF-κB is determined as follows: firefly normalized to renilla and then stimulated divided by unstimulated, scaled to pcDNA empty vector (100%). Data shown are the averages from 4 replicates total from 3 independent experiments. Log-transformed results were analyzed by one-way ANOVA (P < 0.0001), and select empty vector-ORF pairs were analyzed by two-tailed unpaired t test. ***, P < 0.001; ****, P < 0.0001.
K4.2, ORF21, ORF31, and ORF50 inhibit the NF-κB response to FSL-1 in a dose-dependent manner.
We next utilized HEK293T cells and a Flag-TLR2 construct for reporter assays, in which cells were cotransfected with Flag-TLR2, pNF-κB-Luc, pTK-RL, and various amounts of KSHV ORFs. The DNA quantity was equalized using empty pcDNA vector. First, we determined the NF-κB response upon ORF expression. K4.2, ORF21, ORF31, ORF50, and K10.5 did not strongly induce NF-κB; induction was at most 4-fold (Fig. 9A). In comparison, ORF74 in this assay induced approximately 30-fold NF-κB induction (data not shown). We then determined the inhibition of the NF-κB response upon FSL-1 stimulation. As seen in HEK293 TLR2-YFP cells, K4.2, ORF21, ORF31, and ORF50 inhibited the response to FSL-1 in Flag-TLR2-transfected HEK293T cells in a dose-dependent manner (Fig. 9B). K10.5 did not strongly inhibit the FSL-1 response in Flag-TLR2-expressing cells (Fig. 9B).
FIG 9.

ORF21 and ORF50 strongly inhibit TLR2 and TLR4 signaling. (A, B) HEK293T cells were cotransfected with pNF-κB-Luc, pTK-RL, Flag-TLR2, and empty pcDNA vector or increasing concentrations (1×, 2×, or 4×) of the indicated KSHV ORF-Myc. At 18 h posttransfection, cells were unstimulated or stimulated for 6 h with the TLR2 agonist FSL-1 and then lysed in passive lysis buffer. (C, D) HEK293 TLR4-MD2-CD14 cells were cotransfected with pNF-κB-Luc, pTK-RL, and empty pcDNA vector or increasing concentrations (1×, 2×, or 4×) of the indicated KSHV ORF-Myc. At 18 h posttransfection, cells were unstimulated or stimulated for 9 h with the TLR4 agonist LPS and then lysed in passive lysis buffer for measurement of luciferase activity. (A, C) NF-κB response in unstimulated cells, normalized to renilla and scaled to empty vector. (B, D) Fold induction of NF-κB in response to FSL-1 (B) or LPS (D), determined as follows: firefly normalized to renilla and then stimulated divided by unstimulated. Data shown are averages with standard deviations from 3 or 4 replicates total in two independent experiments (A, B) or duplicates from each of 3 independent experiments (C, D). Transformed results were analyzed by one-way ANOVA followed by Dunnett's multiple comparison test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (E) Flag and Myc immunoblotting was used to verify Flag-TLR2 and ORF-Myc expression levels in whole-cell RIPA lysates. Actin was used as a loading control.
We also determined the response to LPS in HEK293 TLR4-MD2-CD14 cells transfected with ORFs identified in TLR2 assays. ORF74 was included in TLR4 studies, as it has been previously shown to downregulate TLR4 expression (17). First, we analyzed the induction of NF-κB in transfected, unstimulated TLR4 cells. Induction was similar to that observed in Flag-TLR2-transfected HEK293T cells, and the TLR4 control ORF74 induced the strongest NF-κB response (Fig. 9C). We next looked at the TLR4-dependent NF-κB response to LPS. We found that ORF21, ORF31, ORF50, and ORF74 inhibited the response to LPS, particularly at the highest concentrations of plasmid used (Fig. 9D).
ORF50 reduces TLR2 and TLR4 protein expression.
Next, we determined protein levels of ORFs and TLR2 in HEK293T cells cotransfected with empty vector or Myc-tagged K4.2, ORF21, ORF31, ORF50, or K10.5 and Flag-TLR2. In whole-cell RIPA lysates, we were able to detect ORF21, ORF31, ORF50, and K10.5 by immunoblotting, but K4.2, expected to run at a size of 18 kDa, was not detected (Fig. 9E). However, its inhibitory effect on TLR signaling suggests it is expressed. Notably, Flag-TLR2 expression was very difficult to detect in the presence of ORF50. While Flag-TLR2 levels with K4.2, ORF21, and ORF31 were similar to that of empty vector (within 30%), no intensity for a Flag-TLR2 band in the ORF50 lysate could be determined using ImageJ (Fig. 9E). We analyzed lysates from multiple independent experiments which included RIPA lysates, as well as samples from the luciferase assay described in Fig. 9A and B, and observed in all cases that Flag-TLR2 protein expression was strongly reduced in the presence of ORF50 (data not shown). Expression of the highest ORF concentrations for both unstimulated and stimulated luciferase samples was verified by immunoblotting for Fig. 9A to D (data not shown). Data were similar to that shown in Fig. 9E.
We then wanted to determine if the reduction observed for Flag-TLR2 in ORF50-expressing cells was specific to TLR2, promoter or tag dependent, or if ORF50 generally inhibits expression of many cellular and cotransfected proteins. We cotransfected ORF31, ORF50, and K10.5 in a 3:1 ratio with several expression constructs of proteins involved in innate immunity. We first looked at coexpression of ORFs with Flag-NEMO in pCMV-Tag2B, which contains a CMV promoter similarly to the Flag-TLR2 expression vector (Fig. 10A). We did not observe an inhibitory effect of ORF50 on Flag-NEMO expression. Compared to empty vector, NEMO expression was similar in the presence of K10.5 and ORF50 (within 30%). We also looked at Flag-MAVS in pEF, which contains an E1Falpha promoter. In the presence of ORF50, Flag-MAVS expression levels were very similar to those in empty vector-transfected cells, with a reduction of only 12% (Fig. 10A).
FIG 10.
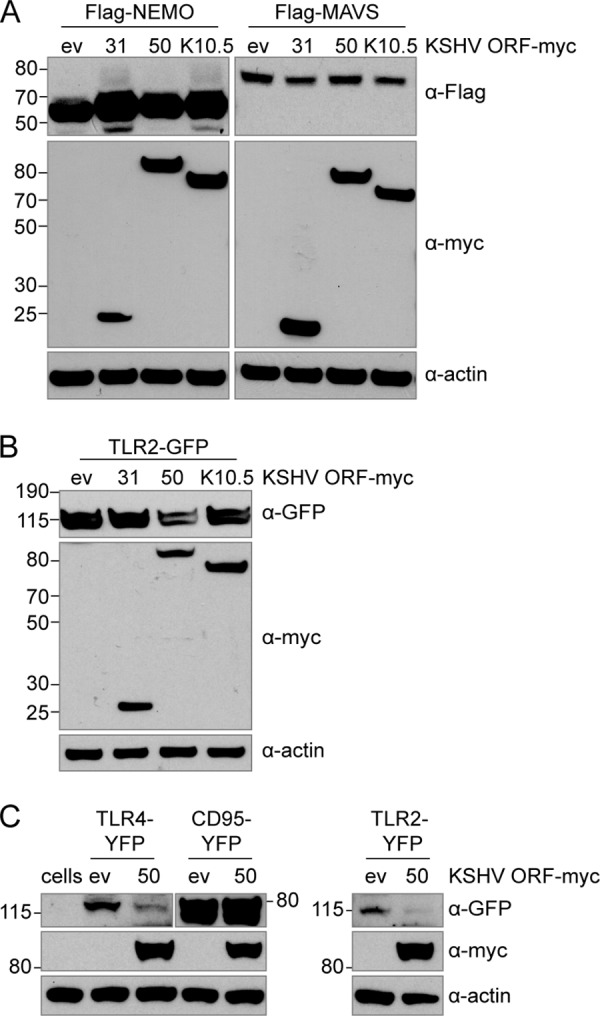
ORF50 specifically affects protein levels of TLR2 and TLR4. HEK293T cells were cotransfected with Myc-tagged KSHV ORFs (ORF31, ORF50, or K10.5) and empty pcDNA vector and Flag-tagged MAVS, Flag-tagged NEMO (A), or TLR2-GFP (B). HEK293T cells were cotransfected with ORF50 or empty pcDNA vector and TLR4-YFP, CD95-YFP, or TLR2-YFP. (C) Nontransfected cells were used as a negative control. Protein levels at 24 h posttransfection in whole-cell lysates were determined by immunoblotting using anti-Flag (A), anti-GFP ab290 (B), or anti-GFP D5.1 (C) and anti-Myc and anti-actin (A, B, C) antibodies.
Next, we looked at expression of coexpressed TLR2-GFP, which is in the retroviral vector pMSCV. Expression of this construct is controlled by the 5′ long terminal repeat region from the murine stem cell PCC4 cell-passaged myeloproliferative sarcoma virus (PCMV). We observed a similar effect to that observed with Flag-TLR2. TLR2-GFP was expressed well in the presence of empty vector, ORF31, and K10.5 (Fig. 10B). While those intensities differed by less than 10%, in contrast, TLR2-GFP was reduced over 50% in ORF50-transfected samples, although transfected cells looked healthy (Fig. 10B). These results indicate that the effect of ORF50 on TLR2 protein expression levels is not promoter or tag dependent and also is not due to general inhibition of protein expression in ORF50-expressing cells.
We further analyzed whether ORF50 has a similar effect on TLR4 expression. When we coexpressed ORF50 with TLR4-YFP or TLR2-YFP, we observed less TLR protein expression compared to that in empty vector-transfected cells (Fig. 10C). TLR4-YFP levels were reduced over 80% compared to that of the empty vector, and TLR2-YFP levels were reduced more than 85% compared to that of the empty vector. The effect appears specific for TLR2 and TLR4, since expression of the type I transmembrane protein CD95-YFP was minimally affected by ORF50 coexpression. For CD95-YFP, an increase of 7.5% in the presence of ORF50 compared to empty vector was determined by ImageJ analysis (Fig. 10C).
ORF50 disrupts plasma membrane localization of TLR2 and TLR4 but not CD95.
We then determined the subcellular localization of TLR2-GFP in live HEK293T cells in the presence of empty pcDNA vector or K4.2, ORF21, ORF31, ORF50, or K10.5. As Myc expression cannot be determined in live cells, we used the ER marker cherry-KDEL as a cotransfection marker. With pcDNA, we observed distinct plasma membrane localization of TLR2-GFP as expected (Fig. 11A). There was also TLR2-GFP in small vesicles and the ER, as would be expected based on TLR2 trafficking. For cells cotransfected with K4.2, ORF31, or K10.5 and TLR2-GFP, we observed a distribution of TLR2-GFP similar to that with empty vector (Fig. 11A). ORF21 strongly affected cellular morphology as previously described (40), but we did not observe an obvious change in TLR2-GFP localization (Fig. 11A). Interestingly, in ORF50-cotransfected cells, the GFP signal was clearly relocalized to an overall cellular distribution (Fig. 11A). ORF-Myc, TLR2-GFP, and actin protein levels of samples transfected in parallel was determined and was similar to Fig. 10B; K4.2 was not detected by immunoblotting (data not shown).
FIG 11.

In ORF50-expressing cells, TLR2 and TLR4 no longer localize to the plasma membrane. HEK293T cells were cotransfected with the indicated plasmids and images were obtained 24 h posttransfection in live cells by confocal microscopy. Cherry-KDEL (red) was used as a marker for cotransfection and the endoplasmic reticulum and included in all transfections. Cells were cotransfected with TLR2-GFP and either empty pcDNA vector, K4.2, ORF21, ORF31, ORF50, or K10.5 (A) or with combinations of TLR4-YFP, TLR2-YFP, or CD95-YFP and empty pcDNA vector or ORF50 (B). Scale bar, 10 μm.
To determine if the effect observed on TLR2 localization is specific to TLR2, we also examined the localization of TLR4-YFP in cells cotransfected with cherry-KDEL and either empty pcDNA vector or ORF50. The YFP signal was very low and sensitive to photobleaching, but we were able to observe clear plasma membrane localization of YFP (Fig. 11B, top). In ORF50-cotransfected cells, the YFP signal was redistributed to an overall cellular distribution (Fig. 11B, top). The same was observed for TLR2-YFP (Fig. 11B, middle). To determine if the relocalization of TLR2 and TLR4 is due to a general effect on plasma membrane receptors, we determined if ORF50 also affects the localization of CD95-YFP. In cells cotransfected with empty pcDNA vector, CD95-YFP, and cherry-KDEL, CD95-YFP was localized to the plasma membrane and ER (Fig. 9B, bottom). The distribution of CD95-YFP was not altered in ORF50-transfected cells (Fig. 11B, bottom). Our results indicate that ORF50 specifically targets TLR2 and TLR4 and that ORF50 does not have a general inhibitory effect on coexpressed type I transmembrane proteins.
DISCUSSION
Gammaherpesviruses are detected by multiple TLRs. So far, it has been shown that MHV68 is detected by TLR2 and TLR9, and KSHV is detected by TLR3, TLR4, and TLR9 (16–18, 20, 21). In primary and immortalized BMDMs, the induction of the proinflammatory cytokines TNF-α and IL-6 in response to MHV68 is extremely limited. We furthermore found that MHV68 abrogated TLR-induced proinflammatory cytokine production for multiple TLR agonists in primary and immortalized macrophages. In our time course assay, we observed moderate inhibition as early as 4 h postinfection, which increased by 8 h. Analysis of protein levels of TLR2-GFP and TLR9-GFP indicated that at the time points chosen, potentially overexpressed levels of TLR2-GFP and TLR9-GFP were not directly targeted for substantial degradation by MHV68. It is possible that insufficient MHV68 protein is produced to target such TLR levels or that the TLR-GFP fusions are very stable. Unfortunately, the antibodies to murine TLR2, TLR4, and TLR9 that we tested were poorly reactive or nonspecific, as validated using immortalized BMDMs from the respective TLR-KO mice (data not shown).
Nevertheless, while we cannot exclude that endogenous levels of TLRs are reduced upon MHV68 infection, it is reasonable to also consider that MHV68 may target downstream effectors since we observe inhibition of the response to TLR1/2, -2/6, -4, -7, and -9 agonists. Herpesviral proteins are known to target cellular proteins for degradation (41, 42). By analyzing endogenous protein levels of a variety of TLR signaling pathway components in MHV68-infected BMDMs, we did not find evidence for MHV68-induced degradation. Unlike the betaherpesvirus MCMV, which targets NEMO (IKKγ) for degradation and thus strongly reduces NEMO expression levels, thereby shutting down TLR signaling (41), infection with MHV68 only minimally affected protein levels of the TLR signaling components tested.
MHV68 is the murine homolog of the human gammaherpesviruses, and so we also determined whether the ability to modulate TLR signaling is conserved in human KSHV. We examined the ability of KSHVLYT to inhibit TLR signaling. KSHV undergoes a brief period of lytic replication before establishing latency, but it is difficult to separate the two pathways experimentally. Reactivation from latency is commonly achieved using chemical stimuli, for example phorbol 12-myristate 13-acetate (PMA). However, PMA itself initiates NF-κB activation (43), which confounds signaling analysis. The use of lytic KSHV circumvents this problem and allows analysis of early events in lytic KSHV infection.
We observed that, like its murine homolog MHV68, KSHV also inhibits TLR signaling. While KSHV is detected by TLR4 (17), the TLR4 response to LPS in nondifferentiated THP-1 cells is poor, so we were unable to examine the ability of KSHV to inhibit TLR4 signaling in this system. TLR2 has not been previously shown to be modulated by KSHV, but it does play a role in detection of MHV68 (16), so we chose to study the ability of KSHV to modulate TLR2 signaling. KSHV-infected THP-1 cells responded poorly to the TLR2 agonist FSL-1 compared to uninfected cells. One possible explanation for our observations is that KSHV actively downregulates TLR2 signaling to minimize the interferon and proinflammatory cytokine responses to additional viral or bacterial pathogens, as a strong innate immune response could otherwise lead to the destruction of the KSHV-infected cell. Another option is that TLR2 does play a role in detection of KSHV, and the virus therefore actively abrogates the pathway during KSHV infection.
Because TNF-α is induced via the transcription factor NF-κB, we then analyzed the ability of KSHV ORFs to modulate TLR2-activated NF-κB-induced luciferase production and found that the lytic ORFs K4.2, ORF21, ORF31, and ORF50 inhibit TLR2 signaling. ORF21, ORF31, and ORF50 also strongly inhibited TLR4 signaling. None have been previously identified to contribute to TLR2 or TLR4 modulation, and they have not previously been found to activate NF-κB (38) nor to modulate NF-κB signaling downstream of other pathways. K4.2 is an immediate early protein which has been recently found to interfere with immunoglobulin secretion and to contribute to lytic replication (44). ORF21 localizes to the cytoplasm of transfected HeLa cells (24) and is a thymidine kinase homolog with poor thymidine kinase activity, which also affects cellular morphology (40, 45). KSHV ORF31 localizes to the cytoplasm (24) and has not been extensively characterized, but its MHV68 homolog is required for lytic replication. The amino acid sequences are 31% identical, but KSHV ORF31 can rescue MHV68 ORF31-null virus, suggesting that MHV68 and KSHV ORF31 proteins are functional homologs (46).
KSHV ORF50 encodes the replication and transcription activator RTA. Expression of RTA/ORF50 is necessary and sufficient to initiate lytic replication (47). In our study, RTA/ORF50 did not induce an NF-κB response and strongly inhibited the response to the TLR2 agonist FSL-1 and the TLR4 agonist LPS. Coexpression analysis of RTA/ORF50 and TLR2 showed that RTA/ORF50 disrupts plasma membrane localization of TLR2 and TLR4 and strongly reduces expression of TLR2 and TLR4. RTA/ORF50 has not been previously shown to target TLR2 or TLR4. However, RTA/ORF50 has been found to be a ubiquitin E3 ligase which can target IRF7 to the proteasome for degradation and thus suppress IFN-β promoter activation (48). Interestingly, RTA/ORF50 has also been described to degrade the TLR3 and TLR4 adaptor protein Toll–IL-1 receptor (TIR) domain-containing adaptor-inducing IFN-β (TRIF), although no direct interaction between RTA/ORF50 and TRIF could be shown (49). It is unclear whether RTA mediates TRIF degradation directly or indirectly. RTA/ORF50 may be modulating expression of TLR2 and TLR4 via its ubiquitin E3 ligase activity. It is also possible that RTA/ORF50 modulates expression of TLR2 and TLR4 via transcriptional regulation.
TLR3 and TLR4 rely on TRIF to induce the type I IFN response (50, 51). TLRs 2 and 4 as well as TRIF are localized mainly to the plasma membrane and cytosol, respectively, whereas RTA/ORF50 predominantly localizes to the nucleus. At this stage, we cannot draw conclusions about the underlying mechanism of RTA/ORF50-induced TLR degradation. It is possible that ORF50 may prevent TLR2 and TLR4 trafficking to the cell surface by degradation immediately after synthesis in the ER, or it may induce degradation after the TLRs have already trafficked to the plasma membrane. It is unclear how a predominantly nuclear protein may have such a function. However, protein synthesis occurs in the cytoplasm, so RTA/ORF50 must be there at least transiently. Also, expression levels of RTA/ORF50 in the cytosol may either be below the detection level by microscopy, or ORF50 may exert its effect on TLR2/4 expression through an indirect mechanism via nuclear activation of an unknown cellular protein that mediates TLR degradation. Coimmunoprecipitation experiments between ORF50 and TLRs may answer this question. Furthermore, it is possible that the degradation of the TLR3/TLR4 adaptor protein TRIF reported by Ahmad et al. (49) may be related to the effect we observe on TLR4 itself. If the receptor is degraded, its adaptor may face the same fate. In combination with our results, it is interesting to hypothesize that RTA/ORF50 has a ubiquitous role in regulating multiple innate immune pathways during KSHV lytic infection.
Both KSHV and MHV68 inhibited TLR2 signaling during infection of macrophages. RTA/ORF50 may modulate TLR2 signaling during infection by specifically targeting TLR2. We examined TLR2-GFP and TLR9-GFP levels during MHV68 infection of immortalized BMDMs and did not observe a strong effect on TLR expression at the time points chosen. There are several possible explanations for this apparent discrepancy. One is that an effect on TLRs occurs only at specific times during infection. It is also possible that TLR9-GFP and TLR2-GFP expression in immortalized BMDMs is very stable, so that we are unable to see an effect in our assay, or that MHV68 protein levels are insufficient to counteract the amount of TLR2 or TLR9. Another possibility is that while KSHV and MHV68 both modulate TLR2 signaling, they use different mechanisms to do so. Further studies will help answer some of these questions.
The bacterial ligands of TLR2 and TLR4 have been widely characterized, and while viral ligands have been suggested for both, they are not as well understood. TLR4 recognizes bacterial lipopolysaccharide. TLR4 has also been implicated in immune recognition of the negative-stranded RNA respiratory syncytial virus (RSV) fusion (F) protein (52, 53). However, additional viral ligands have not been identified so far. TLR2 detects bacterial components such as lipoproteins and peptidoglycans. In addition, TLR2 is thought to detect viral components from multiple viruses (reviewed in reference 54), including either or both of the HCMV surface glycoproteins gB and gH (55). Therefore, TLR2 and TLR4 may be targeted by ORF50/RTA to prevent detection of nascent virions or to prevent induction of the interferon and/or proinflammatory cytokine response upon viral or bacterial superinfection.
NF-κB plays an essential role in the KSHV life cycle, and it is becoming increasingly clear that KSHV carefully manipulates NF-κB transcription and a variety of TLR signaling pathways. Latently infected lymphocytes show high levels of NF-κB activity, and treatment of these cells with an NF-κB inhibitor induces lytic reactivation (56). NF-κB inhibition is required for reactivation of KSHV from latency. However, activation of TLR7/8 signaling can also induce reactivation (19). KSHV encodes both inhibitors and activators of NF-κB transcription (38). The lytic proteins encoded by K4.2 and ORFs 21, 31, and 50 may contribute to control of TLR signaling and inhibition of NF-κB activity during lytic replication. It is conceivable that KSHV and other gammaherpesviruses may also utilize TLR signaling manipulation to moderate the host cell environment. Secondary viral or bacterial infection could lead to a strong interferon or proinflammatory cytokine response, and inhibition of TLR signaling by the gammaherpesviruses may be a mechanism through which they are able to successfully replicate and establish latency.
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft (SFB900 to M.M.B.), Virtual Institute VISTRIE (VH-VI-424 to M.M.B.), and the Initiative and Networking Fund of the Helmholtz Association (VH-NG-637 to M.M.B.).
We thank Christine Standfuß-Gabisch for excellent technical assistance.
Footnotes
Published ahead of print 4 June 2014
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. 10.1126/science.7997879 [DOI] [PubMed] [Google Scholar]
- 2.Dittmer DP, Damania B. 2013. Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)—an update. Curr. Opin. Virol. 3:238–244. 10.1016/j.coviro.2013.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaskovic D, Stancekova M, Svobodova J, Mistrikova J. 1980. Isolation of five strains of herpesviruses from two species of free living small rodents. Acta Virol. 24:468. [PubMed] [Google Scholar]
- 4.Barton E, Mandal P, Speck SH. 2011. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu. Rev. Immunol. 29:351–397. 10.1146/annurev-immunol-072710-081639 [DOI] [PubMed] [Google Scholar]
- 5.Knipe DM, Howley PM. 2013. Fields virology, 6th ed, vol II Wolters Kluwer, Philadelphia, PA [Google Scholar]
- 6.Virgin HW, IV, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71:5894–5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bechtel JT, Liang Y, Hvidding J, Ganem D. 2003. Host range of Kaposi's sarcoma-associated herpesvirus in cultured cells. J. Virol. 77:6474–6481. 10.1128/JVI.77.11.6474-6481.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Budt M, Hristozova T, Hille G, Berger K, Brune W. 2011. Construction of a lytically replicating Kaposi's sarcoma-associated herpesvirus. J. Virol. 85:10415–10420. 10.1128/JVI.05071-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384. 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 10.Broz P, Monack DM. 2013. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 13:551–565. 10.1038/nri3479 [DOI] [PubMed] [Google Scholar]
- 11.Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim YM. 2007. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J. Cell Biol. 177:265–275. 10.1083/jcb.200612056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. 2008. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 452:234–238. 10.1038/nature06726 [DOI] [PubMed] [Google Scholar]
- 13.Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL. 2008. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat. Immunol. 9:1407–1414. 10.1038/ni.1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, Barton GM. 2008. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature 456:658–662. 10.1038/nature07405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Avalos AM, Kirak O, Oelkers JM, Pils MC, Kim YM, Ottinger M, Jaenisch R, Ploegh HL, Brinkmann MM. 2013. Cell-specific TLR9 trafficking in primary APCs of transgenic TLR9-GFP mice. J. Immunol. 190:695–702. 10.4049/jimmunol.1202342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michaud F, Coulombe F, Gaudreault E, Kriz J, Gosselin J. 2010. Involvement of TLR2 in recognition of acute gammaherpesvirus-68 infection. PLoS One 5:e13742. 10.1371/journal.pone.0013742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lagos D, Vart RJ, Gratrix F, Westrop SJ, Emuss V, Wong PP, Robey R, Imami N, Bower M, Gotch F, Boshoff C. 2008. Toll-like receptor 4 mediates innate immunity to Kaposi sarcoma herpesvirus. Cell Host. Microbe 4:470–483. 10.1016/j.chom.2008.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.West J, Damania B. 2008. Upregulation of the TLR3 pathway by Kaposi's sarcoma-associated herpesvirus during primary infection. J. Virol. 82:5440–5449. 10.1128/JVI.02590-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregory SM, West JA, Dillon PJ, Hilscher C, Dittmer DP, Damania B. 2009. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. U. S. A. 106:11725–11730. 10.1073/pnas.0905316106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guggemoos S, Hangel D, Hamm S, Heit A, Bauer S, Adler H. 2008. TLR9 contributes to antiviral immunity during gammaherpesvirus infection. J. Immunol. 180:438–443. 10.4049/jimmunol.180.1.438 [DOI] [PubMed] [Google Scholar]
- 21.West JA, Gregory SM, Sivaraman V, Su L, Damania B. 2011. Activation of plasmacytoid dendritic cells by Kaposi's sarcoma-associated herpesvirus. J. Virol. 85:895–904. 10.1128/JVI.01007-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobs SR, Gregory SM, West JA, Wollish AC, Bennett CL, Blackbourn DJ, Heise MT, Damania B. 2013. The viral interferon regulatory factors of Kaposi's sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by Toll-like receptor 3. J. Virol. 87:798–806. 10.1128/JVI.01851-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pezda AC, Penn A, Barton GM, Coscoy L. 2011. Suppression of TLR9 immunostimulatory motifs in the genome of a gammaherpesvirus. J. Immunol. 187:887–896. 10.4049/jimmunol.1003737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sander G, Konrad A, Thurau M, Wies E, Leubert R, Kremmer E, Dinkel H, Schulz T, Neipel F, Sturzl M. 2008. Intracellular localization map of human herpesvirus 8 proteins. J. Virol. 82:1908–1922. 10.1128/JVI.01716-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. 2008. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9:847–856. 10.1038/ni.1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberson SM, Walker WS. 1988. Immortalization of cloned mouse splenic macrophages with a retrovirus containing the v-raf/mil and v-myc oncogenes. Cell Immunol. 116:341–351. 10.1016/0008-8749(88)90236-5 [DOI] [PubMed] [Google Scholar]
- 27.Steer B, Adler B, Jonjic S, Stewart JP, Adler H. 2010. A gammaherpesvirus complement regulatory protein promotes initiation of infection by activation of protein kinase Akt/PKB. PLoS One 5:e11672. 10.1371/journal.pone.0011672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. 2006. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected]. Nat. Cell Biol. 8:398–406. 10.1038/ncb1384 [DOI] [PubMed] [Google Scholar]
- 29.Reinehr R, Gorg B, Hongen A, Haussinger D. 2004. CD95-tyrosine nitration inhibits hyperosmotic and CD95 ligand-induced CD95 activation in rat hepatocytes. J. Biol. Chem. 279:10364–10373. 10.1074/jbc.M311997200 [DOI] [PubMed] [Google Scholar]
- 30.Adler H, Messerle M, Wagner M, Koszinowski UH. 2000. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J. Virol. 74:6964–6974. 10.1128/JVI.74.15.6964-6974.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 86:9708–9720. 10.1128/JVI.01019-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mathys S, Schroeder T, Ellwart J, Koszinowski UH, Messerle M, Just U. 2003. Dendritic cells under influence of mouse cytomegalovirus have a physiologic dual role: to initiate and to restrict T cell activation. J. Infect. Dis. 187:988–999. 10.1086/368094 [DOI] [PubMed] [Google Scholar]
- 33.Scheibe E, Lienenklaus S, May T, Magalhaes VG, Weiss S, Brinkmann MM. 2013. Measurement of mouse cytomegalovirus-induced interferon-beta with immortalized luciferase reporter cells. Methods Mol. Biol. 1064:355–366. 10.1007/978-1-62703-601-6_25 [DOI] [PubMed] [Google Scholar]
- 34.Dong X, Feng H, Sun Q, Li H, Wu TT, Sun R, Tibbetts SA, Chen ZJ, Feng P. 2010. Murine gamma-herpesvirus 68 hijacks MAVS and IKKbeta to initiate lytic replication. PLoS Pathog. 6:e1001001. 10.1371/journal.ppat.1001001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takashiba S, Van Dyke TE, Amar S, Murayama Y, Soskolne AW, Shapira L. 1999. Differentiation of monocytes to macrophages primes cells for lipopolysaccharide stimulation via accumulation of cytoplasmic nuclear factor kappaB. Infect. Immun. 67:5573–5578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shakhov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV. 1990. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J. Exp. Med. 171:35–47. 10.1084/jem.171.1.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collart MA, Baeuerle P, Vassalli P. 1990. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol. Cell. Biol. 10:1498–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konrad A, Wies E, Thurau M, Marquardt G, Naschberger E, Hentschel S, Jochmann R, Schulz TF, Erfle H, Brors B, Lausen B, Neipel F, Sturzl M. 2009. A systems biology approach to identify the combination effects of human herpesvirus 8 genes on NF-kappaB activation. J. Virol. 83:2563–2574. 10.1128/JVI.01512-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Latz E, Visintin A, Lien E, Fitzgerald KA, Monks BG, Kurt-Jones EA, Golenbock DT, Espevik T. 2002. Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the Toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J. Biol. Chem. 277:47834–47843. 10.1074/jbc.M207873200 [DOI] [PubMed] [Google Scholar]
- 40.Gill MB, Murphy JE, Fingeroth JD. 2005. Functional divergence of Kaposi's sarcoma-associated herpesvirus and related gamma-2 herpesvirus thymidine kinases: novel cytoplasmic phosphoproteins that alter cellular morphology and disrupt adhesion. J. Virol. 79:14647–14659. 10.1128/JVI.79.23.14647-14659.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fliss PM, Jowers TP, Brinkmann MM, Holstermann B, Mack C, Dickinson P, Hohenberg H, Ghazal P, Brune W. 2012. Viral mediated redirection of NEMO/IKKgamma to autophagosomes curtails the inflammatory cascade. PLoS Pathog. 8:e1002517. 10.1371/journal.ppat.1002517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Lint AL, Murawski MR, Goodbody RE, Severa M, Fitzgerald KA, Finberg RW, Knipe DM, Kurt-Jones EA. 2010. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. J. Virol. 84:10802–10811. 10.1128/JVI.00063-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang MS, Chen BC, Yu MT, Sheu JR, Chen TF, Lin CH. 2005. Phorbol 12-myristate 13-acetate upregulates cyclooxygenase-2 expression in human pulmonary epithelial cells via Ras, Raf-1, ERK, and NF-kappaB, but not p38 MAPK, pathways. Cell Signal. 17:299–310. 10.1016/j.cellsig.2004.07.008 [DOI] [PubMed] [Google Scholar]
- 44.Wong LY, Brulois K, Toth Z, Inn KS, Lee SH, O'Brien K, Lee H, Gao SJ, Cesarman E, Ensser A, Jung JU. 2013. The product of Kaposi's sarcoma-associated herpesvirus immediate early gene K4.2 regulates immunoglobulin secretion and calcium homeostasis by interacting with and inhibiting pERP1. J. Virol. 87:12069–12079. 10.1128/JVI.01900-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cannon JS, Hamzeh F, Moore S, Nicholas J, Ambinder RF. 1999. Human herpesvirus 8-encoded thymidine kinase and phosphotransferase homologues confer sensitivity to ganciclovir. J. Virol. 73:4786–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jia Q, Wu TT, Liao HI, Chernishof V, Sun R. 2004. Murine gammaherpesvirus 68 open reading frame 31 is required for viral replication. J. Virol. 78:6610–6620. 10.1128/JVI.78.12.6610-6620.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866–10871. 10.1073/pnas.95.18.10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu Y, Wang SE, Hayward GS. 2005. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22:59–70. 10.1016/j.immuni.2004.11.011 [DOI] [PubMed] [Google Scholar]
- 49.Ahmad H, Gubbels R, Ehlers E, Meyer F, Waterbury T, Lin R, Zhang L. 2011. Kaposi sarcoma-associated herpesvirus degrades cellular Toll-interleukin-1 receptor domain-containing adaptor-inducing beta-interferon (TRIF). J. Biol. Chem. 286:7865–7872. 10.1074/jbc.M110.191452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. 2003. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424:743–748. 10.1038/nature01889 [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. 2003. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science 301:640–643. 10.1126/science.1087262 [DOI] [PubMed] [Google Scholar]
- 52.Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ, Finberg RW. 2000. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 1:398–401. 10.1038/80833 [DOI] [PubMed] [Google Scholar]
- 53.Haynes LM, Moore DD, Kurt-Jones EA, Finberg RW, Anderson LJ, Tripp RA. 2001. Involvement of Toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 75:10730–10737. 10.1128/JVI.75.22.10730-10737.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. 2011. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 11:143–154. 10.1038/nri2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boehme KW, Guerrero M, Compton T. 2006. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J. Immunol. 177:7094–7102. 10.4049/jimmunol.177.10.7094 [DOI] [PubMed] [Google Scholar]
- 56.Brown HJ, Song MJ, Deng H, Wu TT, Cheng G, Sun R. 2003. NF-kappaB inhibits gammaherpesvirus lytic replication. J. Virol. 77:8532–8540. 10.1128/JVI.77.15.8532-8540.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]