

ABSTRACT
A hallmark cell response to influenza A virus (IAV) infections is the phosphorylation and activation of c-jun N-terminal kinase (JNK). However, so far it is not fully clear which molecules are involved in the activation of JNK upon IAV infection. Here, we report that the transfection of influenza viral-RNA induces JNK in a retinoic acid-inducible gene I (RIG-I)-dependent manner. However, neither RIG-I-like receptors nor MyD88-dependent Toll-like receptors were found to be involved in the activation of JNK upon IAV infection. Viral JNK activation may be blocked by addition of cycloheximide and heat shock protein inhibitors during infection, suggesting that the expression of an IAV-encoded protein is responsible for JNK activation. Indeed, the overexpression of nonstructural protein 1 (NS1) of certain IAV subtypes activated JNK, whereas those of some other subtypes failed to activate JNK. Site-directed mutagenesis experiments using NS1 of the IAV H7N7, H5N1, and H3N2 subtypes identified the amino acid residue phenylalanine (F) at position 103 to be decisive for JNK activation. Cleavage- and polyadenylation-specific factor 30 (CPSF30), whose binding to NS1 is stabilized by the amino acids F103 and M106, is not involved in JNK activation. Conclusively, subtype-specific sequence variations in the IAV NS1 protein result in subtype-specific differences in JNK signaling upon IAV infection.
IMPORTANCE Influenza A virus (IAV) infection leads to the activation or modulation of multiple signaling pathways. Here, we demonstrate for the first time that the c-jun N-terminal kinase (JNK), a long-known stress-activated mitogen-activated protein (MAP) kinase, is activated by RIG-I when cells are treated with IAV RNA. However, at the same time, nonstructural protein 1 (NS1) of IAV has an intrinsic JNK-activating property that is dependent on IAV subtype-specific amino acid variations around position 103. Our findings identify two different and independent pathways that result in the activation of JNK in the course of an IAV infection.
INTRODUCTION
Infection of cells with viruses leads to the activation of a variety of signaling cascades. Some of these signaling events represent a cellular response to fight the invading virus; others are virally induced and were found to support virus replication. The activation of c-jun N-terminal kinase (JNK), also known as stress-activated protein kinase (SAPK), along with that of other mitogen-activated protein (MAP) kinases, occurs during the course of many virus infections. This includes infection by Epstein-Barr virus (1), herpes simplex virus (2), reovirus (3), Kaposi's sarcoma virus (4), and influenza A virus (IAV) (5, 6). In the case of IAV, it has been shown that activation of JNK can exert virus-supportive and antiviral functions (7, 8). However, it is still unclear which molecular triggers mediate the phosphorylation and activation of JNK.
The detection of an invading virus by cellular receptors is required to trigger an effective antiviral innate immune response culminating in the upregulation of type I interferon (IFN). Cells express pattern recognition receptors (PRRs) that detect invariant molecular structures shared by pathogens of various origins (pathogen-associated molecular patterns, PAMPs) (9). Toll-like receptors (TLRs) 3, 7, 8, and 9, transmembrane proteins localized at the endosomal and cytoplasmic membranes, have been recognized as PRRs that sense distinct types of virus-derived nucleic acids and activate signaling cascades that result in the induction of type I IFNs (10, 11). MyD88 is a universal adaptor protein, as it is used by all known TLRs (except TLR 3) to activate downstream transcription factors such as NF-κB. Additionally, retinoic acid-inducible gene I (RIG-I)-like receptors have been identified as cytosolic sensors for intracellular viral RNAs containing triphosphate termini (12).
Specifically, RIG-I has been shown to be involved in IAV-mediated beta IFN (IFN-β) upregulation. RIG-I as well as Mda5 activates the antiviral response through associating with the recently identified adaptor protein MAVS (mitochondrial antiviral signaling protein, also known as IPS-1, VISA, or CARDIF), a CARD domain-containing protein that resides in the mitochondrial membrane and that is known to be essential for antiviral innate immunity (13, 14).
IFN-α/β is usually induced within hours after viral infection, a process that requires multiple regulatory and transcriptional factors. Critical transcription factors which have been shown to be involved in regulating IFN-β transcription include IRF-3, AP1, and NF-κB (15, 16). AP1 is activated by JNK. Since induction of IFN-α/β expression requires AP1 activation, the activation of JNK/AP1 has been considered part of the antiviral response (7). However, the inhibition of JNK using a chemical inhibitor resulted in decreased virus replication, suggesting that JNK also has a virus-supportive action (8).
Viral proteins interact with cellular signaling pathways that result in inhibition of antiviral, or stimulation of virus-supportive, mechanisms. In particular, nonstructural protein 1 (NS1) of IAV, a nonessential virulence factor, has multiple accessory functions during viral infection (17). The major role ascribed to NS1 has been its inhibition of host immune responses, especially the limitation of both IFN production and the antiviral effects of some IFN-induced proteins (18). NS1 from IAV strain A/Puerto Rico/8/34 (H1N1) (PR8) was shown to antagonize the double-stranded-RNA-induced activation of JNK (19). NS1 also modulates other important events during the viral replication cycle and in the general host-cell physiology (19, 20). Recently, it was observed that some avian IAV subtypes, such as A/fowl pox virus (FPV)/Bratislava/79 (H7N7) (FPV) and A/FPV/Rostock/34 (H7N1), show stronger JNK activation than human H1N1 subtypes (18, 21).
Here, we demonstrate that the detection of naked IAV RNA by RIG-I results in the activation of JNK but that neither the RIG-I/MAVS pathway nor the MyD88/TLR pathway is involved in JNK activation upon genuine IAV infection. Instead, the nonstructural protein NS1 activates JNK, depending on a subtype-specific sequence variation at amino acid position 103.
MATERIALS AND METHODS
(i) Plasmids, small interfering RNAs (siRNAs), transfections, and isolation of viral RNA.
Plasmids expressing Mda5 (pEF Bos Mda5wt and helicase/CARD deletion mutants) and RIG-I (pCAGGS Flag RIG-I wt, pCAGGS Flag RIG-IC, and pCAGGS Flag RIG-IN) were kindly provided by M. Gale and A. Garcia Sastre, respectively. The MAVS expression plasmids are described in the work of Tang and Wang (22). The expression of the proteins driven by the indicated plasmids was verified by Western blotting using antibody either against the corresponding Flag tag or against wild-type proteins.
The following siRNAs were purchased (Qiagen, Germany): siRNAs for the Mda5 target sequence (human IFIH1-7), CAGAACTGACATAAGAATCAA; RIG-I target sequence (human DDX58-10), TTCTACAGATTTGCTCTACTA; MAVS target sequence (human VISA-2 [MAVS]), TTGCTGAAGACAAGACCTATA; human VISA-1 (MAVS), TACTAGCATGGTGCTCACCAA; MyD88 (human MyD88-5), AACTGGAACAGACAAACTATC; human MyD88-10, CTTGATTGCCTTACAAAGTTA; human MyD88-2, AGGACCCTAAATCCAATAGAA; and human MyD88-11, CAGGACCAGCTGAGACTAAGA.
Transfections of cells with plasmids and siRNA were performed using Lipofectamine 2000 (Invitrogen, Germany) according to the manufacturer's protocol. Viral and cellular RNAs were transfected into A549 or HEK293 cells using DOTAP {N-[1-(2,3-dioleoyloxy)]-N,N,N trimethylammonium propane methyl sulfate} according to the manufacturer's protocol (Roth GmbH, Germany).
Viral genomic RNA was isolated from cells which were infected with the avian influenza virus strain A/FPV/Bratislava/79. One or 2 days postinfection, cell supernatants were collected and centrifuged at 2,500 × g to remove cell debris. The resulting supernatant was centrifuged at 100,000 × g for 2 h. Subsequently, the viral pellet was resuspended. RNA was purified either with TRIzol (Invitrogen, Germany) or with Qiagen RNeasy columns according to the manufacturer's protocol.
(ii) Viruses, cells, and viral infections.
The highly pathogenic avian influenza viruses A/FPV/Bratislava/79 (H7N7), A/FPV/Rostock/34 (H7N1), and SC35M (H7N7) or the human subtypes A/Wilson Smith N (WSN)/1933 (H1N1) (WSN), A/Puerto Rico/8/34 (H1N1) (PR8), A/Panama/2007/99 (H3N2), A/Wisconsin/67/2005 (H3N2), and A/Hamburg/4/09 (H1N1v) were used for infection of human lung epithelial (A549) cells and human embryonic kidney (HEK293) cells. Reassortant viruses were generated according to the method of Mostafa et al. and Hoffmann et al. (23, 24).
To inactivate virus particles, the virus-containing supernatant was spread on a 15-cm petri dish and exposed to UV light (254-nm wavelength, 500-mJ/cm2 Bio-Link BLX 254 irradiation system; Labortechnik GmbH, Germany) at room temperature. The cover of the petri dish was removed when samples were exposed to the UV light, and the petri dish was placed on a beaker to reduce the distance to the UV lamp to 3 cm. The infectivity of the irradiated virus was tested immediately by inoculation of Madin-Darby canine kidney (MDCK) cells.
Cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% heat-inactivated fetal calf serum (FCS). Cells were washed with phosphate-buffered saline (PBS) and infected at the multiplicity of infection of 5 (MOI=5) in PBS-BA (PBS containing 0.2% bovine serum albumin [BSA], 1 mM MgCl2, 0.9 mM CaCl2, 100 U/ml penicillin, 0.1 mg/ml streptomycin) for 30 min at 37°C. The inoculum was aspirated, and cells were incubated with DMEM-BA (medium containing 0.2% BSA and antibiotics). To assess the number of infectious progeny virus particles (PFU) in cell culture supernatants, MDCK cells were used for titration of virus-containing samples.
DF-1 is a continuously growing cell line of chicken embryo fibroblasts (25). DF-1 cells were propagated in DMEM supplemented with FCS (10%), nonessential amino acids (1%), glutamine (1%), and pyruvate (1%).
Mda5- and MyD88-deficient (−/−) mouse embryonal fibroblasts (MEFs) were generated from 12.5-day-old embryos of the corresponding gene-deficient mouse according to standard procedures. Mice were maintained in individually ventilated cages according to animal health regulations approved by the Bezirksregierung Münster, Germany (Az 8.87-50.10.36.09.007). Immortalized MEFs deficient for RIG-I (RIG-I−/−) and immortalized MAVS−/− MEFs were provided by W. Barchet, Bonn, Germany, and Z. J. Chen, Dallas, TX, USA, respectively.
(iii) Western blots.
For Western blotting, cells were lysed on ice with radioimmunoprecipitation assay lysis buffer (25 mM Tris, pH 8.0, 137 mM NaCl, 10% glycerol, 0.1% sodium dodecyl sulfate, 0.5% natrium deoxycholate, 1% NP-40, 2 mM EDTA, pH 8.0, 5 mg/ml leupeptin, 0.2 mM Pefablock, 5 µg/ml aprotinin, 1 mM sodium vanadate, and 5 mM benzamidine) for 30 to 60 min. Lysates were adjusted with respect to protein concentration and subjected to denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequent Western blotting. Phosphorylated JNK was detected using an activation state-specific anti phospho-JNK (pT183/pY185) antibody (BD Biosciences) which recognizes the activated phosphorylated forms of JNK1 and JNK2. Loading controls were performed with a pan-ERK2 antibody (Santa Cruz Biotechnology). Commercially available antibodies were used to detect Mda5, RIG-I, MyD88 (all from Enzo Life Sciences), MAVS (Bethyl Laboratories), IAV M1 protein (AbD Serotec), and JNK (cell signaling). Monoclonal NS1 (clone NS1-23-1; developed at the IMV, Münster, Germany) antibody was used to detect IAV NS1. Protein bands were visualized using a standard enhanced-chemiluminescence reaction and horseradish peroxidase-coupled secondary antibodies. Western blotting was performed at least three times, and representative blots are shown in the figures.
(iv) Immunoprecipitation.
For immunoprecipitation, cells were lysed on ice with Triton lysis buffer (TLB; 20 mM Tris-HCl, pH 7.4, 137 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA, 50 mM sodium glycerophosphate, 20 mM sodium pyrophosphate, 5 mg ml−1 leupeptin, 5 mg ml−1 aprotinin, 0.2 mM Pefabloc, 1 mM sodium vanadate, and 5 mM benzamidine) for 30 min. Cell lysates were processed as described above. For immunoprecipitation input controls, lysates were directly subjected to SDS-PAGE and to Western blotting. Anti-myc antibody was coupled to protein A or G agarose (Roche, Germany) to precipitate myc-tagged NS1. Anti-flag antibody M2 (Sigma, Germany) was used to detect Flag-tagged CPSF30.
(v) Site-directed mutagenesis.
Site-directed mutagenesis was performed using Phusion polymerase (Thermo Scientific, Germany) and phosphorylated primers according to the manufacturer's protocol. Primers were purchased from MWG Biotech, Germany.
All experiments with infectious virus were performed according to German regulations. Experiments involving biosafety level 3 (BSL3) highly pathogenic avian influenza A viruses were performed in a BSL3 containment laboratory approved by the local authorities (RP, Giessen, Germany).
RESULTS
(i) Overexpression of RIG-I, Mda5, and MAVS induces JNK activation.
IAV infection of cells induces JNK phosphorylation within 4 h after infection (Fig. 1A). In the same time course, the intracellular RNA sensor RIG-I mediates the early phase of type I interferon (IFN) induction upon infection (11–13). Using A/Puerto Rico/8/34, we earlier demonstrated that accumulation of viral RNA leads to an activation of JNK (6). This prompted us to analyze whether RIG-I-like receptors are able to induce JNK activity. RIG-I as well as Mda5 is a so-called CARD domain protein. The CARD domain is regarded as the signal-transmitting domain, which interacts with the corresponding CARD domain of the MAVS protein, whereas the helicase domain is thought to represent the RNA-binding domain (11). In A549 cells, overexpression of wild-type RIG-I and Mda5 as well as of the respective CARD domains resulted in a pronounced JNK phosphorylation, while the helicase domains had no effect (Fig. 1B, lanes 1 to 6). Additionally, transfection with a MAVS-overexpressing plasmid also led to JNK phosphorylation in both cell lines. JNK phosphorylation induced by MAVS overexpression was dependent on the mitochondrial localization of MAVS, since deletion of the MAVS transmembrane domain prevented the activation of JNK (Fig. 1B, 9th lane). These results confirm the hypothesis that RIG-I induces JNK upon IAV infection via viral-RNA detection.
FIG 1.
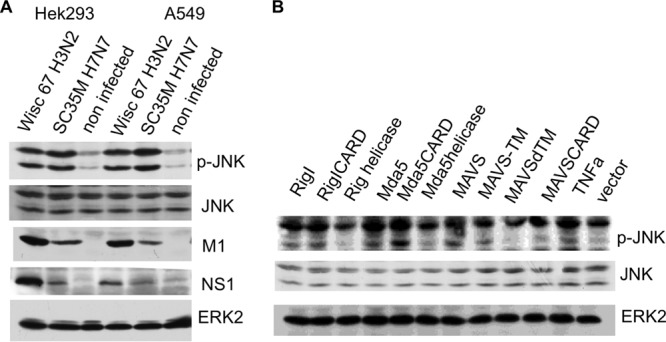
JNK is activated upon IAV infection and by overexpression of RIG-I-like receptor. (A) A549 and HEK293 cells were infected with the human IAV strain A/Wisconsin (Wisc)/67/2005 (H3N2) and avian IAV SC35M (H7N7) at an MOI of 5. After 4 h, cells were harvested and lysates were subjected to an SDS-PAGE and subsequently analyzed by Western blotting with antibodies against the phosphorylated active form of JNK1 and JNK2 (p-JNK), JNK, NS1, and M1. ERK2 served as a loading control. (B) Cells were transfected with expression plasmids as indicated. Cell lysates were harvested 24 h after transfection and analyzed by Western blotting with an anti-phospho-JNK antibody (p-JNK); ERK2 blots served as a loading control. RIG-I, Mda5, and MAVS, plasmids expressing wild-type proteins; RIG-I CARD, RIG-I CARD domain; RIG-I helicase, RIG-I helicase domain; MAVS-TM, MAVS aa 1 to 510 with the transmembrane domain of bcl-xl added; MAVSdTM, MAVS aa 1 to 510 with the transmembrane domain deleted; MAVS CARD, MAVS aa 1 to 98; TNFa, tumor necrosis factor alpha-treated cells.
(ii) Transfection of viral RNA into cells leads to RIG-I-dependent JNK phosphorylation.
RIG-I is known to induce the antiviral response via the recognition of the 5′ triphosphate termini of viral RNA. Additionally, the 3′ untranslated regions of influenza genomic sequences were shown to be 5′ppp-independent ligands for RIG-I (26). To investigate the role of RIG-I in IAV-mediated JNK activation, we suppressed the expression of RIG-I and Mda5 via siRNA and subsequently transfected IAV genomic RNA into cells. Figure 2 demonstrates that JNK is activated by transfection of viral RNA and that downregulation of RIG-I expression, but not of Mda5 expression, inhibits JNK activation by viral-RNA transfection (Fig. 2).
FIG 2.
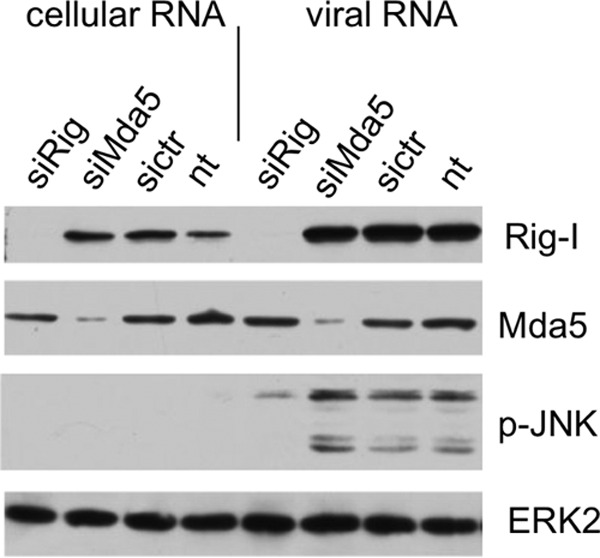
The activation of JNK upon viral-RNA transfection is RIG-I dependent. A549 cells were transfected with siRNA targeting RIG-I (siRig) or Mda5 (siMda5). Subsequently, the cells were transfected with viral RNA or cellular RNA, harvested after 4 h, and analyzed by Western blotting. sictr, control siRNA; nt, not transfected with siRNA.
(iii) IAV infection-induced JNK activation is independent of RIG-I, Mda5, MAVS, and MyD88.
Next we investigated whether RIG-I provokes JNK activation not only by overexpression or viral-RNA transfection but also during a genuine IAV infection. We therefore transfected cells with siRNA targeting RIG-I and control siRNA and subsequently infected these cells with IAV FPV (H7N7) or left them uninfected. When we finally assayed for JNK activation, we surprisingly detected a RIG-I-independent activation of JNK (Fig. 3A), illustrating that RIG-I is dispensable for JNK activation during IAV infection. Thus, JNK is activated primarily in a RIG-I-independent manner, while the viral-RNA sensory pathway does not seem to play a major role. To further investigate whether other known PRR receptor pathways are involved in IAV-induced JNK activation, we suppressed expression of RIG-I, Mda5, MAVS, and MyD88 and subsequently infected these cells with IAV FPV (H7N7). As shown in Fig. 3A and B, virus-induced activation of JNK was not inhibited when the expression of RIG-I, Mda5, MAVS, or MyD88 was knocked down. Moreover, RIG-I-, Mda5 (or MAVS)-, or MyD88-deficient mouse embryonal fibroblasts (MEFs) were infected with IAV and subsequently analyzed for JNK activation. Again, genetic deficiency of neither RIG-I, Mda5, MAVS, nor MyD88 impaired the activation of JNK upon IAV infection, indicating that the respective PRR pathways are not essential for IAV-induced JNK phosphorylation (Fig. 3C).
FIG 3.

IAV infection-mediated JNK activation is independent of RIG-I like receptors and of MyD88-dependent TLRs. (A) A549 cells were transfected with siRNA targeting RIG-I, Mda5, and MAVS, as indicated. Cells were infected at an MOI of 5 with A/FPV/Bratislava/79 (FPV [H7N7]) 48 h after siRNA transfection. Subsequently, cell lysates were harvested 4 h postinfection and analyzed by Western blotting. p-JNK, Western blot with an anti phospho-JNK antibody; ERK2, control blot using anti-ERK2 antibody; Mda5, RIG-I, and MAVS, Western blots using antibodies specific to Mda5, RIG-I, and MAVS, respectively; nt, nontreated; C, control siRNA; 1 and 2, two different siRNAs targeting MAVS (MAVS siRNA 2 was not effective in suppressing MAVS expression); 1+2, MAVS siRNA 1 and 2 mixed. (B) Identical experiments were conducted using four different MyD88 siRNAs. nt, nontreated; C, control siRNA. (C) To further exclude any role of these genes in IAV-mediated JNK activation, MEFs deficient (−/−) for RIG-I, Mda5, MAVS, or MyD88 and the corresponding wild-type MEFs (+/+) were either infected with FPV/Bratislava/79 (FPV) (MOI of 5) or left uninfected. Cell lysates were harvested 4 h postinfection and analyzed by Western blotting using an anti-phospho-JNK antibody; ERK2 blots served as a loading control (ERK2). (D) Chicken DF-1 cells are reported to be devoid of a functional RIG-I (27). (A) DF-1 cells were infected with A/FPV/Bratislava/79 (FPV [H7N7]) and analyzed by Western blotting using anti-phospho-JNK antibodies. As a positive control, DF-1 cells were incubated with anisomycin (aniso), which is a potent JNK activator. As indicated, the anisomycin-positive control experiment and the infection experiment were performed in duplicate.
Recently, it was shown that chickens are devoid of functional RIG-I, providing a plausible explanation for their increased susceptibility to influenza viruses compared to that of ducks (27). Because of this RIG-I deficiency, we infected chicken DF-1 cells with FPV (H7N7). Figure 3D shows that JNK was again readily phosphorylated in DF-1 cells. As a control for whether JNK can be activated and detected in these cells, we used anisomycin as a strong JNK-activating compound. Our data suggested that neither RIG-I -like receptors nor MyD88-dependent TLRs are involved in IAV-mediated JNK activation.
(iv) Functional protein synthesis is essential for IAV infection-mediated JNK activation.
Infection of cells with UV-inactivated IAV did not provoke phosphorylation of JNK (Fig. 4A), suggesting that ongoing viral replication is essential for JNK activation. Next, we tried to interfere with virus-induced JNK activation by blocking diverse steps in the virus life cycle. Cells were infected with IAV FPV (H7N7) and subsequently incubated with diverse inhibitors. In essence, all inhibitors that block viral protein synthesis or act prior to this step also inhibit JNK phosphorylation. Specifically, bafilomycin and cycloheximide prevented JNK activation and viral protein synthesis (Fig. 4C). Bafilomycin is an inhibitor of vacuolar-type H+-ATPases and thus inhibits the acidification of lysosomes, resulting in an abortive infection (28), whereas cycloheximide directly blocks protein synthesis. The inhibitory potential of cycloheximide was evident only in the early phase of IAV infection. As soon as viral protein synthesis was initiated, JNK became increasingly phosphorylated (Fig. 4D). Geldanamycin and 17-AAG, both inhibitors of heat shock protein 90 (hsp90), were also able to prevent JNK phosphorylation (Fig. 4B), although the inhibitor 17-AAG does not completely block viral protein synthesis. Viral proteins, including those of influenza virus, have been shown to require hsp90 for their correct folding, assembly, and maturation (29, 30), and thus hsp90 inhibitors putatively prevent correct folding of viral polypeptides.
FIG 4.
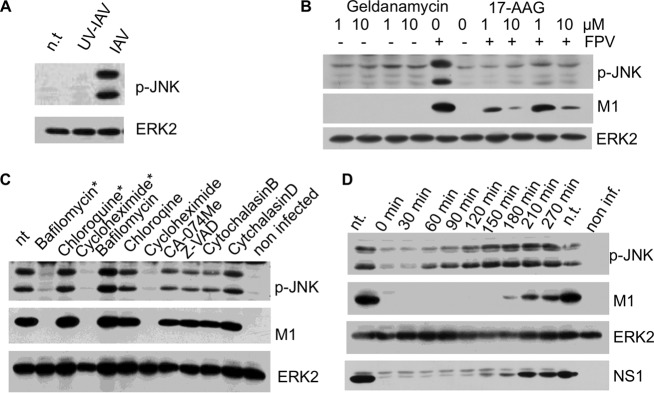
The expression of functional viral proteins is needed for IAV-induced JNK activation. (A) A/FPV/Bratislava/79 (H7N7) (IAV) was inactivated by UV light. Cells were infected with native and UV-treated IAV and analyzed by Western blotting. (B) Cells were infected with A/FPV/Bratislava/79 (FPV [H7N7]), subsequently incubated with inhibitors of the heat shock protein hsp90 (geldanamycin, 17-AAG), and analyzed via Western blotting. (C) Cells were incubated with FPV/Bratislava/79 (H7N7) for 30 min, and subsequently inhibitors were added as indicated. After 4 h, cells were harvested and analyzed via Western blotting. nt, nontreated. * indicates samples that were additionally treated with the indicated inhibitor 20 min before infection. (D) Cells were infected with FPV/Bratislava/79 (H7N7) (MOI of 5) by incubating them for 30 min with the virus-containing supernatant. After removal of the supernatant, cycloheximide was added at the indicated time points postinfection and harvested 270 min postinfection. nt, nontreated; non inf., noninfected.
Together, these data let us speculate that expression of properly folded functional viral proteins is needed for infection-induced RIG-I-independent JNK activation.
(v) IAV NS1 proteins of specific subtypes induce JNK activation.
To identify viral proteins whose expression may induce JNK activity, we transfected expression vectors encoding viral proteins of IAV (H7N7) into cells. We observed a pronounced JNK activation only in cells expressing the NS1 protein of this strain. However, comparison of NS segments (Fig. 5A) and NS1 proteins (Fig. 5B) of different viral origins revealed that subtype-specific differences determine the ability to activate JNK. NS1 of A/FPV/Bratislava (H7N7) and NS1 of A/FPV/Rostock (H7N1), as well as NS1s of the Vietnam (H5N1), Thailand (H5N1), and SC35M (H7N7) strains, were able to activate JNK, whereas the NS1 of PR8 (H1N1) and of WSN (H1N1) did not. Infection of cells with the corresponding IAVs confirmed these results. In addition, NS2 and NS1 of influenza B virus were not able to activate JNK.
FIG 5.
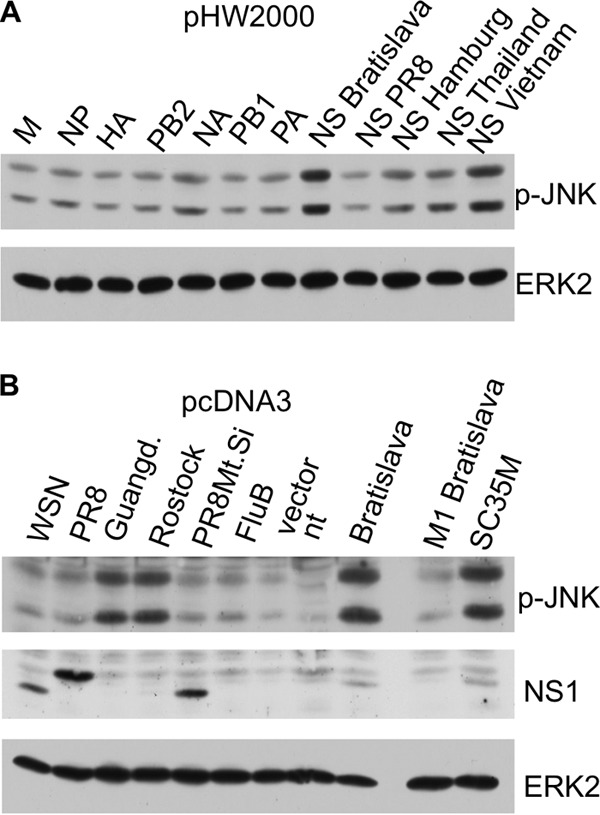
IAV NS1 expression induces JNK activation. (A) pHW2000- and pMPccdB-based plasmids encoding the eight genomic segments of A/FPV/Bratislava/79 were transfected into A549 cells. Additionally, the pHW2000 plasmids containing NS segments of A/Puerto Rico/8/34 (H1N1) (PR8), A/Hamburg/4/09 (H1N1) (Hamburg), A/Thailand/1(Kan-1)/04 (H5N1) (Thailand), and A/Vietnam/1203/04 (H5N1) (Vietnam) were transfected into cells. After 24 h, cells were harvested and analyzed by Western blotting. M, matrix protein; NP, nuclear protein; HA, hemagglutinin; PB2, polymerase B2; NA, neuraminidase; PB1, polymerase B1; PA, polymerase A. (B) pcDNA3 plasmids expressing NS1s from IAVs of various subtypes were transfected into cells, and phosphorylation of JNK was analyzed by Western blotting. In the cases of the Guangdong and Rostock viruses, either the monoclonal NS1 antibody could not detect the subtype-specific NS1 protein or expression was too low to be detected. Guangd., NS1 from A/Goose/Guangdong/3/97 (H5N1); Rostock, A/FPV/Rostock/34 (H7N1); WSN, A/WSN/1933 (H1N1); PR8, A/Puerto Rico/8/34 (H1N1); FluB, influenza B virus; PR8Mt.Si, A/Puerto Rico/8/34/Mount Sinai (H1N1); Bratislava, NS1 from FPV/Bratislava/79 (H7N7); M1 Bratislava, M1 from FPV/Bratislava/79 (H7N7); SC35M, SC35M (H7N7).
Next, we generated reassortant IAVs by reverse genetics to verify the divergent potentials of different NS1 proteins to activate JNK. This also served to verify whether the JNK-activating property is independent of the viral background. FPV/Rostock (H7N7) containing an NS segment of PR8 (H1N1) was unable to activate JNK upon infection, whereas infection with WSN (H1N1) containing an NS segment from FPV/Rostock (H7N1) or FPV/Bratislava (H7N7) readily leads to activation of JNK (Fig. 6A).
FIG 6.
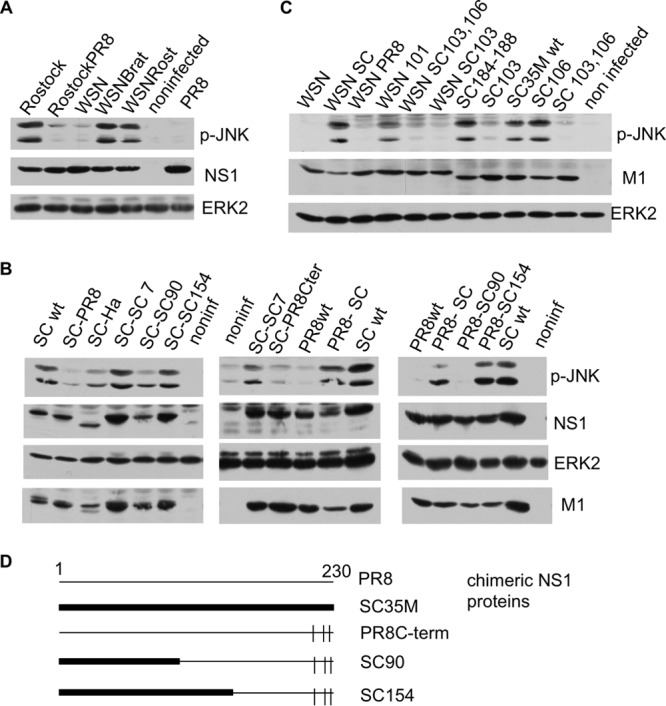
JNK activation depends on amino acid position 103 of the SCM35M NS1 protein. (A) Cells were infected with IAV A/FPV/Rostock/34 (Rostock), A/WSN/1933 (WSN), and A/Puerto Rico/8/34 (PR8) and with reassortant viruses, such as A/FPV/Rostock/34 containing an NS segment from A/Puerto Rico/8 (RostockPR8) and IAV WSN/1933 containing an NS segment from A/FPV/Bratislava/79 (WSNBrat) or from A/FPV/Rostock/34 (WSNRost). Subsequently, cell lysates were analyzed by Western blotting as described in the text. (B) Cells were infected with recombinant IAVs for 4 h and analyzed by Western blotting. SC wt, wild-type SC35M; SC-PR8, SC35M containing the NS segment from A/Puerto Rico/8; SC-Ha, SC35M containing the NS segment from A/Hamburg/4/09; SC-SC 7, SC35M with an SC35M NS segment mutated at seven amino acid positions into the corresponding amino acids of PR8, namely, S3P, F22V, I81M, S114P, P215T, R225G, and E228R; SC-SC90, SC35M containing a chimeric NS segment including amino acids 1 to 90 of SC35M, followed by part of A/Puerto Rico/8, and with the C-terminal amino acid positions 215, 224, and 227 mutated into the corresponding amino acids of SC35M; SC-SC154, SC35M containing a chimeric NS segment that is the same as SC90 except that amino acids 1 to 154 are those encoded by SC35M; PR8wt, IAV Puerto Rico/8/34 containing the NS segment of SC35M; SC-PR8Cter, SC35M IAV containing the NS segment of PR8 mutated at amino acid positions 215, 224, and 227 into the corresponding amino acids of SC35M; PR8-SC, PR8 containing the NS segment of SC35M; PR8-SC90, PR8 with the NS segment of SC90; PR8-SC154, PR8 with the NS segment of SC154. (Right) Schematic representation of chimeric NS SC90 and SC154 segments (see panel D). (C) Cells were infected with recombinant IAVs and analyzed as described above. WSN, A/WSN/1933; WSN SC, WSN with the NS segment of SC35M; WSN PR8, WSN with the NS segment of PR8; WSN 101, WSN with an NS segment containing H101D; WSN SC103, 106, WSN with SC35M NS F103S M106I; WSN SC103, WSN with SC35M NS1 F103S; SC184-188, SC35M with NS with a mutation of GLEWN at positions 184 to 188 of RFKRY; SC103, SC35M with NS F103S; SC106, SC35M with NS M106I; SC103, 106, SC35M with NS F103S M106I. (D) Schematic representation of the chimeric NS1 proteins described for panel B.
Comparing the amino acid sequences of JNK-activating versus non-JNK-activating NS1 proteins, we initially identified 7 amino acids (aa) (positions 3, 22, 81, 114, 215, 225, and 228) as possible candidates responsible for JNK activation (Table 1). Mutation of the three amino acids at positions 215, 224, and 227 of the C terminus of PR8 (H1N1) NS1 into the corresponding amino acids of SC35M (H7N7) NS1 failed to convert the NS1 of PR8 (H1N1) into a JNK-activating NS segment. Similarly, all other mutations of single and multiple amino acids at positions 3, 22, 81, 114, 215, 225, and/or 228 failed to convert the activating SC35M NS segment into a nonactivating one (Fig. 6C). We finally constructed chimeric PR8/SC35M NS segments encoding terminal aa 215 to 230 and aa 1 to 154 (SC154) or aa 1 to 90 (SC90) by using SC35M and the middle part of PR8. Viral infection of cells with recombinant SC35M encoding the SC154 NS1 strongly activated JNK, while SC90 NS1-encoding SC35M did not (Fig. 6B), suggesting that the region between positions 90 and 154 in NS1 mediates JNK activation.
TABLE 1.
Comparison of the amino acid sequences ofthe NS1 proteins of JNK-activating and nonactivating IAV subtypesa

Alignment of amino acid sequences of NS1s of two JNK-activating viruses, IAV SC35M (H7/N7) (SC35M) and A/FPV/Rostock/34 (H7N1) (Rostock), and two nonactivating viruses, A/Puerto Rico/8/34 (H1N1) (PR8) and A/WSN/1933 (H1N1) (WSN). Positions that have been mutagenized are marked. Seven amino acids (positions 3, 22, 81, 114, 215, 225, and 228) were initially identified but were experimentally disproved as being involved in JNK activation. Below the amino acid sequences, the amino acids around position 103 of the NS1 proteins of all IAVs used in this study are shown. Wisco, Wisconsin.
(vi) Phenylalanine at position 103 of SC35M NS1 is decisive for JNK activation.
Since position 114 had been mutagenized before and was shown not to be involved in JNK activation (see above), we envisaged positions F103 and M106 in this capacity, although the nonactivating NS1 of WSN encodes amino acids at these positions identical to those of the activating NS1 proteins of the avian subtypes. Indeed, SC35M NS1 with the double mutation F103S M106I failed to activate JNK, as did the single mutant SC35M NS1 F103S. However, SC35M NS1 M106I was still able to activate JNK, suggesting that amino acid position 103 is primarily decisive for JNK activation by SC35M NS1 (Fig. 6C).
We then asked why the NS1 of WSN, which encodes amino acids identical to those of SC35M at positions 103 (F) and 106 (M), does not activate JNK. We speculated that the histidine (H) at position 101 (instead of aspartic acid [D]) in the direct neighborhood of aa 103 (Table 1) might distort the structure of the motif in such a way that JNK can no longer be activated. Indeed, mutation of this neighboring H at position 101 into D converts the NS1 of WSN from a nonactivating into a JNK-activating NS1 (Fig. 6C).
To demonstrate that our finding concerning the importance of F at position 103 is not restricted to SC35M, we mutated NS1 of avian influenza virus A/Thailand/1(KAN-1)/2004 (H5N1) at position 98 (which corresponds to position 103 of SC35M because of a 5-aa deletion [Table 1]). A/Thailand/1 (KAN-1) NS1 activates JNK in both an avian and a human background, whereas the A/Thailand/1 (KAN-1) NS1 F98S mutant fails to activate JNK in both backgrounds.
In other naturally occurring, mainly avian, IAVs, position 103 of NS1 encodes L or Y instead of F or S. Appropriate SC35M mutants showed that L and Y at position 103 in SC35M, in contrast to amino acid S, do not significantly attenuate JNK activation (Fig. 7A).
FIG 7.
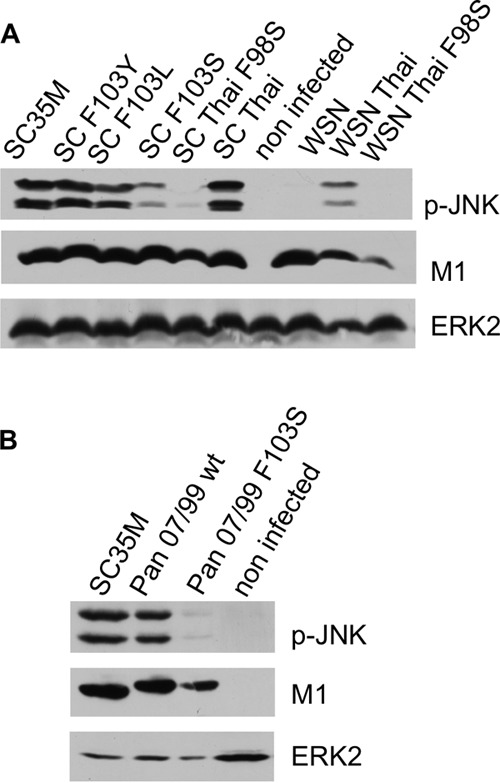
Serine at position 103 of NS1 is not compatible with JNK activation. Cells were infected with recombinant IAVs for 4 h and analyzed by Western blotting. (A) SC35M, wild-type SC35M; SC F103Y, SC35M encoding NS1 F103Y; SCF103L, SC35M encoding NS1 F103L; SC F103S, SC35M encoding NS1 F103S; SC35 Thai, SC35M encoding the NS segment of A/Thailand/1 (KAN-1); SC Thai F98S, SC35M encoding the NS segment of A/Thailand/1 (Kan-1) with F98S; WSN, A/WSN/1933; WSN Thai, A/WSN/1933 encoding the NS segment of Thailand/1 (KAN-1); WSN Thai F98S, A/WSN/1933 encoding the NS segment of Thailand/1 (KAN-1) with F98S. (B) Pan 07/99 wt, wild-type A/Panama 2007/99; Pan 07/99 F103S, A/Panama 2007/99 encoding the NS1 F103S mutant.
To verify that our finding holds true with respect to human IAVs other than PR8 and WSN, which encode an F at position 103 of NS1, we mutated NS1 of human IAV Panama/2007/99 (H3N2). Whereas wild-type IAV Panama 2007 activates JNK as SC35M does, the mutated virus encoding NS1 F103S fails to activate JNK (Fig. 7B).
(vii) CPSF30 is not involved in NS1-induced JNK activation.
The amino acids F103 and M106 are known to stabilize the binding of NS1 to cleavage and polyadenylation factor 30 (CPSF30) (31). Binding of the NS1 protein to CPSF30 prevents CPSF binding to the RNA substrate and inhibits 3′-end cleavage and polyadenylation of host pre-mRNAs. Thus, the NS1 protein selectively inhibits the nuclear export of cellular, but not viral, mRNAs (31, 32). We asked whether the activation of JNK relies on the inhibition of cellular RNA processing by the binding of NS1 to CPSF30. Immunoprecipitation experiments confirmed that the double mutant SC35M NS1 F103S M106I and the single mutant NS1 F103S do not bind or bind much more weakly to CPSF30 than wild-type NS1 or the C-terminal effector domain of SC35M NS1 (Fig. 8). A quadruple mutant, NS1 with GLEWN at positions 184 to 188 changed to RFKRY, destroyed the main NS1-CPSF30 binding motif of NS1 and was completely unable to interact with CPSF30 (Fig. 8) (31–33); it was used to generate a recombinant IAV SC35M virus. This recombinant virus containing NS1 with the mutations at positions 184 to 188 activates JNK (Fig. 6C), indicating that activation of JNK is independent of CPSF30 binding.
FIG 8.
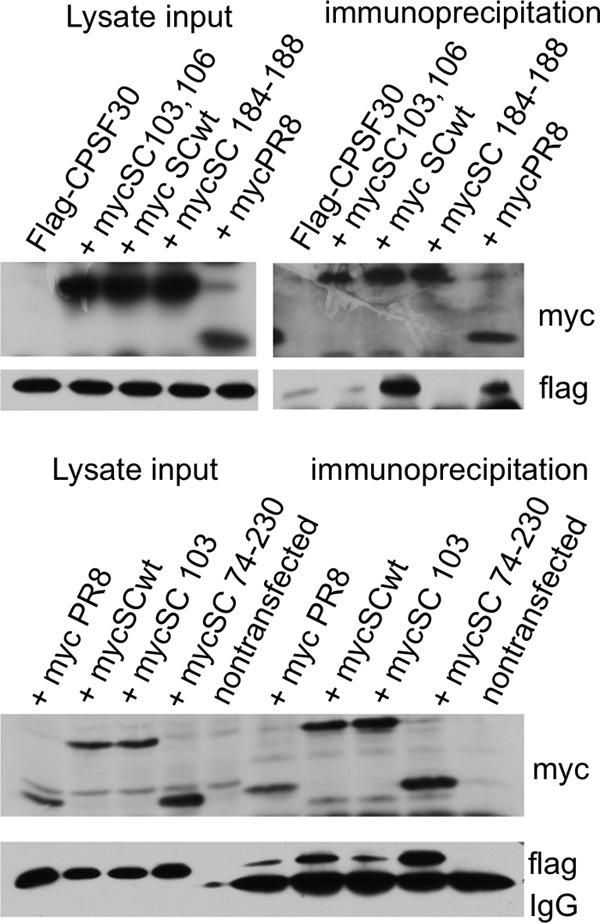
Cells were transfected with Flag-tagged CPSF30 and various myc-tagged NS1 expression constructs. Lysates were immunoprecipitated using a monoclonal anti-myc antibody. Flag-CPSF30, Flag-tagged CPSF30; mycSC103, 106, 6×-myc-tagged SC35M NS1 F103S M106I; myc SCwt, 6×-myc-tagged SC35M NS1; mycSC 184-188, 6×-myc-tagged SC35M NS1 with a mutation of GLEWN at positions 184 to 188 to RFKRY; mycPR8, 1×-myc-tagged PR8 NS1; mycSC103, 6×-myc-tagged SC35M NS1 F103S; mycSC 74-230, 6×-myc-tagged NS1 with amino acid positions 74 to 230 (effector domain).
DISCUSSION
The PRR RIG-I is indispensable for the type I IFN response to many single-stranded RNA viruses. These include negative-stranded viruses of the orthomyxovirus (such as IAV) and paramyxovirus (such as measles, mumps, and Sendai virus) families and positive-stranded viruses like hepatitis C or Japanese encephalitis virus (34). Additionally, we earlier observed that the accumulation of viral RNA triggers the activation of JNK (7). Thus, we initially regarded RIG-I as the prime candidate for serving as the sensor for the signaling cascade that results in IAV-induced JNK activation. Indeed, our assumption was affirmed by the observation that overexpressed RIG-I, Mda5, and MAVS activate JNK. This finding proves a link between the JNK cascade and the RIG-I/MAVS pathway. JNK activation via MAVS overexpression has also been shown earlier (13). Here we could further demonstrate that the activation of JNK by viral RNA is RIG-I dependent, suggesting that in this activation scenario, JNK is located downstream of RIG-I.
Expression of the wild-type RIG-I and Mda5 proteins as well as their CARD domains results in activation of JNK. MAVS activates JNK only when it is associated with the mitochondrial membrane. Thus, the ability of the RIG-I, Mda5, MAVS, and their deletion mutants to phosphorylate JNK correlates well with their potential to activate the IRF3- and IFN-β promoters. Upregulation of IFN and JNK phosphorylation occur concomitantly within 2 to 4 h after IAV infection. Both observations suggest that JNK activation may be part of the antiviral signaling response.
Although we could demonstrate that viral RNA activates JNK via RIG-I, we here provide abundant experimental evidence that JNK activation upon a genuine IAV infection occurs independently of the RIG-I/Mda5/MAVS pathway, suggesting that viral-RNA-mediated JNK activation does not substantially contribute to JNK activity induced by IAV. We could also exclude the possibility that MyD88-dependent Toll-like receptors (TLRs) play a role in IAV-mediated JNK activation (Fig. 3). Thus, although viral RNA results in RIG-I-mediated JNK activation, in the context of IAV infection, JNK is activated by a mechanism that is dominant over and independent of the RIG-I pathway.
In search of the JNK-activating factor, we observed that the inhibition of protein synthesis directly after IAV infection prevents JNK activation. Moreover, inhibitors of the chaperone hsp90 achieved the inhibition of JNK by activation. hsp90 is known to be nearly universally required for viral protein homeostasis. Numerous viral proteins have been shown to require hsp90 for their folding, assembly, and maturation (29, 30), suggesting that a functional viral protein is responsible for IAV-mediated JNK activation. Indeed, we could show that NS1 overexpression is sufficient for JNK activation. NS1 is a multifunctional protein that performs a plethora of activities which contribute toward efficient virus replication and virulence during infection (17). Earlier we reported that the IAV NS1 protein inhibits activation of Jun N-terminal kinase presumably by antagonizing viral-RNA-induced activation. These data were based upon experiments using the NS1 protein from IAV/Puerto Rico/8/34 (H1N1) (7). Our data shown here confirmed that JNK is indeed only weakly activated by IAVs containing an NS segment from PR8 and WSN, both representing the H1N1 subtype. In these cases, solely the viral-RNA/RIG-I pathway possibly activates JNK, and thus JNK activation is indirectly counteracted by the ability of NS1 to inhibit RIG-I. However, NS segments from other IAV subtypes can strongly activate JNK. Hence, the origin of the NS1 protein plays a decisive role. Mutational analysis revealed that the amino acid phenylalanine (F) at position 103 is involved and essential for the activation of JNK. The importance of F103 could be verified in avian Thailand/1 (KAN-1) (H5N1) NS1 and in human Panama 2007/99 (H3N2) NS1. Similarly, the nonactivating NS1 protein from WSN could be converted into a JNK-activating NS protein by mutating a single amino acid at position 101 (H101D), possibly because the histidine at position 101 structurally prevents the F103 from exerting its JNK-activating function. Surprisingly, we were unable to convert NS1 of PR8 into a JNK-activating NS1 protein even in an avian background, suggesting that position 103 is essential for JNK activation but may not be sufficient.
The different modes of JNK activation in the course of an IAV infection remind us of the IAV-dependent activation of other signaling molecules, such as phosphoinositol 3-kinase (PI3K) (35, 36), which results in virus-supporting as well as antiviral effects. PI3K is first activated due to virion attachment, while activation via pathogen recognition receptor-mediated PI3K signaling may contribute to host innate immune defenses (37). In addition, the viral NS1 protein directly binds and activates PI3K to enhance efficient virus replication. Moreover, NS1-mediated activation of PI3K depends on the IAV subtype, similar to NS1-mediated JNK activation (38).
Position 103 of the NS1 protein is known to stabilize the binding of CPSF30 to NS1 (31–33). We confirmed that changing F103 results in a strong reduction of CPSF30 binding to NS1. The cellular 30-kDa subunit of the cleavage and polyadenylation specificity factor (CPSF30) is required for the 3′-end processing of cellular pre-mRNAs. Binding of NS1 thus inhibits the production of all cellular mRNAs, including IFN mRNAs. We speculated that JNK activation might be a consequence of CPSF30-NS1 interaction and the subsequent shutdown of cellular mRNA processing. Our experiments disproved this hypothesis in that mutational analysis showed that JNK activation via NS1 F103 is independent of CPSF30 binding. In view of our data, it might be necessary to think over reports investigating functions of amino acid position 103 of IAV NS1, including those that deal with CPSF30, because each mutation changing the amino acid at position 103 potentially affects both JNK activation and CPSF30 binding.
It is known that NS1 proteins of different IAV subtypes may alter replication efficiency, cell tropism, and host range in a given IAV background (39–43). However, the underlying molecular mechanisms often remain obscure. In accordance with our study, virus-specific differences in host cell signaling, including differential JNK activation, have recently been observed (44). Moreover, NS1 proteins of two avian IAVs differentially inhibit the double-stranded-RNA-induced activation of the AP1 promoter (45). The differential activation of JNK may contribute to some of these subtype-specific effects of NS1.
Of the 2,284 seasonal IAVs isolated from humans since 1933, 2,276 (99.6%) contain F103 (32, 46). Only four seasonal viruses, like the H5N1 HK97 virus, encode an NS1 protein with L instead of the consensus F at position 103 (47). Only five seasonal viruses encode an NS1 protein with a hydrophilic amino acid (S) at position 103, namely, three viruses isolated in 1934 to 1936 (including influenza A/Puerto Rico/8/34), one virus isolated in 1954 (A/Leningrad/54), and one virus isolated in 1976 (A/New Jersey/76) (32). The absence of such viruses since 1976 shows that IAVs encoding NS1 proteins with S103 are selected against during replication in humans (32). In avian IAVs, F103 seems to be less dominant, since L at position 103 has been identified in more than 70% of the avian H9N2 or H6N1 (32) viruses. Additionally, Y at position 103 can also be found, although to a lesser extent. The selection pressure for F103 seems to be less strong in avian viruses; however, this putatively low selection pressure does not rely on a lack of NS1 F103-mediated JNK activation in avian cells, since the JNK was well activated upon IAV infection in chicken fibroblasts (Fig. 4D).
We further showed that L and Y at position 103 is compatible with JNK activation in human cells, at least in the SC35M background, suggesting that PR8 differs with respect to position 103 of NS1 and represents an outstanding subtype with respect to JNK activation.
In summary, we present strong evidence that the activation of the RIG-I/MAVS pathway by viral RNA, which is essential for establishing an antiviral interferon-mediated response, leads to an activation of JNK. Nonetheless, RIG-I/MAVS-mediated signaling is not responsible for JNK activation during an ongoing IAV infection. The activation of JNK at later stages of IAV infection depends on the expression of an NS1 protein of specific subtypes. Here, F at position 103 appears to be essential for the JNK-activating property of NS1. It is the first report that directly links the activation of a MAP kinase with the expression of the IAV NS1 protein. The latter observation and the selective pressure on F at position 103 in human IAV NS1 further suggest that activation of JNK represents an IAV-supportive function, at least in humans.
ACKNOWLEDGMENTS
We thank M. Gale, A. Garcia Sastre, C. Y. Wang, E. D. Tang, and K. A. Tang for providing expression plasmids. We further thank Z. J. Chen for his permission to use MAVS−/− MEFs and M. Colonna, W. Barchet, T. Vogl, and S. Akira for providing the Mda5−/− mouse and the MyD88−/− mouse or cells.
This study was generously supported by different grants from the Deutsche Forschungsgemeinschaft to C.E. and S.L. (grant EH235/1-2 and graduate school grants GRK1409 and SFB1009) and by the German FluResearchNet, a nationwide research network on zoonotic influenza sponsored by the Ministry of Education and Research (BMBF). The IMV is a partner of the Cells-in-Motion Cluster of Excellence at the University of Muenster.
Footnotes
Published ahead of print 28 May 2014
REFERENCES
- 1.Eliopoulos AG, Blake SM, Floettmann JE, Rowe M, Young LS. 1999. Epstein-Barr virus-encoded latent membrane protein 1 activates the JNK pathway through its extreme C terminus via a mechanism involving TRADD and TRAF2. J. Virol. 73:1023–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McLean TI, Bachenheimer SL. 1999. Activation of c-Jun N-terminal kinase by herpes simplex virus type 1 enhances viral replication. J. Virol. 73:8415–8426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clarke P, Meintzer SM, Widmann C, Johnson GL, Tyler KL. 2001. Reovirus infection activates JNK and the JNK-dependent transcription factor c-Jun. J. Virol. 75:11275–11283. 10.1128/JVI.75.23.11275-11283.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamza MS, Reyes RA, Izumiya Y, Wisdom R, Kung HJ, Luciw PA. 2004. ORF36 protein kinase of Kaposi's sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J. Biol. Chem. 279:38325–38330. 10.1074/jbc.M400964200 [DOI] [PubMed] [Google Scholar]
- 5.Kujime K, Hashimoto S, Gon Y, Shimizu K, Horie T. 2000. p38 mitogen-activated protein kinase and c-jun-NH2-terminal kinase regulate RANTES production by influenza virus-infected human bronchial epithelial cells. J. Immunol. 164:3222–3228. 10.4049/jimmunol.164.6.3222 [DOI] [PubMed] [Google Scholar]
- 6.Ludwig S, Ehrhardt C, Neumeier ER, Kracht M, Rapp UR, Pleschka S. 2001. Influenza virus-induced AP-1-dependent gene expression requires activation of the JNK signaling pathway. J. Biol. Chem. 276:10990–10998. 10.1074/jbc.M009902200 [DOI] [PubMed] [Google Scholar]
- 7.Ludwig S, Wang X, Ehrhardt C, Zheng H, Donelan N, Planz O, Pleschka S, García-Sastre A, Heins G, Wolff T. 2002. The influenza A virus NS1 protein inhibits activation of Jun N-terminal kinase and AP-1 transcription factors. J. Virol. 76:11166–11171. 10.1128/JVI.76.21.11166-11171.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nacken W, Ehrhardt C, Ludwig S. 2012. Small molecule inhibitors of the c-Jun N-terminal kinase (JNK) possess antiviral activity against highly pathogenic avian and human pandemic influenza A viruses. Biol. Chem. 393:525–534. 10.1515/hsz-2011-0270 [DOI] [PubMed] [Google Scholar]
- 9.Janeway CA, Jr, Medzhitov R. 2002. Innate immune recognition. Annu. Rev. Immunol. 20:197–216. 10.1146/annurev.immunol.20.083001.084359 [DOI] [PubMed] [Google Scholar]
- 10.Akira S, Takeda K. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4:499–511. 10.1038/nri1391 [DOI] [PubMed] [Google Scholar]
- 11.Yoneyama M, Fujita T. 2009. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 227:54–65. 10.1111/j.1600-065X.2008.00727.x [DOI] [PubMed] [Google Scholar]
- 12.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. 2005. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23:19–28. 10.1016/j.immuni.2005.04.010 [DOI] [PubMed] [Google Scholar]
- 13.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682. 10.1016/j.cell.2005.08.012 [DOI] [PubMed] [Google Scholar]
- 14.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988. 10.1038/ni1243 [DOI] [PubMed] [Google Scholar]
- 15.Maniatis T, Falvo JV, Kim TH, Kim TK, Lin CH, Parekh BS, Wathelet MG. 1998. Structure and function of the interferon-beta enhanceosome. Cold Spring Harb. Symp. Quant. Biol. 63:609–620. 10.1101/sqb.1998.63.609 [DOI] [PubMed] [Google Scholar]
- 16.Honda K, Yanai H, Takaoka A, Taniguchi T. 2005. Regulation of the type I IFN induction: a current view. Int. Immunol. 17:1367–1378. 10.1093/intimm/dxh318 [DOI] [PubMed] [Google Scholar]
- 17.Hale BG, Randall RE, Ortín J, Jackson D. 2008. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89:2359–2376. 10.1099/vir.0.2008/004606-0 [DOI] [PubMed] [Google Scholar]
- 18.Ehrhardt C, Seyer R, Hrincius ER, Eierhoff T, Wolff T, Ludwig S. 2009. Interplay between influenza A virus and the innate immune signaling. Microbes Infect. 1:81–87. 10.1016/j.micinf.2009.09.007 [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Robb NC, Lenz E, Wolff T, Fodor E, Pleschka S. 2010. NS reassortment of an H7-type highly pathogenic avian influenza virus affects its propagation by altering the regulation of viral RNA production and antiviral host response. J. Virol. 84:11323–11335. 10.1128/JVI.01034-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma W, Brenner D, Wang Z, Dauber B, Ehrhardt C, Högner K, Herold S, Ludwig S, Wolff T, Yu K, Richt JA, Planz O, Pleschka S. 2010. The NS segment of an H5N1 highly pathogenic avian influenza virus (HPAIV) is sufficient to alter replication efficiency, cell tropism, and host range of an H7N1 HPAIV. J. Virol. 84:2122–2133. 10.1128/JVI.01668-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hrincius ER, Wixler V, Wolff T, Wagner R, Ludwig S, Ehrhardt C. 2010. CRK adaptor protein expression is required for efficient replication of avian influenza A viruses and controls JNK-mediated apoptotic responses. Cell. Microbiol. 12:831–843. 10.1111/j.1462-5822.2010.01436.x [DOI] [PubMed] [Google Scholar]
- 22.Tang ED, Wang CY. 2009. MAVS self-association mediates antiviral innate immune signaling. J. Virol. 83:3420–3248. 10.1128/JVI.02623-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mostafa A, Kanrai P, Ziebuhr J, Pleschka S. 2013. Improved dual promoter-driven reverse genetics system for influenza viruses. J. Virol. Methods 193:603–610. 10.1016/j.jviromet.2013.07.021 [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. 2000. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. U. S. A. 97:6108–6113. 10.1073/pnas.100133697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barber MR, Aldridge JR, Jr, Webster RG, Magor KE. 2010. Association of RIG-I with innate immunity of ducks to influenza. Proc. Natl. Acad. Sci. U. S. A. 107:5913–5918. 10.1073/pnas.1001755107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis WG, Bowzard JB, Sharma SD, Wiens ME, Ranjan P, Gangappa S, Stuchlik O, Pohl J, Donis RO, Katz JM, Cameron CE, Fujita T, Sambhara S. 2012. The 3′ untranslated regions of influenza genomic sequences are 5′PPP-independent ligands for RIG-I. PLoS One 7(3):e32661. 10.1371/journal.pone.0032661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barber MR, Aldridge JR, Jr, Webster RG, Magor KE. 2010. Association of RIG-I with innate immunity of ducks to influenza. Proc. Natl. Acad. Sci. U. S. A. 107:5913–5918. 10.1073/pnas.1001755107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ochiai H, Sakai S, Hirabayashi T, Shimizu Y, Terasawa K. 1995. Inhibitory effect of bafilomycin A1, a specific inhibitor of vacuolar-type proton pump, on the growth of influenza A and B viruses in MDCK cell. Antiviral Res. 27:425–430. 10.1016/0166-3542(95)00040-S [DOI] [PubMed] [Google Scholar]
- 29.Geller R, Taguwa S, Frydman J. 2012. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim. Biophys. Acta 1823:698–706. 10.1016/j.bbamcr.2011.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang C, Yang Y, Zhou X, Yang Z, Liu X, Cao Z, Song H, He Y, Huang P. 2011. The NS1 protein of influenza A virus interacts with heat shock protein Hsp90 in human alveolar basal epithelial cells: implication for virus-induced apoptosis. Virol. J. 8:181. 10.1186/1743-422X-8-181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Twu KY, Noah DL, Rao P, Kuo RL, Krug RM. 2006. The CPSF30 binding site on the NS1A protein of influenza A virus is a potential antiviral target. J. Virol. 80:3957–3965. 10.1128/JVI.80.8.3957-3965.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nemeroff ME, Barabino SM, Li Y, Keller W, Krug RM. 1998. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′end formation of cellular pre-mRNAs. Mol. Cell 1:991–1000. 10.1016/S1097-2765(00)80099-4 [DOI] [PubMed] [Google Scholar]
- 33.Das K, Ma LC, Xiao R, Radvansky B, Aramini J, Zhao L, Marklund J, Kuo RL, Twu KY, Arnold E, Krug RM, Montelione GT. 2008. Structural basis for suppression of a host antiviral response by influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 105:13093–13098. 10.1073/pnas.0805213105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramos HJ, Gale M., Jr 2011. RIG-I like receptors and their signaling crosstalk in the regulation of antiviral immunity. Curr. Opin. Virol. 1:167–176. 10.1016/j.coviro.2011.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ehrhardt C, Ludwig S. 2009. A new player in a deadly game: influenza viruses and the PI3K/Akt signalling pathway. Cell. Microbiol. 11:863–871. 10.1111/j.1462-5822.2009.01309.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ehrhardt C, Marjuki H, Wolff T, Nürnberg B, Planz O, Pleschka S, Ludwig S. 2006. Bivalent role of the phosphatidylinositol-3-kinase (PI3K) during influenza virus infection and host cell defence. Cell. Microbiol. 8:1336–1348. 10.1111/j.1462-5822.2006.00713.x [DOI] [PubMed] [Google Scholar]
- 37.Hrincius ER, Dierkes R, Anhlan D, Wixler V, Ludwig S, Ehrhardt C. 2011. Phosphatidylinositol-3-kinase (PI3K) is activated by influenza virus vRNA via the pathogen pattern receptor Rig-I to promote efficient type I interferon production. Cell. Microbiol. 13:1907–1919. 10.1111/j.1462-5822.2011.01680.x [DOI] [PubMed] [Google Scholar]
- 38.Ayllon J, Hale BG, García-Sastre A. 2012. Strain-specific contribution of NS1-activated phosphoinositide 3-kinase signaling to influenza A virus replication and virulence. J. Virol. 86:5366–5370. 10.1128/JVI.06722-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spesock A, Malur M, Hossain MJ, Chen LM, Njaa BL, Davis CT, Lipatov AS, York IA, Krug RM, Donis RO. 2011. The virulence of 1997 H5N1 influenza viruses in the mouse model is increased by correcting a defect in their NS1 proteins. J. Virol. 85:7048–7058. 10.1128/JVI.00417-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma W, Brenner D, Wang Z, Dauber B, Ehrhardt C, Högner K, Herold S, Ludwig S, Wolff T, Yu K, Richt JA, Planz O, Pleschka S. 2010. The NS segment of an H5N1 highly pathogenic avian influenza virus (HPAIV) is sufficient to alter replication efficiency, cell tropism, and host range of an H7N1 HPAIV. J. Virol. 84:2122–2133. 10.1128/JVI.01668-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z, Robb NC, Lenz E, Wolff T, Fodor E, Pleschka S. 2010. NS reassortment of an H7-type highly pathogenic avian influenza virus affects its propagation by altering the regulation of viral RNA production and antiviral host response. J. Virol. 84:11323–11335. 10.1128/JVI.01034-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noronha JM, Liu M, Squires RB, Pickett BE, Hale BG, Air GM, Galloway SE, Takimoto T, Schmolke M, Hunt V, Klem E, García-Sastre A, McGee M, Scheuermann RH. 2012. Influenza virus sequence feature variant type analysis: evidence of a role for NS1 in influenza virus host range restriction. J. Virol. 86:5857–5866. 10.1128/JVI.06901-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lam WY, Yeung AC, Chan PK. 2011. Apoptosis, cytokine and chemokine induction by non-structural 1 (NS1) proteins encoded by different influenza subtypes. Virol. J. 8:554. 10.1186/1743-422X-8-554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sutejo R, Yeo DS, Myaing MZ, Hui C, Xia J, Ko D, Cheung PC, Tan BH, Sugrue RJ. 2012. 2012. Activation of type I and III interferon signaling pathways occurs in lung epithelial cells infected with low pathogenic avian influenza viruses. PLoS One 7(3):e33732. 10.1371/journal.pone.0033732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Munir M, Zohari S, Belák S, Berg M. 2012. Double-stranded RNA-induced activation of activating protein-1 promoter is differentially regulated by the non-structural protein 1 of avian influenza A viruses. Viral Immunol. 25:79–85. 10.1089/vim.2011.0059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Macken C, Lu H, Goodman J, Boykin L. 2001. In Osterhaus NC, Hampson AW. (ed), Options for the control of influenza IV, vol IV, p 103–106 Elsevier Science, Amsterdam, Netherlands [Google Scholar]
- 47.Kuo RL, Krug RM. 2009. Influenza a virus polymerase is an integral component of the CPSF30-NS1A protein complex in infected cells. J. Virol. 83:1611–1616. 10.1128/JVI.01491-08 [DOI] [PMC free article] [PubMed] [Google Scholar]