

ABSTRACT
Sequence differences in the EBNA-2 protein mediate the superior ability of type 1 Epstein-Barr virus (EBV) to transform human B cells into lymphoblastoid cell lines compared to that of type 2 EBV. Here we show that changing a single amino acid (S442D) from serine in type 2 EBNA-2 to the aspartate found in type 1 EBNA-2 confers a type 1 growth phenotype in a lymphoblastoid cell line growth maintenance assay. This amino acid lies in the transactivation domain of EBNA-2, and the S442D change increases activity in a transactivation domain assay. The superior growth properties of type 1 EBNA-2 correlate with the greater induction of EBV LMP-1 and about 10 cell genes, including CXCR7. In chromatin immunoprecipitation assays, type 1 EBNA-2 is shown to associate more strongly with EBNA-2 binding sites near the LMP-1 and CXCR7 genes. Unbiased motif searching of the EBNA-2 binding regions of the differentially regulated cell genes identified an ETS-interferon regulatory factor composite element motif that closely corresponds to the sequences known to mediate EBNA-2 regulation of the LMP-1 promoter. It appears that the superior induction by type 1 EBNA-2 of the cell genes contributing to cell growth is due to their being regulated in a manner different from that for most EBNA-2-responsive genes and in a way similar to that for the LMP-1 gene.
IMPORTANCE The EBNA-2 transcription factor plays a key role in B cell transformation by EBV and defines the two EBV types. Here we identify a single amino acid (Ser in type 1 EBV, Asp in type 2 EBV) of EBNA-2 that determines the superior ability of type 1 EBNA-2 to induce a key group of cell genes and the EBV LMP-1 gene, which mediate the growth advantage of B cells infected with type 1 EBV. The EBNA-2 binding sites in these cell genes have a sequence motif similar to the sequence known to mediate regulation of the EBV LMP-1 promoter. Further detailed analysis of transactivation and promoter binding provides new insight into the physiological regulation of cell genes by EBNA-2.
INTRODUCTION
Epstein-Barr virus (EBV) is the main cause of infectious mononucleosis in Western countries and also contributes to several types of human cancer. Some of these diseases vary in incidence dramatically in different parts of the world, and it is possible that natural variation in EBV contributes to differences in disease incidence. It has recently been proposed that differences in the properties of an EBV strain isolated from a Chinese nasopharyngeal carcinoma might contribute to its role in that disease (1). Mutations in the EBNA-3B gene linked to diffuse large B cell lymphoma also support the idea that variation in EBV sequence may be linked to human disease (2).
Type 1 and type 2 strains are the main natural functional variation known in Epstein-Barr virus (3, 4). The types are defined by sequence differences in the EBNA-2 gene, but there are also linked sequence changes in the EBNA-3 family of genes (3). Although both virus types activate human B cells upon infection, type 1 strains are much better than type 2 strains at transforming human B cells into proliferating lymphoblastoid cell lines (LCLs) (5). Type 1 is the main EBV type prevalent all over the world, but type 2 EBV is frequently found in sub-Saharan Africa, where it can be as abundant as type 1 EBV. At present the biological significance of the two types of EBV is not understood, and there is no specific link of these types to human disease. However, the difference in growth transformation remains the clearest example of the functional variation of EBV. Most genes of EBV have a low level of natural sequence variation (less than 5% at the amino acid level), but type 1 EBNA-2 and type 2 EBNA-2 are only 56% identical at the amino acid level. Replacement of the EBNA-2 gene in type 2 EBV with the type 1 EBNA-2 gene confers a type 1 transformation efficiency on the type 2 strain (6), showing that EBNA-2 is the gene functionally important for the growth transformation phenotype.
EBNA-2 is a transcription factor for viral and cell genes transcribed by RNA polymerase II but does not bind DNA directly. EBNA-2 forms a complex with cell DNA-binding proteins, which target it to promoters. The best characterized of these cell proteins is RBP-Jκ (also known as CSL), but PU.1 (7, 8), ATF/CRE (9), and EBF1 binding sites (10) have also been shown to mediate EBNA-2 function at certain promoters. Early studies of EBNA-2 function focused on viral promoters or artificial promoters where the binding sites are close to the transcription start site (TSS), but recent chromatin immunoprecipitation (ChIP) sequencing (ChIP-seq) studies in human B cell lines (10, 11) have shown that EBNA-2 binding sites at cellular gene targets are predominantly located far away from gene TSSs, at distances up to 100 kb up- or downstream.
To investigate why type 1 EBV is more efficient at promoting LCL growth, we previously made chimeras of the type 1 and type 2 EBNA-2 genes and tested them for the ability to maintain growth of an LCL conditionally dependent on transfected EBNA-2. The results (12) showed that sequences from the C-terminal part of type 1 EBNA-2 were sufficient to confer the maintenance of LCL growth when swapped into type 2 EBNA-2. This part of type 1 EBNA-2 contains the arginine-glycine (RG) repeat, conserved region 7 (CR7), and the transactivation domain (TAD). We also identified cell and viral genes that are differentially regulated by the EBNA-2 types (13). The EBV LMP-1 gene is induced more rapidly and strongly by type 1 EBNA-2 (13), and LMP-1 is known to be required for growth transformation by EBV. About 300 cell genes are induced directly by type 1 EBNA-2 (14), but only about 10 of these are regulated differentially, and all 10 are more strongly induced by type 1 EBNA-2. The most differentially regulated gene is CXCR7, which was shown to be essential for the proliferation of LCLs (13).
In the study described in this paper, we mapped the part of type 1 EBNA-2 responsible for the superior growth maintenance properties more precisely. Remarkably, changing a single amino acid (S442D) from serine in type 2 EBNA-2 to the aspartate found in type 1 EBNA-2 confers a type 1 growth phenotype in the LCL growth maintenance assay. This amino acid is located in the TAD of type 1 EBNA-2 and confers a stronger transactivation function and increased binding to some sites at differentially regulated genes. We identify a sequence element that is enriched in the EBNA-2 binding regions of the LMP-1 promoter and cell promoters which are differentially regulated by the EBNA-2 types but not in the EBNA-2 binding sites of genes that are regulated equally by type 1 and type 2 EBNA-2. This motif includes both a PU.1 (Spi-1) and interferon regulatory factor (IRF) binding sequence. It closely resembles the ETS-IRF composite element (EICE) in the IgL(λ) gene enhancer and a similar sequence that mediates PU.1 activation of the LMP-1 promoter. This element may therefore confer the differential effects of type 1 and type 2 EBNA-2 on both LMP-1 and cell gene activation.
MATERIALS AND METHODS
Cell culture.
EREB2.5 cells (15) contain an estrogen receptor (ER)–EBNA-2 fusion protein regulated by β-estradiol and were maintained as described previously (12). Daudi is an EBV-positive Burkitt's lymphoma (BL) cell line (16), and BJAB is an EBV-negative B lymphoma line (17). Daudi/pHEBoMT–EBNA-2 cell lines were grown in RPMI 1640 medium (Gibco, United Kingdom) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS) and 0.3 mg/ml hygromycin B (Roche, Germany). HEK293 cells were cultured in Dulbecco's modified Eagle medium (DMEM; Gibco) supplemented with 10% FBS and antibiotics. Primary B cells were purified from buffy coat residues by negative selection using RosetteSEP human B cell enrichment cocktail (Stemcell Technologies, Canada) and infected with the B95-8 strain of EBV as described previously (12).
Plasmids and cloning.
A QuikChange II site-directed mutagenesis kit (Stratagene, United Kingdom) was used to introduce the SD431-432HN, EEP434-436PEA, F438I, S442D, D442S, or G460Y site-directed mutation into type 1 EBNA-2, type 2 EBNA-2, or chimera 6 EBNA-2 (E2C6) in pBS–EBNA-2 plasmids (12). The modified EBNA-2 sequences were then transferred into the p294 plasmid as described previously (12).
Inducible expression of EBNA-2 was obtained from the metallothionein (MT) promoter in the new pHEBoMT–EBNA-2 series of plasmids. The MT–EBNA-2 expression cassette was inserted into pHEBo (18) as a NotI fragment, having converted the BamHI site of pHEBo to a NotI site using an oligonucleotide. The MT–EBNA-2 expression cassette was assembled using the metallothionein promoter from pMEP4 as an XhoI-HindIII fragment linked to the HindIII-BglII EBNA-2 gene from pSG5–EBNA-2. The type 2 and type 2 S442D alleles of EBNA-2 had also been introduced into pSG5–EBNA-2 by swapping an EcoRI-BglII fragment from the corresponding pBS-E2 plasmids (12), after converting their original SacI site to BglII.
GAL4 DNA-binding domain (DBD)–EBNA-2 TAD fusions were obtained by PCR amplification of EBNA-2 TADs (amino acids 426 to 463 of type 1 EBNA-2) in the pBS–EBNA-2 series of plasmids using primers flanked by BamHI and NotI restriction sites. These EBNA-2 TAD BamHI-NotI fragments were cloned into pcDNA3.1-GAL4 DBD (Addgene).
Transfections.
The EREB2.5 growth assay was performed as described previously (12). For HEK293 cell transfections, 3 × 105 cells were transfected with 4 μg plasmid DNA in 6-well plates using Lipofectamine 2000 (Invitrogen, United Kingdom). For the TAD luciferase reporter assays, 2 × 106 BJAB cells were transfected using a Neon system (Invitrogen). A mixture of 500 ng pFR-Luc reporter plasmid (Agilent, United Kingdom), 10 ng pRL-CMV Renilla plasmid, and 200 ng pcDNA3.1-GAL4 DBD fused to the EBNA-2 TAD was used. Electroporation conditions were 1,200 V, a 20-ms pulse width, and 2 pulses. Transfected cells were transferred into 2 ml RPMI 1640 medium supplemented with 10% FBS and antibiotics and assayed after 24 h.
To generate Daudi/pHEBoMT–EBNA-2 stable cell lines, 2 × 106 cells were transfected with 6 μg pHEBoMT–EBNA-2 plasmid using the Neon transfection system (Invitrogen) set at 1,400 V with a 30-ms pulse width and 1 pulse. Transfected cells were selected with 300 μg/ml hygromycin B.
Immunoblotting antibodies.
Cell samples were lysed and analyzed by Western immunoblotting as described previously (12). Detection of EBNA-2 was with PE2 antibody (Dako), detection of β-actin was with AC-74 antibody (Sigma, United Kingdom), detection of LMP-1 was with CS.1-4 antibody (Dako), and detection of GAL4 DBD was with RK5C1 antibody (Santa Cruz).
Luciferase assays.
At 24 h after transfection, BJAB cells were lysed in 1× passive lysis buffer supplied in the dual-luciferase reporter assay kit (Promega, United Kingdom). Firefly luciferase and Renilla luciferase activities were measured for each cell lysate. Renilla luciferase activity was determined to normalize the values of the firefly luciferase activity for transfection and extraction efficiency.
RNA extraction and quantitative reverse transcription-PCR.
Total cell RNA was extracted from treated cells using an RNeasy kit (Qiagen, United Kingdom). cDNA was prepared using a Protoscript Moloney murine leukemia virus first-strand cDNA synthesis kit (New England BioLabs, United Kingdom) and random primers. For CXCR7 RNA, quantitative PCR (qPCR) was carried out with primers GCAGCCAGCAGAGCTCACAGTTG (forward) and TGGGCATGTTGGGACACATCACC (reverse), with GAPDH (glyceraldehyde-3-phosphate dehydrogenase) used as a normalization control. mRNA levels were quantified using the ΔΔCT threshold cycle (CT) method.
5′ RACE.
EREB2.5 cells were washed twice and resuspended at 5 × 105 cells/ml in RPMI 1640 medium supplemented with 10% FBS and antibiotics. Cells were treated with or without 1 μM β-estradiol (Sigma) for 4 days. RNA was extracted, and 5′ rapid amplification of cDNA ends (RACE) was then performed using a 5′ RACE system (Invitrogen). The gene-specific primers for CXCR7 used were CAAGATGTAGCACTGCGTGTCATAGCC and AGAGCAGGACGCTTTTGTTGGGC. 5′ RACE products were cloned, sequenced, and mapped onto the hg19 human genome sequence using the BLAT program.
ChIP.
ChIP-qPCR assays for EBNA-2 were carried out as described previously (19–21) using chromatin from Daudi/pHEBoMT–EBNA-2 cell lines. Cells at 5 × 105/ml were treated for 24 h with 4 to 10 μM cadmium chloride (CdCl2) before chromatin preparation. A nonspecific IgG immunoprecipitation was used as a negative control in all ChIP experiments. The ChIP-qPCR primers used in this study were as follows: for CXCR7 ChIP-seq model-based analysis of ChIP-seq (MACS) peak 16, forward primer AGATCGGTAGTTGGATGGGTTT and reverse primer CTCATGGTTCTATGTCCTCACCA, and for the CXCR7 ChIP-seq MACS peak 16 nonbinding region, forward primer CACACGAAGGCTGGAGTAGT and reverse primer AGCATGAGAGGAGTGTTGACC; for CXCR7 ChIP-seq MACS peak 13, forward primer TGTGTGTGTCTGCCTGTGG and reverse primer TGGCTGCAGACTTGCATTAT, and for the CXCR7 ChIP-seq peak 13 nonbinding region, forward primer CCATCAGTGAATGGTGGTCA and reverse primer TGCCGTGTAACATGGAAGG; for the LMP-1 ChIP binding region, forward primer AGAGGAGGAGAAGGAGAGCAA and reverse primer CCTGAGGATGGAACACGAC, and for the LMP-1 ChIP nonbinding region, forward primer GGACACGCTCCTTCTTGG and reverse primer ACTGGCTGGATTCTACGCTACT; and for the CCL3 ChIP binding region, forward primer GCTGGAGAGTTCATGCACAG and reverse primer TTCTCCTGTGAGTGTGAAGAGG, and for the CCL3 ChIP nonbinding region, forward primer GAGGTATGCTGATTGATTGTGAA and reverse primer CTACCTTCTCAGCCAGATTATATGC.
Motif searching.
ChIP sequencing data from Mutu III cells (11) were used to identify significant EBNA-2 binding sites (MACS, P < 10−7) around nine previously identified differentially regulated cell gene loci and nine equally regulated gene loci (13). Any binding peak within the gene or upstream or downstream from the gene, but not within an adjacent gene, was included in the analysis. DNA sequences from the EBNA-2 binding sites at differentially regulated cell genes plus the LMP-1 promoter region from positions −320 to +1 (a total of 58 sequences) were then subjected to unbiased analysis by MEME-ChIP for noncentrally enriched motifs of between 6 and 12 nucleotides (22). DNA sequences from the binding sites at equally regulated cell genes (a total of 80 sequences) were analyzed in parallel.
RESULTS
Fine mapping of an amino acid sequence that mediates superior growth properties of type 1 EBNA-2.
Our previous chimera mapping of the type 1 EBNA-2 sequences responsible for its superior growth-promoting properties in EREB2.5 cells was extended, focusing on the transactivation domain (TAD). The loss of EREB2.5 growth maintenance with a type 2 TAD compared to the preservation of growth maintenance with a type 1 TAD (compare chimeras C6 and C7 in Fig. 1A) showed that some type 1 amino acids in this region must be required. There are relatively few amino acid differences in this part of the protein, so it was feasible to mutate each significantly different amino acid either individually or in groups (Fig. 1A). These chimeras were then tested as described before (12) by transfection into EREB2.5 cells, measuring the ability to maintain cell growth in the absence of β-estradiol (Fig. 1B). The results showed that mutant S442D rescued the growth maintenance ability of chimera C6 to become equivalent to that of chimera C7. The other mutations tested did not rescue cell growth. This shows that S442D is the only significant amino acid difference in the TAD region for this assay. The EBNA-2 proteins were all expressed with the expected size on a Western blot (Fig. 1C).
FIG 1.
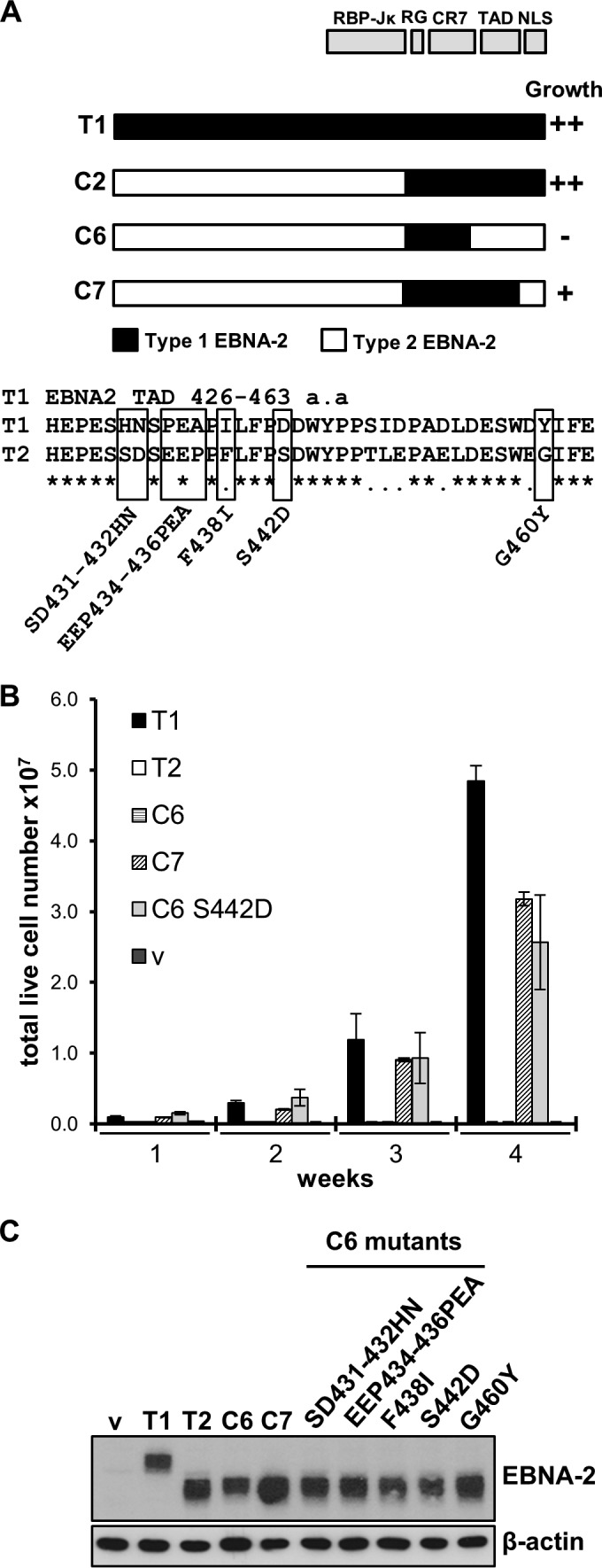
S442D in the EBNA-2 chimera C6 background complements the deficiency of type 2 EBNA-2 in the EREB2.5 growth assay. (A) Growth phenotypes of EBNA-2 chimeras in the EREB2.5 growth assay from previous results (12) are shown for reference. The positions of the RBP-Jκ association, RG, CR7, nuclear localization signal (NLS), and TAD domains of EBNA-2 are indicated. The protein sequence of the TAD is shown below for type 1 EBNA-2 and type 2 EBNA-2. The five mutations indicated were made separately in EBNA-2 chimera C6. a.a, amino acids. (B) Live cell counts for the EREB2.5 growth assay with the S442D EBNA-2 chimera C6 mutant compared to those with chimeras C6 and C7. The averages of the data from at least 4 experiments, each with duplicate transfections, are shown; error bars indicate standard deviations. The other mutants tested, SD431-432HN, EEP434-436PEA, F438I, and G460Y, in EBNA-2 chimera C6 gave no cell growth in this assay (data not shown). (C) Western blot of protein extracts from EREB2.5 cells transfected with type 1 (T1), type 2 (T2), chimera C6 and C7, and C6 mutant EBNA-2 proteins. Cells were harvested 5 days after transfection, and proteins were extracted by radioimmunoprecipitation assay lysis. Type 1 EBNA-2 migrates at 85 kDa, and type 2 EBNA-2 migrates at 75 kDa. β-Actin was monitored as a loading control. v, empty vector.
Although chimeras C7 and C6 S442D both had similar activity in the EREB2.5 cell growth assay, they were quantitatively not as effective as type 1 EBNA-2 or chimera C2. The contribution of S442D was investigated further by making this single amino acid change in type 2 EBNA-2 without any other changes (Fig. 2A). The reciprocal amino acid change of D442S was also made in type 1 EBNA-2. With this minimal disruption of the whole protein structure, the S442D point mutant converted type 2 EBNA-2 to be as effective as type 1 EBNA-2 in the EREB2.5 growth assay (Fig. 2B). The reciprocal point change of D442S abolished the activity of type 1 EBNA-2 in the EREB2.5 assay. Again, these EBNA-2 proteins were all expressed with the expected size on a Western blot (Fig. 2C). The results show that the aspartate at position 442 is essential for type 1 EBNA-2 function in the cell growth assay and the S442D point change is sufficient to convert type 2 EBNA-2 to be as effective as type 1 EBNA-2 in this assay.
FIG 2.
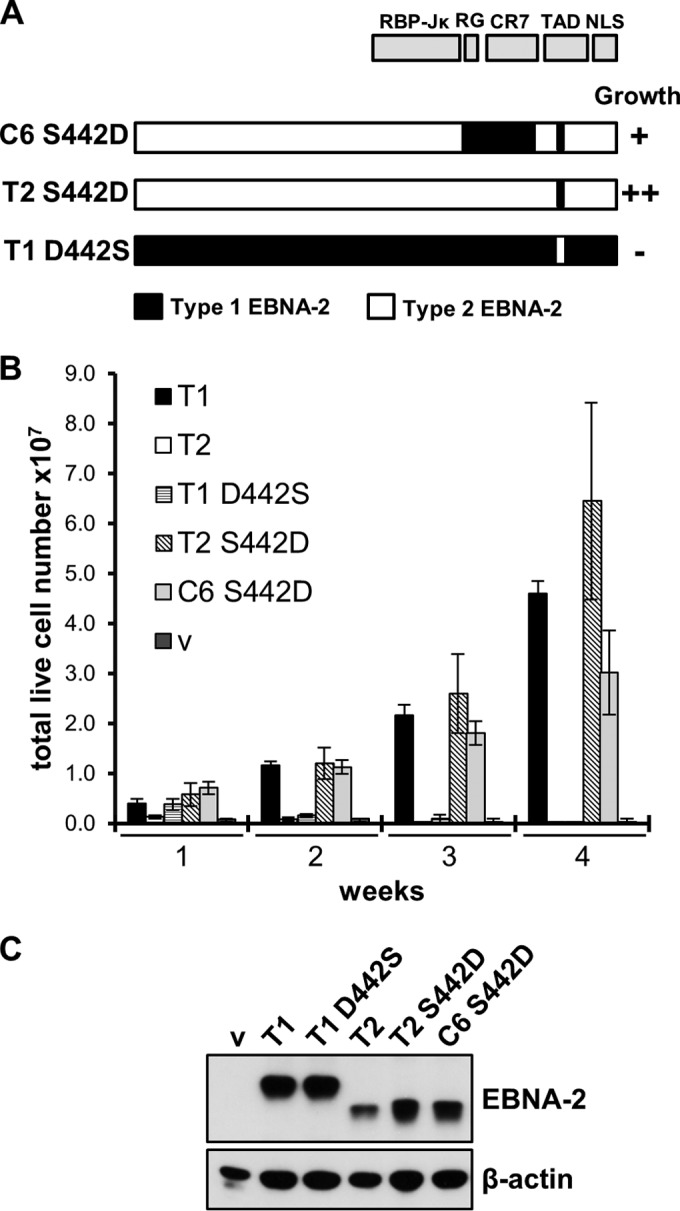
S442D in type 2 EBNA-2 is sufficient to sustain LCL proliferation in the EREB2.5 assay. (A) Cartoon similar to that in Fig. 1A showing the structures of type 2 S442D EBNA-2 and type 1 D442S EBNA-2. (B) Cell counts, determined as described in the legend to Fig. 1B, for the EREB2.5 cell growth assay of the indicated plasmids expressing wild-type type 1, type 2, type 1 D442S, type 2 S442D, and C6 S442D EBNA-2 and an empty vector (v) 1 to 4 weeks after transfection. (C) Western blot analysis of protein extracts from EREB2.5 cells transfected with the indicated EBNA-2 expression plasmids, as described in the legend to Fig. 1C.
The stronger activation domain function of type 1 EBNA-2 is mediated by aspartate 442.
Since Asp 442 lies in the TAD of EBNA-2, we tested whether S442D might give a higher transactivation function. The TADs were fused to the GAL4 DNA-binding domain and tested for the ability to induce expression of a synthetic promoter containing GAL4 DNA-binding sites in a transient-transfection reporter assay in BJAB cells (Fig. 3A). This system had been used for the original identification of the TAD of type 1 EBNA-2 (23). The activity of the type 1 EBNA-2 TAD was about 2-fold higher than that of the type 2 EBNA-2 TAD (Fig. 3B). S442D increased type 2 EBNA-2 transactivation activity so that it was close to that of type 1, and the reciprocal D442S mutation in type 1 EBNA-2 reduced the activity so that it was similar to that of the type 2 EBNA-2 TAD (Fig. 3B). The other mutations that did not increase LCL growth, as indicated in Fig. 1B, also did not increase TAD activity (Fig. 3B). Aspartate 442 can therefore confer increased activity to the TAD of type 2 EBNA-2 and was unique among the amino acids tested in having this activity.
FIG 3.
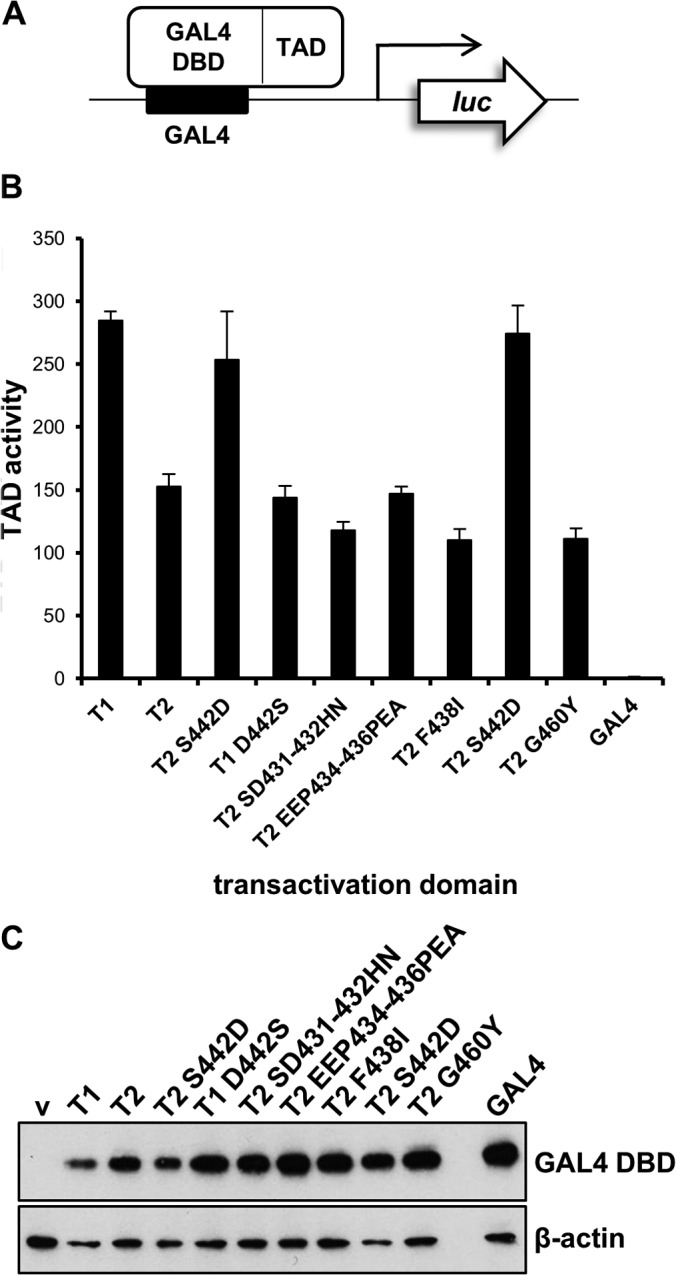
Transactivation domain reporter assay for EBNA-2 mutants. (A) Representation of transactivation domain assay. A firefly luciferase reporter gene can be activated by the GAL4 DBD–EBNA-2 TAD fusion protein being tested. GAL4 DBD binds to GAL4 binding sites in the luciferase reporter plasmid promoter. (B) Transactivation domain assay of EBNA-2 TAD and mutants in BJAB cells 24 h after transfection. TAD and luciferase activity is given relative to that for the empty vector (GAL4) after normalizing for transfection efficiency with a cotransfected Renilla luciferase plasmid. Two independently derived type 2 S442D EBNA-2 TAD plasmids used in this assay are shown. Results are depicted as the means ± standard deviations from 3 independent experiments. (C) Western blot of HEK293 cells transfected with pcDNA3.1-GAL4 DBD plasmids expressing the different TAD fusions. Protein extracts were analyzed by Western blotting using the GAL4 DBD antibody, and β-actin was used as a loading control.
Although they were all expressed from equivalent plasmid structures, it was difficult to control for the equal expression of the GAL4 fusion proteins in the transfected BJAB cells because the proteins were unstable and could not be detected by Western blotting of the BJAB cell extracts. When the same plasmids were transfected into HEK293 cells (which have a much higher transfection efficiency) and extracts were Western blotted for GAL4, all were detected, but only the stable GAL4 part of the fusion protein could be observed (Fig. 3C). The TAD part of the fusion protein was most likely degraded in the cell extracts, leaving only the stable GAL4 DNA-binding domain.
We conclude that a slightly stronger activation domain function of type 1 EBNA-2 is mediated by aspartate 442. This may be a significant factor in the activation of specific genes by EBNA-2 during B cell transformation that results in the superior growth-transforming properties of type 1 EBNA-2.
CXCR7 RNA is rapidly degraded upon EBV infection and then reinduced by EBNA-2.
LMP-1 and CXCR7 are the genes that are the most differentially regulated by type 1 and type 2 EBNA-2 (12), and the mechanism of their regulation is the focus of this study. However, it was noticeable in our previous analysis that uninfected B cells already contain CXCR7 mRNA (12). A more detailed investigation of CXCR 7 mRNA levels during EBV infection has now shown an initial rapid drop followed by a reinduction. At 1 day postinfection with type 1 EBV (strain B95-8), the level was reduced (Fig. 4A), but by 3 days postinfection, the level increased to exceed that of noninfected cells. The initial degradation of CXCR7 RNA appears to result from signal transduction upon viral infection because at day 1 UV-inactivated EBV caused a reduction similar to that for untreated EBV (Fig. 4A). The supernatant left after pelleting of EBV from the virus preparation by ultracentrifugation did not reduce CXCR7 levels, so it appears to be the virus infection that caused the degradation in CXCR7 mRNA. The reduction in CXCR7 mRNA levels was even greater at 4 or 8 h postinfection (Fig. 4B), indicating that the CXCR7 mRNA level at 1 day postinfection was already increasing again, consistent with the expression of EBNA-2 at this time (Fig. 4C). Further evidence for regulation by signal transduction came from a rapid and dramatic reduction in CXCR7 levels in response to tetradecanoyl phorbol acetate (TPA) treatment of the B cells without EBV (Fig. 4A and B). Our previous analysis using B95-8 EBV from a bacterial artificial chromosome rather than the EBV induced from B95-8 cells used here showed a slightly more rapid reinduction of CXCR7 (12). This most likely reflects the different amount of infectious virus used in the experiments. We conclude that the difference in CXCR7 regulation by type 1 and type 2 EBV upon infection of primary B cells (12) seems to lie in a delayed and weaker reinduction of CXCR7 levels after infection by type 2 EBV.
FIG 4.

CXCR7 mRNA is rapidly degraded upon EBV infection and is then reinduced by EBNA-2. (A) Primary B cells were infected with type 1 B95-8 EBV or with UV-inactivated B95-8 EBV (UV B95-8). Separate primary B cell samples were exposed to the supernatant remaining after pelleting of EBV by ultracentrifugation (SUP) or were treated with tetradecanoyl phorbol acetate (TPA). RNA was extracted after 1 or 3 days, cDNA was prepared, and qPCR was performed for CXCR7. The CXCR7 mRNA level normalized to that of GAPDH mRNA is shown relative to the value for mock infection on day 0. Error bars represent standard deviations from at least 3 independent experiments. (B) Primary B cells were infected with B95-8 EBV or treated with TPA as described in the legend to panel A but analyzed for CXCR7 mRNA after 4 or 8 h. (C) Protein extracts from the experiment whose results are shown in panel A were tested for EBNA-2 by immunoblotting (with β-actin as a loading control). We showed previously that EBNA-2 protein expression is first detected between 12 and 16 h after infection (35).
Type 1 EBNA-2 binds to LMP-1 and CXCR7 gene regulatory elements at higher levels.
To determine whether differences in the binding of type 1 and type 2 EBNA-2 to gene regulatory elements also play a role in the increased activation of LMP-1 and CXCR7 by type 1 EBNA-2, we performed ChIP-qPCR analysis using newly generated Daudi cell lines stably transfected with plasmids inducibly expressing EBNA-2. Inducible expression of type 1, type 2, or type 2 S442D EBNA-2 was mediated via the cadmium-responsive metallothionein promoter, and in these constructs, the EBNA-2 polyproline repeat length was equalized in the type 1 and type 2 EBNA-2 alleles to generate EBNA-2 proteins of almost the same size. Following induction, the superior activation of the endogenous LMP-1 gene in the Daudi EBV genome by type 1 EBNA-2 was clearly apparent (Fig. 5A). Consistent with its growth-promoting properties in the EREB2.5 LCL assay, type 2 EBNA-2 with the S442D mutation induced LMP-1 as efficiently as type 1 EBNA-2 in this Daudi cell system (Fig. 5A). The type 2 S442D mutant was also as effective as type 1 EBNA-2 at inducing CXCR7 mRNA (Fig. 5B), whereas the type 2 EBNA-2 showed weak induction in these cells (Fig. 5B). In contrast to primary resting B cells, Daudi cells had a low endogenous level of CXCR7 mRNA prior to the induction by EBNA-2. Several different mRNAs have been reported for CXCR7 (Fig. 6A), so we determined which of these mRNAs is induced by EBNA-2 using a 5′ RACE assay with RNA from EREB2.5 cells (Fig. 5C). Sequencing of the single RACE product (Fig. 5C) demonstrated that the promoter with a TSS at position 237,478,380 on chromosome 2 in the human hg19 genome assembly (highlighted in Fig. 6A) is the TSS induced by EBNA-2. It was this mRNA that was assayed in the qPCR experiment whose results are shown in Fig. 4 and 5B.
FIG 5.
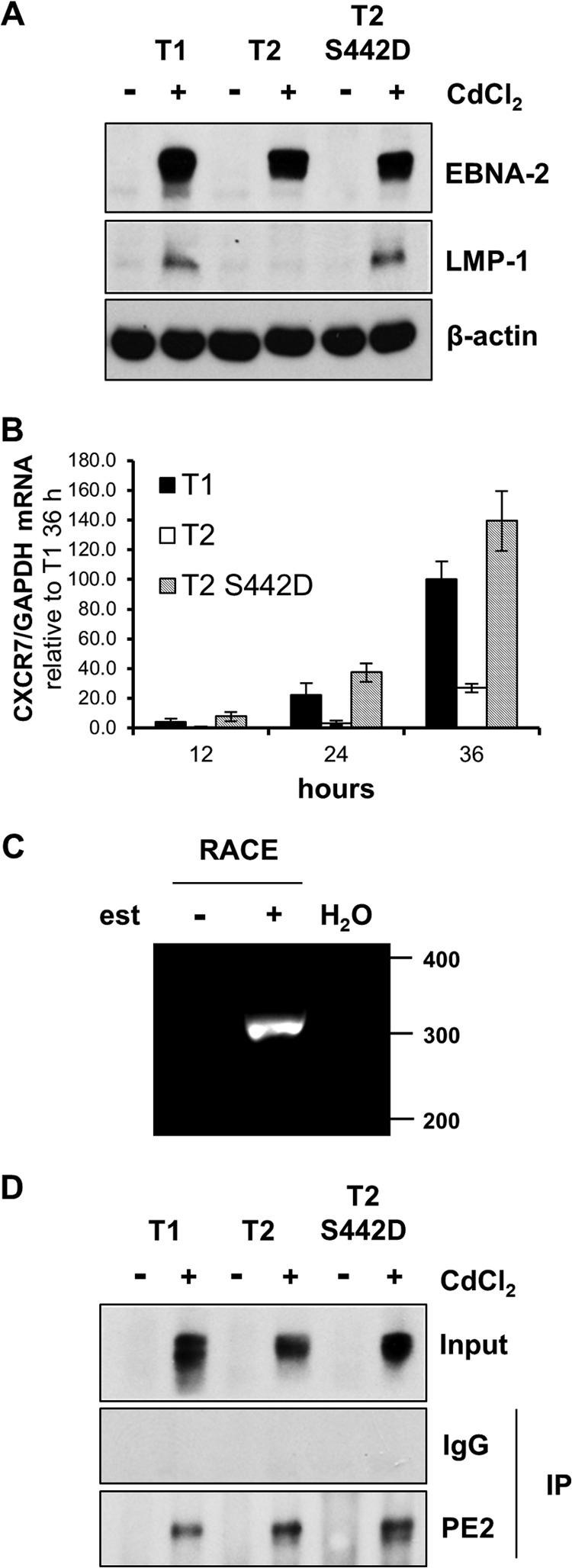
Differential induction of LMP-1 and CXCR7 in Daudi cells with inducible type 1, type 2, or type 2 S442D EBNA-2. (A) Daudi cell lines stably transfected with pHEBoMT plasmids expressing type 1 (T1), type 2 (T2), or type 2 S442D (T2 S442D) EBNA-2 were induced (+) with cadmium chloride (CdCl2) for 24 h or left uninduced (−). Protein samples were analyzed by immunoblotting for EBNA-2, LMP-1, and β-actin (as a loading control). The EBNA-2 polyproline repeat region was equalized in these EBNA-2 plasmids so that all the EBNA-2 proteins were about 75 kDa in these cell lines. (B) The type 1, type 2, and type 2 S442D EBNA-2-expressing cell lines were treated with CdCl2, and total cell RNA was extracted after 12, 24, and 36 h. RNA was converted to cDNA and analyzed by qPCR to quantify CXCR7 mRNA levels. The histograms show the CXCR7 mRNA/GAPDH mRNA ratio normalized to the value for type 1 EBNA-2 at 36 h, which was set equal to 100. Error bars represent standard deviations from at least 3 experiments. (C) 5′ RACE was performed using EREB2.5 cells treated (+) or untreated (−) with β-estradiol (est) in the culture medium. Water (H2O) was used as a negative control for the PCR in the experiment. The single RACE product migrated at about 300 bp on an agarose gel stained with ethidium bromide. (D) Immunoblotting control for the ChIP experiment whose results are shown in Fig. 6. Daudi cell lines were induced (+) with CdCl2 for 24 h or left uninduced (−). Nuclear extracts were prepared, and EBNA-2 ChIP was performed using the PE2 antibody. An unrelated mouse monoclonal IgG antibody was used as a negative control in the ChIP. ChIP samples were then immunoblotted for EBNA-2 (PE2 antibody) to check for approximately equal precipitation of EBNA-2. IP, immunoprecipitation.
FIG 6.

EBNA-2 ChIP binding at CXCR7, LMP-1, and CCL3 promoter elements. (A) ChIP-seq data for type 1 EBNA-2 binding sites at the CXCR7 gene locus in an LCL (GM12878) and in the Mutu III BL cell line using methods described previously (11). The 5′ RACE experiment showed the middle CXCR7 isoform (highlighted) to be the CXCR7 RNA induced by EBNA-2. The y axis displays the number of sequence reads per million background-subtracted reads. Filled boxes, significant peaks of EBNA-2 binding (MACS, <10−7) in Mutu III cells (numbered 1 to 17 from left to right); asterisks, MACS peak locations of the motifs identified by MEME-ChIP that contribute to the EICE consensus (Table 1); chr2, chromosome 2. (B to E) ChIP-qPCR was carried out on nuclei from cells for which the results are shown in Fig. 5D using primers specific to regions known to be bound and not bound by type 1 EBNA-2 in ChIP-seq for which the results are shown in panel A. (B) CXCR7 peak 16 gene locus; (C) CXCR7 peak 13 gene locus; (D) LMP-1 gene locus; (E) CCL3 gene locus. qPCR values for EBNA-2 ChIP (PE2) and the IgG control are given relative to the ChIP input (in percent). Results are shown as the means ± standard deviations from 3 independent ChIP experiments. ns, not significant; **, P < 0.01; ***, P < 0.005.
The sequences required for EBNA-2 activation of the LMP-1 promoter are close to the transcription start site and have been mapped in detail previously (7–9, 24, 25). Recent ChIP sequencing analyses of EBNA-2 binding sites in the Mutu III BL cell line and in an LCL (10, 11) have shown that most EBNA-2 binding sites in the human genome are located far away from gene TSSs at distances up to 100 kb up- or downstream. Using Mutu III ChIP sequencing data, we identified 17 peaks of EBNA-2 binding (MACS, <10−7) upstream and downstream from the confirmed CXCR7 gene TSS (Fig. 6A and Table 1).
TABLE 1.
Thirty-one sites found by MEME in the EBNA-2 binding regions of genes that are differentially regulated by type 1 and type 2 EBNA-2 and contribute to the enriched consensus motif
| Gene (peak)a | Strandb | P valuec | Site sequence |
||
|---|---|---|---|---|---|
| 5′ Flanking | Consensus | 3′ Flanking | |||
| MARCKS (peak 1) | + | 1.44E−07 | TGGAGTCTCT | TTTCACTTCCTC | TTGTCAAACC |
| MARCKS (peak 15) | + | 6.66E−07 | TCCTTCCCCT | TTTTTCTTCCTC | TATCTACCTA |
| MARCKS (peak 14) | + | 1.61E−06 | GGCCTGCCTT | TCTTACTTTCTC | AGCAATTTAA |
| LMP-1d | − | 2.35E−06 | AACACACGCT | TTCTACTTCCCC | TTTCTACGCT |
| MARCKS (peak 15) | + | 2.35E−06 | CGTTCTACTT | TTTCACTTCCTG | TTTTGTTCCT |
| MARCKS (peak 6) | − | 2.35E−06 | CAATTTCAAA | TTCTACTTCCTT | |
| FCRL2 (peak 6) | − | 2.35E−06 | AGTGGCACTA | TTCTACTTCCTT | T |
| CXCR7 (peak 12) | − | 1.17E−05 | CATTCTCAGC | TTTCGCTTTCTT | AAGAAAAGAA |
| MARCKS (peak 3) | + | 1.61E−05 | TCAGTTCTAT | TTCTGCTTTCTT | AGTGTCTTCA |
| ADAMDEC1 (peak 1)e | − | 1.75E−05 | TGAGCCTATG | TCTCGCTTCCTG | CTGGGGTTTA |
| HES1 (peak 2) | − | 1.75E−05 | GGGTGGGATC | TCATACTTCCTC | CCTGAGCTAG |
| MARCKS (peak 16) | − | 2.73E−05 | TCTCAAACAG | TTTTATTTTCTG | CTCATTGTAG |
| CCL3L3 (peak 1) | − | 3.08E−05 | GACAACAATA | TCTCACGTCCTC | TCACTGTGGA |
| MARCKS (peak 15) | + | 3.08E−05 | TCCTCCTTCCCC | TTTTTTCTTC | |
| HES1 (peak 6) | − | 3.57E−05 | TTTTTCTTTCCG | CTTTGGTCTT | |
| FCRL2 (peak 5) | − | 3.92E−05 | CTTCATTCTC | TTTCTTTTCCTT | TAGCAAGAGT |
| FCRL2 (peak 1) | − | 4.31E−05 | GCCCTGATTG | TTCTGCTTTCAC | ATACACCTGC |
| MARCKS (peak 4) | − | 4.68E−05 | TCCATGTCCT | TTCAGCTTCCCC | ACAAATGGCC |
| CXCR7 (peak 12) | − | 5.76E−05 | A | TTTTTCATTCTC | AGCTTTCGCT |
| MARCKS (peak 3) | − | 6.19E−05 | AATAACTGTC | TCTACCTTTCTC | GAATTGTCTA |
| CXCR7 (peak 1) | + | 6.19E−05 | TAACACCCCC | TCCCATTTCCCC | CAGTAACGGT |
| CXCR7 (peak 12) | + | 6.73E−05 | ATTCGGGGTT | TTTTCTTTTCTT | AAGAAAGCGA |
| CXCR7 (peak 4) | + | 7.39E−05 | ATTGGG | TTTGCCTTTCTC | TAGTTCTCTA |
| CXCR7 (peak 16) | − | 8.49E−05 | GCTCTCATGG | TTCTATGTCCTC | ACCATTCCA |
| CXCR7 (peak 12) | − | 9.21E−05 | CTTTCTCTAC | TTTCACTTCATT | GTAAA |
| ZAP70 (peak 1) | − | 1.16E−04 | ACCAACACAG | TTTCATTTCCAG | ATTTCTTTCC |
| CXCR7 (peak 13) | − | 1.67E−04 | AACCCACATC | TCTCTCTCCCCC | GTGCCACCCC |
| FCRL2 (peak 7) | − | 1.78E−04 | GGTTCTTGCT | TTTTTCTTCTAC | TACAACTGAT |
| MARCKS (peak 2) | + | 1.90E−04 | GTGTTTCATT | TCTAACATTCTC | CTGACCTGCC |
| CXCR7 (peak 8) | + | 2.52E−04 | GGAAACAATG | TTATACTTCTCC | TTTTCAG |
| CXCR7 (peak 7) | + | 4.52E−04 | CGATGCGATG | TTTTGCTCCCGT | ACCAGAATTT |
EBNA-2 binding sites identified by ChIP sequencing analysis in the Mutu III BL cell line with MACS with P values of <10−7 annotated by peak number across the gene locus.
+, the motif is found in the DNA sequence provided; −, the motif is found in the reverse complement of the sequence.
The P value for a site was computed from the match score for the site with the position-specific scoring matrix for the enriched motif.
The LMP1 promoter sequence from positions +1 to −320 was also included in the sequence set.
ADAMDEC1 peak 1 did not meet the MACS significance cutoff but was included on the basis of ChIP-qPCR evidence of EBNA-2 binding.
ChIP-qPCR assays were carried out following 24 h of CdCl2 induction of EBNA-2, as time course experiments revealed (data not shown) that this was the optimum time to observe differential effects of type 1 and type 2 EBNA-2. Western blot analysis demonstrated that similar levels of type 1, type 2, and type 2 S442D EBNA-2 were precipitated by the EBNA-2 PE2 monoclonal antibody in ChIP samples (Fig. 5D). ChIP-qPCR analysis detected significantly increased binding of type 1 EBNA-2 compared to type 2 EBNA-2 at two representative EBNA-2 binding sites (marked MACS peaks 16 and 13 in Fig. 6A): at the CXCR7 locus (Fig. 6B and C) and at the LMP-1 promoter (Fig. 6D). In contrast, the binding of both type 1 and type 2 EBNA-2 to a site at the equally regulated CCL3 gene was equivalent, indicating gene specificity (Fig. 6E). In these assays, the binding of type 2 S442D EBNA-2 at both the LMP-1 promoter and the binding site at CXCR7 peak 16 was stronger than that of type 2 EBNA-2 but weaker than that of type 1 EBNA-2 (Fig. 6B and D). Binding of type 2 EBNA-2 at CXCR7 peak 13 was not increased by the S442D mutation (Fig. 6C).
Our data therefore suggest that a reduced transactivation function and a reduced gene-specific binding of type 2 EBNA-2 contribute to the impaired activation of specific viral and cell genes by type 2 EBNA-2. In combination, these effects likely result in the impaired growth-transforming function of type 2 EBNA-2. The S442D mutation in type 2 EBNA-2 restores the transactivation function and partially rescues the reduced gene-specific binding observed for type 2 EBNA-2 at some binding sites.
An EICE is enriched in EBNA-2 binding regions of genes that are differentially regulated by EBNA-2 types.
EBNA-2 does not bind directly to DNA in a sequence-specific manner but accesses DNA sites by associating with a cellular partner protein. RBP-Jκ (CSL) is the best characterized of these, although EBNA-2 has also been shown to interact with PU.1 (Spi-1) in vitro and pull down the protein from extracts (7). In fact, in addition to an RBP-Jκ site, binding sites for PU.1 and EBF1 in the LMP-1 promoter are also required for activation of LMP-1 expression by EBNA-2 (7, 10, 25).
To identify sequences that might characterize the EBNA-2 response elements of genes that are more strongly induced by type 1 EBNA-2 than type 2 EBNA-2, we used MEME-ChIP to perform unbiased motif searching on EBNA-2 binding peaks identified from ChIP sequencing experiments carried out in Mutu III BL cells (11). For this analysis, we identified significant EBNA-2 binding peaks around a set of nine previously identified (13) differentially regulated genes (CXCR7, ADAMDEC1, IL1B, MARCKS, CCL3L3, HES1, ZAP70, FCRL2, IL-4) and nine equally regulated genes (GCET2 [GCSAM], CD69, BATF, FCRL3, CCR7, HEY1, CCL3, RUNX3, DTX1). The LMP-1 promoter region from positions +1 to −320 was also included in the analysis of differentially regulated gene regulatory elements. This analysis returned only one significantly enriched motif in the EBNA-2 binding sites from differentially regulated genes (Fig. 7) and no enriched motifs for the equally regulated gene binding sites. Remarkably, this unbiased analysis identified a consensus motif that occurs 31 times in the set of binding sites analyzed from differentially regulated genes, including one occurrence in the LMP-1 promoter (Table 1). In fact, the motif found in the LMP-1 promoter is the site previously shown to bind PU.1 and a factor previously termed LBF4 (7) (Table 1). This element represents an overlapping binding site for ETS (e.g., PU.1/Spi-1) and IRF transcription factors (EICE), the best-known example of which is located in the IgL(λ) enhancer (5′-AAAAGGAAGTGAAACCA-3′) (26). The GGAA motif represents a core ETS binding site and is combined with an overlapping IRF site (AAXXGAAA). The enriched motif identified in our analysis closely resembles the IgL(λ) enhancer site (Fig. 7). The remaining 30 examples of the enriched motif in the differentially regulated cell gene EBNA-2 binding regions distributed as follows: MARCKS (n = 11), FCRL2 (n = 4), ADAMDEC1 (n = 1), CCL3L3 (n = 1), HES1 (n = 2), ZAP70 (n = 1), and CXCR7 (n = 10). Ten motifs were identified at the CXCR7 locus distributed across seven of the EBNA-2 binding peaks identified by ChIP sequencing analysis (Fig. 6A and Table 1), including CXCR7 MACS peaks 13 and 16 that were analyzed in the ChIP experiment whose results are shown in Fig. 6B and C. These data therefore indicate that a reduced ability of type 2 EBNA-2 to activate gene expression via this motif may be a key factor that leads to the reduced transforming ability of type 2 EBV.
FIG 7.
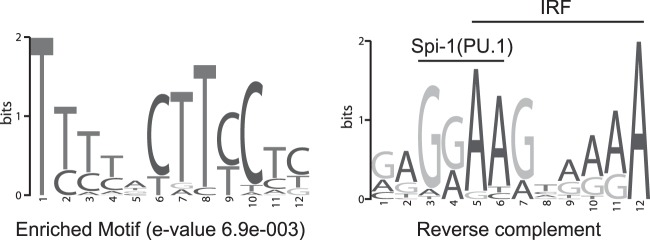
Enriched motif generated by MEME-ChIP analysis of EBNA-2 binding sites at nine differentially regulated cell genes and the EBV LMP-1 promoter shown in standard form (left) and as the reverse complement (right). The vertical scale in bits represents the relative frequency of each base as a fraction of 2. The overlapping PU.1 and IRF motifs that constitute an EICE are indicated. Details of the matching sites are shown in Table 1.
DISCUSSION
The results presented in this paper indicate that EBNA-2 regulation of the differentially regulated cell promoters is likely to be similar to that of LMP-1 and involve regulation through a motif that includes a binding site for ETS family members, such as PU.1 and IRF proteins. A combination of the higher transactivation domain activity mediated by D442 in type 1 EBNA-2, the greater binding of type 1 EBNA-2 at specific gene regulatory elements, and interaction with cell factors that may mediate increased binding is likely to result in the increased expression of these genes and the superior growth of cells containing type 1 EBNA-2.
In future work, we hope to identify the biochemical composition of the complexes that form at these EBNA-2 binding sites and also mutate the sites to demonstrate directly that they mediate the gene regulation by EBNA-2. Since acidic amino acids are frequently present in TADs (27), it is perhaps not surprising that D442 should increase TAD activity, but a physiological phosphomimetic mechanism for the S442D change seems unlikely because the serine in type 2 EBNA-2 does not lie in any known consensus phosphorylation site protein sequence (28). At present it may seem surprising that an amino acid change in the transactivation domain can mediate gene-specific effects and increase the level of EBNA-2 bound in the ChIP assays whose results are shown in Fig. 6B and D. However, the domains of function of EBNA-2 at these sites may differ from those known from EBNA-2 interaction with RBP-Jκ, and additional protein interactions are likely to contribute to the levels of binding observed at promoters in the cell or viral chromosome.
Although PU.1 is able to bind the LMP-1 PU.1 site (7), there is no direct evidence that PU.1 (Spi-1) is the cell factor which mediates EBNA-2 binding at cell genes with motifs similar to the motif present in the LMP-1 promoter. It is possible that EBNA-2 interacts directly with PU.1, since a glutathione S-transferase–type 1 EBNA-2 fragment (amino acids 310 to 376) binds to in vitro-translated mouse PU.1 and was also able to deplete PU.1 from a human B cell extract (7), but we saw no requirement for the region from amino acids 310 to 376 from type 1 EBNA-2 in our chimera growth assays. The role of D442 in type 1 EBNA-2 would therefore not appear to be indirectly contributing to an interaction with PU.1. It is, however, possible that this residue contributes to the binding of type 1 EBNA-2 to another factor that can also activate gene expression via this site, such as an IRF protein that also binds the composite element or another member of the ETS family.
Some protein interactions of EBNA-2 have already been mapped to the TAD region, but these do not seem to have been mapped to the exact region on which we have focused. Amino acids 448 to 471 were found to be sufficient to mediate the interaction between type 1 EBNA-2 and the Tfb1/p62 subunit of TFIIH (29), and W458 of B95-8 type 1 EBNA-2 (equivalent to W454 of the W91 isolate [30]) was required for interaction with p300 and PCAF (31). The presence of the additional acidic amino acid in the TAD of type 1 EBNA-2 could, more likely, contribute to the characteristic relatively nonspecific protein-protein interaction function of TADs and thereby stabilize the assembly of transcription complexes with coactivators, such as histone acetyltransferases and the transcription machinery. The specific involvement of the EICE motif that we have identified could reflect a particular need for the binding and assembly of a stable ETS/IRF complex at the LMP-1 and cell genes that are activated less well by type 2 EBNA-2 lacking this residue. PU.1 is known to be an important pioneer factor in opening up chromatin sites (32), so there may be specific features of the chromatin around these genes that would explain a higher dependency on the factors that associate with EICEs.
In the study described in this paper, we tested the function of EBNA-2 alleles in the context of type 2 EBNA leader protein (EBNA-LP) and EBNA-3 genes. We previously considered whether there could be a type-specific cooperation with EBNA-LP but could find no evidence for that (12). Since EBNA-2 and EBNA-3 family proteins are frequently found at the same chromosomal locations in BL ChIP sequencing and can compete for binding sites (11), it remains to be determined whether type-specific EBNA-3 effects could also contribute to the virus phenotype. In the future, it will be important also to test the S442D mutation in a recombinant EBV strain to determine whether it can reconstitute the process of B cell transformation, but we have already shown the effects of the S442D change in two different assays in EREB2.5 cells and in Daudi cells, in which it modulated the expression of LMP-1 and CXCR7, as was seen with type-specific virus infection (12). It would also be interesting to mutate the EICE motifs that we have identified in some of the cell gene promoters to test directly whether they mediate the differential regulation of gene expression by type 1 and type 2 EBNA-2 observed. This might be done using new clustered regularly interspaced short palindromic repeat or transcription activator-like effector nuclease techniques (33), but the fact that a large number of EBNA-2 binding sites have been identified at many of the target gene loci and the strong possibility of redundancy in these elements would make this a lengthy and technically challenging undertaking.
It is interesting that a single amino acid in EBNA-2 determines the growth maintenance phenotype when there are so many amino acid differences between type 1 and type 2 EBNA-2. From an evolutionary point of view, once the DNA sequence is sufficiently different to prevent homologous recombination, the variants tend to persist in the population, so it is not necessary for all the amino acid differences to be subject to selection to maintain the characteristic type 1 and type 2 EBNA-2 sequences. The biological significance of type 1 and type 2 EBV is still not understood; some possibilities are that their origin might relate to the evolutionary history of EBV, to major histocompatibility complex selection, or to the very high levels of other infectious diseases prevalent in sub-Saharan Africa. There is some evidence that type 1 EBV is better at transforming resting B cells than activated B cells (34), so it is possible that a difference in EBNA-2 could allow the persistence of type 2 in populations with a chronically activated immune system. Type 2 EBV has also very recently been reported to be able to infect T cells (R. Rochford, personal communication), so some of the sequence differences may relate to other phenotypes that are not apparent in the B cell system that we have studied. However, the difference in B cell growth transformation efficiency remains the clearest example of functional variation in Epstein-Barr virus types.
In this paper, we have shown that a combination of the higher transactivation domain activity mediated by D442, the greater binding of type 1 EBNA-2 at differentially regulated gene loci, and a potential influence on cell factors that interact with ETS-IRF composite elements that may mediate the greater binding is likely to determine the increased expression of these genes and the superior growth of cells infected with viruses containing type 1 EBNA-2.
ACKNOWLEDGMENTS
M.J.M. was funded by a Ph.D. studentship from the Biotechnology and Biological Sciences Research Council, and Leukemia and Lymphoma Research supported M.J.W. (project grant 12035). L.C. was supported by a Gordon Piller studentship from Leukemia and Lymphoma Research.
Footnotes
Published ahead of print 21 May 2014
REFERENCES
- 1.Tsai MH, Raykova A, Klinke O, Bernhardt K, Gartner K, Leung CS, Geletneky K, Sertel S, Munz C, Feederle R, Delecluse HJ. 2013. Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep. 5:458–470. 10.1016/j.celrep.2013.09.012 [DOI] [PubMed] [Google Scholar]
- 2.White RE, Ramer PC, Naresh KN, Meixlsperger S, Pinaud L, Rooney C, Savoldo B, Coutinho R, Bodor C, Gribben J, Ibrahim HA, Bower M, Nourse JP, Gandhi MK, Middeldorp J, Cader FZ, Murray P, Munz C, Allday MJ. 2012. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J. Clin. Invest. 122:1487–1502. 10.1172/JCI58092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kieff E, Rickinson A. 2007. Epstein-Barr virus, p 2603–2654 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 4.Tzellos S, Farrell PJ. 2012. Epstein-Barr virus sequence variation—biology and disease. Pathogens 1:156–174. 10.3390/pathogens1020156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rickinson AB, Young LS, Rowe M. 1987. Influence of the Epstein-Barr virus nuclear antigen EBNA 2 on the growth phenotype of virus-transformed B cells. J. Virol. 61:1310–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen JI, Wang F, Mannick J, Kieff E. 1989. Epstein-Barr virus nuclear protein 2 is a key determinant of lymphocyte transformation. Proc. Natl. Acad. Sci. U. S. A. 86:9558–9562. 10.1073/pnas.86.23.9558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johannsen E, Koh E, Mosialos G, Tong X, Kieff E, Grossman SR. 1995. Epstein-Barr virus nuclear protein 2 transactivation of the latent membrane protein 1 promoter is mediated by J.kappa and PU.1. J. Virol. 69:253–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sjoblom A, Jansson A, Yang W, Lain S, Nilsson T, Rymo L. 1995. PU box-binding transcription factors and a POU domain protein cooperate in the Epstein-Barr virus (EBV) nuclear antigen 2-induced transactivation of the EBV latent membrane protein 1 promoter. J. Gen. Virol. 76:2679–2692. 10.1099/0022-1317-76-11-2679 [DOI] [PubMed] [Google Scholar]
- 9.Sjoblom A, Yang W, Palmqvist L, Jansson A, Rymo L. 1998. An ATF/CRE element mediates both EBNA2-dependent and EBNA2-independent activation of the Epstein-Barr virus LMP1 gene promoter. J. Virol. 72:1365–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao B, Zou J, Wang H, Johannsen E, Peng CW, Quackenbush J, Mar JC, Morton CC, Freedman ML, Blacklow SC, Aster JC, Bernstein BE, Kieff E. 2011. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc. Natl. Acad. Sci. U. S. A. 108:14902–14907. 10.1073/pnas.1108892108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McClellan MJ, Wood CD, Ojeniyi O, Cooper TJ, Kanhere A, Arvey A, Webb HM, Palermo RD, Harth-Hertle ML, Kempkes B, Jenner RG, West MJ. 2013. Modulation of enhancer looping and differential gene targeting by Epstein-Barr virus transcription factors directs cellular reprogramming. PLoS Pathog. 9:e1003636. 10.1371/journal.ppat.1003636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancian L, Bosshard R, Lucchesi W, Karstegl CE, Farrell PJ. 2011. C-terminal region of EBNA-2 determines the superior transforming ability of type 1 Epstein-Barr virus by enhanced gene regulation of LMP-1 and CXCR7. PLoS Pathog. 7:e1002164. 10.1371/journal.ppat.1002164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lucchesi W, Brady G, Dittrich-Breiholz O, Kracht M, Russ R, Farrell PJ. 2008. Differential gene regulation by Epstein-Barr virus type 1 and type 2 EBNA2. J. Virol. 82:7456–7466. 10.1128/JVI.00223-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spender LC, Lucchesi W, Bodelon G, Bilancio A, Karstegl CE, Asano T, Dittrich-Breiholz O, Kracht M, Vanhaesebroeck B, Farrell PJ. 2006. Cell target genes of Epstein-Barr virus transcription factor EBNA-2: induction of the p55alpha regulatory subunit of PI3-kinase and its role in survival of EREB2.5 cells. J. Gen. Virol. 87:2859–2867. 10.1099/vir.0.82128-0 [DOI] [PubMed] [Google Scholar]
- 15.Kempkes B, Spitkovsky D, Jansen-Durr P, Ellwart JW, Kremmer E, Delecluse HJ, Rottenberger C, Bornkamm GW, Hammerschmidt W. 1995. B-cell proliferation and induction of early G1-regulating proteins by Epstein-Barr virus mutants conditional for EBNA2. EMBO J. 14:88–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein E, Klein G, Nadkarni JS, Nadkarni JJ, Wigzell H, Clifford P. 1968. Surface IgM-kappa specificity on a Burkitt lymphoma cell in vivo and in derived culture lines. Cancer Res. 28:1300–1310 [PubMed] [Google Scholar]
- 17.Klein G, Lindahl T, Jondal M, Leibold W, Menezes J, Nilsson K, Sundstrom C. 1974. Continuous lymphoid cell lines with characteristics of B cells (bone-marrow-derived), lacking the Epstein-Barr virus genome and derived from three human lymphomas. Proc. Natl. Acad. Sci. U. S. A. 71:3283–3286. 10.1073/pnas.71.8.3283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yates J, Warren N, Reisman D, Sugden B. 1984. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc. Natl. Acad. Sci. U. S. A. 81:3806–3810. 10.1073/pnas.81.12.3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bark-Jones SJ, Webb HM, West MJ. 2006. EBV EBNA 2 stimulates CDK9-dependent transcription and RNA polymerase II phosphorylation on serine 5. Oncogene 25:1775–1785. 10.1038/sj.onc.1209205 [DOI] [PubMed] [Google Scholar]
- 20.Palermo RD, Webb HM, West MJ. 2011. RNA polymerase II stalling promotes nucleosome occlusion and pTEFb recruitment to drive immortalization by Epstein-Barr virus. PLoS Pathog. 7:e1002334. 10.1371/journal.ppat.1002334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McClellan MJ, Khasnis S, Wood CD, Palermo RD, Schlick SN, Kanhere AS, Jenner RG, West MJ. 2012. Downregulation of integrin receptor-signaling genes by Epstein-Barr virus EBNA 3C via promoter-proximal and -distal binding elements. J. Virol. 86:5165–5178. 10.1128/JVI.07161-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Machanick P, Bailey TL. 2011. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27:1696–1697. 10.1093/bioinformatics/btr189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen JI, Kieff E. 1991. An Epstein-Barr virus nuclear protein 2 domain essential for transformation is a direct transcriptional activator. J. Virol. 65:5880–5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fahraeus R, Jansson A, Sjoblom A, Nilsson T, Klein G, Rymo L. 1993. Cell phenotype-dependent control of Epstein-Barr virus latent membrane protein 1 gene regulatory sequences. Virology 195:71–80. 10.1006/viro.1993.1347 [DOI] [PubMed] [Google Scholar]
- 25.Laux G, Adam B, Strobl LJ, Moreau-Gachelin F. 1994. The Spi-1/PU.1 and Spi-B ets family transcription factors and the recombination signal binding protein RBP-J.kappa interact with an Epstein-Barr virus nuclear antigen 2 responsive cis-element. EMBO J. 13:5624–5632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eisenbeis CF, Singh H, Storb U. 1995. Pip, a novel IRF family member, is a lymphoid-specific, PU.1-dependent transcriptional activator. Genes Dev. 9:1377–1387. 10.1101/gad.9.11.1377 [DOI] [PubMed] [Google Scholar]
- 27.Hope IA, Mahadevan S, Struhl K. 1988. Structural and functional characterization of the short acidic transcriptional activation region of yeast GCN4 protein. Nature 333:635–640. 10.1038/333635a0 [DOI] [PubMed] [Google Scholar]
- 28.Ubersax JA, Ferrell JE., Jr 2007. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8:530–541. 10.1038/nrm2203 [DOI] [PubMed] [Google Scholar]
- 29.Chabot PR, Raiola L, Lussier-Price M, Morse T, Arseneault G, Archambault J, Omichinski JG. 2014. Structural and functional characterization of a complex between the acidic transactivation domain of EBNA2 and the Tfb1/p62 subunit of TFIIH. PLoS Pathog. 10:e1004042. 10.1371/journal.ppat.1004042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen JI, Wang F, Kieff E. 1991. Epstein-Barr virus nuclear protein 2 mutations define essential domains for transformation and transactivation. J. Virol. 65:2545–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Grossman SR, Kieff E. 2000. Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc. Natl. Acad. Sci. U. S. A. 97:430–435. 10.1073/pnas.97.1.430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghisletti S, Natoli G. 2013. Deciphering cis-regulatory control in inflammatory cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368:20120370. 10.1098/rstb.2012.0370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaj T, Gersbach CA, Barbas CF., III 2013. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31:397–405. 10.1016/j.tibtech.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aman P, Gordon J, Lewin N, Nordstrom M, Ehlin-Henriksson B, Klein G, Carstensson A. 1985. Surface marker characterization of EBV target cells in normal blood and tonsil B lymphocyte populations. J. Immunol. 135:2362–2367 [PubMed] [Google Scholar]
- 35.Spender L, Cannell E, Hollyoake M, Wensing B, Gawn J, Brimmell M, Packham G, Farrell P. 1999. Control of cell cycle entry and apoptosis in B lymphocytes infected by Epstein-Barr virus. J. Virol. 73:4678–4688 [DOI] [PMC free article] [PubMed] [Google Scholar]