

ABSTRACT
Neonatal immune responses to infection and vaccination are biased toward TH2 at the cost of proinflammatory TH1 responses needed to combat intracellular pathogens. However, upon appropriate stimulation, the neonatal immune system can induce adult-like TH1 responses. Here we report that a new class of vaccine adjuvant is especially well suited to enhance early life immunity. The GVI3000 adjuvant is a safe, nonpropagating, truncated derivative of Venezuelan equine encephalitis virus that targets dendritic cells (DCs) in the draining lymph node (DLN) and produces intracellular viral RNA without propagating to other cells. RNA synthesis strongly activates the innate immune response so that in adult animals, codelivery of soluble protein antigens induces robust humoral, cellular, and mucosal responses. The adjuvant properties of GVI3000 were tested in a neonatal BALB/c mouse model using inactivated influenza virus (iFlu). After a single immunization, mice immunized with iFlu with the GVI3000 adjuvant (GVI3000-adjuvanted iFlu) had significantly higher and sustained influenza virus-specific IgG antibodies, mainly IgG2a (TH1), compared to the mice immunized with antigen only. GVI3000 significantly increased antigen-specific CD4+ and CD8+ T cells, primed mucosal immune responses, and enhanced protection from lethal challenge. As seen in adult mice, the GVI3000 adjuvant increased the DC population in the DLNs, caused activation and maturation of DCs, and induced proinflammatory cytokines and chemokines in the DLNs soon after immunization, including gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), granulocyte colony-stimulating factor (G-CSF), and interleukin 6 (IL-6). In summary, the GVI3000 adjuvant induced an adult-like adjuvant effect with an influenza vaccine and has the potential to improve the immunogenicity and protective efficacy of new and existing neonatal vaccines.
IMPORTANCE The suboptimal immune responses in early life constitute a significant challenge for vaccine design. Here we report that a new class of adjuvant is safe and effective for early life immunization and demonstrate its ability to significantly improve the protective efficacy of an inactivated influenza virus vaccine in a neonatal mouse model. The GVI3000 adjuvant delivers a truncated, self-replicating viral RNA into dendritic cells in the draining lymph node. Intracellular RNA replication activates a strong innate immune response that significantly enhances adaptive antibody and cellular immune responses to codelivered antigens. A significant increase in protection results from a single immunization. Importantly, this adjuvant also primed a mucosal IgA response, which is likely to be critical for protection during many early life infections.
INTRODUCTION
The World Health Organization (WHO) estimates approximately 2 million deaths in neonatal and infant humans (<1 year of age) every year worldwide due to acute infections caused by a limited number of pathogens (1). The availability of effective early life vaccines against those agents would have a significant impact on disease burden in neonates and infants, who are especially vulnerable to infectious diseases and in whom the immune responses generated by most currently available early life vaccines are suboptimal. The need for effective early life vaccines is especially important in resource-poor countries, where the period immediately after childbirth is often the only point of contact with the health care system.
A major obstacle in the development of early life vaccines is that the neonatal immune system is geared more toward a TH2 response at the cost of TH1 responses needed to combat intracellular pathogens (2, 3). The neonatal antibody response to conventional subunit and live attenuated vaccine antigens is of limited magnitude and duration, and CD8+ T-cell responses also are reduced compared to adults (4–6). The predisposition of the neonatal immune system toward a TH2 response is caused by the suboptimal innate immune response, with delayed maturation of neonatal dendritic cells (DCs) and limited production of inflammatory cytokines, which leads to inefficient antigen presentation and stimulation of naive T cells (7–10). Therefore, many vaccines that are effective in adults are poorly immunogenic in early life, hence requiring multiple booster immunizations (5).
Influenza viruses cause millions of annual infections worldwide, with up to 40,000 deaths reported in the United States alone (11). Newborns and infants are at higher risk for influenza-related mortality because of their immature immune systems, which can often lead to severe viral pneumonitis or bacterial superinfection (12). Protection from influenza viruses, like most respiratory viruses, is optimally conferred through virus-specific antibodies, such as IgG and IgA. However, antibody-mediated protection does not fully protect against heterologous strain infection due to the variability in the surface glycoproteins (13). On the other hand, T-cell epitopes are highly conserved across influenza virus strains, and a robust T-cell response can induce broader protection (14). Currently available influenza vaccines—tetravalent inactivated (TIV) or live attenuated (LAIV)—are not recommended for use in infants who are less than 6 months old (15). While LAIV immunization has been more effective in young children (6 to 11 months) compared to TIV immunization, it has been associated with safety concerns, such as increased rate of wheezing and hospitalization (16, 17). Therefore, the development of inactivated influenza vaccines, with improved efficacy in infants less than 6 months of age is urgently needed. In this study, we chose inactivated influenza virus as a model antigen.
Studies have shown that upon appropriate stimulation, the otherwise “immature” neonatal immune system has the capacity to induce adult-like TH1 immune responses and protect against infections (2). The best example of a neonatal TH1 vaccine is the Mycobacterium tuberculosis bacillus Calmette-Guerin (BCG) vaccine against tuberculosis, which induces strong, adult-like gamma interferon (IFN-γ) responses and a protective TH1 response in newborns. The WHO has recommended the BCG vaccine for immunization immediately after birth (18). An increased interest in developing and testing TH1-inducing adjuvants for early life immunizations has resulted in a number of studies in human cord blood cells and in neonatal animal models, using cytokines and ligands of Toll-like receptors (TLRs). Stimulation of CD4+ T cells from human umbilical cord blood with recombinant interleukin 12 (IL-12) resulted in the induction of adult-like IFN-γ responses (19). TLR7/8 agonists, R848, or TLR8 agonist VTX-294 showed TH1 responses, indicated by production of cytokines tumor necrosis factor alpha (TNF-α), IL-1β, and IL-12 secreted by antigen-presenting cells (APCs) from newborn cord blood (20, 21). CpG, a TLR9 agonist, can induce adult-like DC and T-cell activation, while failing to induce adult-like antibody (Ab) responses (22). IC31 (Intercell AG), a two-component adjuvant consisting of an antibacterial peptide and the TLR9 agonist ODN1a, when combined with a pneumococcal conjugate vaccine, enhances protective immunity in neonatal mice (23). A synthetic water-in-oil emulsion-based adjuvant, CRL-8941 (24), and complete Freund's adjuvant (CFA) (25), induced adult-like TH1 immune responses in neonatal mice, indicated by induction of antigen-specific IgG2a antibodies (an indicator of a TH1 response in BALB/c mice) and/or IFN-γ-secreting splenocytes. However, CRL-8941 was reported by the authors to cause “significant local toxicity” in neonatal mice, while CFA is universally toxic. Therefore, there is a continued need for safe and effective adjuvants that can promote strong, balanced, and protective newborn immune responses to new and existing vaccines. The two adjuvants currently approved as components of human vaccines, alum and MF59, are both inducers of TH2-polarized immune responses (26). Therefore, an adjuvant promoting a TH1 response might be useful in inducing a balanced vaccine response by the already TH2-biased neonatal immune system.
In this study, we investigated the effectiveness of a new class of adjuvants in early life vaccination. The novel adjuvant GVI3000 is derived from an alphavirus genome. Venezuelan equine encephalitis (VEE) virus is a positive-sense, single-stranded RNA virus representative of the alphavirus genus. Its ∼11.5-kb RNA genome encodes four nonstructural proteins and three structural proteins, the latter expressed from a 26S subgenomic promoter. Although the adjuvant properties of attenuated VEE virus have been suggested for some time (27, 28), our group has developed and characterized propagation-incompetent VEE replicon particles with strong adjuvant activity (29), and analogous adjuvants have been derived from Semliki Forest virus (30) and Sindbis virus (R. Mikkelsen and R. E. Johnston, unpublished data). VEE adjuvant particles have been referred to as nVRP (29, 31) or GVI3000 (32), and they contain a truncated VEE virus genome that includes the 5′ and 3′ untranslated regions (UTRs) and the nonstructural genes but lack the 26S subgenomic promoter and the structural protein genes. When inoculated into adult mice by subcutaneous, intradermal, or intramuscular routes with a number of soluble (ovalbumin [OVA] or keyhole limpet hemocyanin [KLH]) or inactivated particulate antigens (norovirus, poliovirus, or influenza viruses), these adjuvant particles can enhance humoral, cellular, and mucosal immune responses (29, 31–33). Additionally, enhanced protection against influenza and dengue viruses was demonstrated in a macaque model (34, 35). Upon subcutaneous immunization of mice, GVI3000 particles enter Langerhans cells (LC) at the site of inoculation where viral RNA replication and LC activation presumably occurs (36). These infected immune cells then rapidly migrate to the draining lymphoid node(s) (DLN). Alternatively, GVI3000 particles can migrate directly to DLNs, where they preferentially target DCs (36, 37). In both human- and mouse-derived DCs, in vitro studies have shown an upregulation of costimulatory molecules as well as secretion of proinflammatory cytokines, such as type I IFN, IL-6, and TNF after infection (37–39). In adult mice, cytokine secretion was observed systemically and in the DLNs as soon as 6 h after GVI3000 inoculation (unpublished observation). Tonkin et al. (40) demonstrated that DCs infected ex vivo with GVI3000, when inoculated into adult BALB/c mice, were sufficient to enhance in vivo systemic, mucosal, and cellular responses. Furthermore, the immune cells most frequently targeted and recruited to the draining lymph node were monocyte-derived inflammatory DCs.
On the basis of the properties of the GVI3000 adjuvant, we predicted that it might be well suited as a TH1-polarizing vaccine adjuvant to enhance neonatal immunization. We hypothesize that GVI3000 can induce a sufficiently strong DC activation to overcome the neonatal limitation to induce a Th1 response, hence promoting an “adult-like immune response,” shifting the neonatal immune system to a more balanced TH1-TH2 response, inducing mucosal and T-cell responses, and improving vaccine efficacy in neonates.
In this study, we used a 7-day-old BALB/c neonatal mouse model, which has been reported to more closely resemble the stage of immune maturation and immune-function limitations in human neonates (5). We immunized 7-day-old and adult mice with inactivated influenza virions (iFlu) in the presence of GVI3000 adjuvant and compared the effect of the adjuvant on induced adaptive and innate immune responses after a single immunization of the two age groups. Antigen-specific antibody responses were significantly enhanced by the adjuvant and lasted for at least several months. iFlu with the GVI3000 adjuvant (GVI3000-adjuvanted iFlu) was also able to induce antigen-specific T cells in the DLNs of immunized neonates and primed a mucosal IgA response. The addition of the GVI3000 adjuvant to the iFlu vaccine resulted in a significant increase in protection from a lethal challenge. Furthermore, as observed in adults, inflammatory DCs were the most frequently recruited and targeted immune cells in the DLNs. The proinflammatory innate immune response profile was similar to that in immunized adult mice, indicating that GVI3000 exploits similar pathways in both neonates and adults and can overcome any shortcomings in the neonatal immune system. These findings support the conclusion that codelivery of GVI3000 and an inactivated vaccine can significantly improve immune responses and protection against challenge in neonatal mice.
MATERIALS AND METHODS
Cells.
Vero81 cells were obtained from the American Type Culture Collection (ATCC). The Vero81 cells were maintained at 37°C in Dulbecco's modified Eagle's medium (DMEM) with nutrient mixture F12 medium supplemented with 10% fetal calf serum, penicillin-streptomycin (100 U/ml), and streptomycin (100 μg/ml) in the presence of 5% CO2. L929 mouse fibroblast cells (ATCC CCL-1) were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), penicillin-streptomycin (100 U/ml), and 0.29 mg/ml of l-glutamine. Chicken red blood cells (CRBCs) were obtained from Charles River Laboratories (catalog no. 10100767) and stored at 4°C for hemagglutination inhibition assays.
Viruses and vaccine antigens.
Live influenza A/PR/8/34 virus (catalog no. 10100374) and formalin-inactivated influenza virus A/PR/8/34 (iFlu) (catalog no. 10100782) were obtained from Charles River Laboratories and stored at −80°C until use.
Adjuvant VEE replicon particles (GVI3000).
The production of GVI3000 adjuvant particles, also known as nVRP, has been described previously (31, 41, 42). Briefly, in vitro-transcribed VEE replicon RNA genome, together with two helper RNAs—expressing viral structural genes in trans—were coelectroporated into Vero81 cells. GVI3000 particles were collected in the culture medium 24 h after electroporation. These particles contain only replicon RNA and exclude helper RNAs, which lack the virus-specific packaging signal, rendering these particles propagation incompetent. Cytopathic effect testing was performed on each production batch to confirm the absence of detectable propagation-competent virus that could arise as a contaminant due to RNA recombination. The adjuvant particles were concentrated by ultracentrifugation through a 20% sucrose cushion, suspended in phosphate-buffered saline (PBS) containing 1% human serum albumin, and stored at −80°C until use. The titers of GVI3000 infectious particles were then determined by infection of Vero cells as measured by immunofluorescence staining of VEE nonstructural proteins, using polyclonal sera from mice immunized with VEE nonstructural protein 2.
Two modalities of VEE replicon RNA were packaged in this study: (i) GVI3000 replicon RNA, which encodes the 5′ and 3′ untranslated regions (UTRs) flanking the viral nonstructural genes but lacks the sequence between the nsP4 stop codon (5 nucleotides [nt] before the 26S mRNA transcription start site) and the beginning of the 118-nt 3′ UTR (31); (ii) GVI3000-GFP replicon RNA, which encodes the viral nonstructural protein genes and also the sequence for green fluorescent protein (GFP) under the control of the viral 26S subgenomic promoter. GVI3000-GFP was used as reporter particles to identify GVI3000-infected cells. All known cis-acting signals for RNA replication were included in the GVI3000 genomes. All replicon particles were packaged using wild-type VEE (V3000) envelope sequences (43).
Mice and immunizations.
Pregnant and adult BALB/c mice were purchased from Charles River and were housed at the Global Vaccines Inc. animal facility. All studies were carried out in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals (44). All animal protocols were approved by the Global Vaccines, Inc. Institutional Animal Care and Use Committees (IACUC) prior to performing experiments. Pregnant mice were closely monitored for date and approximate time of delivery.
Seven days after birth, neonatal mice were injected in both rear footpads with PBS or one of the following formulations: iFlu alone, iFlu mixed with alum (Alhydrogel [2%]; Invivogen) or iFlu mixed with GVI3000. The immunization volume was 10 μl (total) (5 μl in each rear footpad). Mice were immunized once unless otherwise stated. Adult control mice (6 to 8 weeks old) were immunized in both rear footpads (10 μl/footpad) after being anesthetized by intraperitoneal (i.p.) injection with a 4/1 mixture (vol/vol) of ketamine (50 μg/g of body weight) and xylazine (15 μg/g of body weight). Mice were observed daily for a week for any adverse reaction to the immunization.
Flu challenge.
The mouse flu challenge model used here has been described elsewhere (45). Three weeks after neonatal immunization, mice were challenged with 106 50% egg infectious doses (EID50) of influenza virus strain PR/8/34 (Charles River, MA). The challenge dose represented the lowest dose that was 100% lethal in control mice immunized with diluent, and it was optimized in a separate experiment. The virus was diluted in a total volume of 50 μl, and 25 μl was introduced into each naris under light isoflurane anesthesia. Mice were observed for 11 days for weight, morbidity, and mortality. The animals were monitored for clinical signs of disease, including ruffling, hunching, signs of dehydration (concave dorsal surface), and reduced spontaneous mobility. To assess protection from postchallenge influenza virus replication in the nose, mice were neonatally immunized, and 5 weeks later, mice were challenged intranasally with 106 EID50 of influenza virus strain PR/8/34. At 48 h postchallenge, mice were euthanized, and nasal tissues were harvested from the tip of the nose to the anterior of the eye sockets, as described before (29, 46). Nasal tissue was collected in 10% FBS medium and centrifuged, followed by storage in TRIzol at −80°C until RNA isolation.
RNA isolation and real-time PCR.
RNA extraction from nasal tissues was performed using the PureLink RNA minikit (Ambion) according to the manufacturer's protocol. RNA samples were then treated with DNase using the Turbo DNA-free kit (Ambion), following the manufacturer's protocol. Quantification of influenza virus strain PR8 hemagglutinin (HA) negative-strand RNA levels in the nasal tissues was performed by reverse transcription-PCR (RT-PCR) as has been described previously (47). Briefly, cDNAs were prepared using reverse transcription with oligo(dT). RT-PCR was performed using TaqMan gene expression assays (Applied Biosystems) and gene-specific primers and probes (Life Technologies). The samples were then run on an ABI Prism 7000 real-time PCR system. During each reaction, a cycle threshold (CT) value for the target gene of interest was generated, which was then normalized to the CT value of the housekeeping gene (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]) yielding a ΔCT value. The ΔCT value of the nVRP-infected sample was then subtracted from the ΔCT value of the antigen-only sample, resulting in a ΔΔCT value. Fold increase in mRNA expression in nVRP group relative to the expression level in antigen-only group were determined by using the formula 2ΔΔCT.
The HA-specific primers (forward [ACTGGACCTTGCTAAAACCC] [starting at nt 772]; reverse [CATTGATGCGTTTGAGGTGATG]) and probes containing a 5' fluorescent reporter (6-FAM), a 3' quencher (IABkFQ), and an internal quencher (ZEN) (/6-FAM/CCCAAAGCC/ZEN/TCTACTCAGTGCGAA A/3IABkFQ/) for RT-PCR were designed and obtained from Integrated DNA Technologies (IDT). Additionally, PR8 HA negative-strand RNA standards were prepared from the influenza virus stock used for infection and run on each sample plate. For the analysis, CT value from each sample was plotted on the standard curve to obtain PR8 viral titer. All CT values were normalized to the 18S housekeeping gene.
Analysis of antigen-specific IgG in serum and IgA in fecal extracts by ELISA.
Mouse serum samples were collected at 3, 6, 9, and 15 weeks after immunization and stored at −20°C. For preparation of fecal extracts, fecal pellets collected 10 to 14 days after the booster immunization were suspended at 0.2 g/ml and disrupted by vortex mixing at 4°C in PBS containing 10% goat serum and 1× protease inhibitors (catalog no. 11873580001; Roche). Samples were centrifuged, and supernatants were filtered through 0.22-μm filters. Antigen-specific IgG and IgA antibodies were detected by enzyme-linked immunosorbent assay (ELISA) on 96-well high binding plates (Thermo Scientific) coated with 4 μg/ml iFlu in PBS. Sera and fecal extracts were added to plates in serial dilutions. Antigen-specific antibodies were detected with horseradish peroxidase-conjugated antibodies specific for mouse IgG (Southern Biotech) or mouse IgA (Southern Biotech) followed by the addition of SureBlue (3,3′,5,5′-tetramethylbenzidine [TMB] microwell peroxidase substrate; KPL) for 30 min. Endpoint titers were determined as the last sample dilution that generated an optical density at 450 nm (OD450) reading of greater than 0.2. For determination of total IgA levels in fecal extracts, 96-well plates were coated with 0.4 μg/ml rabbit anti-mouse IgA (Invitrogen), ELISAs were performed as described above, and a standard curve was generated from dilutions of purified murine IgA (Sigma). This standard curve was used to determine the concentration of both antigen-specific and total IgA in fecal extracts.
HAI.
Influenza virus-specific hemagglutination inhibition assay (HAI) has been described elsewhere (48). Complement was inactivated by heating the serum sample at 56°C for 30 min. Mouse serum was then preadsorbed with 1% suspension of chicken red blood cells resulting in a 1:5 dilution. Briefly, four hemagglutination units (HAU) of influenza virus strain PR8 in 50 μl was mixed with 2-fold serial dilutions of preadsorbed mouse serum (50 μl per dilution) and incubated at room temperature for 30 min (starting dilution 1:10). Fifty microliters of a 1% suspension of CRBCs was then added to each well. In serum dilutions with anti-HA antibody, virus was bound by antibody and was not available to agglutinate the CRBCs. The maximal reciprocal serum dilution where agglutination was completely inhibited is reported as the HAI titer.
IFN-αβ bioassay.
The levels of biologically active type I IFN in mouse sera were determined using an interferon bioassay as previously described (49). Briefly, L929 mouse fibroblasts were seeded in 96-well plates. Mouse serum samples, including the standards, were acidified to a pH of 2.0 overnight, then neutralized to a pH of 7, and added to cells in serial 2-fold dilutions. After 24-h incubation, 105 PFU of encephalomyocarditis virus (EMCV) was added to each well. Twenty-four hours postinfection, cell viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma), and the absorbance was read on a microplate reader at 570 nm. To obtain type I IFN value in international units per milliliter (IU/ml), the absorbance from each sample was compared to a dilution series of IFN-α standard (catalog no. NR-3076; BEI) present in each plate.
Surface staining of cells from draining lymph nodes (DLNs).
At the indicated time points, both draining popliteal lymph nodes from each animal were harvested, combined, and homogenized through 40-μm cell strainers. Cells were washed, counted by using a hemocytometer, and stained at 4°C for the desired surface receptors with a selection of the following fluorochrome-conjugated antibodies specific for CD3, CD4, CD8, CD11b, CD11c, CD40, CD80, CD86, major histocompatibility complex class II (MHCII) (eBioscience), Ly-6G and Ly-6C (BD Bioscience) in PBS containing 1% bovine serum albumin (1% BSA/PBS). After the cells were stained, they were washed and then fixed in 2% paraformaldehyde in PBS for 15 min at room temperature. The cells were analyzed on an Accuri C6 flow cytometer (BD).
IFN-γ ELISPOT assay.
IFN-γ enzyme-linked immunospot (ELISPOT) assay was performed to measure influenza virus PR8 HA-specific IFN-γ-secreting CD4 and CD8 cells in the popliteal draining lymph nodes and spleens of immunized mice. Mouse IFN-γ capture antibody (R&D Systems) was incubated on nitrocellulose membrane plates (96 well; Millipore) overnight at 4°C. Before the addition of cells, the plates were washed and blocked for at least 2 h with complete RPMI 10 (10% fetal bovine serum) at 37°C. Single-cell suspensions in RPMI 10 (2.5 × 105 or 5 × 105 cells per well) from either popliteal draining lymph node or spleen were then added to the plates. Additionally, feeder cells (2.5 × 105 cells per well) from a naive adult BALB/c mouse spleen were added to all wells. Finally, CD4 (HA from amino acids 110 to 120 [HA 110-120]; SFERFEIFPKE) and CD8 (HA 518-526; IYSTVASSL) immunodominant HA peptides from AnaSpec were added to the appropriate cells for 36 h. The cells were removed from plates and washed, a biotinylated mouse IFN-γ detection antibody (R&D Systems) was added to the plates, and the plates were incubated for another 36 h at 4°C. Membranes were again washed, incubated with a streptavidin-alkaline phosphatase conjugate for 2 h at room temperature. The plates were washed, and spots were developed following the addition of 5-bromo-4-chloro-3-indolylphosphate (BCIP)/Nitro Blue Tetrazolium (NBT) substrate. The plates were scanned by Cellular Technologies Ltd. (Shaker Heights, OH). Assay background values were obtained from sample wells with no peptide/protein stimulation and were subtracted from the values obtained with the influenza virus HA peptide. Data are normalized to the number of antigen-specific IFN-γ-secreting cells per 106 cells.
TCID50 assay.
Virus titers in the lungs were determined by 50% tissue culture infectious dose (TCID50) assay on MDCK cells. Lungs collected on day 3 postchallenge were homogenized, serially diluted 10-fold, and then added in quadruplicate to cells in a 96-well plate. After 48 h of incubation, an influenza hemagglutination assay using 5% chicken red blood cells was performed on the medium from each well. TCID50 titers were determined using the Reed and Muench calculator (50).
Multiplex analysis for cytokine expression in the DLNs.
To determine cytokine levels in the DLNs of mice immunized with iFlu alone or with GVI3000, the draining popliteal lymph nodes from each mouse were harvested, combined, and placed in 100 μl of PBS containing 1× protease inhibitors (catalog no. 11873580001; Roche). The lymph nodes were mechanically homogenized with a pestle, followed by centrifugation at 4°C. Supernatant was transferred to another tube and frozen on dry ice. Cytokine levels in the samples were determined by Luminex-based multiplex assay. For one experiment, the levels of 18 cytokines were tested using the Milliplex MAP mouse cytokine/chemokine magnetic bead panel (EMD Millipore, Billerica, MA). The assay was performed by the Center for Gastrointestinal Biology and Disease's Immunotechnologies Core at the University of North Carolina at Chapel Hill. Multianalyte profiling was performed on a Bio-Plex 200 system with an XY platform and high-throughput fluidics (Bio-Rad Laboratories, Hercules, CA). Calibration and validation microspheres for classification and reporter readings, as well as optics and fluidics verification, were also obtained from Bio-Rad. The instrument's sheath fluid was prepared from concentrate obtained from Luminex (Luminex Corporation, Austin, TX). Fluorescence data were acquired using Bio-Plex Manager 6.0 software, and all cytokines were successfully detected. Data analysis was performed using the same software, and a best-fit standard curve was obtained using either four- or five-parameter regression, from which the sample concentrations were derived. Cytokines that were undetectable were assigned a value of half of the lowest limit of detection as listed in the Milliplex MAP mouse cytokine/chemokine magnetic bead panel kit manual.
Statistical analysis.
Antibody titers and cytokine values were evaluated for statistically significant differences by either the analysis of variance (ANOVA) or Mann-Whitney test (GraphPad Prism). Statistical significance is reported as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
RESULTS
GVI3000 safely enhances humoral immune responses to inactivated flu virus (iFlu) in neonatal mice.
To determine the adjuvant effect of GVI3000 on humoral responses to iFlu in neonatal mice, we assessed the magnitude, duration, and quality of serum antibodies and induction of fecal antibody responses by iFlu alone or formulated with the GVI3000 adjuvant. Seven-day-old BALB/c mice from one litter received a single immunization with 2 μg of iFlu mixed with GVI3000 (105 IU), while mice from a second litter were immunized with 2 μg of iFlu only. The mice were monitored daily for local reactogenicity, morbidity, and mortality. No adverse effects of immunization with iFlu alone or with GVI3000 (iFlu-GVI3000) were observed. Over the past several years, approximately 2,000 adult mice and over 350 neonatal mice have been immunized with GVI3000 adjuvant without any observable signs of adverse reaction (29, 31, 32, 40, 46, 51, 52; D. Tonkin, B. Steil, and P. Jorquera, personal communications). Influenza virus-specific IgG ELISA antibody titers in the iFlu-only group were modest and did not increase between 6 and 15 weeks postimmunization (Fig. 1A). In the GVI3000-adjuvanted group, the titers were significantly higher for all time points tested compared to the iFlu-only group. From week 6 to week 15, the titers in GVI3000-adjuvanted mice continued to increase significantly (P < 0.05) compared to week 3 (Fig. 1A). Additionally, week 15 titers were significantly higher than week 6 titers. The influenza virus-specific IgG2a titers (indicator of TH1 response) in the GVI3000-adjuvanted immunization group were significantly higher at 3, 6, and 15 weeks (Fig. 1B), while the IgG1 (indicator of TH2 response) titers were comparable between the groups (Fig. 1C), suggesting that the GVI3000 adjuvant-mediated enhanced IgG response was predominantly in the IgG2a subclass. We performed our studies in the BALB/c strain of mice, although previous results from our lab have shown that adult C57BL/6, 129 Sv/Ev, and other strains of mice produce a similar TH1-biased response upon GVI3000 immunization.
FIG 1.

GVI3000 is an effective adjuvant to iFlu after a single dose in mice immunized as neonates. Three to five 7-day-old BALB/c mice per experimental group were primed through footpad (f.p.) injection with iFlu (2 μg) with 105 IU of GVI3000 (black bars) or without GVI3000 (white bars). (A to C) Three, 6, 9, and 15 weeks after the immunization, influenza virus-specific IgG (A) and IgG1 (B) and IgG2a (C) antibodies were measured in serum by ELISA. (D) GVI3000 induces mucosal immune response in neonatally primed mice. Five or six 7-day-old BALB/c mice were primed through f.p. injection with iFlu (1 μg) only or with 105 IU of GVI3000. Ten weeks after the mice were primed, all groups were boosted with iFlu (1 μg) alone. Ten days after the boost, antigen-specific IgA were measured in fecal extracts by ELISA. The amount of IgA produced is presented as nanograms of influenza virus-specific IgA per microgram of total IgA. (E) Antibodies with influenza virus hemagglutination inhibition activity were measured by hemagglutination inhibition assay (HAI) at 6 weeks after the immunization. All data are presented as means plus standard errors of the means (SEM) (error bars). Values that are significantly different from the value for iFlu alone by the Mann-Whitney test are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.01.
Previous studies have suggested the important role mucosal immunity plays in protecting against influenza virus, which primarily infects and provokes inflammation at respiratory mucosal sites (53). A unique feature of the GVI3000 adjuvant is its ability to induce—in adult mice—an antigen-specific mucosal IgA antibody response after a nonmucosal route of immunization (29, 46). We chose to measure the IgA response in the fecal pellets of immunized mice, as IgA detection in fecal pellets is often a more reliable and unambiguous measure of mucosal response than examining nasal or tracheal lavage fluid. Additionally, it is known that mice have a common mucosal system, in which responses at one mucosal tissue likely mirrors those in other mucosal sites (54). Published studies of adult BALB/c mice have shown that induction of antigen-specific IgA in GVI3000-adjuvanted mice was significantly enhanced in fecal extracts, similar to the increase in the nasal epithelium (29, 51). To address whether GVI3000 can prime the induction of antigen-specific mucosal IgA in early life immunizations, neonatal mice were primed with iFlu alone or with iFlu plus GVI3000, and 10 weeks later, they were boosted with iFlu antigen only. The amount (nanogram of influenza virus-specific IgA/microgram of total IgA) of influenza virus-specific IgA antibody found in fecal materials 10 days after the boost was measured by ELISA (Fig. 1D). Presenting the IgA data as nanogram of influenza virus-specific IgA/microgram of total IgA allows for normalization of the fecal pellet size, which can vary between the groups of mice, genders, and within the same group sampled at different times, making it a reliable and accurate measure (55). While the signal for iFlu-only induction of IgA was at the background level, a significantly larger amount of influenza virus-specific IgA was observed in mice that had GVI3000 adjuvant in the prime. Importantly, the amount of influenza virus-specific IgA in the adjuvanted group was not significantly lower than that found in adult mice (1.59 ng in neonates compared to 1.74 ng influenza virus-specific IgA per μg total IgA in adults), when mice from both age groups were compared in the same experiment (data not shown).
To further assess the quality of the influenza virus-specific antibodies being induced, we measured the ability of the antibodies in the sera to inhibit hemagglutination of chicken red blood cells using an HA binding inhibition assay (HAI). We included an experimental group adjuvanted with alum (Alhydrogel, 1:5), a well-characterized benchmark adjuvant, formulated as indicated by the manufacturer. Six weeks after immunization, HAI titers in the GVI3000-adjuvanted group (geometric mean titer [GMT] = 24) were significantly higher than those in the iFlu-only group (GMT = 10). The HAI titer in the alum-adjuvanted group (GMT = 16) was not significantly different from either GVI3000 or no-adjuvant groups (Fig. 1E). At other time points (3 and 15 weeks), we did not observe any difference in HAI titers between the immunization groups (data not shown).
In summary, GVI3000 adjuvant significantly enhanced antibody responses to iFlu in neonatally immunized mice. Enhanced serum antibodies were mostly of the IgG2a class and had HAI activity. While antibody titers continued to increase during the 15 weeks of study, these titers were of much lower magnitude than those induced by the same vaccine regime in adult mice (unpublished observation). While this was true for serum antibodies after one dose, the ability of the adjuvant to prime a mucosal response seemed as effective in neonates as in adults.
GVI3000 adjuvant induces antigen-specific CD4 and CD8 T cells after a single immunization in neonatal mice.
While the contribution of T-cell immunity to protection against influenza remains a matter of debate, the flu model was utilized to determine whether GVI3000 can enhance the induction of antigen-specific T cells in neonatal mice. We chose to examine the T-cell responses in the DLNs (popliteal) as opposed to the more distal LNs (such as mediastinal). T-cell responses are present in distal LNs at reduced but still detectable levels relative to the DLN responses. In these initial neonatal studies of immunity induced by GVI3000, it was prudent to first evaluate responses in the DLNs where we anticipate the most robust response, with the expectation that immunity detected in one tissue will correlate with immune memory throughout the animal. Groups of three 7-day-old mice were immunized in both rear footpads with iFlu alone (2 μg) or with iFlu mixed with GVI3000 (105 IU). In this experiment, a group adjuvanted with alum was also included. Eight days after immunization, animals were sacrificed, and single-cell suspensions were prepared from the pooled (two per mouse) popliteal DLNs. Cells were evaluated for the presence of IFN-γ-secreting CD8 and CD4 T cells following stimulation with immunodominant HA peptides. We chose to detect HA-specific T cells instead of T cells to other antigens (such as nucleoprotein [NP]), as we were not sure which proteins, other than HA, were intact in the inactivated influenza virus antigen. Furthermore, based on our experience, adult BALB/c mice are capable of inducing T cells specific for multiple antigens after a multivalent immunization, indicating that the T-cell response will most likely be not limited to HA antigen only. Animals receiving iFlu alone produced undetectable to very low levels of influenza virus-specific CD4+ and CD8+ T cells. However, iFlu formulated with GVI3000 adjuvant resulted in a significant increase of both CD4+ and CD8+ T cells (Fig. 2). Although the alum group had low but detectable amounts of CD4+ T cells and CD8+ T cells, they were significantly lower than the GVI3000-immunized group. Taken together, these results strongly suggest that the GVI3000 adjuvant can promote the induction of a CD8+ T-cell response in the TH2-polarized neonatal immune system.
FIG 2.
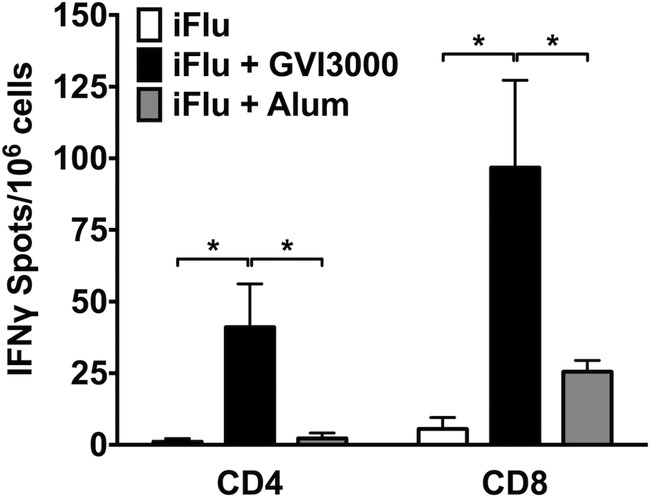
GVI3000 adjuvant promotes influenza virus-specific cellular immune response in DLNs from neonatal mice after a single neonatal immunization. Three 7-day-old BALB/c mice were primed in both rear footpads (f.p.) with iFlu (2 μg) in the presence or absence of 105 IU of GVI3000. As a control adjuvant, three mice from one litter were immunized with alum (1:2 by volume). Eight days after immunization, both popliteal draining lymph nodes from each mouse were harvested and pooled. DLN cells from each experimental group were stimulated with immunodominant CD4 and CD8 HA peptides followed by IFN-γ release ELISPOT assay. Data were normalized to the mock-stimulated positive spots for each experimental group and graphed as the number of IFN-γ-positive spots per 106 DLN cells. Data are presented as means plus SEM. Values that are significantly different (P < 0.05) from the value for iFlu alone by the Mann-Whitney test are indicated by a bar and asterisk.
Neonatal mice immunized with GVI3000-adjuvanted iFlu vaccine have reduced morbidity and mortality and reduced early postchallenge virus titers in nose and lungs.
Two independent flu challenge experiments were performed. In the first experiment, four litters of mice were immunized on day 7 after birth with either PBS (n = 8), iFlu only (n = 5), iFlu with GVI3000 (105 IU) (n = 5), or iFlu formulated with alum (1:5) (n = 7). Three weeks after immunization, the mice received a lethal challenge by intranasal inoculation with 106 EID50 of influenza virus A/PR/8/34. The mice were monitored daily for 11 days for weight loss, morbidity, and mortality. Figure 3A shows the survival curves. All the PBS-immunized control mice succumbed to the challenge. All mice that received a single GVI3000-adjuvated immunization were protected. Three out of 7 mice that received alum-adjuvanted iFlu antigen survived, while 2 out of 5 mice in the group immunized with iFlu only survived. A repeat challenge experiment using a different stock of live influenza virus was done with three immunization groups: PBS control (n = 6), iFlu only (n = 11), and iFlu with GVI3000 (n = 11). In this experiment, mice were monitored for 25 days. No protection was observed in PBS-immunized control mice or iFlu-only-immunized mice, while 7 out of 11 mice survived in the GVI3000-adjuvanted group (Fig. 3B). Taken together, the two experiments demonstrate that the GVI3000-adjuvanted vaccine outperforms vaccine alone but that the vaccine adjuvanted with alum was no more protective than vaccine alone. Postchallenge weight change relative to the starting weight is shown in Fig. 3C to F for the first challenge experiment. In the GVI3000 adjuvant group, one mouse showed no weight loss, although the remaining animals lost ∼12% of their starting weight on average before recovering completely; none of the mice showed any overt clinical signs of disease. In the group given alum, the three mice that survived the challenge showed ruffling and hunching. Taken together, these results demonstrate that GVI3000-adjuvanted vaccine mediated significant protection from influenza virus-induced morbidity and mortality in neonates.
FIG 3.

GVI3000-adjuvanted iFlu protects neonatally immunized mice from a lethal influenza virus challenge. Neonatal mice were immunized with iFlu only (n = 5) or with either GVI3000 (105 IU) (n = 5) or alum (1:5) (n = 7). Included in the challenge were PBS-immunized mice (n = 8). All mice were intranasally challenged with a lethal dose of 106 EID50 of influenza A/PR/8/34 virus at 3 weeks after immunization. (A to F) Mice were monitored for mortality (A and B) and weight (C to F) for 7 days after the challenge.
The effect of neonatal immunization on replication of challenge virus in nasal tissue was detected at 48 h postchallenge by real-time PCR. There was a significant decrease in virus titer in the GVI3000- and alum-adjuvanted groups compared to the iFlu-only-immunized group when mice were challenged 5 weeks after neonatal immunization (Fig. 4A). Additionally, we measured virus titers in the lung on day 3 postchallenge. Mice that received the GVI3000-adjuvanted vaccine showed no detectable TCID50 titers (level of detection = 43.1 TCID50/ml), which was a reduction of over 2 orders of magnitude compared to the titers from groups immunized with PBS (P < 0.01), iFlu only (P < 0.01), and iFlu plus alum (P < 0.05) (Fig. 4B). Although the titers in the alum-adjuvanted mice were significantly higher than those of GVI3000-adjuvanted mice, they were significantly lower than those of iFlu-immunized mice (P < 0.05).
FIG 4.
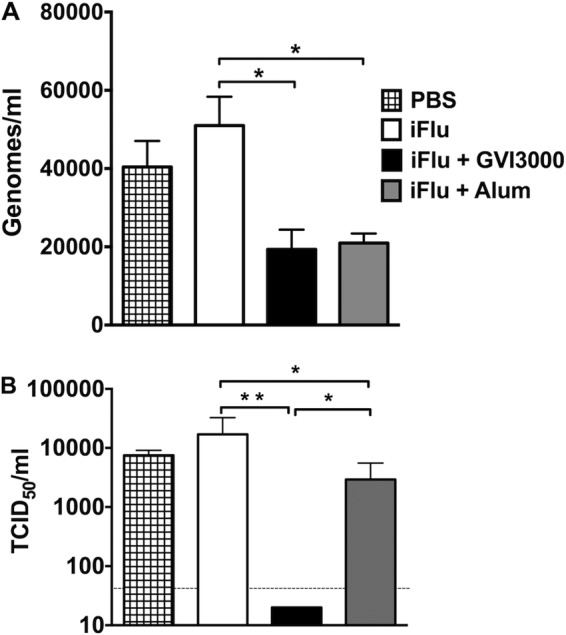
Viral titers in nasal tissues and lungs of challenged mice. Groups of five 7-day-old mice were immunized with either iFlu (1 μg) alone or with an adjuvant: GVI3000 (105 IU) or alum (Alhydrogel [1:5]). An additional PBS-immunized group was included. Five weeks after the single immunization, mice were challenged intranasally with 106 EID50 of influenza A/PR/8/34 virus. (A) Mice were euthanized 48 h postchallenge, and nasal tissues were collected in TRIzol. Following RNA extraction, real-time PCR was done with PR8 HA primer/probes. CT values were plotted on PR8 HA standard curve to calculate virus titer. (B) Mice were euthanized 3 days postchallenge, and their lungs were harvested. After homogenization of lung tissues, TCID50 assay was performed on MDCK cells. Cell supernatants were collected after 48 h of incubation, and hemagglutination assay was performed with 5% chicken red blood cells. TCID50/ml values were calculated using the Reed-Muench method (50). All data are presented as means plus SEM. Values that are significantly different by the Mann-Whitney test are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.01.
These results are consistent with the survival data shown in Fig. 3 and suggest that although alum provides comparable protection early in the nose, GVI3000 adjuvant is better at preventing replication in the lungs and preventing morbidity and mortality.
GVI3000 primarily targets DCs and promotes recruitment of multiple immune cell types into the DLNs of neonatal mice.
GVI3000 adjuvant induces recruitment and infection of APCs in the DLNs of adult mice. Although DCs are the primary targets of GVI3000, other immune cells are also targeted, and it is likely that the targeting of DCs and other APCs play a role in the adjuvant effect in adult mice (40, 46). Further investigation led to the findings that inflammatory DCs, although rare in the DLNs during steady state, were rapidly recruited to the site of inflammation and were the subsets that had the largest fold increase in immune cell population (40).
Given that the neonatal immune response is limited in part by a reduced number of immune cells, one important question was to determine the targets of GVI3000 in neonatal mice. Groups of five mice were immunized in both footpads with PBS, iFlu (1 μg) alone, or iFlu together with GVI3000 expressing GFP (GVI3000-GFP; 105 IU). GVI3000-GFP is identical to GVI3000 except that a subgenomic promoter driving the expression of GFP has been added downstream of NSP4. As GVI3000 cannot propagate to surrounding cells, and as infection of and RNA replication in the target cells must occur to allow GFP expression, cells containing GFP unequivocally identify GVI3000 target cells that support RNA replication (29). At 12 h postinfection (hpi), both popliteal DLNs were harvested from each neonatal mouse, combined, and homogenized. First, we measured the cellularity of the DLN, or total number of cells per lymph node by flow cytometry, using the volume analyzed by an Accuri C6 flow cytometer (BD) and back-calculating the number of cells in the starting sample. Neonates immunized with iFlu mixed with GVI3000-GFP had significantly higher DLN cellularity than neonatal mice immunized with iFlu alone (Fig. 5A). Previously published results in adult BALB/c mice showed a similar 2- to 3-fold increase in DLN cellularity in the presence of GVI3000 (40). Infiltrating immune cells were identified by antibody staining for the following surface markers: conventional DCs (cDCs) (CD11c+ CD11b−), inflammatory DCs (iDCs) (CD11c+ Ly6Chi) (Fig. 5B), macrophages (CD11b+ CD11c−), and neutrophils (Ly6G+). Except for the cDC population, which did not differ significantly between the groups, all immune cell types measured (iDCs, neutrophils, and macrophages) showed significant population increases at 12 hpi (Fig. 5C). The same was true for immune cell populations (B and T cells) at 24 hpi (data not shown). Inflammatory DCs showed the most significant increase (11-fold increase; P = 0.0079), and a similar increase was observed at 12, 18, and 24 hpi (data not shown).
FIG 5.

GVI3000 enhances inflammation in the DLNs of neonatal mice. Five 7-day-old BALB/c mice were immunized in both f.p. with PBS only or with iFlu (1 μg) in the presence of 105 IU of VEE replicon particles labeled with GFP (VRP-GFP) or absence of VRP-GFP. At 12 hpi, both popliteal DLNs from each neonatal mouse were harvested, combined, and manually disrupted into a single-cell suspension. (A) DLN cellularity was assessed by using the volume analyzed by an Accuri C6 flow cytometer (BD) and back-calculating the starting sample amount. (B) Representative histogram for inflammatory dendritic cell gating. (C) Using fluorescent antibody staining, immune cells were stained for DCs (CD11c+), inflammatory DCs (CD11c+ Ly6Chi), neutrophils (Ly6G+), and macrophages (CD11c− CD11b+). The total number of each immune cell present in the DLN sample is shown in the graph. Neut, neutrophils; Macs, macrophages. (D) Cells that were positive for GFP expression were then gated for the surface markers of different immune cells. In panels A and C, the data are presented as means plus SEM. Values that are significantly different by the Mann-Whitney test are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Values that are not significantly different (ns) are indicated.
To determine which cell populations were targeted by GVI3000 in the neonates, GFP-positive cells were quantified in the DLNs at 18 hpi. Although GFP expression was observed in several different immune cell types, iDCs were the predominant target of GVI3000 replication (Fig. 5D). Up to two-thirds of all cells expressing GFP were iDCs. Conventional DCs, macrophages, and neutrophils each represented 6 to 8% of all infected cells. Studies of adult BALB/c mice showed a similar infection pattern of cell populations (40). Approximately 11% of the GFP-positive cells were unidentified. Previous reports and unpublished observations in adult mice suggest that these unidentified cells may be T cells, B cells, and NK cells (40).
GVI3000 adjuvant activates neonatal dendritic cells.
The activation and maturation of infected and bystander DCs was identified by measuring the early in vivo expression of MHCII and the costimulatory molecules CD40, CD80, and CD86. Three groups of four neonatal mice were immunized with PBS, iFlu (1 μg) alone, or iFlu with GVI3000-GFP (105 IU). GVI3000-GFP served as an adjuvant to iFlu and as an expression vector for GFP. This marker was used to distinguish the activation state of infected versus bystander DCs in the GVI3000-adjuvanted group. Both DLNs were harvested from each mouse, homogenized, and stained at 18 hpi. After the cells were gated for inflammatory DCs, they were further gated for GFP expression and analyzed for MHCII, CD40, CD80, and CD86 (Fig. 6). The expression level of all activation/costimulatory markers (indicated by mean fluorescence intensity [MFI]) observed in the iFlu-only-immunized group did not differ from the PBS-immunized group. There were no differences in the MFI of activation/costimulatory markers between the iDCs of the antigen-only group and the GFP-negative iDCs in GVI3000-GFP-immunized group (Fig. 6A to D). However, iDCs that were GFP positive (i.e., GVI3000 infected) had significantly higher MHCII, CD40, CD80, and CD86 MFIs. The trend was similar for cDCs, with MHCII, CD80, and CD86 showing significantly more GFP-positive cells than GFP-negative cells and cDCs in the antigen-only-immunized group (data not shown). We believe that the GFP-positive cells detected in this experiment were due to direct infection by GVI3000-GFP and not due to phagocytosis of infected cells. It has been previously demonstrated in adult BALB/c mice that GVI3000-GFP infection leads to uniform GFP signaling in the cytoplasm instead of punctate GFP signaling, which would have indicated phagocytosis (36; L. White, unpublished observations).
FIG 6.

Dendritic cells are activated in VRP-GFP-adjuvanted mice. Three groups of four mice each were immunized with PBS, iFlu (2 μg) only, or iFlu (2 μg) with VRP-GFP (105 IU). Immune cells from the DLNs (two per mice per pool) were harvested and stained at 18 hpi for inflammatory DCs (CD11c+ Ly6Chi). The VRP-GFP-adjuvanted group had both GFP-positive (GFP +ve) and GFP-negative (GFP -ve) DCs. (A to D) Inflammatory DCs were gated for MHCII (A), CD40 (B), CD80 (C), and CD86 (D) surface markers. Data are presented as the mean fluorescence intensity (MFI) ± SEM. Values that are significantly different by the Mann-Whitney test are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
In summary, as seen in adult mice, GVI3000 targets and activates neonatal iDCs and cDCs. Bystander iDCs were not activated at 18 h in the DLNs of neonates receiving GVI3000 adjuvant. This seems to be different to what was reported ex vivo in bone marrow-derived DCs from adult mice, where bystander DCs were also activated (38).
GVI3000 induces several inflammatory cytokines in neonatal mice early after immunization.
Studies of adult mice have shown that the GVI3000 adjuvant can significantly enhance the inflammatory cytokine/chemokine response after immunization (31). These effector molecules appear within a few hours and return to basal levels within 24 to 36 h after exposure to GVI3000. It has been suggested that human neonatal plasmacytoid DCs are incapable of inducing type I IFNs similar to adult levels (56). Systemic type I IFN was induced to a high level in serum by 6 hpi in neonatal mice immunized with GVI3000-adjuvanted flu vaccine (Fig. 7). Furthermore, the response remains significantly higher than in antigen-only-immunized mice at least until 24 hpi. By 36 hpi, the level of type I IFN had ebbed to near basal levels. The kinetics of type I IFN induction is similar for the two antigens tested (iFlu and Fluzone 2011 vaccine). The levels and kinetics of these IFN responses were similar to those observed in control adult BALB/c mice (data not shown), indicating that 7-day-old mice are capable of adult-like type I IFN innate immune responses to GVI3000-adjuvanted antigens, including both rapid induction and return to basal levels.
FIG 7.
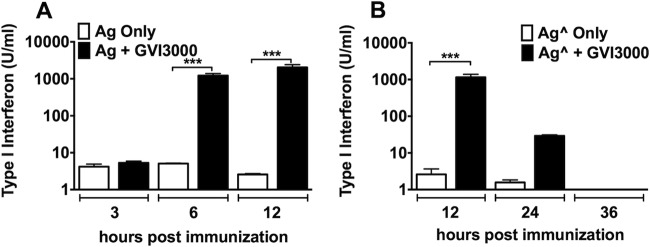
GVI3000 induces systemic type I interferon immune response in neonatal mice. At least four 7-day-old BALB/c mice were immunized by injecting the f.p. with Ag (iFlu; 1 μg) (A) or Aĝ (Fluzone; 1 μg) (B) alone or with 105 IU of GVI3000. At 3, 6, 12, 24, and 36 hpi, mice were terminally bled. Type I IFN response was measured by IFN bioassay. Data are presented as the mean plus SEM. Values that are significantly different by the Mann-Whitney test are indicated by bars and asterisks as follows: ***, P < 0.001.
To measure local induction of other inflammatory cytokines in immunized neonatal mice, an 18-plex assay was performed measuring levels of cytokine/chemokine proteins in the popliteal DLN 12 h after footpad immunization (Table 1). Several cytokines had 20- to 1,500-fold increases in the GVI3000-adjuvanted group compared to the group immunized with antigen only, including IFN-γ, interferon-inducible protein of 10 kDa (IP-10), macrophage chemoattractant 1 (MCP-1), and monokine induced by IFN-γ (MIG). Another group of cytokines, including granulocyte colony-stimulating factor (G-CSF), macrophage inflammatory protein 1β (MIP-1β), and RANTES (regulated on activation, normal T cell expressed and secreted) also had higher levels (5- to 15-fold) at 12 hpi. Adult DLNs analyzed at the same time showed a very similar trend, qualitatively and quantitatively, with granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-1β, and MIP-1β also showing >5-fold increases.
TABLE 1.
Cytokine levels in the DLNs of neonatal mice at 12 hpi
| Cytokine | Mean cytokine level (pg/ml ± SEM) in mice immunized with the following: |
Fold increase over iFlua | ||
|---|---|---|---|---|
| PBS | iFlu | iFlu + GVI3000 | ||
| G-CSF | 28.84 ± 1.55 | 29.92 ± 1.92 | 173.23 ± 7.66 | 5.79 |
| GM-CSF | 6.90 ± 0.57 | 11.21 ± 3.72 | 36.02 ± 1.64 | 3.21 |
| IFN-γ | 0.60 ± 0.00 | 0.60 ± 0.00 | 917.16 ± 102.16 | 1,528.60* |
| IL-1β | 4.10 ± 0.51 | 11.99 ± 1.72 | 25.78 ± 0.96 | 2.15 |
| IL-2 | 28.11 ± 1.71 | 30.44 ± 0.47 | 33.25 ± 1.18 | 1.09 |
| IL-4 | 0.26 ± 0.06 | 0.41 ± 0.10 | 1.54 ± 0.13 | 3.76 |
| IL-5 | 1.02 ± 0.52 | 4.94 ± 1.83 | 18.37 ± 0.91 | 3.72 |
| IL-6 | 32.21 ± 11.14 | 51.72 ± 2.81 | 148.99 ± 4.67 | 2.88 |
| IL-10 | 7.63 ± 4.20 | 9.60 ± 3.15 | 20.48 ± 2.20 | 2.13 |
| IL-12p70 | 2.40 ± 0.00 | 2.40 ± 0.00 | 2.40 ± 0.00 | 1.00 |
| IP-10 | 79.55 ± 7.78 | 99.66 ± 11.41 | 2731.05 ± 69.26 | 27.40* |
| MCP-1 | 8.07 ± 2.87 | 11.02 ± 3.55 | 230.22 ± 18.52 | 20.89* |
| MIG | 122.99 ± 30.75 | 73.29 ± 5.66 | 7338.89 ± 227.30 | 100.13* |
| MIP-1α | 11.77 ± 0.94 | 18.13 ± 0.98 | 55.51 ± 4.89 | 3.06 |
| MIP-1β | 7.02 ± 0.95 | 8.63 ± 0.90 | 134.45 ± 12.81 | 15.58 |
| MIP-2 | 75.43 ± 1.71 | 70.57 ± 2.70 | 96.66 ± 3.63 | 1.37 |
| RANTES | 7.02 ± 0.93 | 5.63 ± 1.05 | 39.78 ± 3.14 | 7.07 |
| TNF-α | 1.60 ± 0.00 | 1.60 ± 0.00 | 5.24 ± 0.66 | 3.28 |
Values that are statistically significantly different (P < 0.05) from the value for the group immunized with iFlu are indicated by an asterisk.
DISCUSSION
The data presented here demonstrate the feasibility of a new class of adjuvants for early life vaccination. The nonpropagating alphavirus replicon particle adjuvant GVI3000 was safe in neonatal mice, and when codelivered with iFlu antigen, enhanced multiple effector functions of the neonatal immune system, including antigen-specific IgG2a antibodies, mucosal IgA, and CD4+ and CD8+ T cells, indicating a shift from a TH2-polarized immune response to a balanced TH1/TH2 immune response that potentiated a significant increase in protection from a lethal influenza virus challenge. Many of the innate immune response cytokines and chemokines induced by GVI3000 in adult mice were also induced in neonatal animals. As in adults, DCs in neonatal mice were targeted and activated by the adjuvant particles, and a high proportion of inflammatory cells were recruited to the DLN. The relevances of these findings are severalfold. First, it is the first report, to our knowledge, to show a protective immune response to influenza in a neonatal animal model, suggesting that the alphavirus adjuvants presumptively could be used in infants. Second, a single immunization with the GVI3000-adjuvanted vaccine was sufficient to protect in this model, which represents a significant improvement over existing early life vaccines that require multiple immunizations, including current vaccines for influenza. This is especially relevant in poor countries, where birth and early life are often the only opportunities for patient contact with health care providers. Third, GVI3000 activates neonatal innate immunity and enhances all arms of the adaptive immune system, including the humoral, cellular, and mucosal compartments.
It is well established from studies of animal models and humans that the immune response to vaccination in neonates is weaker than that of adults (2, 5, 57, 58). Vaccination in neonates is characterized by (i) serum antibody titers that are lower in magnitude (59, 60) and of shorter duration (61, 62), (ii) APCs that show reduced upregulation of costimulatory molecules (63), (iii) T cells that exhibit a TH2 bias, secreting high levels of IL-4 and low levels of IFN-γ (2, 24, 64–66), and (iv) significantly limited cytotoxic T lymphocyte (CTL) responses to vaccines (reviewed in reference 5). In this study, we confirmed the age-related deficiencies in neonatal mice relative to adult mice in different areas of the adaptive and innate immune responses. After a single immunization with iFlu antigen, IgG Ab titers and HAI titers in neonates were significantly lower than those induced in adult mice. In addition, upon vaccination with antigen only, neonatal mice showed approximately 20-fold-fewer cells in the popliteal DLN than adult mice, including reduced numbers of iDCs, cDCs, neutrophils, and macrophages. Furthermore, the neonatal response to antigen alone was characterized by lower expression of most cytokines in a panel of 18 inflammatory cytokines and chemokines examined. Other deficiencies observed in the neonatal responses to iFlu, such as minimal to undetectable induction of CD4+ or CD8+ T cells and no mucosal IgA priming, were also present in the adult responses, and therefore, these deficiencies were in part determined by the nature of the antigen.
Although the overall magnitude of the serum Ab titers in the GVI3000-adjuvanted groups was still lower in neonates than in adults, the GVI3000 adjuvant was responsible for a significant increase in influenza virus-specific IgG antibodies, especially IgG2a, at 3, 6, and 15 weeks, and HAI Abs at 6 weeks after a single immunization. As seen in adult mice, GVI3000 enhanced influenza virus-specific IgG2a Abs (TH1). A similar result was obtained when neonatal mice were immunized with a GVI3000-adjuvanted inactivated dengue virus serotype 4 vaccine. This result was in clear contrast with the mainly IgG1 increase (TH2) observed when the dengue virus antigen was used as an adjuvant with alum (unpublished results), highlighting the qualitative differences between the two adjuvants. In the mucosal compartment, antigen-specific IgA was induced after two immunizations only when GVI3000 was present in the priming inoculation, with titers that were similar to those induced in mice primed as adults. Since we have previously demonstrated that unadjuvanted iFlu in adult mice does not induce mucosal IgA after one or two immunizations (J. M. Thompson and R. E. Johnston, unpublished data), our data suggest that the induction of mucosal IgA in neonatally primed mice is due to the primary immunization and not to the antigen-only boost. The present study assumes that the fecal IgA was produced at the mucosal surface (67), as has been observed with iFlu in adults (32, 46). Therefore, the GVI3000 adjuvant may be of particular benefit in neonatal vaccines against mucosal pathogens. In the neonatal TH2-polarized immune response, antigen-specific, IFN-γ-producing T cells are poorly induced by immunization with nonreplicating antigens (5). GVI3000 was able to overcome this deficiency after one immunization.
The presence of GVI3000 also resulted in improvement of protection in the intranasal lethal challenge model. However, in the second challenge experiment (Fig. 3B), not only the GVI3000-adjuvanted group but also the control unadjuvanted group had a higher number of lethal infections than in the first challenge experiment (Fig. 3A), implying that the second challenge was more stringent than the first one. This can be explained, as the two independent challenge experiments were performed with different challenge stocks (live influenza virus PR8) obtained separately from the same commercial vendor (Charles River, MA), and it is not unexpected that minor differences in virus titers may result in differences in virulence. Our results show that while GVI3000-adjuvanted neonatal mice had significantly improved protection compared to mice immunized with iFlu only, the use of alum as an adjuvant did not improve protection, despite similar functional HAI antibody titers at 3 weeks postimmunization. This implies that the improved protection mediated by GVI3000 may be attributed to immune effectors other than HAI antibodies. It has been shown previously that antibodies are effective in protection from homologous influenza virus challenge in adult mice (68–70). It is likely that in neonates, due to the weaker antibody response to iFlu (even with adjuvant), those antibody levels may not be sufficient for complete protection. Under these circumstances, and as suggested by our data, cellular and mucosal immune responses, if induced, could play a significant role in early life protection.
One of the reported deficiencies of the neonatal immune system is the limited quantity of immune cells available compared to adults (2, 71–75). Consistent with this, our observations show that the cellularity of the popliteal DLN of an unimmunized 7-day-old BALB/c mouse or an age-matched animal immunized with antigen only was lower than that for a 6-week-old adult mouse. In GVI3000 adjuvant-inoculated neonatal mice, we observed a significant 2- to 3-fold increase of DLN cellularity, which was similar to the percentage increase in cellularity induced by GVI3000 in adult mice reported by Tonkin et al. (40). Additionally, we observed that the total number of iDCs in GVI3000-adjuvanted neonatal DLNs accounted for approximately 8% of the total number of cells in the DLN, while it was <2% in the adult DLN (40). The robust recruitment and targeting of iDCs suggest an important role for these cells in the mediation of the GVI3000 adjuvant effect. DCs have been shown to be sufficient to mediate the GVI3000 adjuvant effect (40), but it is possible that other cell types complement the role of iDCs. We therefore cannot definitively point to a specific cell type that mediates the immune enhancement driven by GVI3000, but we predict that the ability of GVI3000 to trigger cell recruitment in neonates is an important component of this outcome.
We hypothesized that, as in adult mice, the adjuvant effect of GVI3000 in neonates was associated with the ability to (i) target and activate neonatal DCs and (ii) induce a robust TH1 innate immune response. Previous in vitro studies using VEE replicon particles (VRP) expressing GFP to infect adult human (38) and murine bone marrow-derived DCs (BMDCs) (37) showed an upregulation in costimulatory molecules CD40, CD80, and CD86. Additional studies using VRP-transduced human DCs showed DC maturation, secretion of proinflammatory cytokines, and significant expansion of antigen-specific T cells (38). In our in vivo system, we observed that both conventional and inflammatory DCs had significantly higher expression of MHCII, CD40, CD80, and CD86 after administration of GVI3000. However, this increase was observed only in DCs that were GVI3000 positive (GFP positive), but not in the “bystander DCs.” These results indicate that the infected DCs are the primary driver of the adjuvant effect of GVI3000 in the neonates. This finding is in contrast to what was described in in vitro studies of adult DCs, where both GVI3000-infected and bystander DCs showed upregulation of those markers (38). We can speculate that the absence of activation of bystander DCs in neonates at the time points examined may contribute to the reduced magnitude of the immune responses in neonates compared to adults. On the other hand, we cannot rule out the possibility that the “bystander” DCs would show a significant increase in expression of maturation and costimulatory molecules if examined at later times, after 18 h, as was observed in vitro at 24 h postinfection of human DCs (38). Further studies are needed to better understand the kinetics of DC maturation and expression of costimulatory molecules in vivo and its effect on T-cell response.
Our second hypothesis was that the significantly improved humoral and cellular GVI3000 adjuvant effect was associated with a robust TH1 cytokine response initiated early after immunization. Consistent with this hypothesis, a rapid, strong, and self-limited innate immune response was observed locally and systemically upon GVI3000 immunization, based on the rapid cytokine induction. The innate immune responses were consistent even when other antigens were used (data not shown), indicating that the GVI3000 adjuvant was primarily responsible for the enhanced response.
Our results indicate that GVI3000 can induce adult-like systemic and local innate immune responses in neonates. Although type I IFN function is not required for the adjuvant activity of GVI3000 in adult mice (51), it is an indicator of GVI3000 RNA replication and of a robust innate immune response. The observation that neonatal mice had adult-level type I IFN responses to GVI3000 indicates that the adjuvant may overcome the impairment in type I IFN induction observed in newborn cord blood (76, 77). Additionally, the type I IFN response was systemic, peaking at 12 h postimmunization and falling to the background level by 36 h. This demonstrates that the GVI3000-mediated inflammatory response is transient, reducing any chance of prolonged inflammation or toxicity. One of the reasons for immunizing neonatal mice in the footpad was the ability to investigate the process of inflammation in a single DLN. The results of cytokine expression analysis in the DLN further supported our second hypothesis in that the enhanced cellular immune response is preceded by a TH1-promoting innate immune response. As with systemic type I IFN expression, a similar pattern of peak expression of most cytokines at 12 hpi was observed with gradual reduction to the background level by 36 hpi (data not shown).
IFN-γ is a hallmark of a TH1 response; however, its production is impaired in newborns, as evidenced by the lack of IFN-γ induction in neonatal mice after subcutaneous injection of lipopolysaccharide (LPS) (78). BCG vaccination is one of the few examples of vaccines that can induce adult-like IFN-γ as has been demonstrated in CD4 T cells of 2-month-old human newborns that had been immunized at birth after stimulation with a mycobacterial purified protein derivative (79). IFN-γ protein expression was rapidly upregulated in neonatal mice after GVI3000 inoculation, which induced an adult-like response (adult data not shown). The IFN-γ-responsive chemokine IP-10 (chemokine [C-X-C motif] ligand 10 [CXCL10]) (80) was significantly upregulated in GVI3000-adjuvanted neonatal mice to a level similar to that in adult animals. As observed in adult mice, GVI3000 did not induce IL-12 protein expression, even though this cytokine is strongly associated with induction of IFN-γ and a TH1 response. Therefore, further investigation is needed to elucidate the role of IL-12, or lack thereof, in the GVI3000 adjuvant mode of action. Other cytokines that were upregulated but do not have a clearly defined role in TH1 responses were MCP-1, MIG, and MIP-1β. Overall, the cytokine expression pattern between neonates and adults was quite similar, indicating the following. (i) Neonates are capable of mounting an adult-like innate immune response. (ii) The mechanism whereby the GVI3000 adjuvant induces innate immune response may be similar in neonates and adults.
Improved technologies are urgently needed for vaccines against several infectious diseases, especially those affecting children in developing countries. This study demonstrates the potential of GVI3000 for early life vaccination.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant 5UO1-AI078060 awarded to L.J.W. by the National Institute of Allergy and Infectious Diseases (NIAID) (grant title “A tetravalent dengue vaccine based on Alphavirus replicons”). In addition, this work received support from grant NIAID 5R01-AI088250-04 awarded to R.E.J. (grant title “Role of innate immune responses in the activity of an Alphavirus based adjuvant”).
We thank Melissa Mattocks and Jill Whitley for alphavirus replicon particle production, Alex Schafer for RT-PCR reagents, Alan Whitmore for excellent technical help, Lance Blevins and Ellen Young for help with the TCID50 assay, and members of Global Vaccines Inc. for helpful discussions. Murine interferon alpha (MuIFN-α) (NR-3076) was obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH.
R.E.J. was a coinventor of the alphavirus adjuvant technology at the University of North Carolina and may receive a portion of any associated royalty payments to the university.
Footnotes
Published ahead of print 4 June 2014
REFERENCES
- 1.World Health Organization. 2014. World health statistics. World Health Organization, Geneva, Switzerland: http://apps.who.int/iris/bitstream/10665/112738/1/9789240692671_eng.pdf?ua=1 [Google Scholar]
- 2.Adkins B, Leclerc C, Marshall-Clarke S. 2004. Neonatal adaptive immunity comes of age. Nat. Rev. Immunol. 4:553–564. 10.1038/nri1394 [DOI] [PubMed] [Google Scholar]
- 3.Zaghouani H, Hoeman CM, Adkins B. 2009. Neonatal immunity: faulty T-helpers and the shortcomings of dendritic cells. Trends Immunol. 30:585–591. 10.1016/j.it.2009.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Langrish CL, Buddle JC, Thrasher AJ, Goldblatt D. 2002. Neonatal dendritic cells are intrinsically biased against Th-1 immune responses. Clin. Exp. Immunol. 128:118–123. 10.1046/j.1365-2249.2002.01817.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siegrist CA. 2001. Neonatal and early life vaccinology. Vaccine 19:3331–3346. 10.1016/S0264-410X(01)00028-7 [DOI] [PubMed] [Google Scholar]
- 6.Willems F, Vollstedt S, Suter M. 2009. Phenotype and function of neonatal DC. Eur. J. Immunol. 39:26–35. 10.1002/eji.200838391 [DOI] [PubMed] [Google Scholar]
- 7.de Brito CA, Goldoni AL, Sato MN. 2009. Immune adjuvants in early life: targeting the innate immune system to overcome impaired adaptive response. Immunotherapy 1:883–895. 10.2217/imt.09.38 [DOI] [PubMed] [Google Scholar]
- 8.Futata EA, Fusaro AE, de Brito CA, Sato MN. 2012. The neonatal immune system: immunomodulation of infections in early life. Expert Rev. Anti Infect. Ther. 10:289–298. 10.1586/eri.12.9 [DOI] [PubMed] [Google Scholar]
- 9.PrabhuDas M, Adkins B, Gans H, King C, Levy O, Ramilo O, Siegrist CA. 2011. Challenges in infant immunity: implications for responses to infection and vaccines. Nat. Immunol. 12:189–194. 10.1038/ni0311-189 [DOI] [PubMed] [Google Scholar]
- 10.Prendergast AJ, Klenerman P, Goulder PJ. 2012. The impact of differential antiviral immunity in children and adults. Nat. Rev. Immunol. 12:636–648. 10.1038/nri3277 [DOI] [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention. 2010. Estimates of deaths associated with seasonal influenza—United States, 1976–2007. MMWR Morb. Mortal. Wkly. Rep. 59:1057–1062 http://www.cdc.gov/mmwr/pdf/wk/mm5933.pdf [PubMed] [Google Scholar]
- 12.Barnard DL. 2009. Animal models for the study of influenza pathogenesis and therapy. Antiviral Res. 82:A110–A122. 10.1016/j.antiviral.2008.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stamboulian D, Bonvehi PE, Nacinovich FM, Cox N. 2000. Influenza. Infect. Dis. Clin. North Am. 14:141–166. 10.1016/S0891-5520(05)70222-1 [DOI] [PubMed] [Google Scholar]
- 14.Kohlmeier JE, Woodland DL. 2009. Immunity to respiratory viruses. Annu. Rev. Immunol. 27:61–82. 10.1146/annurev.immunol.021908.132625 [DOI] [PubMed] [Google Scholar]
- 15.Labella AM, Merel SE. 2013. Influenza. Med. Clin. North Am. 97:621–645. 10.1016/j.mcna.2013.03.001 [DOI] [PubMed] [Google Scholar]
- 16.Belshe RB, Edwards KM, Vesikari T, Black SV, Walker RE, Hultquist M, Kemble G, Connor EMCAIV-T Comparative Efficacy Study Group. 2007. Live attenuated versus inactivated influenza vaccine in infants and young children. N. Engl. J. Med. 356:685–696. 10.1056/NEJMoa065368 [DOI] [PubMed] [Google Scholar]
- 17.Zangwill KM, Belshe RB. 2004. Safety and efficacy of trivalent inactivated influenza vaccine in young children: a summary for the new era of routine vaccination. Pediatr. Infect. Dis. J. 23:189–197. 10.1097/01.inf.0000116292.46143.d6 [DOI] [PubMed] [Google Scholar]
- 18.Marchant A, Goetghebuer T, Ota MO, Wolfe I, Ceesay SJ, De Groote D, Corrah T, Bennett S, Wheeler J, Huygen K, Aaby P, McAdam KP, Newport MJ. 1999. Newborns develop a Th1-type immune response to Mycobacterium bovis bacillus Calmette-Guerin vaccination. J. Immunol. 163:2249–2255 [PubMed] [Google Scholar]
- 19.Wu CY, Demeure C, Kiniwa M, Gately M, Delespesse G. 1993. IL-12 induces the production of IFN-gamma by neonatal human CD4 T cells. J. Immunol. 151:1938–1949 [PubMed] [Google Scholar]
- 20.Dowling DJ, Tan Z, Prokopowicz ZM, Palmer CD, Matthews MA, Dietsch GN, Hershberg RM, Levy O. 2013. The ultra-potent and selective TLR8 agonist VTX-294 activates human newborn and adult leukocytes. PLoS One 8:e58164. 10.1371/journal.pone.0058164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy O, Suter EE, Miller RL, Wessels MR. 2006. Unique efficacy of Toll-like receptor 8 agonists in activating human neonatal antigen-presenting cells. Blood 108:1284–1290. 10.1182/blood-2005-12-4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kovarik J, Bozzotti P, Love-Homan L, Pihlgren M, Davis HL, Lambert PH, Krieg AM, Siegrist CA. 1999. CpG oligodeoxynucleotides can circumvent the Th2 polarization of neonatal responses to vaccines but may fail to fully redirect Th2 responses established by neonatal priming. J. Immunol. 162:1611–1617 [PubMed] [Google Scholar]
- 23.Olafsdottir TA, Lingnau K, Nagy E, Jonsdottir I. 2009. IC31, a two-component novel adjuvant mixed with a conjugate vaccine enhances protective immunity against pneumococcal disease in neonatal mice. Scand. J. Immunol. 69:194–202. 10.1111/j.1365-3083.2008.02225.x [DOI] [PubMed] [Google Scholar]
- 24.Barrios C, Brandt C, Berney M, Lambert PH, Siegrist CA. 1996. Partial correction of the TH2/TH1 imbalance in neonatal murine responses to vaccine antigens through selective adjuvant effects. Eur. J. Immunol. 26:2666–2670. 10.1002/eji.1830261118 [DOI] [PubMed] [Google Scholar]
- 25.Forsthuber T, Yip HC, Lehmann PV. 1996. Induction of TH1 and TH2 immunity in neonatal mice. Science 271:1728–1730. 10.1126/science.271.5256.1728 [DOI] [PubMed] [Google Scholar]
- 26.Kenney RT, Edelman R. 2003. Survey of human-use adjuvants. Expert Rev. Vaccines 2:167–188. 10.1586/14760584.2.2.167 [DOI] [PubMed] [Google Scholar]
- 27.Craig CP, Reynolds SL, Airhart JW, Staab EV. 1969. Alterations in immune responses by attenuated Venezuelan equine encephalitis vaccine. I. Adjuvant effect of VEE virus infection in guinea pigs. J. Immunol. 102:1220–1227 [PubMed] [Google Scholar]
- 28.Howard RJ, Craig CP, Trevino GS, Dougherty SF, Mergenhagen SE. 1969. Enhanced humoral immunity in mice infected with attenuated Venezuelan equine encephalitis virus. J. Immunol. 103:699–707 [PubMed] [Google Scholar]
- 29.Thompson JM, Whitmore AC, Konopka JL, Collier ML, Richmond EM, Davis NL, Staats HF, Johnston RE. 2006. Mucosal and systemic adjuvant activity of alphavirus replicon particles. Proc. Natl. Acad. Sci. U. S. A. 103:3722–3727. 10.1073/pnas.0600287103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hidmark AS, Nordstrom EK, Dosenovic P, Forsell MN, Liljestrom P, Karlsson Hedestam GB. 2006. Humoral responses against coimmunized protein antigen but not against alphavirus-encoded antigens require alpha/beta interferon signaling. J. Virol. 80:7100–7110. 10.1128/JVI.02579-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tonkin DR, Jorquera P, Todd T, Beard CW, Johnston RE, Barro M. 2010. Alphavirus replicon-based enhancement of mucosal and systemic immunity is linked to the innate response generated by primary immunization. Vaccine 28:3238–3246. 10.1016/j.vaccine.2010.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steil BP, Jorquera P, Westdijk J, Bakker WA, Johnston RE, Barro M. 2014. A mucosal adjuvant for the inactivated poliovirus vaccine. Vaccine 32:558–563. 10.1016/j.vaccine.2013.11.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LoBue AD, Thompson JM, Lindesmith L, Johnston RE, Baric RS. 2009. Alphavirus-adjuvanted norovirus-like particle vaccines: heterologous, humoral, and mucosal immune responses protect against murine norovirus challenge. J. Virol. 83:3212–3227. 10.1128/JVI.01650-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carroll TD, Matzinger SR, Barro M, Fritts L, McChesney MB, Miller CJ, Johnston RE. 2011. Alphavirus replicon-based adjuvants enhance the immunogenicity and effectiveness of Fluzone in rhesus macaques. Vaccine 29:931–940. 10.1016/j.vaccine.2010.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White LJ, Sariol CA, Mattocks MD, Wahala MPBW, Yingsiwaphat V, Collier ML, Whitley J, Mikkelsen R, Rodriguez IV, Martinez MI, de Silva A, Johnston RE. 2013. An alphavirus vector-based tetravalent dengue vaccine induces a rapid and protective immune response in macaques that differs qualitatively from immunity induced by live virus infection. J. Virol. 87:3409–3424. 10.1128/JVI.02298-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacDonald GH, Johnston RE. 2000. Role of dendritic cell targeting in Venezuelan equine encephalitis virus pathogenesis. J. Virol. 74:914–922. 10.1128/JVI.74.2.914-922.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moran TP, Burgents JE, Long B, Ferrer I, Jaffee EM, Tisch RM, Johnston RE, Serody JS. 2007. Alphaviral vector-transduced dendritic cells are successful therapeutic vaccines against neu-overexpressing tumors in wild-type mice. Vaccine 25:6604–6612. 10.1016/j.vaccine.2007.06.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moran TP, Collier M, McKinnon KP, Davis NL, Johnston RE, Serody JS. 2005. A novel viral system for generating antigen-specific T cells. J. Immunol. 175:3431–3438. 10.4049/jimmunol.175.5.3431 [DOI] [PubMed] [Google Scholar]
- 39.Nishimoto KP, Laust AK, Wang K, Kamrud KI, Hubby B, Smith JF, Nelson EL. 2007. Restricted and selective tropism of a Venezuelan equine encephalitis virus-derived replicon vector for human dendritic cells. Viral Immunol. 20:88–104. 10.1089/vim.2006.0090 [DOI] [PubMed] [Google Scholar]
- 40.Tonkin DR, Whitmore A, Johnston RE, Barro M. 2012. Infected dendritic cells are sufficient to mediate the adjuvant activity generated by Venezuelan equine encephalitis virus replicon particles. Vaccine 30:4532–4542. 10.1016/j.vaccine.2012.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis NL, Caley IJ, Brown KW, Betts MR, Irlbeck DM, McGrath KM, Connell MJ, Montefiori DC, Frelinger JA, Swanstrom R, Johnson PR, Johnston RE. 2000. Vaccination of macaques against pathogenic simian immunodeficiency virus with Venezuelan equine encephalitis virus replicon particles. J. Virol. 74:371–378. 10.1128/JVI.74.1.371-378.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pushko P, Parker M, Ludwig GV, Davis NL, Johnston RE, Smith JF. 1997. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology 239:389–401. 10.1006/viro.1997.8878 [DOI] [PubMed] [Google Scholar]
- 43.Davis NL, Powell N, Greenwald GF, Willis LV, Johnson BJ, Smith JF, Johnston RE. 1991. Attenuating mutations in the E2 glycoprotein gene of Venezuelan equine encephalitis virus: construction of single and multiple mutants in a full-length cDNA clone. Virology 183:20–31. 10.1016/0042-6822(91)90114-Q [DOI] [PubMed] [Google Scholar]
- 44.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC [Google Scholar]
- 45.Davis NL, Brown KW, Johnston RE. 1996. A viral vaccine vector that expresses foreign genes in lymph nodes and protects against mucosal challenge. J. Virol. 70:3781–3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson JM, Nicholson MG, Whitmore AC, Zamora M, West A, Iwasaki A, Staats HF, Johnston RE. 2008. Nonmucosal alphavirus vaccination stimulates a mucosal inductive environment in the peripheral draining lymph node. J. Immunol. 181:574–585. 10.4049/jimmunol.181.1.574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schafer A, Brooke CB, Whitmore AC, Johnston RE. 2011. The role of the blood-brain barrier during Venezuelan equine encephalitis virus infection. J. Virol. 85:10682–10690. 10.1128/JVI.05032-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cottey R, Rowe CA, Bender BS. 2001. Influenza virus. Curr. Protoc. Immunol. Chapter 19:Unit 19.11. 10.1002/0471142735.im1911s42 [DOI] [PubMed] [Google Scholar]
- 49.Cruz CC, Suthar MS, Montgomery SA, Shabman R, Simmons J, Johnston RE, Morrison TE, Heise MT. 2010. Modulation of type I IFN induction by a virulence determinant within the alphavirus nsP1 protein. Virology 399:1–10. 10.1016/j.virol.2009.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27:493–497 [Google Scholar]
- 51.Thompson JM, Whitmore AC, Staats HF, Johnston R. 2008. The contribution of type I interferon signaling to immunity induced by alphavirus replicon vaccines. Vaccine 26:4998–5003. 10.1016/j.vaccine.2008.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thompson JM, Whitmore AC, Staats HF, Johnston RE. 2008. Alphavirus replicon particles acting as adjuvants promote CD8+ T cell responses to co-delivered antigen. Vaccine 26:4267–4275. 10.1016/j.vaccine.2008.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holmgren J, Czerkinsky C. 2005. Mucosal immunity and vaccines. Nat. Med. 11(Suppl 4):S45–S53. 10.1038/nm1213 [DOI] [PubMed] [Google Scholar]
- 54.Mestecky J. 1987. The common mucosal immune system and current strategies for induction of immune responses in external secretions. J. Clin. Immunol. 7:265–276. 10.1007/BF00915547 [DOI] [PubMed] [Google Scholar]
- 55.Elson CO, Ealding W, Lefkowitz J. 1984. A lavage technique allowing repeated measurement of IgA antibody in mouse intestinal secretions. J. Immunol. Methods 67:101–108. 10.1016/0022-1759(84)90089-9 [DOI] [PubMed] [Google Scholar]
- 56.Stetson DB, Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381. 10.1016/j.immuni.2006.08.007 [DOI] [PubMed] [Google Scholar]
- 57.Adkins B, Bu Y, Guevara P. 2002. Murine neonatal CD4+ lymph node cells are highly deficient in the development of antigen-specific Th1 function in adoptive adult hosts. J. Immunol. 169:4998–5004. 10.4049/jimmunol.169.9.4998 [DOI] [PubMed] [Google Scholar]
- 58.Li L, Lee HH, Bell JJ, Gregg RK, Ellis JS, Gessner A, Zaghouani H. 2004. IL-4 utilizes an alternative receptor to drive apoptosis of Th1 cells and skews neonatal immunity toward Th2. Immunity 20:429–440. 10.1016/S1074-7613(04)00072-X [DOI] [PubMed] [Google Scholar]
- 59.Giuliano M, Mastrantonio P, Giammanco A, Piscitelli A, Salmaso S, Wassilak SG. 1998. Antibody responses and persistence in the two years after immunization with two acellular vaccines and one whole-cell vaccine against pertussis. J. Pediatr. 132:983–988. 10.1016/S0022-3476(98)70395-6 [DOI] [PubMed] [Google Scholar]
- 60.Tiru M, Hallander HO, Gustafsson L, Storsaeter J, Olin P. 2000. Diphtheria antitoxin response to DTP vaccines used in Swedish pertussis vaccine trials, persistence and projection for timing of booster. Vaccine 18:2295–2306. 10.1016/S0264-410X(99)00539-3 [DOI] [PubMed] [Google Scholar]
- 61.Pihlgren M, Friedli M, Tougne C, Rochat AF, Lambert PH, Siegrist CA. 2006. Reduced ability of neonatal and early-life bone marrow stromal cells to support plasmablast survival. J. Immunol. 176:165–172. 10.4049/jimmunol.176.1.165 [DOI] [PubMed] [Google Scholar]
- 62.Pihlgren M, Schallert N, Tougne C, Bozzotti P, Kovarik J, Fulurija A, Kosco-Vilbois M, Lambert PH, Siegrist CA. 2001. Delayed and deficient establishment of the long-term bone marrow plasma cell pool during early life. Eur. J. Immunol. 31:939–946. [DOI] [PubMed] [Google Scholar]
- 63.Marshall-Clarke S, Tasker L, Parkhouse RM. 2000. Immature B lymphocytes from adult bone marrow exhibit a selective defect in induced hyperexpression of major histocompatibility complex class II and fail to show B7.2 induction. Immunology 100:141–151. 10.1046/j.1365-2567.2000.00035.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adkins B. 1999. T-cell function in newborn mice and humans. Immunol. Today 20:330–335. 10.1016/S0167-5699(99)01473-5 [DOI] [PubMed] [Google Scholar]
- 65.Kovarik J, Siegrist CA. 1998. Optimization of vaccine responses in early life: the role of delivery systems and immunomodulators. Immunol. Cell Biol. 76:222–236. 10.1046/j.1440-1711.1998.00746.x [DOI] [PubMed] [Google Scholar]
- 66.Siegrist CA. 2007. The challenges of vaccine responses in early life: selected examples. J. Comp. Pathol. 137(Suppl 1):S4–S9 [DOI] [PubMed] [Google Scholar]
- 67.Grewal HM, Karlsen TH, Vetvik H, Ahren C, Gjessing HK, Sommerfelt H, Haneberg B. 2000. Measurement of specific IgA in faecal extracts and intestinal lavage fluid for monitoring of mucosal immune responses. J. Immunol. Methods 239:53–62. 10.1016/S0022-1759(00)00171-X [DOI] [PubMed] [Google Scholar]
- 68.Chen J, Fang F, Li X, Chang H, Chen Z. 2005. Protection against influenza virus infection in BALB/c mice immunized with a single dose of neuraminidase-expressing DNAs by electroporation. Vaccine 23:4322–4328. 10.1016/j.vaccine.2005.03.035 [DOI] [PubMed] [Google Scholar]
- 69.Chen Z, Matsuo K, Asanuma H, Takahashi H, Iwasaki T, Suzuki Y, Aizawa C, Kurata T, Tamura S. 1999. Enhanced protection against a lethal influenza virus challenge by immunization with both hemagglutinin- and neuraminidase-expressing DNAs. Vaccine 17:653–659. 10.1016/S0264-410X(98)00247-3 [DOI] [PubMed] [Google Scholar]
- 70.Galarza JM, Latham T, Cupo A. 2005. Virus-like particle vaccine conferred complete protection against a lethal influenza virus challenge. Viral Immunol. 18:365–372. 10.1089/vim.2005.18.365 [DOI] [PubMed] [Google Scholar]
- 71.Dadaglio G, Sun CM, Lo-Man R, Siegrist CA, Leclerc C. 2002. Efficient in vivo priming of specific cytotoxic T cell responses by neonatal dendritic cells. J. Immunol. 168:2219–2224. 10.4049/jimmunol.168.5.2219 [DOI] [PubMed] [Google Scholar]
- 72.Dakic A, Shao QX, D'Amico A, O'Keeffe M, Chen WF, Shortman K, Wu L. 2004. Development of the dendritic cell system during mouse ontogeny. J. Immunol. 172:1018–1027. 10.4049/jimmunol.172.2.1018 [DOI] [PubMed] [Google Scholar]
- 73.Garcia AM, Fadel SA, Cao S, Sarzotti M. 2000. T cell immunity in neonates. Immunol. Res. 22:177–190. 10.1385/IR:22:2-3:177 [DOI] [PubMed] [Google Scholar]
- 74.Muthukkumar S, Goldstein J, Stein KE. 2000. The ability of B cells and dendritic cells to present antigen increases during ontogeny. J. Immunol. 165:4803–4813. 10.4049/jimmunol.165.9.4803 [DOI] [PubMed] [Google Scholar]
- 75.Sun CM, Fiette L, Tanguy M, Leclerc C, Lo-Man R. 2003. Ontogeny and innate properties of neonatal dendritic cells. Blood 102:585–591. 10.1182/blood-2002-09-2966 [DOI] [PubMed] [Google Scholar]
- 76.Aksoy E, Albarani V, Nguyen M, Laes JF, Ruelle JL, De Wit D, Willems F, Goldman M, Goriely S. 2007. Interferon regulatory factor 3-dependent responses to lipopolysaccharide are selectively blunted in cord blood cells. Blood 109:2887–2893 [DOI] [PubMed] [Google Scholar]
- 77.De Wit D, Olislagers V, Goriely S, Vermeulen F, Wagner H, Goldman M, Willems F. 2004. Blood plasmacytoid dendritic cell responses to CpG oligodeoxynucleotides are impaired in human newborns. Blood 103:1030–1032 [DOI] [PubMed] [Google Scholar]
- 78.Cusumano V, Mancuso G, Genovese F, Cuzzola M, Carbone M, Cook JA, Cochran JB, Teti G. 1997. Neonatal hypersusceptibility to endotoxin correlates with increased tumor necrosis factor production in mice. J. Infect. Dis. 176:168–176. 10.1086/514019 [DOI] [PubMed] [Google Scholar]
- 79.Vekemans J, Amedei A, Ota MO, D'Elios MM, Goetghebuer T, Ismaili J, Newport MJ, Del Prete G, Goldman M, McAdam KP, Marchant A. 2001. Neonatal bacillus Calmette-Guerin vaccination induces adult-like IFN-gamma production by CD4+ T lymphocytes. Eur. J. Immunol. 31:1531–1535. [DOI] [PubMed] [Google Scholar]
- 80.Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. 2002. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J. Immunol. 168:3195–3204. 10.4049/jimmunol.168.7.3195 [DOI] [PubMed] [Google Scholar]