

ABSTRACT
HIV-1 utilizes the cellular protein LEDGF/p75 as a chromosome docking and integration cofactor. The LEDGF/p75 gene, PSIP1, is a potential therapeutic target because, like CCR5, depletion of LEDGF/p75 is tolerated well by human CD4+ T cells, and knockout mice have normal immune systems. RNA interference (RNAi) has been useful for studying LEDGF/p75, but the potent cofactor activity of small protein residua can be confounding. Here, in human cells with utility for HIV research (293T and Jurkat), we used transcription activator-like effector nucleases (TALENs) to completely eradicate all LEDGF/p75 expression. We performed two kinds of PSIP1 knockouts: whole-gene deletion and deletion of the integrase binding domain (IBD)-encoding exons. HIV-1 integration was inhibited, and spreading viral replication was severely impaired in PSIP1−/− Jurkat cells infected at high multiplicity. Furthermore, frameshifting the gene in the first coding exon with a single TALEN pair yielded trace LEDGF/p75 levels that were virologically active, affirming the cofactor's potency and the value of definitive gene or IBD exon segment deletion. Some recent studies have suggested that LEDGF/p75 may participate in HIV-1 assembly. However, we determined that assembly of infectious viral particles is normal in PSIP1−/− cells. The potency of an allosteric integrase inhibitor, ALLINI-2, for rendering produced virions noninfectious was also unaffected by total eradication of cellular LEDGF/p75. We conclude that HIV-1 particle assembly and the main ALLINI mechanism are LEDGF/p75 independent. The block to HIV-1 propagation in PSIP1−/− human CD4+ T cells raises the possibility of gene targeting PSIP1 combinatorially with CCR5 for HIV-1 cure.
IMPORTANCE LEDGF/p75 dependence is universally conserved in the retroviral genus Lentivirus. Once inside the nucleus, lentiviral preintegration complexes are thought to attach to the chromosome when integrase binds to LEDGF/p75. This tethering process is largely responsible for the 2-fold preference for integration into active genes, but the cofactor's full role in the lentiviral life cycle is not yet clear. Effective knockdowns are difficult because even trace residua of this tightly chromatin-bound protein can support integration cofactor function. Here, in experimentally useful human cell lines, we used TALENs to definitively eradicate LEDGF/p75 by deleting either all of PSIP1 or the exons that code for the integrase binding domain. HIV-1 replication was severely impaired in these PSIP1 knockout cells. Experiments in these cells also excluded a role for LEDGF/p75 in HIV-1 assembly and showed that the main ALLINI mechanism is LEDGF/p75 independent. Site-specific gene targeting of PSIP1 may have therapeutic potential for HIV-1 disease.
INTRODUCTION
The cellular chromatin-associated protein LEDGF/p75 (where LEDGF is lens epithelium-derived growth factor), a product of the PSIP1 gene, is a lentiviral integration cofactor. Originally described as a transcriptional coactivator (1), the protein is considered the main chromosome attachment factor for the viral preintegration complex (PIC) (2–6; for reviews, see references 7 and 8). In current models, LEDGF/p75 is envisioned to tether the PIC to the site of its subsequent integration, thus facilitating integration and strongly contributing to the approximately 2-fold preference for the virus to integrate into active genes (9). The key intermolecular tether forms when LEDGF/p75 binds to a host chromosome through a set of elements in its N terminus (10–13) and to the integrase (IN) dimer through the integrase-binding domain (IBD) in its C terminus (14, 15).
The LEDGF IBD binds to the HIV-1 IN multimer by making key hydrogen bonding and hydrophobic contacts with the V-shaped pocket at the IN catalytic core domain (CCD) dimer interface as well as by establishing polar interactions with the N-terminal domain of another dimer (16–19). Well-characterized single amino acid IBD mutations that disrupt IN binding are known, e.g., IBD D366A/N (16, 17).
RNA interference (RNAi) against LEDGF/p75 has been useful but problematic in practice. The protein is tightly attached, throughout the cell cycle, to one of the two reactants in the HIV-1 integration process (chromosomal DNA) (3, 15). In human CD4+ T cell lines, maximally stringent RNAi-mediated knockdown of LEDGF/p75 sufficient to reduce it to an undetectable level in the Triton X-resistant, DNase- and salt-extractable chromatin-bound (S2) fraction (11) was required to demonstrate significant impairment of HIV-1 infection, and this technique helped elicit its cofactor role in integration (4). In such cells and in Psip1 knockout (KO) mouse embryonic fibroblasts, approximately 5- to 10-fold inhibition localized to the early phase of HIV-1 replication has been observed (4, 6).
Among the HIV-1 dependency factors, LEDGF/p75 stands out in being used by all lentiviruses across the primate, ungulate, and feline groups (and by no other retroviruses in the other six genera), indicating consistent selection pressure during the evolution of the lentiviral genus (20–22). This unusual pan-lentiviral dependency factor usage is the case despite the lack of conservation of specific amino acid side chains in IN dimer clefts of the various lentiviral integrase proteins (22). There is as yet insufficient explanation for the centrality of the protein to lentiviral biology, and the contribution of the protein to sustained systemic replication and pathogenesis in vivo is unknown. An isoform of the protein, LEDGF/p52, is produced by alternative splicing; it shares the N-terminal 325 amino acids of LEDGF/p75 but lacks the integrase binding domain and plays no known virological role. In this paper, the acronym LEDGF will henceforth refer to the p75 isoform.
Allosteric integrase inhibitors, or ALLINIs, also known as the noncatalytic site IN inhibitor (NCINIs) (23) and LEDGINs (24), were identified as a class by the ability to disrupt the interaction of LEDGF with HIV-1 IN in vitro and thus impair the viral integration step in cells (24). However, a more potent (and apparently main) mechanism of ALLINI action was subsequently identified: disrupting proper particle assembly (23, 25–30). Accumulating evidence suggests that this effect is mediated when the inhibitor binding to the IN dimer interface at the principal LEDGF binding pocket induces enhanced IN multimerization, which results in aberrant particle assembly; the effect is reminiscent of class II IN mutant effects that are known to broadly perturb myriad functions of the Gag-Pol precursor and its protease-derived proteins (26, 27, 31). It is not clear whether this production-phase antiviral effect also involves LEDGF, which is entirely plausible since the drugs and the IBD bind to essentially the same protein interface. Some studies have suggested LEDGF dependence and that LEDGF incorporation into HIV-1 particles occurs and could be needed for normal HIV-1 infectivity (28, 32–34).
It is difficult to answer these questions about the viral biology of LEDGF with the currently available reagents and the paucity of relevant, informative gene knockout cells. RNAi-depleted cells still contain some LEDGF protein, and frequent resorting for coexpressed fluorescent proteins has been required to maintain the optimally mRNA-depleted state (4, 35–37). Mouse Psip1 gene KO cell lines are available and have proved useful (6, 38, 39), but they cannot be used for HIV assembly experiments or for spreading viral replication studies as there are complex species-specific defects in proper assembly (40) and as mouse T cells also have early event blocks (41). A PSIP1 knockout pre-B cell leukemia line (Nalm-6) was generated by homologous recombination (42) but does not represent a normal cellular substrate for HIV-1 replication and is poorly suited to studying viral assembly.
Here, we used transcription activator-like effector nucleases (TALENs) to delete specific segments of the PSIP1 gene from informative human cell lines to address two questions: does LEDGF play a role in HIV-1 assembly, and does the main ALLINI antiviral mechanism involve LEDGF? TALENs are designable site-specific nucleases engineered from fusions of FokI endonuclease to modularly assembled transcription activator-like effectors (TALEs) from Xanthomonas (43–46). These plant pathogens inject TALEs that bind specific host genome DNA sequences and alter cell transcription to the invading bacterium's advantage. TALEs contain conserved 33- to 35-amino-acid repeats that differ mainly at positions 12 and 13 (the repeat variable diresidues, or RVDs). Different RVDs that recognize different DNA base pairs can be assembled in series, and, unlike zinc finger nucleases (ZFNs), the code is reliably applicable without iterative postassembly refinements of the protein. A pair of TALENs is constructed to bind to the left and right of the target sequence, causing a double-strand break (DSB) when the two FokI domains dimerize. Error-prone repair by the cellular nonhomologous end-joining (NHEJ) pathway generates gene-disrupting indels at the target site. Use of two TALEN pairs can produce full deletion of the intervening chromosome DNA (47–50).
We constructed TALENs that can efficiently target PSIP1 in adherent and suspension cells (HT1080, 293T, and Jurkat). We found that while HIV-1 integration and spreading replication were inhibited in these PSIP1-deleted cells in which all possibility of LEDGF expression has been eradicated at the gene level, infectious HIV-1 particle assembly was normal. Use of a single TALEN pair to disrupt the gene by introducing frameshifting indels in the first coding exon yielded only trace LEDGF levels in immunoblotting, but these were virologically active, affirming the need for definitive gene deletion. These results establish definitively that LEDGF does not play a role in particle assembly and that its cofactor role depends on its role in early events. We determined also that the potency of a representative ALLINI to disrupt infectious particle formation is unaffected by total extirpation of LEDGF coding capacity from the cell. We concur with the accumulating evidence (23, 26, 27) that the dominant ALLINI mechanism is independent of LEDGF. These PSIP1 knockout cell lines will be useful for further HIV research and as substrates for further combinatorial gene knockouts.
MATERIALS AND METHODS
TALEN design and assembly.
TALENs were built in the pcGoldy-TALEN scaffold using Golden Gate assembly methods (47, 51, 52). TALEN plasmids were transfected into 293T and HT1080 cells with polyethylenimine (PEI) and into Jurkat cells by electroporation (Neon system; Invitrogen). TALEN pairs X, A, and B bind, respectively, to the following PSIP1 gene sequences, where the spacer region, which is the site of predicted DSBs, is in lowercase: GACTCGCGATTTCAAacctggagacctcatCTTCGCCAAGATGAA, CAATGGATTCTCGACttcaaaggatacatgcTGAGATTAAAAATTC, and CAGGTAATCATGGAAaagtctacaatgttgTATAACAAGTTTAAG. After transfection, substantial gene editing in the bulk population was verified using genomic DNA PCR assays. Then, single-cell cloning was carried out by limiting dilution in 96-well plates, with clones chosen from plates having <15 colonies per plate. Individual cell clones were screened by PCR on genomic DNA and by immunoblotting for LEDGF. Six primers were used for PCR: primer 1 (5′-CAGACGGGCGATTTCTGGCC), primer 2 (5′-GTGCTTTTACACTGCATGTTGC), primer 3 (5′-CCTCATGCTGTCTTTGTTCAGC), primer 4 (5′-GTCTGCCAATAAACCATAGGG), primer 5 (5′-GGATGTGAACAGATGCATTGAG), and primer 6 (5′-CTGACTAACTTTGAATCGCCG). Primer and TALEN pair binding sites are shown in Fig. 1.
FIG 1.
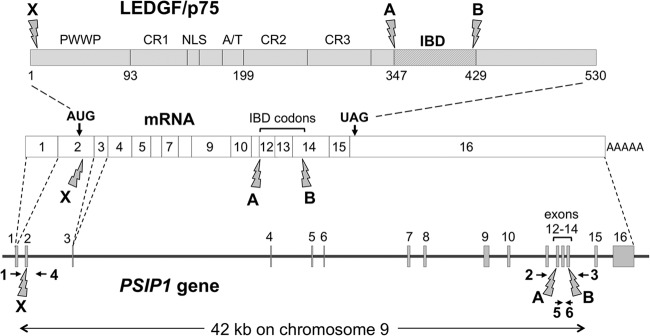
TALEN-mediated targeting of human PSIP1. The human PSIP1 locus on chromosome 9, the spliced LEDGF mRNA, and the LEDGF protein are diagrammed. The 16 exons are numbered in the gene and the mRNA; exon 2 is the first coding exon. Exon and intron lengths are drawn to correct scale. Targeted chromosome DSB sites for TALEN pairs X, A, and B are indicated by lightning bolt symbols at bottom, and the corresponding locations in the mRNA and protein are indicated in the same way. Each of the six TALENs in the three TALEN pairs has a binding site within an exon (exon 2, 12, or 14, respectively). Numbered horizontal arrows indicate six PCR primers used in different permutations for identifying chromosomal deletions. In the protein, the PWWP domain, nuclear localization signal (NLS), charged regions (CR1 to CR3), A/T hook domain, and integrase binding domain (IBD, residues 347 to 429) are labeled.
Cells.
HT1080, 293T, and Jurkat E6 cells were obtained from the American Type Culture Collection (ATCC). Cell clones were maintained in Dulbecco's modified Eagle's medium (DMEM) or RPMI 1640 medium with 10% heat-inactivated fetal calf serum (FCS), penicillin-streptomycin, and l-glutamine. The stable LEDGF knockdown 293T cell line, engineered with lentiviral vector-transduced U6-promoted short hairpin RNAs (shRNAs) that target the LEDGF mRNA (293T-si1340/1428, where the 293T cell line expresses the target sequences at nucleotides [nt] 1340 and 1428) was described previously (20).
Sequencing.
Genomic DNA was isolated from cells using a DNeasy kit (Invitrogen) according to the manufacturer's instructions. PCR products obtained using the primer pair 1 and 4 and the pair 1 and 3 for the whole gene KO and primer pair 2 and 3 for the IBD deletion were cloned (StrataClone Ultra Blunt PCR cloning kit; Stratagene). Eight to 10 independent clones for each product were sequenced.
Immunoblotting.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate, 1% NP-40, 150 mM Tris-HCl, pH 8.0) with protease inhibitors (Complete Mini; Boehringer). The fractionation protocol has been characterized extensively (4, 11, 37). S1 is the Triton X-100-extractable (non-chromatin-bound) fraction. S2 is the Triton X-100-resistant chromatin-bound fraction released by further DNase I- and salt-based extraction; it contains released chromatin-associated proteins. Protein was quantified with the Bradford assay. Twenty micrograms of lysate or S2 fraction was boiled in Laemmli buffer with β-mercaptoethanol for 10 min and then electrophoresed in 10% Tris-HCl gels (Bio-Rad) and transferred overnight to Immobilon P membranes (Millipore). Blocked membranes were incubated overnight with a primary anti-LEDGF monoclonal antibody (611714; BD Biosciences) at a 1:500 dilution or with rabbit anti-LEDGF/p75 (A300-848A; Bethyl Laboratories) at a 1:1,000 dilution and then washed with Tris-buffered saline–Tween 20 (TBST) three times for 7 min each. Afterward, membranes were incubated for 1 h at room temperature with a secondary goat anti-mouse or anti-rabbit horseradish peroxidase (HRP), at 1:4,000. After three 10-min washes with TBST, membranes were incubated in SuperSignal West Pico chemiluminescent substrate (Pierce) for 1 to 2 min and exposed to film. Primary antibodies to green fluorescent protein (GFP) (JL-8; catalogue no. NC9777966) and tubulin (T5168; Sigma) were used at a 1:4,000 dilution.
Vectors and viruses.
HIV-1luc and HIV-1GFP, the vesicular stomatitis virus G protein (VSV-G)-pseudotyped NL4-3R−E−Δ426 luciferase (luc) and GFP reporter viruses, have been described previously as were feline immunodeficiency virus (FIV)-GFP, FIV-luc, and murine leukemia virus (MLV)-GFP (4). HIV-1 NL4-3 was produced by transfection of 293T cells in 175-cm2 flasks with 10 μg of pNL4-3 plasmid DNA using PEI. Particle inputs were normalized by reverse transcriptase (RT) activity or p24 antigen as described previously (53). p24 antigen was measured using a Zeptometrix enzyme-linked immunosorbent assay (ELISA). Mean p24 amounts ± standard deviations (SD) were calculated from duplicate measurements for each sample. Infectivity per ng of p24 was determined by titration on GHOST cells according to the NIH AIDS Research and Reference Reagent Program protocol. For infections of Jurkat cells with p24-normalized viruses, 106 cells were infected in a T25 flask. The cells were washed the next day 5 times with RPMI medium to remove input virus, and a time zero p24 sample was collected. Cultures were maintained in continuously dividing state by splitting 1:5 when needed, and supernatants were sampled every 2 to 4 days for p24 measurements.
Alu-PCR.
Quantitative Alu-PCR for quantification of integrants was done 14 days postransduction with VSV-G-pseudotyped HIV-1luc in two stages, according to the method of Llano et al. (4). In a first stage we amplified between Alu sequences and U3 sequences using 300 nM AluI TCC CAG CTA CTG GGG AGG CTG AGG, 300 nM Alu2 GCC TCC CAA AGT GCT GGG ATT ACA G, and 100 nM U3 AS CTC CGG ATG CAG CTC TCG. We used the following parameters: 12 cycles of 95°C for 10 s, 63°C for 10 s, and 72°C for 2 min 50 s, with a temperature transition rate of 5°C/s. In a second stage we amplified a nested product within U3 using 300 nM sense primer GAA CTA CAC ACC AGG GCC and 300 nM antisense primer CTC CGG ATG CAG CTC TCG. PCR amplification was performed in a Roche LightCycler as follows: 50 cycles of 95°C for 10 s, 65°C for 10 s, and 72°C for 9 s, with a temperature transition rate of 5°C/s. We used human mitochondrial DNA as a loading control. It was quantified using 20 pmol of sense primer GAA TGT CTG CAC AGC CAC TT and 20 pmol of antisense primer TAG AAA GGC TAG GAC CAA AC. PCR amplification and analysis were performed as follows: 40 cycles of 95°C for 10 s, 54°C for 20 s, and 72°C for 30 s, with a temperature transition rate of 5°C/s.
Expression of LEDGF and LANA31-GFP-IBD with retroviral vectors.
The gammaretroviral vector MLV-p75 (4) was used to reexpress LEDGF in PSIP1 gene-deleted cells. An internal ribosome entry site-linked neoR gene is located downstream of the p75 cDNA. Cells were selected and maintained in 0.8 mg/ml G418. PSIP1−/− Jurkat cells were also transduced with lentiviral vectors that encode LANA31-GFP-IBD and LANA31-GFP-IBDD366N. GFP-positive cells were isolated on a FACSAria cell sorter.
Effect of ALLINI-2 on HIV-1 production and infectivity.
A total of 30,000 293T cells were plated per well in 96-well plates, allowed to adhere overnight, and then PEI transfected with 60 ng of HIV-1 GFP transfer vector TSING (4), 60 ng of an HIV-1 packaging plasmid, and 20 ng of a VSV-G-encoding plasmid. After 8 h, cells were washed and treated with ALLINI-2 (26) or an equivalent volume of dimethyl sulfoxide (DMSO). After an additional 48 h, supernatants were filtered (0.45-μm pore size), and titers were determined on fresh 293T cells in medium containing no ALLINI-2 or DMSO. Titers are calculated as GFP-transducing units per ml. Virion reverse transcriptase activities were determined as previously described using a 32P-based assay with an oligo(dT) template (53). Experiments were performed in triplicate, and values are reported as means ± SD.
ALLINI-2 target cell effect.
A total of 30,000 Jurkat cells or 10,000 293T cells were plated in 96-well plates and treated with ALLINI-2 or DMSO for 2 h prior to infection with HIV-1luc. The inhibitor was maintained for 3 more days before luciferase expression was measured. Percent inhibition was calculated as 1 − [lucALLINI-2]/[lucControl]. All infections were performed in triplicate, and the average percent inhibition was graphed ± SD.
RESULTS
Efficient TALEN-mediated gene targeting of human PSIP1.
The human fibrosarcoma cell line HT1080 was transfected with TALEN pair X (Fig. 1A). This pair of TALENs targets exon 2, downstream of the initiator ATG codon. A GFP-expressing plasmid was cotransfected, and GFP-positive cells were enriched with a FACSAria cell sorter. These were then single-cell cloned by limiting dilution 5 days after sorting. Triton-resistant, DNase- and salt-extractable chromatin-bound fractions (S2 fractions) were prepared as previously described (4, 11) from expanded clones and parental cells and immunoblotted for LEDGF (Fig. 2A). The gene targeting was remarkably efficient: 9 of 26 clones were disrupted at both alleles (Fig. 2A). Some clones were mono-allelically disrupted as evidenced by the fainter immunoblot bands in Fig. 2A (e.g., clones 8, 21, 22, and 24). Genomic DNA PCR assays identified a range of frameshifting indels at the targeted chromosomal site in the second exon (Fig. 2B). However, when we challenged the PSIP1−/− HT1080 clones with HIV-1, we observed no consistent reduction in viral infectivity (Fig. 2C). Based on the considerations discussed above, we suspected residual expression of some LEDGF in these cells that was below the detection limit of the immunoblotting. We therefore refined immunoblotting sensitivity, using a different antibody that recognizes the C terminus of the protein. Indeed, the X pair-derived PSIP1−/− HT1080 clones were expressing a comparatively minute amount of LEDGF (Fig. 2D). As the proteins appeared to have molecular masses indistinguishable from wild-type LEDGF on our gels, this expression is likely due to translational read-through or internal ribosome initiation from a downstream in-frame ATG closely adjacent to the indels introduced into exon 2 (Fig. 2E).
FIG 2.

Efficient gene disruption in HT1080 cells. (A) HT1080 cells were transfected with TALEN pair X and then cloned by limiting dilution 5 days later. Chromatin-bound (S2) fractions (4, 11) from the expanded single-cell clones and parental HT1080 cells were immunoblotted for LEDGF. Gene targeting was highly efficient: 9 of 26 clones were knocked out at both alleles (dark arrows, clones 1, 2, 5, 6, 13, 16, 17, 18, and 20). Some clones are −/+ (see fainter immunoblot bands). (B) Genomic DNA PCR with primers 1 and 4 identified a range of frame-shifting indels centered on the TALEN X spacer region in the second exon. The presence of more than two deletion alleles in some cell clones of these diploid cells reflects postcloning retargeting in some of the clone's progeny cells by persisting TALEN proteins, which can bind and generate a second DSB in cases where the initial deletion was confined to the spacer. Some sequences were found in multiple cloned PCR products (cell clone 1, two 11-nt deletions; clone 16, two 23-nt deletions; clone 17, three 13-nt deletions). (C) −/− HT1080 clones were challenged with a single-cycle HIV-1 reporter virus, HIV-1luc. (D) Optimized LEDGF immunoblotting of the −/− clones, with antibody that recognizes the C-terminal domain of LEDGF. (E) Left and right TALEN binding sites (underlined capital letters) and spacer region (site of predicted DSB and indel generation) for TALEN pair X in exon 2. The LEDGF start codon and an in-frame methionine codon downstream from the targeted site are highlighted; the latter is a likely site of secondary translation initiation. RLU, relative light units. Uppercase and underlined nucleotides represent TALEN binding sites. Dashes represent deleted residues. Spacer region and flanking nucleotides are lowercase.
TALEN-mediated deletion of PSIP1 or IBD-encoding exons in 293T and Jurkat cell lines.
To definitively eradicate all possibility of LEDGF protein existence, we then designed two additional TALEN pairs that were used in various permutations with pair X (Fig. 1A). These are pair A and B. The target of TALEN pair A is in exon 12 (the first RVD of the left-hand TALEN in pair A binds to the C in TCA, the codon for Ser 347, which is the first amino acid of the IBD). The target of pair B is in exon 14, with the last RVD in the right-hand TALEN binding to a G located 13 nt upstream of the terminal IBD residue, V429. Thus, chromosomal deletions caused by combined use of the A and B pairs are predicted to remove any functional IBD from the protein.
Clonal Jurkat and 293T cell lines that are −/− for the entire 42-kb gene were generated by cotransfecting TALEN pairs X and B (X+B); clones −/− for only the exons that encode the IBD were similarly generated by cotransfecting TALEN pairs A and B (A+B). For the Jurkat cells, we obtained one X+B pair-generated clone with a deletion of exons 2 to 14 (PSIP1−/−) and two A+B pair-generated clones with deletions of exon 12 to 14 (IBD−/−; clones 1 and 2). For 293T cells we identified one PSIP1−/− and one IBD−/− cell clone (Fig. 3 and 4). Correct deletion of all PSIP1 alleles in the cells was verified at the DNA and protein levels; exact chromosomal joining sites were also determined, confirming deletion of intervening segments between the predicted binding sites of the TALEN pairs (Fig. 3 and 4A to C). Immunoblotting demonstrated complete absence of LEDGF in the 293T cells that have deletions of the whole gene and of the predicted shorter protein in the IBD-deleted cells. Interestingly, for the Jurkat clones, no detectable protein was found in either type of deletion mutant (Fig. 4A, right panel), which might reflect splicing program dysfunction in the IBD−/− deletion mutants.
FIG 3.

PSIP1 chromosome segment excisions generate Jurkat PSIP1−/−, Jurkat IBD−/−, 293T PSIP1−/−, and 293T IBD−/− cell lines. (A) PCR assays with the primers shown in Fig. 1 were performed on genomic DNA from bulk populations of 293 T cells transfected a week earlier with the A+B TALEN pair (left gel) or the X+B TALEN pair (right gel). (B) Same PCR assays as in panel B performed with genomic DNA from bulk populations of Jurkat cells. (C) Final 293T and Jurkat clones obtained with complete deletion of the PSIP1 gene (∼42 kb, three left gels) and the IBD exon-intron segment only (right gel).
FIG 4.

Characterization of Jurkat and 293T KO cells. (A) Immunoblotting for LEDGF protein in cell lysates and S2 fractions from parental cells and TALEN-modified clones; 293T cell data are shown in the left panel, and Jurkat cell data are in the right panel. (B) Sequencing of the NHEJ-generated chromosomal junction sites. PCR primer 1 and 3 products were sequenced for both 293T and Jurkat PSIP1−/− cells. (C) PCR primer 2 and 3 products were sequenced for both 293 T and Jurkat IBD−/− cells. The sequences demonstrate complete deletions between the two sites of DSB of the TALEN pairs used in combination. TALEN spacer segments are underlined and lowercase. Dashes represent the intervening wild-type segments of the indicated lengths (42 kb and 0.5 kb).
Impaired lentiviral infection and integration in PSIP1 knockout cells.
We then challenged the 293T and Jurkat clones with serial dilutions of pseudotyped single-cycle HIV-1 and FIV reporters that express either firefly luciferase or GFP (Fig. 5). Both HIV-1luc (where transcription is directed by the HIV-1 long terminal repeat [LTR]) and FIV vectors (reporters under the transcriptional control of an internal cytomegalovirus [CMV] immediate-early gene promoter) were blocked in the knockout cells compared to parental cells (Fig. 5A, B, D, and E). We observed no difference between the X+B pair-generated clones with deletions of all of PSIP1 or the A-B pair-generated clones with deletion of just the IBD exon-intron segment of the gene; lentiviral infection was equally inhibited in both. In contrast, a gammaretrovirus (MLV-GFP) was unaffected in all cell lines (Fig. 5C and F). This result demonstrates lentiviral specificity. Alu-PCR assays performed at 14 days after infection showed that HIV-1 integration was reduced 8- to 12-fold in Jurkat PSIP1 KO cells (Fig. 5G).
FIG 5.

HIV-1, FIV, and MLV reporter infection and integration analysis. The indicated cells were infected with increasing doses of luciferase- or GFP-encoding single-cycle vectors. (A and B) HIV-1-based vectors. (C and D) FIV-based vectors. (E and F) MLV vectors. For the luciferase assay, cell lysates from equal numbers of cells were assayed for activity 72 h after infection. For GFP, cells were assayed with flow cytometry for GFP at 72 h. Error bars represent standard deviations of duplicate measurements. (G) Alu-PCR results 14 days after infection. RLU, relative light units.
HIV-1 infectivity is rescued in T cells by reexpression of LEDGF or expression of an alternatively tethered IBD (LANA31-GFP-IBD).
To establish specificity further, we reexpressed LEDGF in the Jurkat PSIP1−/− cell line, using a gammaretroviral vector. This resulted in low levels of LEDGF expression (Fig. 6A), perhaps due to silencing of the gammaretroviral LTR in Jurkat cells, but substantial rescue of HIV-1 infectivity (Fig. 6B). A second type of rescue of the gene-deleted state was then carried out to demonstrate dependence on the IBD and dependence on chromosome tethering. LANA31 is a 31-amino-acid peptide that forms the N terminus of Kaposi's sarcoma herpesvirus LANA protein. It binds to the histone 2A/B dimer (54). The expression of this alternatively tethered IBD (LANA31-GFP-IBD) in T cell lines can rescue the loss of infectivity due to LEDGF knockdown (37). Untethered GFP-IBD has dominantly interfering antiviral activity, and we have also shown previously that fusing LANA31 to the N terminus of GFP-IBD switches it from an integration-blocking protein to an integration cofactor that rescues more than two logs of infectivity (37). Here, we expressed LANA31-GFP-IBD in the gene knockout cells using lentiviral vectors. This required high-multiplicity transduction due to the impaired lentiviral vector infectivity produced by the absence of functional LEDGF. LANA31-GFP-IBD expression rescued infectivity in the PSIP1−/− and the IBD−/− lines (Fig. 6C and D). Specificity was further demonstrated by the lack of rescuing activity of the single amino acid mutant, LANA31-GFP-IBDD366N (Fig. 6C and D). Taken together, these data establish that the observed antiviral effects of the TALEN-mediated gene deletions are specifically (i) due to LEDGF, (ii) attributable to loss of IBD binding to IN, and (iii) dependent on the loss of chromosome tethering per se rather than other functions mediated by the N-terminal chromatin binding domain ensemble.
FIG 6.

Reexpression of LEDGF and alternatively tethered IBD (LANA31-GFP-IBD, with and without a D366N mutation. (A) Immunoblots for LEDGF protein. (B and C) The indicated cells were infected with increasing doses of HIV-1luc. (D) Immunoblotting of these cells for LEDGF and LANA31-GFP-IBD proteins using anti-LEDGF and anti-GFP primary antibodies, respectively.
Spreading viral replication is blocked by LEDGF eradication.
We next determined how the complete absence of LEDGF protein influences spreading HIV-1 replication in Jurkat cells. The clones were infected with HIV-1 NL4-3 at multiplicities of infection (MOIs) of 0.1 and 1.0 (Fig. 7A and B). We observed that the effect of the definitive protein-eradicating PSIP1 gene deletions on spreading replication in this CD4+ T cell line was profound, exceeding the replication delays observed after stable shRNA-mediated knockdown in the human CD4+ T cell line SupT1 (4, 37). We followed the Jurkat cell cultures for 82 days. At an MOI of 1.0, the delay in PSIP1−/− cells was 46 days (times to peak HIV-1 NL4-3 replication for Jurkat and Jurkat PSIP1−/− were 10 and 56 days, respectively) (Fig. 7A and B). It was curious, however, that the delay in IBD−/− clone 2 was consistently less. We speculated that IBD−/− clone 2 might express some residual IBD, perhaps enabled by aberrant reintegration of the gene segment of exons 12 to 14 excised by TALEN pairs A and B. We therefore performed PCR for this internal IBD exon-intron genomic segment using primers 5 and 6 (Fig. 1) and found that is indeed the case (Fig. 7C). IBD−/− clone 2 but not clone 1 contains a detectable IBD segment although it is not present in the normal locus position (Fig. 4C); the precise rearrangement involved was not pursued as we wished to focus henceforth on clone 1.
FIG 7.

Replication-competent HIV-1 properties in Jurkat cells. (A and B) The indicated cells were infected with HIV-1 NL4−3 at two different MOIs (0.1 and 1.0). Cells were followed over 82 days, and supernatant p24 antigen was measured every 2 to 4 days. Equal cell surface levels of CD4 and CXCR4 were verified by flow cytometry in the parental and knockout cells prior to infection (data not shown). Cultures were terminated on day 82 after infection. (C) PCR assays with primers 5 and 6 were performed on genomic DNA from the indicated cell lines (primer locations are shown in Fig. 1). (D) Titers of HIV-1 produced in Jurkat and Jurkat PSIP1−/− cells (infectious units [IU] per ng p24).
HIV-1 production and infectious particle assembly are normal in PSIP1−/− cells.
We found that viruses produced by replication in the parental Jurkat cells and the PSIP1 KO cells were equally infectious per ng of p24 (Fig. 7D). Sequencing also showed that there were no mutations in the integrase genes of these viruses (data not shown). As 293T cells are a preferred cell line for HIV-1 assembly and budding experiments, the PSIP1−/− 293T cells allowed us to further test the possibility of participation of LEDGF in late phases of the HIV-1 life cycle. HIV-1GFP viruses produced in IBD−/− 293T cells (Fig. 8A) and PSIP1−/− 293T cells (Fig. 8B) had undiminished infectivities.
FIG 8.
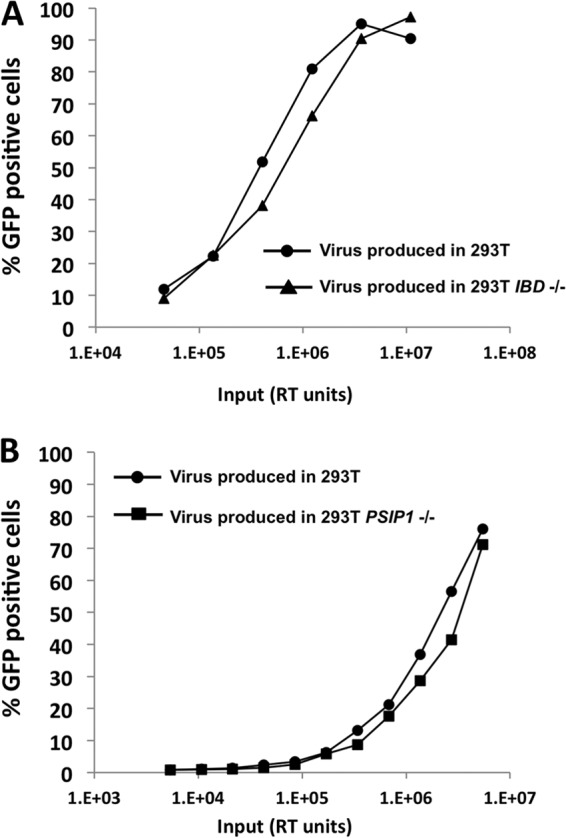
Comparative infectivities of HIV-1GFP viruses produced in parental and gene knockout 293T cells. Titrations of the RT-normalized preparations were done on Supt1 target cells and scored by flow cytometry. (A) Comparison of viral particles produced in 293T and in 293T IBD−/− cells. (B) Comparison of viral particles produced in 293T and in 293T PSIP1−/− cells.
ALLINI potency in blocking HIV-1 infectious particle formation is unaffected by total removal of LEDGF-encoding capacity from the cell.
Structural and biochemical studies have shown that ALLINIs bind to the HIV-1 IN CCD dimer interface at the principal LEDGF binding pocket and induce enhanced IN multimerization, which compromises catalytic activities of IN and prevents LEDGF binding to the preformed stable synaptic complex between the IN tetramer and viral DNA complex (25, 31, 55). Consistent with these in vitro findings ALLINIs blocked a step at or prior to 3′ processing during the early stage of virus replication (55). However, ALLINI antiviral activity was recently determined to primarily reflect inhibition of proper HIV-1 particle maturation (23, 27). Here, using ALLINI-2, an allosteric inhibitor previously described by Feng et al. (26), we found that LEDGF was dispensable for the ability of the inhibitor to impair the late stage of virus replication (Fig. 9A to D). The 50% effective concentrations (EC50s) of ALLNI-2 when added to the producer cells were not significantly different in cells lacking LEDGF (PSIP1−/− cells) or just the C-terminal protein segment containing the IBD (IBD−/− cells) (Table 1). We also determined that although ALLINIs are known to impair HIV-1 infectivity by leading to the production of particles that have reverse transcription defects in target cells, ALLNI-2 did not inhibit formation of a catalytically active reverse transcriptase enzyme in virions (Fig. 9E). The RT activities per ng of p24 for virions produced in the different cells were equivalent in the presence and absence of the inhibitor. This finding is consistent with data showing that proteolytic processing of the Gag-Pol precursor assessed by immunoblotting was unaffected by several allosteric integrase inhibitors (BI-D, GS-A, GS-B, and CXC05045), while the particle morphology was strongly aberrant (23, 27, 28), and that virion endogenous RT activities were not impaired by GS-B (23).
FIG 9.

Effects of adding ALLINI-2 during viral production on the infectivity of HIV-1 particles (late-stage effects). (A to D) Effect of ALLINI-2 (26) versus equal amounts of DMSO carrier on infectivity, represented as infectious units (IU) per ng of p24. (E) Reverse transcriptase activities per ng of p24 for HIV-1 virions produced in the indicated cell lines. The chemical structure of the inhibitor is shown in the lower left corner.
TABLE 1.
Antiviral EC50s
| Cell type | ALLINI-2 EC50 (μM) |
|---|---|
| 293T producer cell | 0.35 ± 0.06 |
| 293T PSIP1−/− producer cell | 0.29 ± 0.08 |
| 293T IBD−/− producer cell | 0.44 ± 0.06 |
| 293T target cell | >10 |
| 293T PSIP1−/− target cell | 4.6 ± 0.51 |
| 293T IBD−/− target cell | 2.86 ± 0.6 |
| Jurkat target cell | >10 |
| Jurkat PSIP1−/− target cell | 0.84 ± 0.08 |
When tested during early events in parental and knockout 293T and Jurkat cells, ALLINI-2 was considerably more potent in either PSIP1−/− or IBD−/− cells than in parental cells (Fig. 10 and Table 1). As previously suggested (27, 29, 39), this enhanced potency may be due to the lack of competing endogenous LEDGF during early stages of the infection process.
FIG 10.
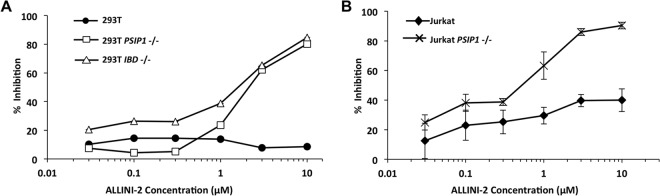
Effects of adding ALLINI-2 during infection (early-stage effects). (A) 293T lines. (B) Jurkat lines. Results are shown as percent inhibition of HIV-1luc luciferase activity. Error bars represent standard deviations of duplicate measurements.
Taken together, our results indicate that the role of LEDGF in the HIV-1 life cycle is confined to early events. LEDGF is dispensable for normal production and maturation of HIV-1. The results also show that the impairment to infectious HIV-1 particle assembly produced by a representative ALLINI is independent of LEDGF.
DISCUSSION
Here, we used TALENS to target the HIV-1 dependency factor LEDGF and generate valuable cell lines for HIV-1 research. Single-site gene disruptions were achieved with high efficiency, with 9 of 26 HT1080 cell clones having bi-allelic NHEJ-generated gene disruptions after a single transfection of TALEN pair X. All three TALEN pairs were effective when used in combinations for chromosomal segment excision (X+B and A+B). Large chromosome segments exceeding 40 kb (comprising most of the gene) and the segment of exons 12 to 14 (IBD-encoding) were excised selectively from PSIP1 in both adherent cells (293T) and CD4+ T cells (Jurkat). The HT1080 and Jurkat E6 cells banked with ATCC are pseudodiploid (normal modal chromosome numbers of 46, with some translocations). 293 and 293T cells are variably hypotriploid, with the few published subsequent karyotypes for 293 (56) and 293T (57) cells still revealing two copies of chromosome 9. Although chromosome 9 ploidy changes could have occurred in the parental cell lines prior to our gene knockouts, the assays we performed on the clonal lines indicate that TALEN-mediated deletion of all alleles was achieved. Sensitive immunoblotting for LEDGF protein and reverse transcription-PCR (RT-PCR) for mRNA were negative, and the DNA analyses demonstrated absence of the targeted deletion segments, as well as diagnostic X-to-B and A-to-B chromosome 9 fusion junctions.
The results in the HT1080 cells (Fig. 2) reinforce our previous findings that a fractionally minute amount of cellular LEDGF is sufficient to provide HIV-1 integration cofactor function (4). The definitive chromosome 9 gene segment excision knockouts, which extirpate all possibility of any LEDGF protein that can bind IN, in two cell lines that are heavily used for viral production and spreading replication studies, respectively, provide important research reagents with novel utility to the field. Our results further provide definitive evidence in two key areas. First, we conclude that LEDGF plays no detectable role in the assembly phase of the HIV-1 life cycle. Second, our results add to the accumulating recent evidence that ALLINIs act most potently during the assembly phase of the life cycle to disrupt infectious particle formation. Although there is heightened sensitivity to the early-phase ALLINI-2 effect in PSIP1−/− and IBD−/− target cells, presumably due to the lack of competing cellular LEDGF, the EC50s are still significantly higher than the EC50s for the late-phase effect (Table 1).
While it has been suggested that the assembly-phase effects of allosteric integrase inhibitors may be partly LEDGF dependent and also that LEDGF incorporation into HIV-1 particles may occur and be needed for normal HIV-1 infectivity (28, 32–34), we show here that reductions in particle infectivity triggered by ALLINI-2 are LEDGF independent as they were fully preserved in PSIP1 gene-deleted human cells.
The RT activities of HIV-1 particles produced in the presence of ALLINI-2 were unaffected despite the severe decrease in particle infectivity (Fig. 9E). Normal virion endogenous RT activities (23) and unperturbed Gag-Pol processing patterns (27, 28) have been reported with other ALLINIs even when particle core morphology determined by electron microscopy is grossly aberrant. Thus, our data are consistent with evidence that these inhibitors likely affect viral core maturation steps that occur subsequent to Gag-Pol and Gag protein proteolysis and that may be sensitive to improper IN multimerization (27).
Analyses of spreading HIV-1 replication in the Jurkat knockout cell lines show a substantial delay in HIV-1 propagation, even at high MOIs (Fig. 7). The substantial block to spreading replication at MOIs of 1.0 and 0.1 in this CD4+ T cell line exceed the replication delays previously observed after stable shRNA-mediated knockdown in other human CD4+ T cell lines. Considered together with the RNAi drift problem and need for periodic resorting of shRNA-depleted cells, the greater viral delay in PSIP1 knockout cells serves both to provide a more useful human CD4+ T cell line for further experimental work and to establish that the role of LEDGF as a cofactor for efficient viral replication is somewhat more substantial than was previously determined. The results in Jurkat IBD−/− clone 2 are interesting. Our initial perplexity at the greater relative permissivity of this cell line to HIV-1 replication than PSIP1−/− Jurkat cells and IBD−/− Jurkat clone 1 prompted us to ask whether the excised IBD exon segment still persisted in some form in clone 2. This was indeed the case. PCR assays using primers that flank the A and B sites were consistently negative, while PCR for sequences inside the A-to-B interval remained positive, suggesting that the excised A-B fragment reinserted in clone 2 in some fashion at an alternate genome position. The exact genomic configuration and the nature and extent of the protein expression from the rearranged segment of exons 12 to 14 detected by primers 5 and 6 are unclear at present. Of the two Jurkat IBD−/− clones, clone 1 is suited for further studies.
While antiretroviral drugs have revolutionized the treatment of HIV-1 disease, they do not result in its cure and are associated with the problems of resistance, persistent immune dysfunction and chronic inflammation, complex metabolic disturbances, accelerated aging phenomena including immunosenescence, and the adherence, toxicity and access problems associated with the necessary lifelong combination drug treatment (58, 59). In the United States, only approximately one-third of those with HIV-1 are currently prescribed antiretroviral therapy (ART), and 25% of these do not achieve effective virological control. Curative treatments for HIV disease are now considered high-priority goals, and two main strategies are envisioned at present: eradication of all latently infected cells and protection of uninfected target cells (60).
The burgeoning site-specific gene targeting armamentarium, including TALENs and CRISPR/Cas systems, is promising for the latter strategy (61). Observations in patients who are naturally CCR5−/− (62), the cure of HIV-1 in a patient with comorbid leukemia by allogeneic bone marrow transplantation from a CCR5−/− donor (63), and recent experiments with ZFN-based CCR5 gene knockouts (64–66) have established CCR5 as the most promising first-line target for curative gene therapy strategies. However, viruses that use the main alternative chemokine receptor CXCR4 are prevalent, and they encounter no restriction in CCR5−/− cells. Our results demonstrate that HIV-1 replication is substantially, though not completely, blocked in normally very permissive PSIP1−/− CD4+ Jurkat T cells (Fig. 5 to 7). This antiviral effect is specific to the loss of the chromosome tethering via the IBD as the IBD D366N mutation abrogated rescue and the LANA31-tethered IBD rescued well (Fig. 6 and 7). Coupled with the observation that the eradication of LEDGF did not produce evident toxicity in these cells (similarly to prior stringently knocked down CD4+ T cell lines) and that Psip1−/− mice completed development and did not have discernible immune system abnormalities (their two problems were poor feeding in the neonatal period, which led to enhanced juvenile mortality, and Hox gene-related skeletal dysmorphisms) (6, 67), the data suggest the possibility that PSIP1 and CCR5 may have potential for combinatorial site-specific nuclease gene targeting in HIV-1 disease. Recent advances in site-specific nuclease technologies have shown the feasibility of multiplex gene targeting (68, 69). Finally, as we noted above, the persistence of LEDGF/p75 dependence throughout the evolution of the lentiviral genus (20–22) may not be sufficiently explained at present. We did not observe mutations in the IN genes of viruses passaged in the knockout Jurkat cells, but the potential for HIV-1 adaptation to loss of LEDGF and the interplay with HRP-2 (15, 39, 42, 70) are interesting questions. Further gene knockouts in these PSIP1−/− or IBD−/− cells may be helpful in addressing these basic research questions.
ACKNOWLEDGMENTS
We are grateful for funding from NIH grant AI77344 and the John H. Tietze Foundation Trust to E.M.P., NIH grant AI110310 to M.K. and J.R.F, and NIH grant AI062520 to M.K.
Footnotes
Published ahead of print 18 June 2014
REFERENCES
- 1.Ge H, Roeder RG. 1994. Purification, cloning, and characterization of a human coactivator, PC4, that mediates transcriptional activation of class II genes. Cell 78:513–523. 10.1016/0092-8674(94)90428-6 [DOI] [PubMed] [Google Scholar]
- 2.Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, De Clercq E, Debyser Z. 2003. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 278:372–381. 10.1074/jbc.M209278200 [DOI] [PubMed] [Google Scholar]
- 3.Maertens G, Cherepanov P, Pluymers W, Busschots K, De Clercq E, Debyser Z, Engelborghs Y. 2003. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem. 278:33528–33539. 10.1074/jbc.M303594200 [DOI] [PubMed] [Google Scholar]
- 4.Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. 2006. An essential role for LEDGF/p75 in HIV integration. Science 314:461–464. 10.1126/science.1132319 [DOI] [PubMed] [Google Scholar]
- 5.Vandekerckhove L, Christ F, Van Maele B, De Rijck J, Gijsbers R, Van den Haute C, Witvrouw M, Debyser Z. 2006. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J. Virol. 80:1886–1896. 10.1128/JVI.80.4.1886-1896.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shun MC, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, Engelman A. 2007. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 21:1767–1778. 10.1101/gad.1565107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelman A, Cherepanov P. 2008. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 4:e1000046. 10.1371/journal.ppat.1000046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poeschla EM. 2008. Integrase, LEDGF/p75 and HIV replication. Cell. Mol. Life Sci. 65:1403–1424. 10.1007/s00018-008-7540-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, Bushman F. 2005. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 11:1287–1289. 10.1038/nm1329 [DOI] [PubMed] [Google Scholar]
- 10.Turlure F, Maertens G, Rahman S, Cherepanov P, Engelman A. 2006. A tripartite DNA-binding element, comprised of the nuclear localization signal and two AT-hook motifs, mediates the association of LEDGF/p75 with chromatin in vivo. Nucleic Acids Res. 34:1653–1665. 10.1093/nar/gkl052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Llano M, Vanegas M, Hutchins N, Thompson D, Delgado S, Poeschla EM. 2006. Identification and characterization of the chromatin binding domains of the HIV-1 integrase interactor LEDGF/p75. J. Mol. Biol. 360:760–773. 10.1016/j.jmb.2006.04.073 [DOI] [PubMed] [Google Scholar]
- 12.Eidahl JO, Crowe BL, North JA, McKee CJ, Shkriabai N, Feng L, Plumb M, Graham RL, Gorelick RJ, Hess S, Poirier MG, Foster MP, Kvaratskhelia M. 2013. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res. 41:3924–3936. 10.1093/nar/gkt074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Nuland R, van Schaik FM, Simonis M, van Heesch S, Cuppen E, Boelens R, Timmers HM, van Ingen H. 2013. Nucleosomal DNA binding drives the recognition of H3K36-methylated nucleosomes by the PSIP1-PWWP domain. Epigenetics Chromatin 6:12. 10.1186/1756-8935-6-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cherepanov P, Devroe E, Silver PA, Engelman A. 2004. Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J. Biol. Chem. 279:48883–48892. 10.1074/jbc.M406307200 [DOI] [PubMed] [Google Scholar]
- 15.Vanegas M, Llano M, Delgado S, Thompson D, Peretz M, Poeschla E. 2005. Identification of the LEDGF/p75 HIV-1 integrase-interaction domain and NLS reveals NLS-independent chromatin tethering. J. Cell Sci. 118:1733–1743. 10.1242/jcs.02299 [DOI] [PubMed] [Google Scholar]
- 16.Cherepanov P, Ambrosio AL, Rahman S, Ellenberger T, Engelman A. 2005. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. U. S. A. 102:17308–17313. 10.1073/pnas.0506924102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cherepanov P, Sun ZY, Rahman S, Maertens G, Wagner G, Engelman A. 2005. Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75. Nat. Struct. Mol. Biol. 12:526–532. 10.1038/nsmb937 [DOI] [PubMed] [Google Scholar]
- 18.Hare S, Shun MC, Gupta SS, Valkov E, Engelman A, Cherepanov P. 2009. A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog. 5:e1000259. 10.1371/journal.ppat.1000259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKee CJ, Kessl JJ, Shkriabai N, Dar MJ, Engelman A, Kvaratskhelia M. 2008. Dynamic modulation of HIV-1 integrase structure and function by cellular LEDGF protein. J. Biol. Chem. 283:31802–31812. 10.1074/jbc.M805843200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Llano M, Vanegas M, Fregoso O, Saenz DT, Chung S, Peretz M, Poeschla EM. 2004. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral pre-integration complexes. J. Virol. 78:9524–9537. 10.1128/JVI.78.17.9524-9537.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Busschots K, Vercammen J, Emiliani S, Benarous R, Engelborghs Y, Christ F, Debyser Z. 2005. The interaction of LEDGF/p75 with integrase is lentivirus-specific and promotes DNA binding. J. Biol. Chem. 280:17841–17847. 10.1074/jbc.M411681200 [DOI] [PubMed] [Google Scholar]
- 22.Cherepanov P. 2007. LEDGF/p75 interacts with divergent lentiviral integrases and modulates their enzymatic activity in vitro. Nucleic Acids Res. 35:113–124. 10.1093/nar/gkl885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balakrishnan M, Yant SR, Tsai L, O'Sullivan C, Bam RA, Tsai A, Niedziela-Majka A, Stray KM, Sakowicz R, Cihlar T. 2013. Non-catalytic site HIV-1 integrase inhibitors disrupt core maturation and induce a reverse transcription block in target cells. PLoS One 8:e74163. 10.1371/journal.pone.0074163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel B, Strelkov SV, De Maeyer M, Chaltin P, Debyser Z. 2010. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 6:442–448. 10.1038/nchembio.370 [DOI] [PubMed] [Google Scholar]
- 25.Kessl JJ, Jena N, Koh Y, Taskent-Sezgin H, Slaughter A, Feng L, de Silva S, Wu L, Le Grice SF, Engelman A, Fuchs JR, Kvaratskhelia M. 2012. Multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 287:16801–16811. 10.1074/jbc.M112.354373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng L, Sharma A, Slaughter A, Jena N, Koh Y, Shkriabai N, Larue RC, Patel PA, Mitsuya H, Kessl JJ, Engelman A, Fuchs JR, Kvaratskhelia M. 2013. The A128T resistance mutation reveals aberrant protein multimerization as the primary mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 288:15813–15820. 10.1074/jbc.M112.443390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jurado KA, Wang H, Slaughter A, Feng L, Kessl JJ, Koh Y, Wang W, Ballandras-Colas A, Patel PA, Fuchs JR, Kvaratskhelia M, Engelman A. 2013. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. U. S. A. 110:8690–8695. 10.1073/pnas.1300703110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desimmie BA, Schrijvers R, Demeulemeester J, Borrenberghs D, Weydert C, Thys W, Vets S, Van Remoortel B, Hofkens J, De Rijck J, Hendrix J, Bannert N, Gijsbers R, Christ F, Debyser Z. 2013. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 10:57. 10.1186/1742-4690-10-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le Rouzic E, Bonnard D, Chasset S, Bruneau JM, Chevreuil F, Le Strat F, Nguyen J, Beauvoir R, Amadori C, Brias J, Vomscheid S, Eiler S, Levy N, Delelis O, Deprez E, Saib A, Zamborlini A, Emiliani S, Ruff M, Ledoussal B, Moreau F, Benarous R. 2013. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology 10:144. 10.1186/1742-4690-10-144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jurado KA, Engelman A. 2013. Multimodal mechanism of action of allosteric HIV-1 integrase inhibitors. Expert Rev. Mol. Med. 15:e14. 10.1017/erm.2013.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christ F, Shaw S, Demeulemeester J, Desimmie BA, Marchand A, Butler S, Smets W, Chaltin P, Westby M, Debyser Z, Pickford C. 2012. Small-molecule inhibitors of the LEDGF/p75 binding site of integrase block HIV replication and modulate integrase multimerization. Antimicrob. Agents Chemother. 56:4365–4374. 10.1128/AAC.00717-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desimmie BA, Humbert M, Lescrinier E, Hendrix J, Vets S, Gijsbers R, Ruprecht RM, Dietrich U, Debyser Z, Christ F. 2012. Phage display-directed discovery of LEDGF/p75 binding cyclic peptide inhibitors of HIV replication. Mol. Ther. 20:2064–2075. 10.1038/mt.2012.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desimmie B, Schrijvers R, Vets S, Demeulemeester J, Proost P, De Rijck J, Bannert N, Christ F, Gijsbers R, Debyser Z. 2013. Incorporation of LEDGF/p75 in viral particles is crucial for HIV infectivity, paper 138. 2013 Meet. Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 34.Desimmie B, Schrijvers R, Demeulemeester J, Borrenberghs D, Weydert C, Thys W, Vets S, Hendrix J, Bannert N, Gijsbers R, Debyser Z. 2013. LEDGINs reveal a role for LEDGF/p75 in late stage HIV replication, paper 102. 2013 Meet. Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 35.Meehan A, Saenz D, Morrison J, Peretz M, Poeschla E. 2011. LEDGF dominant interference proteins demonstrate pre-nuclear exposure of HIV-1 integrase and synergize with LEDGF depletion to destroy viral infectivity. J. Virol. 85:3570–3583. 10.1128/JVI.01295-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miest T, Saenz D, Meehan A, Llano M, Poeschla EM. 2009. Intensive RNAi with lentiviral vectors in mammalian cells. Methods 47:298–303. 10.1016/j.ymeth.2008.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meehan AM, Saenz DT, Morrison JH, Garcia-Rivera JA, Peretz M, Llano M, Poeschla EM. 2009. LEDGF/p75 proteins with alternative chromatin tethers are functional HIV-1 cofactors. PLoS Pathog. 5:e1000522. 10.1371/journal.ppat.1000522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferris AL, Wu X, Hughes CM, Stewart C, Smith SJ, Milne TA, Wang GG, Shun MC, Allis CD, Engelman A, Hughes SH. 2010. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. Natl. Acad. Sci. U. S. A. 107:3135–3140. 10.1073/pnas.0914142107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Jurado KA, Wu X, Shun MC, Li X, Ferris AL, Smith SJ, Patel PA, Fuchs JR, Cherepanov P, Kvaratskhelia M, Hughes SH, Engelman A. 2012. HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic Acids Res. 40:11518–11530. 10.1093/nar/gks913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bieniasz PD, Cullen BR. 2000. Multiple blocks to human immunodeficiency virus type 1 replication in rodent cells. J. Virol. 74:9868–9877. 10.1128/JVI.74.21.9868-9877.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baumann JG, Unutmaz D, Miller MD, Breun SK, Grill SM, Mirro J, Littman DR, Rein A, KewalRamani VN. 2004. Murine T cells potently restrict human immunodeficiency virus infection. J. Virol. 78:12537–12547. 10.1128/JVI.78.22.12537-12547.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schrijvers R, De Rijck J, Demeulemeester J, Adachi N, Vets S, Ronen K, Christ F, Bushman FD, Debyser Z, Gijsbers R. 2012. LEDGF/p75-independent HIV-1 replication demonstrates a role for HRP-2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog. 8:e1002558. 10.1371/journal.ppat.1002558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moscou MJ, Bogdanove AJ. 2009. A simple cipher governs DNA recognition by TAL effectors. Science 326:1501. 10.1126/science.1178817 [DOI] [PubMed] [Google Scholar]
- 44.Bogdanove AJ, Voytas DF. 2011. TAL effectors: customizable proteins for DNA targeting. Science 333:1843–1846. 10.1126/science.1204094 [DOI] [PubMed] [Google Scholar]
- 45.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. 2009. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512. 10.1126/science.1178811 [DOI] [PubMed] [Google Scholar]
- 46.Wood AJ, Lo TW, Zeitler B, Pickle CS, Ralston EJ, Lee AH, Amora R, Miller JC, Leung E, Meng X, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Meyer BJ. 2011. Targeted genome editing across species using ZFNs and TALENs. Science 333:307. 10.1126/science.1207773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carlson DF, Tan W, Lillico SG, Stverakova D, Proudfoot C, Christian M, Voytas DF, Long CR, Whitelaw CB, Fahrenkrug SC. 2012. Efficient TALEN-mediated gene knockout in livestock. Proc. Natl. Acad. Sci. U. S. A. 109:17382–17387. 10.1073/pnas.1211446109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma AC, Lee HB, Clark KJ, Ekker SC. 2013. High efficiency in vivo genome engineering with a simplified 15-RVD GoldyTALEN design. PLoS One 8:e65259. 10.1371/journal.pone.0065259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Piganeau M, Ghezraoui H, De Cian A, Guittat L, Tomishima M, Perrouault L, Rene O, Katibah GE, Zhang L, Holmes MC, Doyon Y, Concordet JP, Giovannangeli C, Jasin M, Brunet E. 2013. Cancer translocations in human cells induced by zinc finger and TALE nucleases. Genome Res. 23:1182–1193. 10.1101/gr.147314.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao A, Wang Z, Hu Y, Wu Y, Luo Z, Yang Z, Zu Y, Li W, Huang P, Tong X, Zhu Z, Lin S, Zhang B. 2013. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 41:e141. 10.1093/nar/gkt464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG, II, Tan W, Penheiter SG, Ma AC, Leung AY, Fahrenkrug SC, Carlson DF, Voytas DF, Clark KJ, Essner JJ, Ekker SC. 2012. In vivo genome editing using a high-efficiency TALEN system. Nature 491:114–118. 10.1038/nature11537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. 2011. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39:e82. 10.1093/nar/gkr218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morrison JH, Guevara RB, Marcano AC, Saenz DT, Fadel HJ, Rogstad DK, Poeschla EM. 2014. Feline immunodeficiency virus envelope glycoproteins antagonize tetherin through a distinctive mechanism that requires virion incorporation. J. Virol. 88:3255–3272. 10.1128/JVI.03814-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856–861. 10.1126/science.1120541 [DOI] [PubMed] [Google Scholar]
- 55.Tsiang M, Jones GS, Niedziela-Majka A, Kan E, Lansdon EB, Huang W, Hung M, Samuel D, Novikov N, Xu Y, Mitchell M, Guo H, Babaoglu K, Liu X, Geleziunas R, Sakowicz R. 2012. New class of HIV-1 integrase (IN) inhibitors with a dual mode of action. J. Biol. Chem. 287:21189–21203. 10.1074/jbc.M112.347534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bylund L, Kytola S, Lui WO, Larsson C, Weber G. 2004. Analysis of the cytogenetic stability of the human embryonal kidney cell line 293 by cytogenetic and STR profiling approaches. Cytogenet. Genome Res. 106:28–32. 10.1159/000078556 [DOI] [PubMed] [Google Scholar]
- 57.Oka Y, Nakajima K, Nagao K, Miura K, Ishii N, Kabayashi H. 2010. 293FT cells transduced with four transcription factors (OCT4, SOX2, NANOG, and LIN28) generate aberrant ES-like cells. J. Stem Cells Regen. Med. 6:149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deeks SG. 2011. HIV infection, inflammation, immunosenescence, and aging. Annu. Rev. Med. 62:141–155. 10.1146/annurev-med-042909-093756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Justice AC. 2010. HIV and aging: time for a new paradigm. Curr. HIV/AIDS Rep. 7:69–76. 10.1007/s11904-010-0041-9 [DOI] [PubMed] [Google Scholar]
- 60.Deeks SG, Autran B, Berkhout B, Benkirane M, Cairns S, Chomont N, Chun TW, Churchill M, Di Mascio M, Katlama C, Lafeuillade A, Landay A, Lederman M, Lewin SR, Maldarelli F, Margolis D, Markowitz M, Martinez-Picado J, Mullins JI, Mellors J, Moreno S, O'Doherty U, Palmer S, Penicaud MC, Peterlin M, Poli G, Routy JP, Rouzioux C, Silvestri G, Stevenson M, Telenti A, Van Lint C, Verdin E, Woolfrey A, Zaia J, Barre-Sinoussi F. 2012. Towards an HIV cure: a global scientific strategy. Nat. Rev. Immunol. 12:607–614. 10.1038/nri3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gaj T, Gersbach CA, Barbas CF., III 2013. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31:397–405. 10.1016/j.tibtech.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang Y, Paxton WA, Wolinsky SM, Neumann AU, Zhang L, He T, Kang S, Ceradini D, Jin Z, Yazdanbakhsh K, Kunstman K, Erickson D, Dragon E, Landau NR, Phair J, Ho DD, Koup RA. 1996. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat. Med. 2:1240–1243. 10.1038/nm1196-1240 [DOI] [PubMed] [Google Scholar]
- 63.Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, Schneider T, Hofmann J, Kucherer C, Blau O, Blau IW, Hofmann WK, Thiel E. 2009. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 360:692–698. 10.1056/NEJMoa0802905 [DOI] [PubMed] [Google Scholar]
- 64.Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, Guschin DY, Rupniewski I, Waite AJ, Carpenito C, Carroll RG, Orange JS, Urnov FD, Rebar EJ, Ando D, Gregory PD, Riley JL, Holmes MC, June CH. 2008. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 26:808–816. 10.1038/nbt1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, Crooks GM, Kohn DB, Gregory PD, Holmes MC, Cannon PM. 2010. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 28:839–847. 10.1038/nbt.1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, Holmes MC, Gregory PD, Ando DG, Kalos M, Collman RG, Binder-Scholl G, Plesa G, Hwang WT, Levine BL, June CH. 2014. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 370:901–910. 10.1056/NEJMoa1300662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sutherland HG, Newton K, Brownstein DG, Holmes MC, Kress C, Semple CA, Bickmore WA. 2006. Disruption of Ledgf/Psip1 results in perinatal mortality and homeotic skeletal transformations. Mol. Cell. Biol. 26:7201–7210. 10.1128/MCB.00459-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carroll D. 2014. Genome engineering with targetable nucleases. Annu. Rev. Biochem. 83:409–439. 10.1146/annurev-biochem-060713-035418 [DOI] [PubMed] [Google Scholar]
- 70.Izumoto Y, Kuroda T, Harada H, Kishimoto T, Nakamura H. 1997. Hepatoma-derived growth factor belongs to a gene family in mice showing significant homology in the amino terminus. Biochem. Biophys. Res. Commun. 238:26–32. 10.1006/bbrc.1997.7233 [DOI] [PubMed] [Google Scholar]