

ABSTRACT
Influenza is a global health concern, causing death, morbidity, and economic losses. Chemotherapeutics that target influenza virus are available; however, rapid emergence of drug-resistant strains is common. Therapeutic targeting of host proteins hijacked by influenza virus to facilitate replication is an antiviral strategy to reduce the development of drug resistance. Nuclear export of influenza virus ribonucleoprotein (vRNP) from infected cells has been shown to be mediated by exportin 1 (XPO1) interaction with viral nuclear export protein tethered to vRNP. RNA interference screening has identified XPO1 as a host proinfluenza factor where XPO1 silencing results in reduced influenza virus replication. The Streptomyces metabolite XPO1 inhibitor leptomycin B (LMB) has been shown to limit influenza virus replication in vitro; however, LMB is toxic in vivo, which makes it unsuitable for therapeutic use. In this study, we tested the anti-influenza virus activity of a new class of orally available small-molecule selective inhibitors of nuclear export, specifically, the XPO1 antagonist KPT-335 (verdinexor). Verdinexor was shown to potently and selectively inhibit vRNP export and effectively inhibited the replication of various influenza virus A and B strains in vitro, including pandemic H1N1 virus, highly pathogenic H5N1 avian influenza virus, and the recently emerged H7N9 strain. In vivo, prophylactic and therapeutic administration of verdinexor protected mice against disease pathology following a challenge with influenza virus A/California/04/09 or A/Philippines/2/82-X79, as well as reduced lung viral loads and proinflammatory cytokine expression, while having minimal toxicity. These studies show that verdinexor acts as a novel anti-influenza virus therapeutic agent.
IMPORTANCE Antiviral drugs represent important means of influenza virus control. However, substantial resistance to currently approved influenza therapeutic drugs has developed. New antiviral approaches are required to address drug resistance and reduce the burden of influenza virus-related disease. This study addressed critical preclinical studies for the development of verdinexor (KPT-335) as a novel antiviral drug. Verdinexor blocks progeny influenza virus genome nuclear export, thus effectively inhibiting virus replication. Verdinexor was found to limit the replication of various strains of influenza A and B viruses, including a pandemic H1N1 influenza virus strain, a highly pathogenic H5N1 avian influenza virus strain, and a recently emerging H7N9 influenza virus strain. Importantly, oral verdinexor treatments, given prophylactically or therapeutically, were efficacious in limiting lung virus burdens in influenza virus-infected mice, in addition to limiting lung proinflammatory cytokine expression, pathology, and death. Thus, this study demonstrated that verdinexor is efficacious against influenza virus infection in vitro and in vivo.
INTRODUCTION
Influenza virus causes seasonal epidemics of mild to severe respiratory illness and occasionally death (1–3). Older individuals, young children, and the immunocompromised are at high risk of serious complications, which may include bacterial pneumonia and exacerbation of chronic medical conditions such as congestive heart failure, asthma, or diabetes (4–6). Vaccines against circulating influenza virus strains are the mainstay of prophylaxis and are annually available; however, there are issues with predicting which influenza virus strains may emerge and preparing sufficient quantities of vaccine and difficulty in storage and administration (7). Moreover, vaccine efficacy is variable and generally low in the populations at greatest risk of complications from influenza virus infection, the young and the elderly (8). Despite increasing vaccination rates, influenza-related hospitalizations are increasing (8, 9). Currently, there are four antiviral drugs licensed for use in the United States, all of which target viral components. Amantadine and rimantadine target the viral M2 protein, binding to the pore, blocking the proton channel, and preventing release of the viral RNA genome into the cytoplasm of the infected cell (10). These adamantines are active against influenza A viruses but not influenza B viruses. Widespread drug resistance has developed in H3N2 viruses (11), and circulating pandemic H1N1 (pH1N1) viruses are resistant to adamantine (12). Resistance to these drugs has resulted in the CDC not recommending their use to treat infection with these influenza virus strains (12). Oseltamivir and zanamivir make up the second class of influenza antivirals, i.e., the neuraminidase (NA) inhibitors. These drugs bind to the active site and block the enzymatic activity of the viral NA, causing the virus to aggregate on the surface of infected cells during virus release, preventing spread of the progeny virus during infection (13). Both influenza A and B viruses are susceptible to NA inhibitors; however, drug resistance has been reported. For this reason, the use of NA inhibitors is not recommended for uncomplicated outpatient cases of influenza where there are no identified risk factors, highlighting the pressing need for novel influenza virus therapeutics (12).
Influenza A virus causes seasonal disease, regular epidemics, and more rarely pandemics. There are two influenza B virus lineages, Yamagata and Victoria, that cocirculate in humans, causing seasonal disease and epidemics (1, 14, 15). The features of influenza A viruses that enable pandemics are thought to include the error-prone RNA-dependent RNA polymerase introducing mutations, antigenic drift in the surface glycoproteins, and adaptation to the host response to infection. The segmented nature of the genome enables reassortment of the genome during coinfection, and there are numerous susceptible species and reservoirs of influenza A virus (14, 15). The rapid and dramatic changes in virus host specificity and antigenicity hinder the development of vaccines in response to pandemics and even reduce the ability to prepare through vaccine stockpiling, further supporting the need for broadly reactive antiviral drugs.
Several studies have recently reported host cellular factors involved in influenza A virus replication (16–24). These factors can either act to suppress virus replication (antiviral factors) or be hijacked by the virus to support its replication (proviral factors). Targeting of these proviral host factors represents a potentially viable therapeutic strategy for the treatment of influenza virus replication. Importantly, because of the stability of host gene targets compared to viruses that rapidly adapt for best fitness in hosts, targeting of host genes also offers an innovative and refractory approach to limiting drug resistance. Since influenza virus replicates in the nuclei of infected cells, targeting of the host machinery required to shuttle viral proteins from the nucleus to the cytoplasm is a viable therapeutic strategy. In this case, following receptor-mediated binding and endocytosis of the influenza virion, the viral M2 proton channel protein mediates virion acidification inside the endosome, which leads to its subsequent fusion and release of virus ribonucleoproteins (vRNPs) containing the negative-stranded viral RNA (vRNA) genome, nucleoprotein (NP), and polymerases (PA, PB1, PB2) into the cell cytoplasm (25). For replication to take place in the cell nucleus, vRNPs are subsequently imported into the nucleus via cellular importin-α (26–28). Following virus RNA replication and transcription in the nucleus, progeny vRNPs are exported to the cytoplasm to allow the packaging of new virus progeny (29–32). Nuclear export of influenza virus vRNPs has been shown to be mediated by host exportin 1 (XPO1) interaction with nuclear export signal (NES)-containing viral nuclear export protein (NEP) associated with vRNPs (29, 32, 33). Using RNA interference (RNAi) screening of a human drug target library, we have identified XPO1 as a host proinfluenza factor. XPO1 silencing resulted in reduced influenza virus replication. Importantly, this mechanism of vRNP nuclear export is conserved in all types and subtypes of influenza virus (30, 33); thus, targeting of this pathway can potentially serve as a broad-spectrum anti-influenza virus therapeutic strategy.
XPO1 is a member of the karyopherin-β superfamily of nuclear transport proteins, which includes 15 different importin and exportin proteins. There are seven NEPs, designated XPO1 through XPO7. XPO1 is the best-characterized nuclear exporter, transporting ∼220 proteins and certain RNA species from the nucleus to the cytoplasm through the nuclear pore complex (34). XPO1 binds to a diverse array of protein cargos through binding of the XPO1 hydrophobic groove and the canonical leucine-rich or 10- to 15-residue-long motifs spanning four or five spaced hydrophobic amino acid domains termed NESs in cargo proteins (35). In the nucleus, XPO1 forms a quaternary complex with the cargo protein, Ran-GTP, and Ran-BP3. The role of Ran-BP3 is to promote nucleotide exchange by Ran-GEF RCC1, as well as to increase the affinity of XPO1 for Ran-GTP and the transported cargo. In the cytoplasm, the complex is dissociated through the combined action of Ran-GAP and Ran-BP1. The natural product leptomycin B (LMB), a prototypical inhibitor of XPO1, has been widely used to study XPO1 functions and the transport of its cargo molecules (34, 36, 37). LMB binds by forming an irreversible covalent bond with cysteine-528 of XPO1 to prevent the nuclear export of XPO1 cargo molecules (36, 38). Importantly, LMB has been shown to block vRNP nuclear export and inhibit influenza virus replication. However, LMB has substantial cytotoxicity in vivo that is potentially due to off-target activity (39) and the irreversible covalent binding of LMB to cysteine-528 of XPO1 (38). Thus, LMB was deemed unsuitable as a therapeutic agent (40).
A new class of orally available selective inhibitors of nuclear export (SINE) was recently developed (41, 42) by using molecular modeling to screen a small virtual library of compounds for activity against the NES groove of XPO1 and specific binding to XPO1 (35, 41, 43–46). However, unlike LMB, SINE compounds form a slowly reversible covalent bond with cysteine-528 of XPO1 (35, 38, 42). Thus, SINE compounds interfere with the nuclear export of NES-bearing proteins, including the NEP of influenza A and B viruses and NES-containing host proteins (47). SINE XPO1 inhibitors show minimal cytotoxicity to normal cells and demonstrated good tolerability in rodents, dogs, and nonhuman primates. In addition, verdinexor displayed single-agent activity in phase I and II clinical trials in client-owned dogs with B- and T-cell lymphomas (48, 49). Another closely related SINE compound, selinexor, is currently in multiple phase I and II studies of human patients with advanced solid and hematological malignancies (such as NCT01607892, NCT01607905, NCT01986348, and NCT02025985, see ClinicalTrials.gov) and is showing evidence of anticancer activity with good tolerability (50–52). In this study, the efficacy of verdinexor against multiple circulating and noncirculating influenza A and B virus strains was evaluated in vitro. Furthermore, its abilities to limit disease pathogenesis and lung virus burdens following challenges with representative strains of circulating influenza A virus subtypes (H1N1 and H3N2) were evaluated in vivo in a mouse model of influenza virus infection. The findings from this study demonstrated that verdinexor is efficacious against the influenza virus strains examined. Furthermore, because of its mechanism of action, it is likely to be efficacious against all strains of influenza virus.
MATERIALS AND METHODS
Cell cultures and influenza virus stocks.
Human type II respiratory epithelial (A549) cells (ATCC CCL-185), Madin-Darby canine kidney (MDCK) cells (ATCC CCL-34), and human embryonic kidney (293T) cells (ATCC CRL-3216) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% heat-inactivated fetal bovine serum (HyClone, Logan, UT) in a 37°C incubator with 5% CO2. Influenza virus strains A/WSN/33 (H1N1), A/California/04/09 (pH1N1), A/California/04/09 (MA-pH1N1; mouse adapted), A/Philippines/2/82-X79 (H3N2), and A/Vietnam/1204/04 (H5N1; highly pathogenic avian influenza virus [HPAIV]), A/mute swan/MI/451072-2/06 (H5N1; low-pathogenicity avian influenza virus [LPAIV]), A/red knot/NJ/0523470/06 (H7N3), A/Anhui/1/2013 (H7N9), B/Florida/04/06, and B/Ohio/01/05 were propagated in 9-day-old embryonic chicken eggs, and titers were determined in MDCK cells as previously described (53, 54). Studies involving influenza A virus strains A/Anhui/1/2013 (H7N9) and A/Vietnam/1204/04 (H5N1; HPAIV) were performed under appropriate biosafety level 3 conditions.
RNAi transfection.
Small interfering RNAs (siRNAs) targeting human XPO1 and MEK and a nontargeting siRNA were used (Dharmacon Thermo Fisher). A549 cells were reverse transfected with siRNA by using DharmaFECT-1 reagent (Dharmacon) as previously described (20). Transfections were carried out for 48 h to allow maximal expression knockdown before cells were infected with influenza virus at a multiplicity of infection (MOI) of 0.001. The level of infectious virus was measured at 48 h postinfection (hpi) by titer determination of A549 cell supernatant on MDCK cells (54). For virus titer determinations, culture supernatants were serially diluted and titers were determined on MDCK cells for 72 h. Hemagglutination (HA) assays were performed with turkey red blood cells and virus-infected MDCK cell supernatant as described previously (54, 55). The HA titer was determined from the highest dilution factor that produced a positive HA reading, and virus titers were calculated as 50% tissue culture infective doses (TCID50s) with the Spearman-Kärber formula (54, 55). In addition, when specified, A549 cell monolayers on culture plates were fixed and analyzed for the presence of influenza virus NP by immunofluorescence staining as described below.
Plasmids and transfections.
Total RNA from cells infected with influenza virus A/WSN/33 (H1N1), A/California/04/09 (pH1N1), and A/Anhui/1/2013 (H7N9) were used in a standard reverse transcription (RT)-PCR to amplify the viral NEP coding sequence. PCR primers were designed to eliminate the native stop codon from the NEP sequence. The NEP coding sequence was ligated to the TA cloning vector pCDNA3.1 (Life Technologies) by following the manufacturer's instructions, resulting in NEP fused in frame with a C-terminal poly-His tag and the V5 epitope. The constructs were verified by DNA sequencing. FLAG-hCRM1 plasmid DNA was obtained from Addgene (56). Plasmid DNA transfections were done with FuGENE HD transfection reagent (Promega).
Immunoblot and coimmunoprecipitation analyses.
Cells were lysed in radioimmunoprecipitation assay buffer (Cell Signaling Technology). For immunoblotting, equivalent protein amounts were diluted in SDS sample buffer (4× buffer contained 40% glycerol, 240 mM Tris HCl [pH 6.8], 8% SDS, 0.04% bromophenol blue, and 5% β-mercaptoethanol), boiled, and resolved by SDS-polyacrylamide gel electrophoresis, followed by immunoblotting. Rabbit polyclonal antibodies against human XPO1 (Santa Cruz Biotechnology), FLAG M2 (Sigma-Aldrich), influenza virus A NS2 (Thermo Scientific), or glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Millipore) were used as the primary antibodies. Horseradish peroxidase-conjugated goat anti-rabbit antibody (Sigma-Aldrich) was used as the secondary antibody. Protein bands were visualized following the addition of SuperSignal West Dura Extended Duration Substrate (Pierce, Thermo Scientific) with the FluorChem-E Western imaging system (ProteinSimple). For coimmunoprecipitation of XPO1, protein complexes were captured on anti-FLAG M2 magnetic beads (Sigma-Aldrich). Bound protein complexes were washed in 1× Tris-buffered saline, eluted with SDS loading buffer, and then subjected to immunoblotting as described above.
In vitro efficacy assays.
Verdinexor (KPT-335) was synthesized by Piramal Pharma Solutions (Aurora, ON, Canada) under a contract from Karyopharm Therapeutics. Verdinexor was dissolved in dimethyl sulfoxide (DMSO). For dose-response virus inhibition experiments, cells were washed once with phosphate-buffered saline prior to the titration of verdinexor with the Hewlett-Packard (HP) D300 Digital Dispenser (Tecan) (57) 2 h before infection. Cells were then infected with influenza virus strains at an MOI of 0.01. At 24 hpi, culture supernatants were collected for virus titer determination in MDCK cells as described above. Percent virus inhibition was calculated relative to that in DMSO-treated cells. Cellular toxicity was determined by measuring adenylate kinase release with the ToxiLight Bioassay kit (Lonza). For in vitro studies, verdinexor was used at a 1 μM concentration and LMB was used at 10 nM as a positive control. Cells were inoculated with influenza virus A/WSN/33 (H1N1) at an MOI of 3, incubated on ice for 30 min to allow virus binding, and subsequently incubated in a 37°C incubator to allow synchronized virus infection. At 7 hpi, cells were fixed for immunofluorescence staining or collected for RNA isolation.
Immunofluorescence staining.
To visualize the subcellular localization of vRNP, cells were stained for the viral NP or M2 protein. A549 cells were fixed with cold methanol-acetone (80:20) for 15 min and incubated with a mouse anti-NP monoclonal (ATCC H16-L10-4R5), a rabbit anti-influenza virus M2 polyclonal (GeneTex), or a rabbit anti-nuclear factor kappa B (NF-κB) p65 monoclonal (Cell Signaling Technology) antibody overnight at 4°C and then incubated for 1 h with an Alexa 488-conjugated goat anti-mouse or an Alexa 546-conjugated goat anti-rabbit (Invitrogen) secondary antibody and counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) as described previously (24). Cells were visualized with the EVOS fluorescent imaging system (Advanced Microscopy Group).
RT-qPCR analyses.
To assess NF-κB-dependent cytokine expression, cDNA was synthesized from total RNA with the Verso cDNA synthesis kit and random hexamers as primers (Thermo Scientific). cDNA were subsequently used for quantitative PCR (qPCR) amplifications with murine Ifng, Tnf, Il1b, Il6, and Gapdh gene-specific primers (PrimerBank, Harvard Medical School) (58) and RT2 SYBR green qPCR master mix (SABiosciences) in an MX3005P thermocycler as previously described (24). For evaluation of subcellular viral RNA distribution, nuclear and cytoplasmic RNAs were isolated with the SurePrep nuclear or cytoplasmic RNA purification kit (Fisher Scientific). For detection of strand-specific viral RNA in cytoplasmic and nuclear fractions, primers specific for influenza virus A segment 5 positive-sense mRNA and negative-sense vRNA containing an additional unrelated 18- to 20-nucleotide tag at the 5′ end for increased specificity to distinguish the different RNA species as described previously (59). Briefly, equal amounts of fractionated RNA were used to synthesize cDNA complementary to the different types of influenza virus A RNA with the Verso cDNA synthesis kit (Thermo Scientific). qPCR analysis was performed with RT2 SYBR green qPCR master mix (SABiosciences) and primer sets specific to the corresponding influenza virus RNA (59) in an MX3005P thermocycler. The abundance of viral RNA was normalized to GAPDH, and its expression relative to that in mock-treated samples was calculated with the 2−ΔΔCT formula.
In vivo efficacy studies.
BALB/c female mice (6 to 8 weeks old) were obtained from the National Cancer Institute. All experiments and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Georgia. All experiments were performed with 10 mice per group and repeated independently at least twice.
For in vivo studies to evaluate lung virus burdens, verdinexor was administered orally by gavage at the doses and time points before or after influenza virus infection indicated in Results. Mice were inoculated intranasally with mouse-adapted influenza virus strain A/California/04/09 (pH1N1) or A/Philippines/2/82-X79 (H3N2) at 10 times the respective MID50 (50% mouse infective dose) of 35 or 75 PFU. Lungs were collected at 96 hpi, homogenized in serum-free DMEM, and serially diluted to assess virus titers on MDCK cells as described above.
For a study of verdinexor pharmacokinetics in plasma, 21 male CD1 mice (3 mice per group per time point, approximately 28 g of body weight) [BK Laboratory Animal Co. Ltd., Shanghai, China; qualification no. SCXK(SH) 2008-0016 12470] were used. Plasma verdinexor concentrations were determined by an ultrahigh-performance liquid chromatography–mass spectrometry method.
Histopathological analyses.
Lungs from four or five mice per group of normal mock-infected mice and diluent- or verdinexor-treated mice infected with 10 50% lethal doses (LD50) of influenza virus A/California/04/09 (mouse-adapted pH1N1) were perfused with 10% (wt/vol) buffered formalin (Fisher Scientific). Formalin-fixed tissues were embedded in paraffin wax, sectioned (4 μm thick), and mounted on glass slides. Tissues were stained with hematoxylin and eosin (H&E) or stained for viral NP by immunohistochemistry (IHC) with a mouse monoclonal antibody as previously described (60) and examined by a board-certified pathologist at the College of Veterinary Medicine at the University of Georgia. Lung pathology was scored on a scale of 0 to 4 as previously described (61). A score of 0 is defined as unremarkable; 1 is defined as minimal changes in the bronchiolar epithelium with minimal perivascular inflammation; 2 is defined as mild, multifocal bronchiolar epithelial changes with perivascular and peribronchiolar inflammation; 3 is defined as moderate, multifocal bronchiolar epithelial changes with perivascular, peribronchiolar, and alveolar inflammation; and 4 is defined as marked, diffuse bronchiolar epithelial changes with perivascular, peribronchiolar, and alveolar inflammation.
Statistical analyses.
Statistical analyses were done with Student's t test, except for the survival study, in which the log rank (Mantel-Cox) test was used. Results are presented as means ± standard errors (SE). P values of ≤0.05 were considered significant.
RESULTS
Knockdown of XPO1 gene expression results in a reduction of influenza virus replication.
To validate the requirement of XPO1 for replication of influenza virus, siRNA transfection was performed to silence XPO1 gene expression. A549 cells were transfected with a nontargeting siRNA (siNEG) or a siRNA targeting human XPO1. Additionally, a siRNA targeting MEK was used as a positive control and the requirement of MEK for influenza virus replication has been previously demonstrated (62). Transfected cells were incubated for 48 h to allow expression knockdown. To validate XPO1 expression silencing, XPO1 siRNA (siXPO1)-transfected cells were harvested at 96 h posttransfection for immunoblot analysis. Cells transfected with siXPO1 displayed an XPO1 protein level >90% lower than that of siNEG-transfected cells (Fig. 1A). Cells were subsequently infected with various strains of influenza A and B viruses at an MOI of 0.001. As evaluated by immunofluorescence staining of viral NP, virus-positive cells were scarce in MEK siRNA (siMEK)- and siXPO1-tranfected cells, while >70% of the cells transfected with siNEG displayed virus-positive staining at 48 h after influenza virus A/WSN/33 (H1N1) infection (96 h after siRNA transfection) (Fig. 1B). To further demonstrate the requirement of XPO1 for influenza virus replication across types and subtypes, cells were infected with various influenza A and B virus strains. Culture supernatant was collected at 48 hpi to measure infectious virus production (Fig. 1C to I). Cells transfected with siXPO1 displayed significantly lower influenza virus A/WSN/33 (H1N1) (P ≤ 0.0001), A/California/04/09 (pH1N1) (P ≤ 0.05), A/Philippines/2/82-X79 (H3N2) (P ≤ 0.0001), A/mute swan/MI/451072-2/06 (H5N1) (P ≤ 0.0001), A/red knot/NJ/1523470/06 (H7N3) (P ≤ 0.0001), A/Anhui/1/2013 (H7N9) (P ≤ 0.01), and/or B/Ohio/01/05 (P ≤ 0.0001) titers than siNEG-transfected cells.
FIG 1.

Silencing of XPO1 gene expression results in reduction of influenza virus replication. A549 cells were transfected with nontargeting siNEG as a negative control, siMEK as a positive control, or siXPO1. (A) Cells were harvested at 96 h posttransfection to evaluate XPO1 protein knockdown efficiency by immunoblot analysis. Densitometry analysis was performed to determine the XPO1 protein level normalized to GAPDH. Percent XPO1 expression in siXPO1-transfected cells was calculated relative to that in siNEG-transfected cells. (B) At 48 h posttransfection, cells were infected at an MOI of 0.001 with influenza virus A/WSN/33 (H1N1) (B, C), A/California/04/09 (pH1N1) (D), A/Philippines/2/82-X79 (H3N2) (E), A/mute swan/MI/451072-2/06 (H5N1) (F), A/red knot/NJ/1523470/06 (H7N3) (G), A/Anhui/1/2013 (H7N9) (H), or B/Ohio/01/05 (Flu B) (I) or at an MOI of 3 with A/WSN/33 (H1N1) (J). At 48 (A) or 8 (J) hpi, cells were fixed, stained for viral NP (green) and nuclei (DAPI; blue), and visualized by fluorescence microscopy. Bars, 200 μm. (B to H) At 48 hpi, culture supernatants were collected for virus titer determination in MDCK cells and TCID50s were calculated with the Spearman-Kärber formula. Data are expressed as the mean ± SE (n = 6). ****, P ≤ 0.0001; **, P < 0.01; *, P < 0.05 (relative to siXPO-transfected cells).
XPO1 has been previously reported to mediate the nuclear export of influenza virus vRNPs (29, 32, 33). To validate this mechanism of the role of XPO1 during influenza virus replication, siRNA-transfected cells were infected with influenza virus A/WSN/33 (H1N1) at an MOI of 3 to allow all of the cells to be infected. As assessed by NP immunostaining, cells transfected with siXPO1 showed nuclear retention of vRNP, while cells transfected with siNEG or siMEK displayed cytoplasmic vRNP localization at 8 hpi (Fig. 1J). These findings further validate the conserved requirement of XPO1 for the replication of influenza A and B viruses by facilitating nuclear export of the vRNP complex.
Verdinexor, a novel SINE, is efficacious against influenza A and B viruses.
The structure of a novel SINE XPO1 inhibitor verdinexor (KPT-335) is shown in Fig. 2A. To determine the toxicity of verdinexor, A549 cells were treated with increasing doses of verdinexor and the values for percent cellular cytotoxicity were determined. The verdinexor concentration that resulted in a median cytotoxicity between the baseline (DMSO) and the maximum observed cytotoxicity was determined by the nonlinear regression method (Fig. 2B; 50% cytotoxic concentration [CC50], 26.8 μM). To assess the efficacy of verdinexor against influenza virus, A549 cells were pretreated with DMSO as a control or with increasing concentrations of verdinexor ranging from 0.1 to 100 μM in one-third-log increments. Two hours posttreatment, cells were subsequently inoculated with the influenza A and B virus strains listed in Table 1. Culture supernatants were collected at 24 hpi for virus titer determination (Fig. 2C to H). Percent virus titers relative to that of the DMSO-treated control were used to calculate the 50% inhibitory concentration (IC50) for each virus by nonlinear regression (Table 1). Selectivity indices (SIs) were calculated to determine the efficacy of verdinexor against influenza virus strains as the ratio of the CC50 to the IC50 for the corresponding strain. Verdinexor was found to have good efficacy against the influenza A and B virus strains tested in the nanomolar range (SI of ≥99), except against the emerging A/Anhui/1/2013 (H7N9) strain, against which it showed some but limited efficacy (SI of 64). These findings demonstrate a relatively broad anti-influenza virus efficacy of verdinexor in vitro, which warranted an evaluation of its in vivo efficacy.
FIG 2.

Verdinexor, a novel SINE, is efficacious against influenza A and B viruses. (A) Structure of verdinexor (KPT-335). (B to H) A549 cells were mock treated (M) or treated with increasing doses of verdinexor for 2 h at 37°C. (B) Cytotoxicity was assessed with the ToxiLight bioassay kit at 24 h posttreatment. The percent cytotoxicity in verdinexor-treated cells was determined relative to that in nontreated (0% cytotoxicity) and lysed (100% cytotoxicity) cells and graphed to evaluate the median cytotoxic concentration. (C to H) Cells were infected with influenza virus A/WSN/33 (H1N1) (C), A/California/04/09 (pH1N1) (D), A/Philippines/2/82-X79 (H3N2) (E), A/mute swan/MI/451072-2/06 (H5N1) (F), A/Anhui/1/2013 (H7N9) (G), or B/Ohio/01/05 (Flu B) (H). Culture supernatants were collected at 24 hpi for virus titer determination in MDCK cells, and experiments were repeated at least twice.
TABLE 1.
In vitro efficacy of verdinexor against influenza A and B virus strains
| Influenza virus strain | Type or subtype | IC50 (μM) | CC50 (μM) | SI |
|---|---|---|---|---|
| A/California/04/09 | H1N1 | 0.20 | 26.8 | 134 |
| A/Philippines/2/82-X79 | H3N2 | 0.04 | 26.8 | 670 |
| A/Vietnam/1204/04 (HPAIV) | H5N1 | 0.27 | 26.8 | 99 |
| A/mute swan/MI/451072-2/06 (LPAIV) | H5N1 | 0.18 | 26.8 | 149 |
| A/red knot/NJ/1523470/06 | H7N3 | 0.06 | 26.8 | 447 |
| A/Anhui/1/2013 | H7N9 | 0.42 | 26.8 | 64 |
| B/Florida/04/06 | B | 0.09 | 26.8 | 298 |
| B/Ohio/01/05 | B | 0.01 | 26.8 | 2,680 |
Verdinexor inhibits nuclear export of vRNP.
XPO1 facilitates the nuclear export of influenza virus vRNP, which is crucial for influenza virus replication in host cells (30, 33). Inhibition of XPO1-dependent nuclear export by its prototypical inhibitor LMB has been shown to result in the accumulation of vRNPs in the nuclei of infected cells, preventing viral packaging and new virus progeny, thereby blocking virus replication (63). Thus, verdinexor was evaluated with A549 cells, which were treated with DMSO, verdinexor (1 μM), or LMB (10 nM). Two hours posttreatment, the cells were infected with influenza virus A/WSN/33 (H1N1) at an MOI of 3. The cells were fixed at 8 hpi, a time point corresponding to a later stage of virus replication, and the localization of viral NP (a component of the vRNP complex) was determined by immunofluorescence staining (Fig. 3A). Similar to LMB-treated cells, verdinexor-treated cells displayed nuclear NP staining at 8 hpi, but DMSO-treated cells did not, suggesting retention of vRNPs in the nuclei of verdinexor- and LMB-treated cells. Furthermore, verdinexor and LMB treatment also altered the localization of viral NS1, another influenza virus protein that is known to possess a nuclear localization signal (NLS) and an NES that shuttle it between the cytoplasm and the nucleus to modulate various host responses in both cell compartments (data not shown). To further evaluate the ability of verdinexor to block vRNA nuclear export, verdinexor-treated A549 cells were harvested at 8 hpi and subjected to subcellular fractionation and RNA isolation to extract cytoplasmic and nuclear RNAs. The relative abundance of negative-sense vRNA and positive-sense mRNA in the cytoplasm and nucleus were evaluated by strand-specific viral RNA RT-qPCR. Cells pretreated with verdinexor demonstrated 56.6-fold enrichment of nuclear negative-sense vRNA relative to that of DMSO-treated cells (P ≤ 0.0001), while the relative abundance of cytoplasmic negative-sense vRNA was significantly (P ≤ 0.001) reduced (Fig. 3B). However, no significant difference in the relative abundance of nuclear and cytoplasmic positive-sense viral mRNA was observed between DMSO- and verdinexor-treated cells (Fig. 3C), demonstrating verdinexor's anti-influenza virus activity by blocking vRNP nuclear export but not viral mRNA.
FIG 3.

Verdinexor treatment resulted in nuclear accumulation of vRNP. A549 cells were pretreated for 2 h with DMSO, verdinexor (1 μM), or LMB (10 nM). Cells were infected with influenza virus A/WSN/33 at an MOI of 3 for 8 h. (A) Cells were fixed and stained for viral NP (green) and nuclei (DAPI; blue) and visualized under a fluorescence microscope. (B, C) Cells were subjected to cytoplasmic-nuclear fractionation, followed by RNA isolation from each fraction. To determine the relative nuclear-cytoplasmic distribution of influenza virus vRNA (B) or mRNA (C), RNA from both fractions was used for RT-qPCR analysis with strand-specific primers for A/WSN/33 segment 5. Viral RNA abundance was normalized to GAPDH, and the abundance relative to that in mock-infected cells was graphed. Data are expressed as the mean ± SE (n = 3). n.s., not significant; ***, P < 0.001; ****, P < 0.0001 (relative to mock-treated infected cells).
To assess whether the vRNP nuclear retention observed in Fig. 3A to C was indeed due to the inhibition of vRNP nuclear-cytoplasmic export in cells treated with verdinexor, viral NP localization was evaluated at later time points after influenza A/WSN/33 (H1N1) virus infection (Fig. 4A). Similar to LMB-treated cells, the majority of verdinexor-treated cells displayed nuclear NP localization that was extended through the 12- and 18-hpi time points, which are past one round of normal virus replication, suggesting blocked vRNP nuclear export, resulting in its nuclear retention. However, the localization of a non-NLS/NES-bearing viral protein, M2, was not affected by treatment with verdinexor or LMB.
FIG 4.

Verdinexor blocks influenza virus vRNP nuclear export by disrupting XPO1-NEP binding. (A, B) A549 cells were pretreated for 2 h with DMSO, verdinexor (1 μM), or LMB (10 nM). Cells were infected with influenza virus A/WSN/33 (H1N1) (A) or A/Anhui/1/2013 (H7N9) (B) at an MOI of 3. Cells were fixed and stained for viral NP (green), M2 (red), and nuclei (DAPI; blue) and visualized under a fluorescence microscope. Bars, 200 μm. (C) 293T cells were transfected with a NEP expression plasmid derived from influenza virus A/WSN/33 (H1N1), A/California/04/09 (pH1N1), or A/Anhui/1/2013 (H7N9). At 48 h posttransfection, cells were mock treated or treated with verdinexor (1 μM) for 6 h. Cells were cotransfected with a FLAG-XPO1 plasmid. Cells were harvested for coimmunoprecipitation (IP) with anti-FLAG affinity beads. Ten percent input and eluate from the beads were resolved by SDS-PAGE and subjected to immunoblot (IB) analyses of FLAG-XPO1 and NEP. Densitometry analysis of coimmunoprecipitated NEP bands was performed, and intensities of verdinexor-treated relative to mock-treated bands for each strain are indicated below the blot.
Verdinexor blocks the nuclear vRNP export of several influenza A virus strains by disrupting XPO1-NEP binding.
Since verdinexor showed incomplete efficacy against influenza virus A/Anhui/1/2013 (H7N9), its ability to block vRNP nuclear export during A/Anhui/1/2013 (H7N9) virus infection was also evaluated and compared to A/WSN/33 (H1N1) infection (Fig. 4B). As with A/WSN/33 (H1N1) infection, cells treated with verdinexor or LMB showed nuclear NP localization, even at late time points postinfection. Localization of viral M2 protein was not affected in verdinexor- or LMB-treated cells. However, A/Anhui/1/2013 (H7N9) was observed to replicate with faster kinetics than A/WSN/33 (H1N1), which may account for the higher concentration of verdinexor required to effectively suppress virus replication (Fig. 2G). Together, these findings demonstrated a broad ability of verdinexor to block influenza virus vRNP nuclear export, including during infection with influenza virus A/Anhui/1/2013 (H7N9), as well as other strains of influenza virus.
To further assess verdinexor's direct action against XPO1 viral cargo protein NEP, its ability to disrupt XPO1-NEP complex formation in the absence of other viral factors was also evaluated. FLAG-tagged XPO1 and NEP from A/WSN/33 (H1N1), A/California/04/09 (pH1N1), or A/Anhui/1/2013 (H7N9) expression plasmids were cotransfected into 293T cells. At 48 h posttransfection, cells were mock treated or treated with verdinexor (1 μM) for 6 h. Cells were harvested for subsequent coimmunoprecipitation analyses with anti-FLAG affinity beads to assess FLAG-XPO1 binding with NEP from various influenza virus A strains (Fig. 4C). FLAG-XPO1 was able to bind to NEP of A/WSN/33 (H1N1), as well as that of A/California/04/09 (pH1N1) and A/Anhui/1/2013 (H7N9). Importantly, verdinexor treatment was able to reduce XPO1-NEP binding in all three of the strains evaluated. These results strengthen the findings on verdinexor's activity to block the nuclear export of the vRNP of various influenza virus A strains by disrupting XPO1 binding to its influenza virus protein cargo, NEP.
Verdinexor is a slowly reversible inhibitor of XPO1-dependent influenza virus replication in vitro and is bioavailable in vivo after oral administration.
Verdinexor is a SINE compound that acts as a slowly reversible inhibitor of XPO1; thus, the length of verdinexor's inhibitory effect against influenza A virus infection was subsequently evaluated. A549 cells were treated with 1 μM verdinexor for 2 h. Subsequently, the treatment medium was removed and replaced with normal growth medium and the cells were incubated for different lengths of time prior to infection with influenza virus A/WSN/33 at an MOI of 0.01. At 48 hpi, cell supernatant was collected for virus titer determination. Treatment of cells up to 24 h prior to infection was as efficacious as treatment at 2 h preinfection (Fig. 5A; P ≤ 0.001 compared to mock-treated cells), while treatment at 36 h prior to infection resulted in a significant virus titer reduction but reduced efficacy (P ≤ 0.05). However, verdinexor treatment at 48 h preinfection did not result in a significant virus titer reduction. These results are in line with the previously reported in vitro half-life (t1/2) of verdinexor of approximately 24 h (48).
FIG 5.
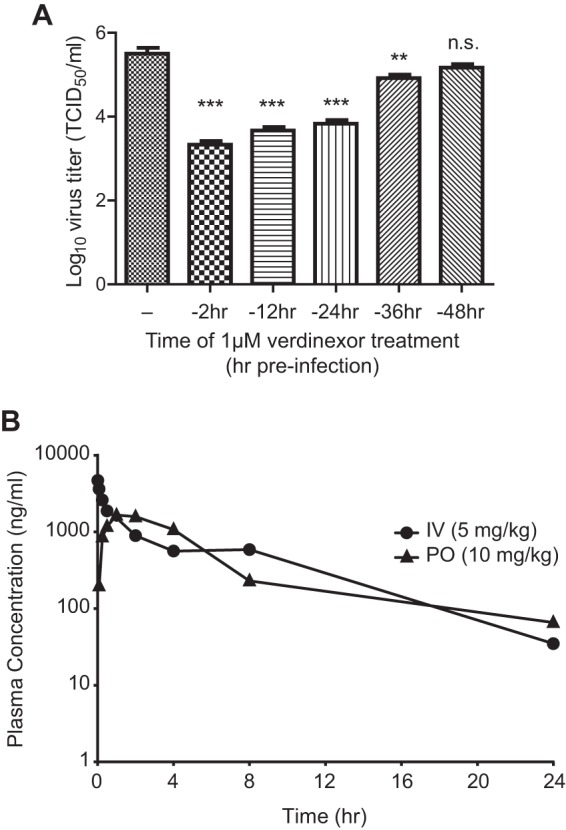
Verdinexor is a slowly reversible inhibitor of XPO1-mediated influenza virus replication in vitro and is bioavailable after oral administration in vivo. (A) A549 cells were mock treated (−) or treated with 1 μM verdinexor for 2 h. Following treatment, the drug-containing medium was removed and replaced with normal growth medium. Cells were incubated for different lengths of time posttreatment, prior to infection with influenza virus A/WSN/33 at an MOI of 0.01. Culture supernatants were collected at 48 hpi for virus titer determination. Data are expressed as the mean ± SE (n = 3). n.s., not significant; **, P < 0.01; ***, P < 0.001 (relative to mock-treated infected cells). (B) For a PK study of p.o. or i.v. administration of verdinexor in mice, verdinexor was given orally by gavage at 10 mg/kg (30 mg/m2) or i.v. at 5 mg/kg (15 mg/m2). Plasma verdinexor concentrations were determined with a validated ultrahigh-performance liquid chromatography–mass spectrometry method, and the mean concentration at each time point postadministration was plotted (n = 3).
To determine verdinexor's suitability for in vivo use against influenza virus infection, its pharmacokinetic (PK) profile in mice was evaluated. The mean plasma verdinexor concentration after intravenous (i.v.) or peroral (p.o.) administration at each time point was assessed (graphed in Fig. 5B). The PK parameters of oral verdinexor administration are summarized in Table 2. On the basis of its PK profile, oral verdinexor treatment was found to be a suitable route of drug administration and its ability to limit influenza virus infection in a mouse model was subsequently investigated.
TABLE 2.
PK parameters of verdinexor in male CD1 mice given a p.o. dose of 10 mg/kg
| PK parametera | Estimated valueb |
|---|---|
| Cmax | 1,660 ng/ml |
| Tmax | 1.00 h |
| Terminal t1/2 | 4.88 h |
| AUClast | 10,300 h · ng/ml |
| AUC∞ | 10,800 h · ng/ml |
| F | 43.5% |
Cmax, peak drug concentration in serum; Tmax, time when Cmax was observed; AUClast, area under plasma drug concentration-time curve from time zero to the time of the last quantifiable concentration; AUC∞, area under plasma drug concentration-time curve from time zero to infinity; F, percent bioavailability.
Mean values for three mice are shown.
Verdinexor reduces the lung influenza virus burden.
Since verdinexor was shown to be effective against influenza virus in vitro, its ability to reduce influenza virus replication in vivo was evaluated. Verdinexor was delivered by gavage because it is bioavailable after oral administration (Fig. 5B and Table 2). To assess the efficacy of prophylactic treatment (outlined in Fig. 6A), mice were pretreated with diluent only or 10 or 20 mg/kg verdinexor for 1 day (1×Rx), 2 days (2×Rx), or 3 days (3×Rx) prior to inoculation with 10 MID50 of mouse-adapted influenza virus strain A/California/04/09 (MA-pH1N1) (Fig. 6B) or 10 MID50 of strain A/Philippines/2/82 (H3N2) (Fig. 6C). Lungs were harvested at 96 hpi and homogenized to evaluate lung virus titers. Compared to diluent-only treatment of mice, prophylactic administration of 10 mg/kg verdinexor at 1 day prior to infection (P ≤ 0.05) significantly reduced lung A/California/04/09 (MA-pH1N1) virus titers, as did treatment with 20 mg/kg given 2 (P ≤ 0.01) or 3 (P ≤ 0.001) days prior to infection. Similarly, prophylactic treatment with 10 mg/kg verdinexor for 2 (P ≤ 0.001) or 3 (P ≤ 0.01) days prior to infection or with 20 mg/kg for 2 (P ≤ 0.001) or 3 (P ≤ 0.001) days prior to infection produced lung A/Philippines/2/82-X79 (H3N2) virus titers significantly lower than those of diluent-only control mice. To assess the therapeutic efficacy of verdinexor (outlined in Fig. 6D), we treated mice with verdinexor at 50 mg/kg (daily total) at 24, 72, or both 24 and 72 hpi as either a single daily dose (1×Rx) or two daily doses (2×Rx of 25 mg/kg each). Control groups of mice received the diluent only or a standard regimen of twice-daily oseltamivir treatment (10 mg/kg/day) for 3 days starting at 24 hpi. Twice-daily verdinexor treatment at 72 hpi (P ≤ 0.05) or treatments at 24 and 72 hpi significantly (P ≤ 0.001) reduced lung A/California/04/09 (MA-pH1N1) virus burdens compared to that of mice that received diluent-only treatment, while oseltamivir treatment did not result in a significant reduction of lung virus titers (Fig. 6E). Similarly, treatment with verdinexor at 24 and 72 hpi (P ≤ 0.001) or twice daily at 72 hpi (P ≤ 0.05) significantly reduced lung influenza virus A/Philippines/2/82-X79 (H3N2) titers compared to those of the diluent-only control group (Fig. 6F). These studies show that verdinexor has in vivo antiviral activity with an ability to reduce lung virus titers that is similar to or better than that of a standard oseltamivir regimen.
FIG 6.

Verdinexor reduces lung influenza A virus burdens. The efficacy of in vivo prophylactic (A to C) and therapeutic (D to F) verdinexor treatment was evaluated. (A to C) Mice were treated orally with diluent only or with 10 or 20 mg/kg verdinexor daily for 3 days (3×Rx), 2 days (2×Rx) or 1 day (1×Rx) prior to infection. (D to F) Mice were treated orally at 24, 72, or both 24 and 72 hpi with 50 mg/kg of verdinexor either as a single dose (1×Rx) or as two doses (2×Rx, 25 mg/kg every 12 h). Oral oseltamivir (10 mg/kg/day for 3 days) and diluent-only treatments were used as positive and negative controls, respectively. Mice were infected with 10 MID50 of A/California/04/09 (mouse-adapted pH1N1) (B, E) or A/Philippines/2/82-X79 (H3N2) (C, F). Lungs were collected at 96 hpi and homogenized for virus titer determination in MDCK cells. Treatments were compared to the diluent-only negative-control group by Student's t test. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (relative to diluent-treated, virus-infected mice).
Verdinexor reduces proinflammatory cytokine expression in the lung.
Influenza virus infection and the lung viral load contribute to disease pathogenesis, which is exacerbated in part by the host immune response to infection. NF-κB signaling has been shown to correlate with the severity of influenza virus infection through the upregulation of proinflammatory cytokine expression (64, 65). Thus, damping of this response can reduce influenza virus-associated disease pathogenesis. It was shown that a SINE XPO1 inhibitor induced nuclear retention, leading to inhibition of NF-κB signaling (42, 66). Indeed, verdinexor treatment resulted in the nuclear accumulation of NF-κB p65 in uninfected A549 cells (Fig. 7A). Thus, the effects of verdinexor treatment on influenza virus-induced proinflammatory cytokine expression were subsequently evaluated in vivo. Mice were treated with the diluent only or administered verdinexor prophylactically (three daily treatments of 20 mg/kg preinfection) and then challenged with 10 MID50 of mouse-adapted influenza virus A/California/04/09 (MA-pH1N1). Lungs were collected at 96 hpi for total RNA isolation, and the expression of key NF-κB-dependent proinflammatory cytokines was evaluated by RT-qPCR. Verdinexor treatment lowered the expression of gamma interferon (Ifng; Fig. 7B, P ≤ 0.05), tumor necrosis factor alpha (Tnf; Fig. 7C; P ≤ 0.01), interleukin-1β (Il1b; Fig. 7D; P ≤ 0.01), and interleukin-6 (Il6; Fig. 7E; P ≤ 0.05) to basal levels. These results show that verdinexor also acts to reduce the expression of proinflammatory cytokines associated with influenza virus infection.
FIG 7.

Verdinexor treatment reduces proinflammatory cytokine expression. (A) A549 cells were mock treated (−) or treated with 1 μM verdinexor (+) for 4 h. Cells were fixed, stained for the NF-κB p65 subunit, and visualized under a fluorescence microscope. Bars, 200 μm. (B to E) Mice were treated orally with diluent only or 20 mg/kg verdinexor daily for 3 days prior to infection with 10 MID50 of mouse-adapted A/California/04/09 (MA-pH1N1). Lungs were collected at 96 hpi, and total RNA was isolated. The expression of Ifng (B), Tnf (C), Il1b (D), and Il6 (E) relative to that of Gapdh was evaluated by RT-qPCR. The data are from four mice per experimental group. *, P ≤ 0.05; **, P ≤ 0.01 (relative to diluent-treated, virus-infected mice).
Verdinexor treatment reduces pulmonary disease pathogenesis and death associated with lethal influenza A virus challenge.
To assess the ability of verdinexor to reduce lung pathology associated with influenza virus infection, mice were prophylactically treated with diluent only or verdinexor (three daily treatments of 20 mg/kg prior to infection) and subsequently mock infected or infected with a lethal dose (10 LD50) of mouse-adapted A/California/04/09 (MA-pH1N1) (the experimental design is illustrated in Fig. 8A). Lungs were harvested at 48 and 96 hpi for assessment of influenza virus NP antigen by IHC (Fig. 8) or stained with H&E for evaluation of pathology (Fig. 9). The level of influenza virus-positive cells in verdinexor-treated mice (Fig. 8D and F) was lower than that in the lungs of diluent-treated mice and appeared to be limited to the bronchial epithelium and minimal in the interstitial epithelium (alveolar septa) (Fig. 8C and E). These findings suggest that verdinexor decreases virus penetration of the lower regions of the respiratory tract and demonstrate verdinexor's ability to limit virus spread in the lungs. No virus-positive cells were detectable in the lungs of mock-infected mice treated with verdinexor (Fig. 8B).
FIG 8.

Verdinexor treatment limits virus spread to the lower regions of the respiratory tract. (A) Experimental design. Mice were mock treated orally with diluent only (−verdinexor; C, E) or treated with 20 mg/kg verdinexor daily for 3 days (+verdinexor; B, D, F) prior to mock infection (B) or infection with 10 LD50 of mouse-adapted A/California/04/09 (MA-pH1N1) (C to F). Lungs were harvested at 48 (C, D) or 96 (B, E, F) hpi, fixed, sectioned, and subjected to IHC with mouse monoclonal antibody to NP, 3,3′-diaminobenzidine chromogen, and hematoxylin counterstain. (B) Lungs from verdinexor-treated but mock-infected mice displayed an absence of viral antigen. (C) At 48 hpi in mock-treated mice infected with influenza virus, there was extensive viral antigen staining of the bronchiolar epithelium, which was sloughing into the lumen. Viral antigen was also observed in the epithelial cells of the alveolar septa. (D) At 48 hpi in verdinexor-treated mice, there was viral antigen staining of the bronchiolar epithelium; however, virus staining was minimal in the interstitium. (E) At 96 hpi in mock-treated mice, much of the bronchiolar wall was ulcerated and there was extensive and intense antigen staining of the bronchiolar epithelium, which had sloughed into the lumen. There was also viral antigen staining of epithelial cells of the alveolar septa and staining of cells in the peribronchiolar inflammation (arrow). (F) At 48 hpi in verdinexor-treated mice, the bronchiolar wall was also ulcerated and there was NP antigen staining of the bronchiolar epithelium; however, viral antigen staining in the alveolar septa was minimal. B, bronchiolar lumen. Bars, 200 μm.
FIG 9.

Verdinexor treatment reduces disease pathology associated with a lethal influenza A virus challenge. Mice were orally mock treated with diluent only (−verdinexor; C, E) or treated with 20 mg/kg verdinexor daily for 3 days (+verdinexor; B, D, F) prior to mock infection (B) or infection with 10 LD50 of mouse-adapted A/California/04/09 (MA-pH1N1) (C to F). Lungs were harvested, fixed, sectioned, and stained with H&E at 48 (C, D) and 96 (B, E, F) hpi for histopathology evaluation. (A) Overall lung pathology was scored on a scale of 0 to 4, where a score of 0 is unremarkable pathology; 1 is minimal changes in the bronchiolar epithelium with minimal perivascular inflammation; 2 is mild, multifocal bronchiolar epithelial changes with perivascular and peribronchiolar inflammation; 3 is moderate, multifocal bronchiolar epithelial changes with perivascular, peribronchiolar, and alveolar inflammation; and 4 is marked, diffuse bronchiolar epithelial changes with perivascular, peribronchiolar, and alveolar inflammation. Data are expressed as the mean ± SE of four or five mice per group. *, P ≤ 0.05; **, P ≤ 0.01 (relative to diluent-treated, virus-infected mice). (B to F) Representative H&E staining is shown. (B) Lungs from verdinexor-treated mice that were mock infected did not show any pathological abnormality and thus served as the baseline control group. Bronchioles had no evidence of epithelial apoptosis and had no cells in the lumen, and there were no parenchymal changes. (C) At 48 hpi in influenza virus-infected mice that received diluent-only treatment, there was extensive apoptotic cell death with sloughing of the dead cells into the bronchiolar lumen. Mild peribronchiolar inflammation was present, but the parenchyma had few changes, except for a few histiocytes in alveoli. (D) At 48 hpi in verdinexor-treated mice infected with influenza virus, the bronchiolar epithelium had areas of apoptotic cell death but there were few parenchymal changes. (E) At 96 hpi, mice that received diluent-only treatment displayed extensive cell death of the bronchiolar epithelium with narrowing of the lumen. Severe peribronchiolar and vascular inflammation was present. Extensive areas of parenchymal inflammation obliterated large areas. (F) At 96 hpi, verdinexor-treated mice displayed a level of bronchiolar epithelium cell death; however, only mild peribronchiolar and perivascular inflammation was observed. The parenchyma had few areas of inflammation that obliterate the alveoli. B, bronchioles; P, parenchyma; V, vessel; arrows, peribronchiolar inflammation. Bars, 200 μm.
Lung pathology was scored, and the results are summarized in Fig. 9A. Influenza virus-challenged mice that received verdinexor had less overall lung pathology than mock-treated mice (P ≤ 0.01 and P ≤ 0.05 for 48 and 96 hpi, respectively). Mice treated with verdinexor but not infected with virus had no detectable pathological changes in their lungs (Fig. 9B) or tracheas (data not shown). Evaluation of histopathology in influenza virus-challenged mice at 48 hpi showed that the bronchial epithelium had areas exhibiting mild to high levels of cell death, areas of hypereosinophilic cytoplasm, and karyorrhexis or karyolysis characteristic of apoptosis, which was confirmed with cleaved caspase-3 IHC, and there was a level of sloughing of the dead cells into the lumen in both the diluent-only- and verdinexor-treated groups. By 96 hpi, there was a higher degree of multifocal ulceration and attenuation of the epithelium, accumulation of cellular debris, and mild inflammation of the mucosa in diluent-treated mice (Fig. 9C and E) than in verdinexor-treated mice (Fig. 9D and F). Notably, the verdinexor-treated mice showed less severe pathology, as indicated by reduced inflammation in the bronchioles and limited parenchymal changes compared to those of diluent-only-treated mice at 48 and 96 hpi. In addition, perivascular and peribronchial inflammation, which consisted predominantly of lymphocytes and neutrophils, was substantially lower in verdinexor-treated mice than in diluent-only-treated mice (arrows). These findings further support the ability of verdinexor to reduce influenza virus-mediated disease pathology and inflammation.
To assess verdinexor's ability to reduce the number of deaths associated with lethal influenza virus infection, mice were infected with a lethal dose (10 LD50) of mouse-adapted A/California/04/09 (MA-pH1N1) and treated orally with diluent only, with verdinexor at 10 or 20 mg/kg every other day (QOD2) at 24 and 72 hpi, or with oseltamivir at 10 mg/kg daily for 3 days. Body weight, clinical signs, and survival were monitored daily for 14 days. Mice that received oral verdinexor treatment at 10 or 20 mg/kg QOD2 or daily oseltamivir treatment were partially protected from death associated with the lethal influenza virus infection (Fig. 10; P ≤ 0.05, P ≤ 0.0001, and P ≤ 0.001, respectively). There was no significant difference in survival between the verdinexor and oseltamivir treatment groups. These findings provide further evidence of the ability of verdinexor to reduce disease and death associated with influenza virus infection, thereby supporting verdinexor as a novel anti-influenza virus drug.
FIG 10.
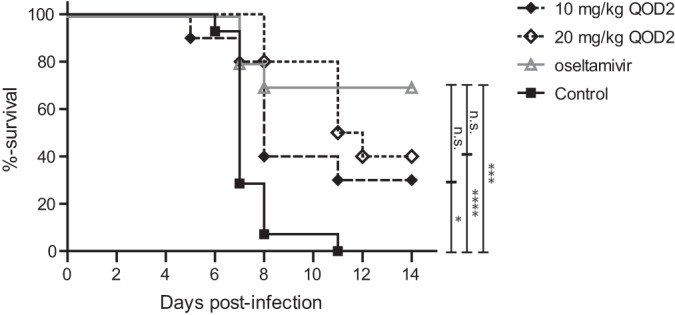
Verdinexor treatment improves survival following a lethal influenza A virus challenge. Mice were infected with 10 LD50 of mouse-adapted A/California/04/09 (MA-pH1N1). Mice were subsequently treated orally with diluent only as a control group or treated with 10 or 20 mg/kg of verdinexor every other day at 24 and 72 hpi (QOD2). A group of mice were treated with oseltamivir (10 mg/kg daily for 3 days) as a treatment control. Survival was evaluated daily for 14 days in accordance with the guidelines of the IACUC of the University of Georgia. Ten mice per treatment group were used for the survival study, and groups were compared by using the log rank (Mantel-Cox) test. *, P ≤ 0.05; ***, P ≤ 0.001; ****, P ≤ 0.0001 (relative to the diluent-treated, virus-infected control group); n.s., not significant (relative to the oseltamivir-treated, virus-infected group).
DISCUSSION
XPO1-mediated nuclear export has been reported to be co-opted by many viruses during various stages of virus replication. Influenza virus uses host XPO1 to mediate the nuclear export of vRNP for subsequent virus assembly, and this mechanism is conserved among influenza A, B, and C viruses (Fig. 1) (30, 33, 63). In addition to influenza virus, other viruses have also been reported to co-opt XPO1, including respiratory syncytial virus (RSV). RSV is one of the major agents of serious lower respiratory tract illness afflicting the young, old, and immunocompromised, with no approved vaccines or antivirals (67). The matrix (M) protein of RSV has been shown to be nuclear at an early stage of RSV replication but cytoplasmic at later time points (68, 69). RSV M protein possesses a NES and has been shown to be transported through XPO1-mediated nuclear export (68). LMB results in the nuclear retention of M and significant reduction of RSV titers in vitro. XPO1 is also implicated in the regulation of dengue virus NS5 protein transport from the nucleus to the cytoplasm. During dengue virus infection, XPO1 inhibition results in altered kinetics of virus production and reduces the induction of interleukin-8 (70). Inhibition of XPO1/Rev-mediated viral RNA transport also arrests human immunodeficiency virus type 1 (HIV-1) transcription, inhibits the production of new HIV-1 virions, and reduces HIV-1 levels (71, 72). Other virus-encoded proteins that are XPO1 cargos include the herpes simplex virus 1 tegument protein (VP13/14 or hUL47), human cytomegalovirus protein pp65, and the severe acute respiratory syndrome coronavirus open reading frame 3b protein (73–75). These findings suggest that targeting of XPO1-mediated export of viral factors represents a novel and broadly effective therapeutic strategy against multiple viruses that co-opt the XPO1 pathway for their replication strategy.
A novel class of SINE compounds has recently been developed by molecular modeling to screen a virtual library of compounds for activity against the NES groove of XPO1 (35, 76). SINE compounds inhibit nuclear-cytoplasmic export by forming a slowly reversible bond with cysteine-528 in the cargo recognition site of XPO1. SINE compounds are smaller than LMB, and a 2.2-Å crystal structure of the XPO1—Ran—Ran-BP1 complex bound to a SINE compound demonstrated that it occupies part of the groove in XPO1 that is usually occupied by the NES but penetrates the groove much more deeply than LMB does to block XPO1-mediated protein export (35). SINE compounds are bioavailable after oral administration (Fig. 5 and Table 2) and were originally developed as therapeutics for various solid and hematologic malignancies, as XPO1 cargo proteins include numerous tumor suppressor proteins (TSPs) such as p53, p73, p21, p27, FOXO1, FOXO3a, FOXO4, PP2A, BRCA1, BRCA2, and E2F4 (35, 41, 77). In normal cells, export of these proteins from the nucleus by XPO1 prevents multiple TSPs from acting in the absence of DNA damage or other oncogenic insults. Thus, SINE compounds can be used to force nuclear retention, accumulation, and functional activation of TSPs to limit oncogenesis (35, 41, 43–46). Among SINE compounds, verdinexor is currently undergoing the final stages of clinical evaluation in dogs with lymphoma (48). Additionally, a related SINE compound, selinexor (KPT-330), is currently undergoing multiple phase II studies with human patients with advanced, relapsed, and refractory cancers (ClinicalTrials.gov NCT01607905, NCT01607892). Preliminary results from these studies demonstrated that selinexor is generally well tolerated, with good exposure upon oral administration and evidence of anticancer activity (50–52).
In this study, a representative SINE compound, verdinexor, was found to be efficacious and inhibit the replication of various influenza A and B virus strains, including the pandemic H1N1 virus, H5N1 HPAIV, and to a lesser extent the emerging H7N9 influenza A virus (Fig. 2 and Table 1). Furthermore, mice treated with verdinexor displayed lower lung virus titers, less disease pathology, and fewer deaths following infection with representative strains of circulating influenza A virus subtypes H1N1 and H3N2 (Fig. 6 and 8 to 10). A low level of cytotoxicity of verdinexor and the compound class in various cell lines was found (Fig. 2C and data not shown), a factor important for progression to a proof-of-concept trial of verdinexor as an antiviral drug candidate. In addition to influenza virus, SINE compounds have also been shown to inhibit the replication of several other viruses, such as HIV (78), hepatitis C virus, and rabies virus (data not shown).
A related SINE compound, KPT-350, is currently undergoing preclinical investigation for its anti-inflammatory activity as an inhibitor of NF-κB (IκB), which has been shown to be transported through an XPO1-mediated mechanism (79, 80). In addition to its role in the inhibition of NF-κB activation in the cytoplasm, IκB has been previously shown to repress postinduction NF-κB activation by displacing NF-κB–DNA binding. Postinduction NF-κB nuclear export is thought to replenish the cytoplasmic pool of inactive NF-κB/IκB complexes for subsequent activation, and their export from the nucleus is reported to be mediated through NES-bearing IκB (79, 81–83). Consequently, efficient nuclear export of IκB is required for the maintenance of a high level of NF-κB activity, which has been previously shown in autoimmune disorders such as systemic lupus erythematosus, multiple sclerosis, and rheumatoid arthritis (84). Treatment with an XPO1 inhibitor causes nuclear accumulation of IκB, leading to subsequent inactivation of NF-κB signaling (79, 80, 85, 86); thus, SINE compounds also act as oral NF-κB inhibitors for the treatment of inflammatory diseases. This NF-κB anti-inflammatory activity is relevant to the present study because of influenza virus-associated immunopathology. In particular, exacerbation of NF-κB signaling has been shown to correlate with the severity of influenza virus infection and the development of acute respiratory distress syndrome, the hallmarks of which are excessive production of proinflammatory cytokines and the resulting immunopathology (64, 65, 87). Thus, damping of NF-κB-dependent cytokine secretion can also result in reduction of influenza virus-associated immunopathology, in addition to SINE's direct effect on influenza virus replication. Indeed, verdinexor-treated mice displayed reduced expression of inflammatory cytokines, inflammation, and disease pathology following influenza virus infection (Fig. 7 to 9). This additive treatment effect can ultimately lead to better overall disease outcomes and survival of influenza virus-infected mice (Fig. 10) and ultimately patients.
As part of a study to assess the feasibility of verdinexor treatment in human patients, Karyopharm Therapeutics had conducted studies evaluating the specificity and potential off-target effects of verdinexor. A large panel of in vitro protein binding assays was performed to evaluate the potential interference of verdinexor with various principal receptor-ligand interactions (peptides, growth factors, ion channels), kinases, and cysteine proteases (including caspases and matrix metalloproteinases) (study 11-8102, Caliper Life Sciences; data not shown). At a concentration of 10 μM, none of the enzymes, receptors, kinases, or cysteine proteases were significantly affected by verdinexor treatment. Given these results, verdinexor is considered to be a highly selective agent against proteins, including proteins that possess an active cysteine residue in their active site, such as cysteine proteases (including caspases) and kinases, with limited potential for off-target effects. Potential cardiac electrophysiology effects of verdinexor were also assessed in an automated non-good laboratory practice in vitro human ether-à-go-go-related gene (hERG) channel current inhibition assay. The IC50 of verdinexor for the hERG potassium current was >30 μM. These studies indicate the selectivity of verdinexor and its safety for potential use in humans.
Together, this study showed that verdinexor is a novel inhibitor of influenza virus infection in vitro and in vivo that warrants further evaluation of its efficacy and safety in a second animal model, such as ferrets, to provide a stepping stone toward future clinical studies.
ACKNOWLEDGMENTS
We thank Paula Brooks for performing the primary RNAi screening study, Cheryl Jones for preparing virus stocks, Les Jones for cloning of the influenza virus NEP expression plasmids, and Caleb White and Emery Register for their technical assistance.
This study was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (HHSN266200700006C), and funding from the Georgia Research Alliance.
Footnotes
Published ahead of print 25 June 2014
REFERENCES
- 1.Fiore AE, Uyeki TM, Broder K, Finelli L, Euler GL, Singleton JA, Iskander JK, Wortley PM, Shay DK, Bresee JS, Cox NJ, Centers for Disease Control and Prevention (CDC) 2010. Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recomm. Rep. 59(RR-8):1–62 http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5908a1.htm [PubMed] [Google Scholar]
- 2.Hoyert DL, Kung H-C, Smith BL. 2005. Deaths: preliminary data for 2003. Natl. Vital Stat. Rep. 53:1–48 http://www.cdc.gov/nchs/data/nvsr/nvsr53/nvsr53_15.pdf [PubMed] [Google Scholar]
- 3.Podewils LJ, Liedtke LA, McDonald LC, Hageman JC, Strausbaugh LJ, Fischer TK, Jernigan DB, Uyeki TM, Kuehnert MJ. 2005. A national survey of severe influenza-associated complications among children and adults, 2003-2004. Clin. Infect. Dis. 40:1693–1696. 10.1086/430424 [DOI] [PubMed] [Google Scholar]
- 4.Block SL. 2004. Role of influenza vaccine for healthy children in the US. Paediatr. Drugs 6:199–209. 10.2165/00148581-200406040-00001 [DOI] [PubMed] [Google Scholar]
- 5.Loeb M. 2004. Pneumonia in the elderly. Curr. Opin. Infect. Dis. 17:127–130. 10.1097/00001432-200404000-00010 [DOI] [PubMed] [Google Scholar]
- 6.Olshaker JS. 2003. Influenza. Emerg. Med. Clin. North Am. 21:353–361. 10.1016/S0733-8627(03)00018-X [DOI] [PubMed] [Google Scholar]
- 7.Fiore AE, Bridges CB, Cox NJ. 2009. Seasonal influenza vaccines. Curr. Top. Microbiol. Immunol. 333:43–82. 10.1007/978-3-540-92165-3_3 [DOI] [PubMed] [Google Scholar]
- 8.McElhaney JE. 2005. The unmet need in the elderly: designing new influenza vaccines for older adults. Vaccine 23(Suppl 1):S10–S25. 10.1016/j.vaccine.2005.04.019 [DOI] [PubMed] [Google Scholar]
- 9.Thompson WW, Shay DK, Weintraub E, Brammer L, Bridges CB, Cox NJ, Fukuda K. 2004. Influenza-associated hospitalizations in the United States. JAMA 292:1333–1340. 10.1001/jama.292.11.1333 [DOI] [PubMed] [Google Scholar]
- 10.Rosenberg MR, Casarotto MG. 2010. Coexistence of two adamantane binding sites in the influenza A M2 ion channel. Proc. Natl. Acad. Sci. U. S. A. 107:13866–13871. 10.1073/pnas.1002051107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bright RA, Medina MJ, Xu X, Perez-Oronoz G, Wallis TR, Davis XM, Povinelli L, Cox NJ, Klimov AI. 2005. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet 366:1175–1181. 10.1016/S0140-6736(05)67338-2 [DOI] [PubMed] [Google Scholar]
- 12.Fiore AE, Fry A, Shay D, Gubareva L, Bresee JS, Uyeki TM. 2011. Antiviral agents for the treatment and chemoprophylaxis of influenza—recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm. Rep. 60:1–24 http://www.cdc.gov/mmwr/preview/mmwrhtml/rr6001a1.htm [PubMed] [Google Scholar]
- 13.Moscona A. 2005. Neuraminidase inhibitors for influenza. N. Engl. J. Med. 353:1363–1373. 10.1056/NEJMra050740 [DOI] [PubMed] [Google Scholar]
- 14.Wright PF, Neumann G, Kawaoka Y. 2007. Orthomyxoviruses, p 1691–1740 In Fields BN, Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 15.Palese P, Shaw ML. 2007. Orthomyxoviridae: the viruses and their replication., p 1647–1689 In Fields BN, Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 16.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254. 10.1016/j.cell.2009.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hao L, Sakurai A, Watanabe T, Sorensen E, Nidom CA, Newton MA, Ahlquist P, Kawaoka Y. 2008. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 454:890–893. 10.1038/nature07151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlas A, Machuy N, Shin Y, Pleissner K-P, Artarini A, Heuer D, Becker D, Khalil H, Ogilvie LA, Hess S, Maurer AP, Muller E, Wolff T, Rudel T, Meyer TF. 2010. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463:818–822. 10.1038/nature08760 [DOI] [PubMed] [Google Scholar]
- 19.König R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, Gao Q, Andrews SE, Bandyopadhyay S, De Jesus P, Tu BP, Pache L, Shih C, Orth A, Bonamy G, Miraglia L, Ideker T, Garcia-Sastre A, Young JA, Palese P, Shaw ML, Chanda SK. 2010. Human host factors required for influenza virus replication. Nature 463:813–817. 10.1038/nature08699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meliopoulos VA, Andersen LE, Brooks P, Yan X, Bakre A, Coleman JK, Tompkins SM, Tripp RA. 2012. MicroRNA regulation of human protease genes essential for influenza virus replication. PLoS One 7(5):e37169. 10.1371/journal.pone.0037169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shapira SD, Gat-Viks I, Shum BOV, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, Hill DE, Regev A, Hacohen N. 2009. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139:1255–1267. 10.1016/j.cell.2009.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sui B, Bamba D, Weng K, Ung H, Chang S, Van Dyke J, Goldblatt M, Duan R, Kinch MS, Li W-B. 2009. The use of random homozygous gene perturbation to identify novel host-oriented targets for influenza. Virology 387:473–481. 10.1016/j.virol.2009.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meliopoulos VA, Andersen LE, Birrer KF, Simpson KJ, Lowenthal JW, Bean AG, Stambas J, Stewart CR, Tompkins SM, van Beusechem VW, Fraser I, Mhlanga M, Barichievy S, Smith Q, Leake D, Karpilow J, Buck A, Jona G, Tripp RA. 2012. Host gene targets for novel influenza therapies elucidated by high-throughput RNA interference screens. FASEB J. 26:1372–1386. 10.1096/fj.11-193466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perwitasari O, Yan X, Johnson S, White C, Brooks P, Tompkins SM, Tripp RA. 2013. Targeting organic anion transporter 3 with probenecid as a novel anti-influenza A virus strategy. Antimicrob. Agents Chemother. 57:475–483. 10.1128/AAC.01532-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whittaker GR. 2001. Intracellular trafficking of influenza virus: clinical implications for molecular medicine. Expert Rev. Mol. Med. 3:1–13. 10.1017/S1462399401002575 [DOI] [PubMed] [Google Scholar]
- 26.Cros JF, Garcia-Sastre A, Palese P. 2005. An unconventional NLS is critical for the nuclear import of the influenza A virus nucleoprotein and ribonucleoprotein. Traffic 6:205–213. 10.1111/j.1600-0854.2005.00263.x [DOI] [PubMed] [Google Scholar]
- 27.Melen K, Fagerlund R, Franke J, Kohler M, Kinnunen L, Julkunen I. 2003. Importin alpha nuclear localization signal binding sites for STAT1, STAT2, and influenza A virus nucleoprotein. J. Biol. Chem. 278:28193–28200. 10.1074/jbc.M303571200 [DOI] [PubMed] [Google Scholar]
- 28.Weber F, Kochs G, Gruber S, Haller O. 1998. A classical bipartite nuclear localization signal on Thogoto and influenza A virus nucleoproteins. Virology 250:9–18. 10.1006/viro.1998.9329 [DOI] [PubMed] [Google Scholar]
- 29.Boulo S, Akarsu H, Ruigrok RW, Baudin F. 2007. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 124:12–21. 10.1016/j.virusres.2006.09.013 [DOI] [PubMed] [Google Scholar]
- 30.Elton D, Simpson-Holley M, Archer K, Medcalf L, Hallam R, McCauley J, Digard P. 2001. Interaction of the influenza virus nucleoprotein with the cellular CRM1-mediated nuclear export pathway. J. Virol. 75:408–419. 10.1128/JVI.75.1.408-419.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nayak DP, Hui EK, Barman S. 2004. Assembly and budding of influenza virus. Virus Res. 106:147–165. 10.1016/j.virusres.2004.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paterson D, Fodor E. 2012. Emerging roles for the influenza A virus nuclear export protein (NEP). PLoS Pathog. 8(12):e1003019. 10.1371/journal.ppat.1003019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paragas J, Talon J, O'Neill RE, Anderson DK, Garcia-Sastre A, Palese P. 2001. Influenza B and C virus NEP (NS2) proteins possess nuclear export activities. J. Virol. 75:7375–7383. 10.1128/JVI.75.16.7375-7383.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ossareh-Nazari B, Gwizdek C, Dargemont C. 2001. Protein export from the nucleus. Traffic 2:684–689. 10.1034/j.1600-0854.2001.21002.x [DOI] [PubMed] [Google Scholar]
- 35.Etchin J, Sun Q, Kentsis A, Farmer A, Zhang ZC, Sanda T, Mansour MR, Barcelo C, McCauley D, Kauffman M, Shacham S, Christie AL, Kung AL, Rodig SJ, Chook YM, Look AT. 2013. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia 27:66–74. 10.1038/leu.2012.219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S. 1999. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. U. S. A. 96:9112–9117. 10.1073/pnas.96.16.9112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weis K. 2002. Nucleocytoplasmic transport: cargo trafficking across the border. Curr. Opin. Cell Biol. 14:328–335. 10.1016/S0955-0674(02)00337-X [DOI] [PubMed] [Google Scholar]
- 38.Sun Q, Carrasco YP, Hu Y, Guo X, Mirzaei H, Macmillan J, Chook YM. 2013. Nuclear export inhibition through covalent conjugation and hydrolysis of leptomycin B by CRM1. Proc. Natl. Acad. Sci. U. S. A. 110:1303–1308. 10.1073/pnas.1217203110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mutka SC, Yang WQ, Dong SD, Ward SL, Craig DA, Timmermans PB, Murli S. 2009. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 69:510–517. 10.1158/0008-5472.CAN-08-0858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newlands ES, Rustin GJ, Brampton MH. 1996. Phase I trial of elactocin. Br. J. Cancer 74:648–649. 10.1038/bjc.1996.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Etchin J, Sanda T, Mansour MR, Kentsis A, Montero J, Le BT, Christie AL, McCauley D, Rodig SJ, Kauffman M, Shacham S, Stone R, Letai A, Kung AL, Thomas Look A. 2013. KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br. J. Haematol. 161:117–127. 10.1111/bjh.12231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lapalombella R, Sun Q, Williams K, Tangeman L, Jha S, Zhong Y, Goettl V, Mahoney E, Berglund C, Gupta S, Farmer A, Mani R, Johnson AJ, Lucas D, Mo X, Daelemans D, Sandanayaka V, Shechter S, McCauley D, Shacham S, Kauffman M, Chook YM, Byrd JC. 2012. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood 120:4621–4634. 10.1182/blood-2012-05-429506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Azmi AS, Aboukameel A, Bao B, Sarkar FH, Philip PA, Kauffman M, Shacham S, Mohammad RM. 2013. Selective inhibitors of nuclear export block pancreatic cancer cell proliferation and reduce tumor growth in mice. Gastroenterology 144:447–456. 10.1053/j.gastro.2012.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ranganathan P, Yu X, Na C, Santhanam R, Shacham S, Kauffman M, Walker A, Klisovic R, Blum W, Caligiuri M, Croce CM, Marcucci G, Garzon R. 2012. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood 120:1765–1773. 10.1182/blood-2012-04-423160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tai YT, Landesman Y, Acharya C, Calle Y, Zhong MY, Cea M, Tannenbaum D, Cagnetta A, Reagan M, Munshi AA, Senapedis W, Saint-Martin JR, Kashyap T, Shacham S, Kauffman M, Gu Y, Wu L, Ghobrial I, Zhan F, Kung AL, Schey SA, Richardson P, Munshi NC, Anderson KC. 2014. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia 28:155–165. 10.1038/leu.2013.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang K, Wang M, Tamayo AT, Shacham S, Kauffman M, Lee J, Zhang L, Ou Z, Li C, Sun L, Ford RJ, Pham LV. 2013. Novel selective inhibitors of nuclear export CRM1 antagonists for therapy in mantle cell lymphoma. Exp. Hematol. 41:67–78e64. 10.1016/j.exphem.2012.09.002 [DOI] [PubMed] [Google Scholar]
- 47.Xu D, Grishin NV, Chook YM. 2012. NESdb: a database of NES-containing CRM1 cargoes. Mol. Biol. Cell 23:3673–3676. 10.1091/mbc.E12-01-0045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.London CA, Bernabe LF, Barnard S, Kisseberth WC, Borgatti A, Henson M, Wilson H, Jensen K, Ito D, Modiano JF, Bear MD, Pennell ML, Saint-Martin J-R, McCauley D, Kauffman M, Shacham S. 2014. Preclinical evaluation of the novel, orally bioavailable selective inhibitor of nuclear export (SINE) KPT-335 in spontaneous canine cancer: results of a phase I study. PLoS One 9(2):e87585. 10.1371/journal.pone.0087585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cejka D, Losert D, Wacheck V. 2006. Short interfering RNA (siRNA): tool or therapeutic? Clin. Sci. 110:47–58. 10.1042/CS20050162 [DOI] [PubMed] [Google Scholar]
- 50.Kuruvilla J, Gutierrez M, Shah B, Gabrail N, Brown P, Stone R, Garzon R, Savona M, Baz R, Mau-Sorensen M, Davids M, Byrd J, Shacham S, Rashal T, Yau CY, McCauley D, Saint-Martin J-R, McCartney J, Landesman Y, Klebanov B, Pond G, Oza A, Kauffman M, Mirza MR. 2013. Preliminary evidence of anti-tumor activity of selinexor (KPT-330) in a phase I trial of a first-in-class oral selective inhibitor of nuclear export (SINE) in patients (pts) with relapsed/refractory non-Hodgkin's lymphoma (NHL) and chronic lymphocytic leukemia (CLL), abstr. 90 In Abstr. 55th Annu. Meet. Am. Soc. Hematol., New Orleans, LA [Google Scholar]
- 51.Mau-Sorensen M, Razak AR, Mahipal A, Mahaseth H, Gerecitano JF, Shacham S, Yau CY, Lassen U, Shields AF, McCauley D, Cooksey J, Tan DSP, Rashal T, Shacham E, Landesman Y, Pond G, Oza AM, Kauffman M, Siu LL, Mirza MR. 2013. Safety and anti-tumor activity of KPT-330, a first-in-class oral XPO1 selective inhibitor of nuclear export—phase I study expanded with colon cancer cohort. Gastrointestinal Cancers Symposium. American Society of Clinical Oncology, San Francisco, CA [Google Scholar]
- 52.Savona M, Garzon R, Brown P, Yee K, Lancet J, Gutierrez M, Gabrail N, Mau-Sorensen M, Baz R, Shacham S, Rashal T, Yau CY, McCauley D, Saint-Martin J-R, McCartney J, Landesman Y, Klebanov B, Pond G, Oza A, Kauffman M, Mirza MR, Stone RM. 2013. Phase I trial of selinexor (KPT-330), a first-in-class oral selective inhibitor of nuclear export (SINE) in patients (pts) with advanced acute myelogenous leukemia (AML), poster 1 In Abstr. 55th Annu. Meet. Am. Soc. Hematol., New Orleans, LA [Google Scholar]
- 53.Woolcock PR. 2008. Avian influenza virus isolation and propagation in chicken eggs. Methods Mol. Biol. 436:35–46. 10.1007/978-1-59745-279-3_6 [DOI] [PubMed] [Google Scholar]
- 54.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27:493–497 [Google Scholar]
- 55.Szretter KJ, Balish AL, Katz JM. 2006. Influenza: propagation, quantification, and storage. Curr. Protoc. Microbiol. 15:Unit 15G.1. 10.1002/0471729256.mc15g01s3 [DOI] [PubMed] [Google Scholar]
- 56.Wang W, Budhu A, Forgues M, Wang XW. 2005. Temporal and spatial control of nucleophosmin by the Ran-Crm1 complex in centrosome duplication. Nat. Cell Biol. 7:823–830. 10.1038/ncb1282 [DOI] [PubMed] [Google Scholar]
- 57.Jones RE, Zheng W, McKew JC, Chen CZ. 2013. An alternative direct compound dispensing method using the HP D300 digital dispenser. J. Lab. Autom. 18:367–374. 10.1177/2211068213491094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X, Spandidos A, Wang H, Seed B. 2012. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 40:D1144–D1149. 10.1093/nar/gkr1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kawakami E, Watanabe T, Fujii K, Goto H, Watanabe S, Noda T, Kawaoka Y. 2011. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA. J. Virol. Methods 173:1–6. 10.1016/j.jviromet.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Driskell EA, Jones CA, Stallknecht DE, Howerth EW, Tompkins SM. 2010. Avian influenza virus isolates from wild birds replicate and cause disease in a mouse model of infection. Virology 399:280–289. 10.1016/j.virol.2010.01.005 [DOI] [PubMed] [Google Scholar]
- 61.Le VL, Courtney CL, Steel J, Compans RW. 2013. Closely related influenza viruses induce contrasting respiratory tract immunopathology. PLoS One 8(9):e76708. 10.1371/journal.pone.0076708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pleschka S, Wolff T, Ehrhardt C, Hobom G, Planz O, Rapp UR, Ludwig S. 2001. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 3:301–305. 10.1038/35060098 [DOI] [PubMed] [Google Scholar]
- 63.Watanabe K, Takizawa N, Katoh M, Hoshida K, Kobayashi N, Nagata K. 2001. Inhibition of nuclear export of ribonucleoprotein complexes of influenza virus by leptomycin B. Virus Res. 77:31–42. 10.1016/S0168-1702(01)00263-5 [DOI] [PubMed] [Google Scholar]
- 64.Bernasconi D, Amici C, La Frazia S, Ianaro A, Santoro MG. 2005. The IκB kinase is a key factor in triggering influenza A virus-induced inflammatory cytokine production in airway epithelial cells. J. Biol. Chem. 280:24127–24134. 10.1074/jbc.M413726200 [DOI] [PubMed] [Google Scholar]
- 65.Josset L, Belser JA, Pantin-Jackwood MJ, Chang JH, Chang ST, Belisle SE, Tumpey TM, Katze MG. 2012. Implication of inflammatory macrophages, nuclear receptors, and interferon regulatory factors in increased virulence of pandemic 2009 H1N1 influenza A virus after host adaptation. J. Virol. 86:7192–7206. 10.1128/JVI.00563-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hamuza M, Landesman Y, Klebanov B, Kauffman M, Shacham S, Endres J, Fox DA, McCauley D. 2013. Novel selective inhibitors of nuclear export attenuate inflammation and prevent bone mineral density loss in multiple preclinical models of rheumatoid arthritis. Arthritis Rheum. 65(Suppl 10):1727 (Abstract.) [Google Scholar]
- 67.Tripp RA. 2004. Pathogenesis of respiratory syncytial virus infection. Viral Immunol. 17:165–181. 10.1089/0882824041310513 [DOI] [PubMed] [Google Scholar]
- 68.Ghildyal R, Ho A, Dias M, Soegiyono L, Bardin PG, Tran KC, Teng MN, Jans DA. 2009. The respiratory syncytial virus matrix protein possesses a Crm1-mediated nuclear export mechanism. J. Virol. 83:5353–5362. 10.1128/JVI.02374-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghildyal R, Ho A, Wagstaff KM, Dias MM, Barton CL, Jans P, Bardin P, Jans DA. 2005. Nuclear import of the respiratory syncytial virus matrix protein is mediated by importin beta1 independent of importin alpha. Biochemistry 44:12887–12895. 10.1021/bi050701e [DOI] [PubMed] [Google Scholar]
- 70.Rawlinson SM, Pryor MJ, Wright PJ, Jans DA. 2009. CRM1-mediated nuclear export of dengue virus RNA polymerase NS5 modulates interleukin-8 induction and virus production. J. Biol. Chem. 284:15589–15597. 10.1074/jbc.M808271200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Daelemans D, Afonina E, Nilsson J, Werner G, Kjems J, De Clercq E, Pavlakis GN, Vandamme A-M. 2002. A synthetic HIV-1 Rev. inhibitor interfering with the CRM1-mediated nuclear export. Proc. Natl. Acad. Sci. U. S. A. 99:14440–14445. 10.1073/pnas.212285299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pollard VW, Malim MH. 1998. The HIV-1 Rev protein. Annu. Rev. Microbiol. 52:491–532. 10.1146/annurev.micro.52.1.491 [DOI] [PubMed] [Google Scholar]
- 73.Sanchez V, Mahr JA, Orazio NI, Spector DH. 2007. Nuclear export of the human cytomegalovirus tegument protein pp65 requires cyclin-dependent kinase activity and the Crm1 exporter. J. Virol. 81:11730–11736. 10.1128/JVI.02760-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Williams P, Verhagen J, Elliott G. 2008. Characterization of a CRM1-dependent nuclear export signal in the C terminus of herpes simplex virus type 1 tegument protein UL47. J. Virol. 82:10946–10952. 10.1128/JVI.01403-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Freundt EC, Yu L, Park E, Lenardo MJ, Xu X-N. 2009. Molecular determinants for subcellular localization of the severe acute respiratory syndrome coronavirus open reading frame 3b protein. J. Virol. 83:6631–6640. 10.1128/JVI.00367-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dong X, Biswas A, Suel KE, Jackson LK, Martinez R, Gu H, Chook YM. 2009. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature 458:1136–1141. 10.1038/nature07975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Turner JG, Dawson J, Sullivan DM. 2012. Nuclear export of proteins and drug resistance in cancer. Biochem. Pharmacol. 83:1021–1032. 10.1016/j.bcp.2011.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boons E, Vanstreels E, Vercruysse T, Shacham S, Landesman Y, Baluglu E, Tamir S, Pannecouque C, Daelemans D. 2013. Replication inhibition by small-molecule inhibitors targeting the HIV Rev-CRM1/XPO1 interaction, abstr 20. Abstr. 26th Int. Conf. Antiviral Res., San Francisco, CA [Google Scholar]
- 79.Huang TT, Kudo N, Yoshida M, Miyamoto S. 2000. A nuclear export signal in the N-terminal regulatory domain of Iκ Bα controls cytoplasmic localization of inactive NF-κ B/Iκ Bα complexes. Proc. Natl. Acad. Sci. U. S. A. 97:1014–1019. 10.1073/pnas.97.3.1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tam WF, Lee LH, Davis L, Sen R. 2000. Cytoplasmic sequestration of Rel proteins by IκBα requires CRM1-dependent nuclear export. Mol. Cell. Biol. 20:2269–2284. 10.1128/MCB.20.6.2269-2284.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Arenzana-Seisdedos F, Thompson J, Rodriguez MS, Bachelerie F, Thomas D, Hay RT. 1995. Inducible nuclear expression of newly synthesized I kappa B alpha negatively regulates DNA-binding and transcriptional activities of NF-kappa B. Mol. Cell. Biol. 15:2689–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arenzana-Seisdedos F, Turpin P, Rodriguez M, Thomas D, Hay RT, Virelizier JL, Dargemont C. 1997. Nuclear localization of I kappa B alpha promotes active transport of NF-kappa B from the nucleus to the cytoplasm. J. Cell Sci. 110:369–378 [DOI] [PubMed] [Google Scholar]
- 83.Lee S-H, Hannink M. 2002. Characterization of the nuclear import and export functions of IκBε. J. Biol. Chem. 277:23358–23366. 10.1074/jbc.M111559200 [DOI] [PubMed] [Google Scholar]
- 84.Brown KD, Claudio E, Siebenlist U. 2008. The roles of the classical and alternative nuclear factor-kappaB pathways: potential implications for autoimmunity and rheumatoid arthritis. Arthritis Res. Ther. 10:212. 10.1186/ar2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O'Connor S, Shumway S, Miyamoto S. 2005. Inhibition of IkappaBalpha nuclear export as an approach to abrogate nuclear factor-kappaB-dependent cancer cell survival. Mol. Cancer Res. 3:42–49 http://mcr.aacrjournals.org/content/3/1/42.long [PubMed] [Google Scholar]
- 86.Zerfaoui M, Errami Y, Naura AS, Suzuki Y, Kim H, Ju J, Liu T, Hans CP, Kim JG, Abd Elmageed ZY, Koochekpour S, Catling A, Boulares AH. 2010. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation. J. Immunol. 185:1894–1902. 10.4049/jimmunol.1000646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Y, Sun H, Fan L, Ma Y, Sun Y, Pu J, Yang J, Qiao J, Ma G, Liu J. 2012. Acute respiratory distress syndrome induced by a swine 2009 H1N1 variant in mice. PLoS One 7(1):e29347. 10.1371/journal.pone.0029347 [DOI] [PMC free article] [PubMed] [Google Scholar]